
Abstract
This chapter presents cytogenetic and molecular pathology of sarcomas, with emphasis on recurrent abnormalities that are characteristic features of individual cancer types. General concepts concerning biology, pathology, diagnostic techniques, and molecular diagnostic applications of cancers are presented in the beginning of the chapter. The second portion presents each of the major sarcoma types, in alphabetical order, with a specific structure: basic pathology, clinical features, molecular genetic pathology, and molecular diagnosis. Most of the tumors presented herein are soft tissue sarcomas, as few recurrent abnormalities have been established to have biologic and diagnostic significance in bone tumors. In contrast to other diseases and cancers presented in this textbook, sarcomas are more commonly associated with cytogenetic changes, as opposed to molecular changes. Nevertheless, a variety of approaches (e.g., classical cytogenetic analysis, fluorescence in situ hybridization, reverse-transcriptase polymerase chain reaction, immunohistochemistry) are presented, with strengths and weaknesses of each for clinical application in each tumor type, as appropriate.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
General Concepts
Definition
-
Bone and soft tissue tumors are neoplasms arising from mesenchymal tissues of the body, which may be either benign or malignant, although most solid tumors for which molecular diagnosis is applied are malignant
-
Malignant bone and soft tissue tumors are referred to as sarcomas, and are classified primarily by the type of tissue (e.g., liposarcoma [adipose], rhabdomyosarcoma [skeletal muscle], chondrosarcoma [cartilage]), rather than by anatomic site
Clinical Features
-
Presentation, treatment, and outcome are heterogeneous, not only between different tumor types, but also within each tumor type. Some very coarse generalizations can be made, but exceptions are very common
-
Presentation, treatment, and prognosis are dependent on disease stage (function of tumor size, local extension, and distant spread), and grade (function of the microscopic features of the tumor cells and the architecture/pattern of their growth)
-
Sarcomas tend to spread to distant sites via blood vessels
-
Bone and soft tissue sarcomas are much less common than carcinomas in the USA (according to American Cancer Society estimates for 2011)
-
∼14,000 new cancers/year out of a total of ∼1,600,000 new cancers/year
-
∼5,500 deaths/year out of a total of ∼570,000 deaths/year
-
-
Primary osseous neoplasms are less common than primary soft tissue cancers (∼3,000/year vs. ∼11,000/year), with a mildly higher lethality rate (∼1,500 deaths/year vs. ∼4,000 deaths/year)
-
Primary neoplasms of bone and soft tissue are more common in children (∼7% of childhood cancers) than in adults (<1%)
Basic Principles
-
Molecular pathology of sarcomas is also heterogeneous, but generally involves alteration(s) in genes encoding proteins critical for regulating cellular growth and proliferation, programmed cell death (apoptosis), differentiation, and/or motility
-
Tumor suppressor genes (TSG) typically inhibit cellular functions such as growth, proliferation, and motility, but may also promote functions such as adhesion, apoptosis, differentiation, and DNA repair
-
TSGs are inactivated in cancer, by diverse mechanisms including deletion, point mutation, promoter methylation, or chromosomal rearrangement
-
Generally, both tumor suppressor alleles are inactivated in a cancer (homozygous), although cancer may arise in association with haploinsufficiency of some TSGs as a result of a single mutant allele
-
Many hereditary cancer syndromes involve an inherited dysfunctional TSG (e.g., RB1 in rtinoblastoma, TP53 in Li–Fraumeni syndrome), and neoplasia result when the second allele is affected by a sporadic mutation (second hit) in a given tissue. These syndromes have dominant inheritance patterns, even though both alleles are mutated in the tumors
-
-
Oncogenes typically promote cellular functions such as growth, proliferation, and motility, but may also inhibit functions such as differentiation, adhesion, DNA repair, and apoptosis
-
Oncogenes are activated in cancer, by diverse mechanisms including polyploidy, polysomy, gene amplification, chromosomal rearrangement, and point mutation
-
Generally, one oncogene allele is altered in a cancer, unless the mechanism of alteration is amplification, polysomy, or polyploidy, in which case multiple copies are present
-
-
Sarcomas usually have characteristic, recurrent chromosomal translocations that have, in some instances, become criteria for the diagnosis of these tumors (see Table 15.1)
Table 15.1 Specific chromosomal translocations in sarcomas -
Some translocation breakpoints interrupt genes directly, resulting in a novel fusion oncogenic protein, while other breakpoints may result in deregulated expression, generally overexpression, of the new fusion gene
-
For example, t(11;22)(q24;q12) in Ewing is sarcoma results in a hybrid protein containing the C terminal DNA-binding domain of FLI1 (11q24) and the N terminal transactivating domain of EWSR1 (22q12), under the regulation of the constitutively expressed EWSR1 promoter. The result is increased transcriptional activation of genes with FLI1 binding sites, leading to increased cell growth and proliferation
-
-
Two common types of genetic alterations have been observed: translocations forming chimeric protein tyrosine kinases (ALK and ETV6–NTRK3) and translocations encoding a chimeric autocrine growth factor (COLA1A–PDGFB)
-
Many translocations have chromosomal variants, where one chromosome band is consistently rearranged, but may be translocated to different chromosome partner regions
-
For example, 22q12 (EWSR1) may be translocated to 11q24 (FLI1), 21q12 (ERG), 7p22 (ETV1), 2q33 (FEV), 17q12 (E1AF), and 20q13 (NFATC2) in Ewing sarcoma
-
Many translocations have molecular variants, where the breakpoints may vary within the involved gene(s), leading to fusion between different exons
-
EWSR1–FLI1 fusions in Ewing is sarcoma commonly fuse exon 7 of EWSR1 to either exon 5 or 6 of FLI1, although many other configurations have been described
-
-
Some genes are involved in different translocations in different type of lesions
-
In addition to the Ewing sarcoma fusions, EWSR1 is fused to WT1 in desmoplastic small round cell tumor, to NR4A3 in extraskeletal myxoid chondrosarcoma to ATF1 in clear cell sarcoma and angiomatoid fibrous histiocytoma, to DDIT3 in myxoid liposarcoma
-
-
-
Although most translocations are unique to a specific sarcoma, some translocations can be seen in different tumor types, including epithelial cancers
-
ETV6–NTRK3 fusions are seen in congenital mesoblastic nephroma, secretory breast carcinoma, and isolated cases of acute myeloid leukemia
-
EWSR1–ATF1 fusions are seen in angiomatoid fibrous histiocytoma, clear cell sarcoma, and hyalinizing clear cell carcinoma of the salivary glands
-
EWSR1–POUF5 fusions are seen in soft tissue myoepithelioma, hidradenoma of the skin, and mucoepidermoid carcinoma of salivary glands
-
ALK rearrangements are seen in anaplastic large cell lymphoma, nonsmall lung carcinoma, and inflammatory myofibroblastic tumor
-
-
-
Numerical abnormalities of whole chromosomes (e.g., trisomy, monosomy) or subchromosomal regions (e.g., amplification) tend to be less specific, secondary alterations in sarcomas
-
Because different sarcomas may require different, specific assays, and sarcomas are sufficiently uncommon that control materials and expertise can be hard to accumulate, few labs offer a comprehensive menu for sarcoma testing
-
Nomenclature for genes is inconsistent, and subject to revision. We have chosen to use the Human Genome Organization (HUGO [sic])-standardized nomenclature, but when each gene is first discussed, we also include, in parentheses, the legacy name used in the original papers
-
For example, when the gene at the chromosome 22 breakpoint in t(11;22) is first presented, it is as EWSR1 (EWS), because EWSR1 is the standard nomenclature according to HUGO, while EWS was the name first given to this gene upon its discovery in Ewing sarcoma. Subsequently in the text, however, this gene will be referred to solely as EWSR1, for simplicity
-
Molecular Diagnostics of Soft Tissue and Bone Tumors
Test Indications
-
Diagnosis, usually as an adjunct to morphology and immunohistochemistry (IHC)
-
Prognosis
-
Theranosis, selection of therapies targeted to specific molecular genetic abnormalities
-
Risk assessment for hereditary cancer syndromes
-
Minimal disease testing, either for monitoring success/failure of therapy or for screening (less well developed for sarcomas)
General Technical Considerations
-
Sampling: cancers are somatic diseases, and the lesion itself must be analyzed, which is likely to require an invasive procedure
-
Less invasive techniques, including small biopsies, fine needle aspiration, brushing, and fluid collection, tend to yield small amounts of cancer cells admixed with benign cells that can interfere with some kinds of analysis
-
-
Fixation: most archived tumor samples are embedded in paraffin after fixation in formalin (FFPE), which cross-links DNA–RNA–proteins, inactivating the proteins and protecting the nucleic acids from digestion
-
DNA isolated from FFPE tissues breaks into pieces roughly 500 bp or smaller during isolation, so techniques that require larger stretches of DNA are unreliable for these samples
-
Some other fixatives (e.g., Zenker, B5) contain heavy metals that inhibit enzymes (e.g., Taq polymerase) used for molecular diagnosis, and other tissue treatments (Bouin, bone decalcifying) contain acids that damage nucleic acids and/or inhibit testing (e.g., fluorescence in situ hybridization or FISH)
-
Samples that are not fixed promptly undergo nucleic acid degradation from ubiquitous nucleases, which impairs analysis, especially for RNA
-
Tissue additives are available that report to preserve nucleic acids if applied at the time of fixation; however, they have yet to garner widespread use
-
-
Heterogeneity: most tumors contain an admixture of the cancer cells with benign cells (see Fig. 15.1), including residual normal tissue infiltrated by the cancer, reactive elements (e.g., lymphocytes, macrophages, and fibroblasts) recruited and/or stimulated to try to contain the cancer, and foci of necrosis. In general, this is more of a problem for carcinomas than for sarcomas, which tend to overgrow tissue in continuous fashion
Fig. 15.1 
A sarcoma showing intratumor heterogeneity. The malignant cells grow in sheets that are intimately intermingled with reactive, benign elements, including inflammatory cells, entrapped soft tissue, and blood vessels. A DNA sample from this section of a sarcoma would contain admixed malignant and benign DNA, which may make certain types of DNA analysis challenging
-
The cancer cells themselves may also be heterogeneous, such that genetic changes in one part of the tumor may not be seen in another part
-
Some techniques are particularly unreliable for heterogeneous samples, and partial purification of the cancer cells (e.g., microdissection or flow cytometric sorting) may be required before analysis. Direct sequence analysis, for example, requires that approximately 20% of the sample DNA contains the mutation in order for it to be reliably detected
-
Mutation screening (e.g., single-stranded conformational polymorphism [SSCP], heteroduplex-mismatch cleavage, denaturing gradient gel electrophoresis [DGGE], DNA analysis by denaturing high performance liquid chromatography [DHPLC], high resolution melting (HRM) analysis) methods are often less affected by heterogeneity, but may yield incomplete information requiring followup with another assay to define the exact molecular variation
-
Allele-specific amplification and hybridization techniques are considerably less affected by heterogeneity, but are restricted to the exact sequence variants tested
-
Heterogeneity is less of a limitation for in situ hybridization (ISH) techniques, where analysis is performed cell by cell with correlative histology to confirm that tumor cells are being analyzed and not the intermingled benign cells
-
IHC can be used to detect molecular abnormalities that alter the level of expression of a protein. IHC is less useful for detecting mutations that alter the function of a protein without changing its abundance, unless the mutation alters a specific epitope recognized by a monoclonal antibody
-

Basic Methodologies
-
Karyotype analysis (Fig. 15.2) provides a general assessment of large chromosomal abnormalities, including numerical abnormalities and large rearrangements
Fig. 15.2 
Classic sarcoma partial GTG-banded karyotypes depicting recurrent chromosomal rearrangements observed in sarcomas
-
Requires fresh tissue
-
Most sarcomas grow well in short-term culture, requiring 3–4 days
-
Cultured cells are arrested in metaphase, then stained, usually with Giemsa (GTG-banding)
-
Admixture of benign stromal cells is a concern
-
Subtle and cryptic translocations can be very difficult to detect by standard GTG-banding, as can small deletions and amplifications
-
-
ISH enables detection of specific chromosomal abnormalities, including subtle or cryptic rearrangements, and small deletions and amplifications
-
Requires a prior knowledge of a suspected aberration
-
Probes hybridize to unique locus-specific DNA sequences, which are usually genomic clones but may be cDNAs, and vary in size from about 1 to 100s of kb
-
May be done with radioactive (ISH), fluorescent (FISH), chromogenic (CISH), or silver (SISH) probes for detection
-
FISH, CISH, and SISH enable rapid detection
-
CISH and SISH enable immediate correlation of the hybridization signal with histopathology, which is particularly valuable for heterogeneous samples
-
The number of colors that can be distinguished with light microscopy limits CISH and SISH; most applications are for numerical alterations (e.g., aneuploidy, amplification)
-
-
FISH, by using different colored probes, enables simultaneous detection of more than one chromosomal region, which is particularly valuable for analyzing multiple abnormalities within a single cell, or for analysis of chromosomal translocations
-
Correlation of FISH signals with histopathology is more difficult
-
-
However, most laboratories do FISH on metaphase spreads from cultured samples or interphase nuclei, and/or CISH on FFPE tissue sections
-
Metaphase FISH has the same sample requirements as conventional karyotyping
-
Interphase FISH can be performed on intact nuclei (nondividing cells) from cytologic samples (touch or smear preparations, fluid cell suspensions), or from whole tissue samples (enzymatic disaggregation or histologic sections)
-
Standard histology (4 μm) sections can miss signals due to sectioning through the nucleus, and require analysis of many more cells
-
50 μm thick sections enable evaluation of intact nuclei and definitive interpretation
-
Poor cell preservation/morphology can impair interpretation
-
-
-
-
Two different types of hybridization strategies (break apart, dual fusion) can be used to detect translocations (see Fig. 15.3)
Fig. 15.3 
Examples of two different types of hybridization strategies for locus-specific probes: (a) A break apart probe shows a FUS gene rearrangement at 16p11 from a t(7;16)(q34;p11), a characteristic abnormality in low grade fibromyxoid sarcoma. Two differently labeled probes (spectrum orange and spectrum green) hybridize to opposite sides of the breakpoint of the FUS gene located at 16p11. The 16p11 FUS region in its normal state would be seen as two immediately adjacent or fused orange + green (yellow) signals. However, if a rearrangement at 16p11 region has occurred, as in this example, separate orange and green signals would be seen. (b) A dual fusion probe shows a BCR/ABL fusion from a t(9;22)(q34;q11.2), a characteristic abnormality in chronic myelocytic leukemia. Two differently labeled probes (spectrum orange and spectrum green) each hybridize near one of the chromosomal breakpoints involved in a translocation. The 9q34 ABL1 and 22q11.2 BCR regions in their normal state would be seen as two separate orange and green signals. However, if a t(9;22) has occurred, as in this example, two immediately adjacent or fused orange + green (yellow) signals would be seen
-
Break apart assay design uses differentially (usually red/green) labeled probes that hybridize to the 5′ and 3′ side, respectively, of one of the breakpoints in a translocation, with sufficient proximity that in normal (nontranslocated) alleles, the two probes overlap optically to give a fused (yellow) signal; however, in a translocation, the red and green signals are separated and detected separately
-
Break apart probe designs offer the greatest sensitivity for detection of rearrangements that consistently involve one chromosomal region (e.g., EWSR1), but which may have many different chromosomal variants; however, this design cannot identify the translocation partner, and therefore cannot distinguish between chromosomal variants
-
Most commercial FISH probe kits are for break apart probe designs
-
-
Dual fusion assay design uses differentially labeled (usually red/green) probes that each hybridize near one of the chromosomal breakpoints involved in a translocation, such that a fused (yellow) signal is seen when the translocation is present, but separate red and green signals are seen in normal (untranslocated) alleles
-
Dual fusion probe designs can distinguish between chromosomal variants of translocations, but require a separate assay for each variant to be tested
-
-
-
No ISH assays can distinguish between molecular variants
-
ISH can also be used to detect gene amplifications and deletions, but scoring systems may vary, and take on greater significance when FISH is performed on 4- or 5-μm sections because of sectioning through the nucleus
-
Polymerase chain reaction (PCR) enables analysis of very small genetic changes, including point mutations, microdeletions/insertions, and molecular variants of translocations
-
Fresh or frozen tissue is optimal, but FFPE tissues work consistently for amplicons less than approximately 500 bp; larger amplicons will amplify inconsistently
-
Simple detection involves slab gel or capillary electrophoresis
-
Sensitivity may be enhanced by a variety of techniques, including real time probe detection, and Southern transfer of the PCR product followed by oligonucleotide probe hybridization
-
-
PCR may be used to prepare DNA for other detection strategies (e.g., direct sequencing, oligonucleotide hybridization, restriction digestion) or mutation screening approaches
-
DNA PCR is generally not used often for translocations because breakpoints are usually in introns, requiring a very large PCR product; reverse transcription-PCR (RT-PCR) is preferable because exon primers can flank breakpoints and give a small enough amplicon for successful analysis
-
“Real time” PCR can be used to quantitate gene dosage
-
Contamination of PCR reactions with PCR products from previous assays is a problem for clinical testing, and countermeasures include
-
Ultraviolet irradiation of consumables to damage carryover PCR products
-
Physical separation of assay setup, amplification, and detection
-
Meticulous cleansing of work benches with bleach
-
Dedicated supplies and reagents for PCR use
-
One-step PCR protocols, as opposed to nested protocols
-
Spinning PCR tubes briefly before opening
-
Closed tube detection systems, such as “real time” PCR
-
Uracil-N-glycosylase endonuclease, used with a dNTP mix containing dUTP in place of dTTP, to destroy carryover PCR product
-
-
-
Direct Sanger dideoxyterminator sequence analysis (see Fig. 15.4) is considered the diagnostic “gold standard” for detection of point mutations and small deletions and insertions
Fig. 15.4 
KIT mutation detection in a gastrointestinal stromal tumor (GIST) by direct sequence analysis. An 18 bp deletion in exon 11 is shown, which results in apparent sequence noise due to superposition of mutant sequence bearing the deletion and wild-type sequence lacking the deletion. By sequencing in both directions, the precise boundaries of the deletion can be determined
-
Amplification is usually required first
-
Intratumor heterogeneity is highly problematic, and most automated sequence analyzers cannot distinguish a mutation from background noise if the mutant allele accounts for less than approximately 20% of the total DNA
-
Sensitivity may be extended considerably by mutant enrichment strategies, including silencing of wild-type alleles with peptide nucleic acid (PMA) or locked nucleic acid (LNA) probes, selective amplification of mutant sequences with lower denaturation temperatures in the PCR (COLD-PCR), or restriction digestion of wild-type sequence between stages of a nested reaction
-
Method of choice for genes with a very wide spectrum of mutations, or for which the mutation spectrum is not fully characterized
-
“Next generation” sequencing technologies that involve massive-scale parallel sequencing of individual template strands hold great promise for multiplexed sequence analysis of specific targets or whole exomes/genomes from samples
-
Because of low throughput and high cost, these technologies are currently best suited for research applications, but are likely to become standard clinical instruments as speed increases and cost decreases
-
-
-
Allele-specific amplification or hybridization enables fast and sensitive diagnosis of specific point mutations
-
The exact mutation must be known; novel mutations will be missed
-
Allele-specific hybridization assays range from simple paper dot blots to massively multiplexed silicon chip microarrays
-
Large-scale analysis of thousands of genes at once can be performed with microarrays to study patterns of gene expression or amplification/deletion, which may be used to classify a solid tumor
-
Currently, however, these techniques are most employed in research studies to discover a more limited number of candidate diagnostic markers for subsequent focused analysis
-
Comparative methods require careful and controversial selection of control samples
-
-
-
RT-PCR (see Fig. 15.5) uses RNA to detect small molecular variations as with DNA PCR, but principal differences include variable template expression and absence of introns
Fig. 15.5 
RT-PCR for JAZF1-SUZ12 (JJAZ1) fusion transcript in endometrial stromal sarcoma. GAPDH is used as a reference gene, to ensure adequate analysis of RNA from cases with no detectable JAZF1-SUZ12 fusion transcript. Cases 1 through 9 show results from several patients with endometrial stromal tumors: cases 1, 4, 5, 7, 8, and 9 are positive, while cases 2, 3, and 6 are negative (photo adapted from a generous gift of Dr. Jeffrey Sklar, Yale University School of Medicine)
-
Distinction of molecular variants is one of the primary indications for RT-PCR
-
Fresh or frozen tissue is optimal; formalin-fixed tissues require a very small (e.g., <200 bp) RT-PCR product for most consistent results, but are often problematic regardless
-
RNA is very susceptible to degradation from ubiquitous RNases, and meticulous technique is required, as are prompt and thorough tissue preservation
-
RT-PCR is prone to contamination in the same way as is PCR, and the same precautions apply, with the addition of “no RT” controls
-
RT-PCR requires different primers for each chromosomal variant, and likely for different molecular variants also
-
“Real time” RT-PCR can be used to quantitate gene expression
-
-
Southern blot hybridization enables detection of chromosomal rearrangements and molecular variants, as well as large intragenic deletions
-
Need sample frozen or fresh tissue
-
Technically challenging, slow, costly, usually radioactive; near obsolete
-
Can distinguish between molecular variants, potentially with a single assay design, but multiple probes and enzymes may be needed, depending on intron size and distance between breakpoints
-
Most useful for alterations spanning too great a distance for simple PCR, yet too small for FISH (e.g., large intragenic deletions)
-
-
IHC is a surgical pathology technique that uses antibodies directed against proteins whose expression is an indication of underlying molecular pathology
-
Antibodies are applied directly to tumor sections on a glass slide, are detected with a chromogenic substrate, and enable rapid assessment of individual tumor cells by a trained pathologist
-
Two broad types of antibodies are used: polyclonal and monoclonal
-
Polyclonal antibodies are developed by inoculating an animal (most often a rabbit) with the target protein; the animal mounts an immune response and antibodies are purified from its serum
-
Typically, polyclonal antibodies have broad and variable specificity and strength, and vary considerably from one lot to another
-
-
Monoclonal antibodies are produced by fusing in vitro the splenocytes from an inoculated animal (typically mouse) with myeloma cells, forming “hybridoma” cells that are separated and grown individually in culture. The hybridoma cells secrete individual immunoglobulins, which can be harvested from culture
-
Typically, monoclonal antibodies are very specific to an individual antigenic epitope, and are very strong. They can be renewed in culture and, therefore, have less lot–lot variation
-
-
-
Most IHC antibodies are directed against a normal protein, and are used to detect abnormalities that change the amount of expression of either a normal protein or an abnormal fusion protein that has the normal antigenic epitope
-
Newer approaches at developing mutant protein-specific antibodies hold promise for distinguishing abnormal proteins from normal proteins. These are most useful in tumors with one or a limited number of molecular variants
-




Specific Sarcomas
Alveolar Rhabdomyosarcoma
-
Basic pathology (see Fig. 15.6)
Fig. 15.6 
Alveolar rhabdomyosarcoma, showing the characteristic pattern of tumor cell growth, with tumor cells adherent to the periphery of, and floating dyscohesively within, alveolar spaces separated by fibrous septae (slide courtesy of Dr. Christopher Fletcher, Brigham and Women’s Hospital)
-
Malignant neoplasm of skeletal muscle, with characteristic histology
-
Nests of small, round, undifferentiated cells separated by thin fibrous septae
-
-
Clinical features
-
Occurs primarily in 10–30-year-old patients
-
20% of all rhabdomyosarcomas
-
Presents most commonly in extremities and perineum
-
Prognosis is poor; worst of all rhabdomyosarcoma variants
-
Treatment is chemotherapy
-
-
Molecular genetic pathology
-
t(2;13)(q35;q14) in approximately 70% of cases (see Fig. 15.2A)
-
PAX3 gene (at 2q35) breakpoint in intron 7
-
FOXO1 (FKHR) gene (at 13q14) breakpoint in intron 1
-
Fusion protein contains the N terminal DNA-binding domain of PAX3 and the C terminal transcription activating domain of FOXO1, leading to oncogenesis through activation of growth and proliferation genes with PAX3 binding sites, including MITF and PDGFB
-
-
t(1;13)(p36;q14) in approximately 10% of cases
-
PAX7 gene (at 1p36) breakpoint in intron 7
-
FOXO1A gene (at 13q14) breakpoint in intron 1
-
-
Fusion gene organization and consequence are analogous to the PAX3–FOXO1 fusion
-
PAX7–FOXO1 is commonly amplified on double minute chromosomes
-
-
20% of cases have neither t(2;13) nor t(1;13)
-
These cases represent a genetically heterogeneous subgroup, with some that have variant translocations involving related members of the PAX and/or FOXO1 gene families (e.g., PAX–NCOA1, PAX3–AFX), and some that truly lack rearrangements or have other types of genetic abnormalities
-
-
-
Molecular diagnostics
-
Test indications
-
Establish diagnosis
-
Prognosis: PAX7–FOXO1 cases are associated with localized lesions and favorable prognosis
-
Minimal residual disease detection after therapy, bone marrow involvement
-
-
Additional technical considerations
-
FISH: commercial break apart probe is available for FOXO1 but cannot distinguish between the two chromosomal variants, t(2;13) and t(1;13)(PAX3–FOXO1 and PAX7–FOXO1)
-
RT-PCR: many different designs have been reported, including oligo dT/random/FOXO1A-specific primers for RT, one-step/two-step/nested amplification, and gel electrophoresis/Southern transfer probe hybridization/real time detection
-
Wild-type FKHR is constitutively expressed and can serve as control transcript
-
Breakpoints are restricted to intron 7 of PAX3/PAX7 and intron 1 of FOXO1, but are variable within these large introns, precluding DNA analysis by PCR
-
Consensus PAX primers can be designed, enabling amplification of both chromosomal variants with one reaction
-
-
Southern blot: multiple probes/digests needed due to large introns (20 kb for PAX genes, 130 kb for FKHR) with varying breakpoints
-
-
Additional interpretive considerations
-
PAX7–FOXO1A is commonly amplified on double minute chromosomes in a subgroup of ARMS
-
Fusion-negative cases have outcomes intermediate between those with PAX7–FOXO1 and PAX3–FOXO1
-
Fusion transcript can be detected in samples in the absence of morphologic evidence of disease, suggesting a role in minimal residual disease testing
-
-

Alveolar Soft Part Sarcoma
-
Basic pathology (see Fig. 15.7)
Fig. 15.7 
Alveolar soft part sarcoma, showing the characteristic alveolar pattern of growth, with well-circumscribed round nests of uniform, eosinophilic, polygonal tumor cells. A reticulin stain would highlight the boundaries of the tumor cell nests, and a PAS stain would demonstrate cytoplasmic crystals
-
Mesenchymal tumor of uncertain histogenesis with distinctive morphology
-
Alveolar nests of tumor cells surrounded by reticulin framework
-
Uniform round cells with single nuclei
-
Periodic acid Schiff (PAS)-positive granular cytoplasmic crystals
-
Characteristic rectangular/rhomboid cytoplasmic crystals seen with electron microscope
-
-
-
Clinical features
-
Often occurs in second or third decade, more frequently in females
-
Extremities (especially, thighs/buttocks) and orbit are most common sites, but also in sites with no skeletal muscle
-
Relatively indolent clinical course, with approximately 50% 10-year survival, but distant metastases are common and most patients die of the disease
-
Tumors are refractory to chemotherapy, and treatment is aggressive surgical excision
-
-
Molecular genetic pathology
-
der(17)t(X;17)(p11.2;q25) in approximately 100% (see Fig. 15.2B)
-
TFE3 gene (at Xp11.2) breakpoints in introns 1 and 2
-
ASPSCR1 (ASPL) gene (at 17q25)
-
Fusion protein contains the N terminus of ASPSCR1 and the C terminus of TFE3, including TFE3 DNA-binding domain, under regulation of ASPSCR1 promoter
-
-
-
Molecular diagnostics
-
Test indications: establish diagnosis
-
Additional technical considerations
-
Karyotype: unbalanced translocation, der(17): the reciprocal translocation partner, der(X), is usually absent
-
FISH: no commercial probes are currently available
-
RT-PCR: not commonly performed, as significance of distinguishing molecular variants (intron 1 or intron 2 TFE3 breakpoints) is unclear
-
IHC: nuclear localization of TFE3 is sensitive and specific, and is the simplest and most widely employed diagnostic method
-
-
Additional interpretive considerations
-
A balanced translocation, t(X;17)(p11.2;q25), involving the same TFE3 and ASPSCR1 genes, is seen in a specific subset of renal adenocarcinomas, in pediatric and young adult patients
-
-

Angiomatoid Fibrous Histiocytoma
-
Basic pathology
-
Tumors can resemble hemangioma, with nests of histiocyte-like cells, hemorrhagic spaces, and chronic inflammatory cells
-
Multiple nonendothelialized pseudovascular spaces with recent and old hemorrhage present
-
-
Clinical features
-
Painless, slow growing, subcutaneous mass, primarily in the extremities
-
Typically affects children/teens
-
Symptoms may include fever, anemia, weight loss
-
Wide local excision is typically sufficient, as the tumor has a very low rate of metastasis
-
-
Molecular genetic pathology
-
t(12;16)(q13;p11) associated with ATF1–FUS fusion
-
FUS1 gene (at 16p11), exon 5
-
ATF1 gene (at 12q13), exon 5
-
-
t(12;22)(q13;q12) associated with EWSR1–ATFI gene fusion
-
EWSR1 gene (at 22q12), exon 7
-
ATF1 gene (at 12q13), exon 5
-
-
t(2;22)(q34;q12) associated with EWSR1–CREB1 gene fusion
-
EWSR1 gene (at 22q12), exon 7
-
CREB1 gene (at 2q34), exon 7
-
Although these fusions were not described initially in AFH, EWSR1–CREB1 fusion may be the most common rearrangement in this tumor
-
-
Each EWSR1 fusion protein retains the bZIP domain mediating DNA-binding and dimerization of CREB1 or ATF1. The kinase inducible domain (KID), which is either excluded or truncated in different forms of EWSR1–ATF1, is not included in EWSR1–CREB1
-
-
Molecular diagnostics
-
Test indications: establish diagnosis
-
Additional technical considerations
-
FISH: commercial break apart probe is available for FUS and EWSR1
-
FISH for EWSR1 cannot distinguish between t(12;22); and t(2;22); clinical significance of this distinction is unclear
-
-
IHC:TFE3 but not MITF-M is overexpressed in the EWSR1–ATF1 positive tumors
-
-
Additional interpretive considerations
-
Both t(12;22) and t(2;22) have also been reported in clear cell sarcoma
-
The t(12;22) has been also reported in hyalinizing clear cell carcinoma of the salivary glands
-
-
Atypical Lipoma/Well-Differentiated Liposarcoma/Dedifferentiated Liposarcoma
-
Basic pathology
-
Low grade neoplasm of adipose tissue with several morphologic subtypes, including lipoma-like (most common), sclerosing, and inflammatory
-
All have mature fat with variably-sized adipocytes and fibromyxoid stroma with spindle cells and focal cellular atypia
-
Lockhern cells have sharply outlined nuclear vacuoles
-
Mitoses and lipoblasts are uncommon
-
Dedifferentiated liposarcoma involves (typically) abrupt transition to high grade area with mitoses (>5 per 10 high-powered fields), nonlipogenic cells, and heterologous elements, including metaplastic bone
-
-
Clinical features
-
Most common type of liposarcoma
-
Primarily affects adults, fourth to sixth decades
-
Typically involves lower limbs, retroperitoneum, paratesticular regions, mediastinum
-
May recur locally
-
Clear surgical margins are critical, especially for sclerosing type
-
Retroperitoneal liposarcomas are difficult to resect and often dedifferentiate
-
-
Dedifferentiation
-
∼10% of cases, more often from retroperitoneal or paratesticular sites
-
Confers aggressive behavior
-
Recurrence 40–80%
-
Metastasis 10–15%
-
Death 30–50%
-
-
-
-
Molecular genetic pathology
-
Supernumerary ring and/or giant marker chromosome(s) in virtually all cases
-
Intratumor variability in size and number
-
Typically involve material from 12q13–q21
-
Oncogenes in these regions include MDM2, CDK4, HMGA2, GLI, CHOP
-
Material from other chromosomes also often involved
-
-
-
Coexisting other numerical/structural abnormalities in ∼30% of cases
-
Mostly appear random, other than
-
Loss of 13q material
-
Telomeric associations involving 11p
-
-
-
A subset has gain of material at 12q15–q24 instead of rings/giant markers
-
Associated with minimal atypia
-
Distinct from 12q13–12q15 balanced translocations seen in simple lipoma
-
-
Similar rings and giant marker chromosomes are also seen in dedifferentiated liposarcoma, though a more complex karyotype is frequently observed
-
-
Molecular diagnosis
-
Test indications: currently unclear
-
Technical considerations
-
Karyotype is typical method, but dedifferentiated liposarcoma areas grow in vitro very poorly
-
FISH: commercial break apart probe is available for MDM2
-
Rings/giant marker chromosomes are negative for centromeric probes
-
-
Clear Cell Sarcoma (Melanoma of Soft Parts)
-
Basic pathology (see Fig. 15.8)
Fig. 15.8 
Clear cell sarcoma (H&E stain), showing characteristic histologic appearance, with nests of tumor cells rimmed by thin fibrous septae. Tumor cells are polygonal with clear or eosinophilic cytoplasm and round-oval uniform nuclei with prominent nucleoli. Immunohistochemical stains for HMB-45 or S100 would demonstrate melanocytic differentiation
-
Mesenchymal neoplasm of uncertain histogenesis, with nests of tumor cells with melanocytic differentiation
-
Positive stain reactions for melanin, S100, HMB45, MITF, and melanosomes evident by electron microscopy
-
Separated by reticulin fibrous septae
-
Cells have uniform cytology, with clear or eosinophilic cytoplasm
-
Melanomyctic markers may not be expressed in clear cell sarcomas that arise in the GI tract
-
-
Clinical features
-
Mostly adolescents, young adults
-
Commonly involves extremities, especially foot and ankle from/near tendons, fascia, aponeuroses, but may arise from a wide range of anatomic sites, gastrointestinal (GI) included
-
Often painful
-
Progression is slow and gradual, but relentless, with a high propensity for regional or distant metastases
-
Little sensitivity to conventional multiagent chemotherapy, and treatment is usually radical resection
-
-
Molecular genetic pathology
-
t(12;22)(q13;q12) in approximately 90% of cases (see Fig. 15.2c)
-
EWSR1 gene (at 22q12) breakpoints in intron 7, 8, or 10
-
ATF1 gene (at 12q13) breakpoints in intron 3 or 4
-
-
Molecular variants
-
Type 1: EWSR1 exon 8—ATF1 exon 4 (85%)
-
Type 2: EWSR1 exon 10—ATF1 exon 5
-
Type 3: EWSR1 exon 7—ATF1 exon 5
-
-
Fusion gene has N terminal transactivating domain of EWS and C terminal leucine zipper dimerization and DNA-binding domains of ATF1, under control of ubiquitously expressed EWS promoter, causing oncogenesis by increased activation of genes bearing ATF1 sites
-
EWSR1–ATF1 soft tissue clear cell sarcomas show consistent melanocytic differentiation, as well as expression of MITF-M transcript
-
t(2;22)(q34;q12) exclusively in tumors of GI tract
-
EWSR1 gene (at 22q12)
-
CREB1 gene (at 2q34)
-
EWSR1–CREB1 tumors lack melanocytic markers
-
-
-
Molecular diagnostics
-
Test indications: establish diagnosis
-
Additional technical considerations
-
FISH: a commercial break apart probe is available for EWSR1, but cannot distinguish between different molecular variants of EWSR1–ATF1, or the chromosomal variant, t(2;22)
-
RT-PCR: can distinguish between different molecular variants of EWSR1–ATF1, but clinical significance of this distinction is unclear
-
-
Additional interpretive considerations
-
GI tract clear cell sarcomas with either EWSR1–ATF1 or EWSR1–CREB1 lack melanocytic markers, in contrast to the EWSR1–ATF1 soft tissue clear cell sarcoma
-
Both t(12;22) and t(2;22) have also been reported in angiomatoid fibrous histiocytoma
-
The t(12;22) has been also reported in hyalinizing clear cell carcinoma of the salivary gland
-
-

Dermatofibrosarcoma Protuberans
-
Basic pathology (see Fig. 15.9)
Fig. 15.9 
Dermatofibrosarcoma protuberans (H&E stain). The tumor consists of a continuous sheet of spindled cells arranged in tight storiform whorls
-
Uncommon neoplasm of low to intermediate malignant potential
-
Composed of noncircumscribed nodular lesions of S100−/CD34+ spindle cells arranged in tight storiform whorls
-
Involving dermis and subcutaneous zones of skin, usually with a thin zone of dermis between the tumor and epidermis
-
Pigmented (Bednar tumor) and myxoid variants exist
-
-
Clinical features
-
Most commonly occurs in ages 30–50 years
-
Trunk and proximal extremities are most common sites
-
Initially grows slowly, with a later phase of rapid growth
-
Frequently recurs locally (50%), even after wide resection (12%), and rarely metastasizes (1–4%)
-
Wide surgical resection is usual treatment
-
A number of clinical studies have shown a high response rate to imatinib therapy in both locally advanced and metastatic lesions
-
-
-
Molecular genetic pathology
-
Supernumerary ring chromosome, derived from a t(17;22)(q22;q13) in most cases
-
COL1A1 gene (at 17q21.31–q22) has many different breakpoints, exons 29 and 32 being slightly more frequently involved
-
PDGFB gene (at 22q13) breakpoint is in intron 1
-
Fusion gene includes nearly the entire PDGFB sequence fused to a variable length of N terminal COL1A1 sequence, under control of the COL1A1 promoter, leading to oncogenesis by constitutive activation of the PDGFB growth signaling
-
The frequency of unbalanced cytogenetic abnormalities suggests a dosage effect or a low level of amplified expression of PDGFB
-
-
-
Molecular diagnostics
-
Test indications: establish diagnosis
-
Additional technical considerations
-
Karyotype: rare variant translocations have been reported
-
FISH: no commercial probes are available, but whole-painted chromosome probes for chromosomes 17 and 22 are useful to identify the presence of chromosomal material of both chromosomes in ring/marker chromosomes
-
RT-PCR: multiple breakpoints in COL1A1 can complicate analysis; most protocols involve nested RT-PCR, with multiple primers spaced throughout COL1A1
-
-
Additional interpretive considerations
-
The same COL1A1–PDGFB fusion has been reported in giant cell fibroblastoma of childhood, but in linear t(17;22), not supernumerary ring chromosomes
-
-

Desmoplastic Round Cell Tumor
-
Basic pathology (see Fig. 15.10)
Fig. 15.10 
Desmoplastic small round cell tumor (H&E stain). The tumor consists of sheets of small tumor cells with little cytoplasm, growing within dense desmoplastic stroma
-
Aggressive, poorly differentiated tumor with characteristic histology
-
Infiltrating nests of small round cells with prominent desmoplasia
-
Evidence of multilineage differentiation including immunoreactivity for keratin, desmin, and neuron-specific enolase
-
Characteristically involves the peritoneum
-
-
Clinical features
-
Mostly in children (boys) and young adults
-
Located almost exclusively on the peritoneal surfaces of the abdomen
-
Very aggressive tumor, with a very poor prognosis: 35% overall progression-free survival at 5 years
-
Treatment is surgery, followed by intensive chemotherapy and radiotherapy
-
-
Molecular genetic pathology
-
t(11;22)(p13;q12) in approximately all cases (see Fig. 15.2d)
-
EWSR1 gene (at 22q12) breakpoint usually in intron 7, but also 8 and 9
-
WT1 gene (at 11p13) breakpoints in intron 7
-
Fusion protein includes N terminal transactivation domain of EWSR1 and the C terminal zinc finger DNA-binding domain of WT1, under control of the EWSR1 promoter, leading to oncogenesis through overexpression and increased activation of WT1 DNA-binding domain
-
Molecular variants
-
EWSR1 exon 7—WT1 exon 8
-
EWSR1 exon 8—WT1 exon 8
-
EWR1S exon 9—WT1 exon 8
-
-
-
-
Molecular diagnostics
-
Test indications
-
Establish diagnosis, as differential diagnosis of small, round blue cell tumors of childhood is broad, and requires ancillary testing to narrow
-
-
Additional technical considerations
-
IHC: antibodies to the carboxy terminus of WT1 show overexpression in these tumors, while antibodies to the amino terminus of WT1 show absence of expression; IHC is sensitive, specific, and simple, and is the most commonly employed diagnostic method
-
FISH: commercial break apart probe is available for EWSR1, but cannot distinguish desmoplastic round cell tumor (EWSR1–WT1) from extraosseous Ewing sarcoma (EWSR1–FLI1 and other EWSR1 fusions), which is often another diagnostic consideration in these patients
-
RT-PCR: can distinguish molecular variants, but this distinction is currently of unknown clinical significance
-
Southern blot: enabled by the short length of WT1 intron 7, which contains the breakpoints
-
-
Additional interpretive considerations: rare chromosomal variants must be further investigated by FISH, RT-PCR, and IHC
-

Endometrial Stromal Sarcomas
-
Basic pathology (see Fig. 15.11)
Fig. 15.11 
Endometrial stromal sarcoma. The tumor consists of a proliferation of bland, small round cells with scant cytoplasm and smooth chromatin, resembling endometrial stroma
-
Uncommon tumor of endometrial stroma, with benign, low grade and high grade variants
-
Grade is based primarily upon extent of infiltration of adjacent myometrium, cytologic pleomorphism, and mitotic activity
-
Cells resemble proliferative phase endometrial stroma, but displace uninvolved benign glandular elements
-
-
Clinical features
-
Primarily affects middle-aged women
-
Patients present with vaginal bleeding, pelvic pain
-
Prognosis is grade-dependent
-
Benign stromal nodules are cured surgically
-
Low grade endometrial stromal sarcomas can recur after surgery (20%), often many years later, and rarely (10%) metastasize
-
High grade endometrial stromal sarcomas are aggressive, with frequent recurrence and metastasis
-
-
-
Molecular genetic pathology
-
t(7;17)(p15;q21) in approximately 50–60% of cases (see Fig. 15.2e)
-
JAZF1 gene (at 7p15)
-
SUZ12 (JJAZ1) gene (at 17q21)
-
Fusion protein contains nearly all of SUZ12, a polycomb group transcriptional repressor, fused to the amino terminal region of JAZF1, under control of the JAZF1 promoter. The mechanism by which this gene fusion induces neoplasia is still being actively investigated
-
-
t(6;7)(p21;p15) in approximately 20% of cases
-
JAZF1 (at 7p15)
-
PHF1 gene (at 6p21)
-
PHF1, like SUZ12, is homologous to a Drosophila zinc finger Polycomb gene
-
-
t(10;17)(q22;p13)
-
FAM22A/B genes (at 10q23.2 and 10q22.3)
-
YWHAE gene (at 17p13)
-
The 14–3–3 oncoprotein results from a t(10;17) genomic rearrangement, leading to fusion between 14–3–3ε (YWHAE) and either of two nearly identical FAM22 family members (FAM22A or FAM22B)
-
The rearrangement results in an inframe fusion between YWHAE (exon 1–5) and one of the two highly homologous genes (FAM22A or FAM22B, exon 2–7)
-
-
-
Molecular diagnostics (see Fig. 15.5)
-
Test indications
-
Establish diagnosis
-
-
Additional technical considerations
-
Karyotype: single cases with other chromosome abnormalities have been reported
-
FISH: no commercial probes are available for t(7;17) and t(10;17)
-
-
Additional interpretive considerations
-
The t(7p15)/JAZF1 positive tumors appear to be more common in low grade endometrial stromal sarcomas of classic histology, but has been reported in cases with high grade histology, and even in occasional mixed tumors (adenosarcoma, carcinosarcoma)
-
The t(10;17) positive tumors appear to be histologically higher grade and clinically more aggressive than JAZF1-rearranged tumors. These tumors display high grade (but nonpleomorphic) round cell histology that is immunophenotypically undifferentiated and frequently includes an admixed low grade spindle cell component with fibrocollagenous/fibromyxoid stroma that is positive for ER, PR, and CD10 immunohistochemically
-
YWHAE rearrangement and JAZF1 rearrangement are mutually exclusive
-
The (10;17)(q22;p13) induced YWHAE–FAM22 genetic fusion seen in endometrial stromal sarcoma is the identical recurrent translocation reported in clear cell sarcoma of the kidney
-
-

Epithelioid Hemangioendothelioma
-
Basic pathology
-
Proliferation of round (epithelioid) cells that typically form cord-like structures embedded within an edematous, proteoglycan-rich, extracellular matrix
-
No specific biomarkers distinguish this tumor from other vascular lesions
-
-
Clinical features
-
Wide age range
-
Affects both genders equally
-
Arises in soft tissue and bone, as well as visceral organs, especially liver and lungs
-
Two prognostic categories, classic and malignant, stratified by mitotic activity and size
-
Treatment is by surgical resection, although multifocal visceral disease may be treated with transplantation
-
-
Molecular genetic pathology
-
t(1;3)(p36.3;q25) in ∼85% of cases
-
CAMTA1 gene (at 1p36.3) breakpoints in exon 8 or 9
-
WWTR1 gene (at 3q25) breakpoint in exon 2, 3, or 4
-
Fusion protein contains the amino terminus of WWTR1 and the carboxy terminus of CAMTA1, under the transcriptional control of the WWRT1 promoter
-
Molecular variants
-
Type 1: WWTR1 exon 4—CAMTA1 exon 8
-
Type 2: WWTR1 exon 4—CAMTA1 exon 9
-
Type 3: WWTR1 exon 2—CAMTA1 exon 9
-
Type 4: WWTR1 exon 3—CAMTA1 exon 9
-
-
-
-
Molecular diagnostics
-
Indications for molecular genetic testing: diagnosis
-
This gene fusion has not been detected in any of the morphological mimics of epithelioid hemangioendothelioma, such as hemangioendothelioma, epithelioid angiosarcoma, or epithelioid sarcoma-like hemangioendothelioma
-
-
Additional technical considerations
-
FISH: no commercial probes are available
-
-
Ewing Sarcoma
-
Basic pathology (see Fig. 15.12)
Fig. 15.12 
Primitive neuroectodermal tumor. This member of the Ewing sarcoma family is an extraosseous tumor composed of sheets of small, uniform, round cells with modest eosinophilic cytoplasm, and occasional cytoplasmic glycogen vacuoles. Focal rosettes may be present
-
Small round blue cell tumor
-
Initially described in bone, but may also involve extraosseous sites
-
Tumors often have large areas of necrosis
-
The cells are small, uniform, and round
-
PAS-positive glycogen granules
-
Immunoreactivity for CD99/O13 antigen (MIC2 gene product)
-
-
Clinical features
-
Most common in children (5–20) and young adults (<30)
-
Usually presents with pain or swelling, but can also present with systemic symptoms
-
Usually involves diaphysis of long bones, originating in medullary cavity and eventually penetrating through cortex to soft tissues
-
Characteristic “onion skin” appearance of cortex on X-rays as tumor lifts periosteum and new bone is laid down
-
Frequently metastasizes
-
Controversy regarding extraosseous lesions, and whether they represent true Ewing sarcoma
-
Peripheral neuroepithelioma also called primitive neuroectodermal tumor, esthesioneuroblastoma (olfactory epithelium), and Askin tumor (chest wall)
-
-
Poor prognosis when treated with surgery and radiotherapy (5-year survival 5–8%), but multiagent chemotherapy has increased 5-year survival to approximately 75%
-
-
Molecular genetic pathology
-
t(11;22)(q24;q12) in 90–95% (see Fig. 15.2f)
-
EWSR1 gene (at 22q12) has multiple breakpoints
-
FLI1 gene (at 11q24) has multiple breakpoints
-
Fusion protein contains carboxy terminal DNA-binding transcriptional activation domain of FLI1 and the amino terminal transactivation domain of EWSR1, under the regulatory control of the ubiquitously expressed EWSR1 promoter. This leads to oncogenesis through upregulation of genes with FLI1 sites
-
Molecular variants (see Fig. 15.13)
Fig. 15.13 
Structure of the EWSR1 and FLI1 genes, and the most common EWSR1-FLI1 fusions seen in Ewing sarcoma. The upper figure shows the structure of the EWSR1 gene, with the most prevalent translocation breakpoints indicated by long arrows (intron 7 and intron 10), and less prevalent breakpoints indicated by short arrows. The next figure shows the FLI1 gene in the same manner, with the most common breakpoints in intron 5 and intron 4. At the bottom are the two most common fusion types, type I (EWSR1 intron 7—FLI1 intron 5) and type II (EWSR1 intron 7—FLI1 intron 4). Note that the figures are not drawn to scale
-
Type I: EWSR1 exon 7—FLI1 exon 6 (∼60%)
-
Type II: EWSR1 exon 7—FLI1 exon 5 (∼20%)
-
Many other variants, but the breakpoint is always downstream of EWSR1 exon 7 and upstream of FLI1 exon 9
-
-
-
Chromosomal variants are seen in 5–10% of cases, all involving EWSR1 with different partner genes
-
t(21;22)(q12;q12) with EWSR1–ERG fusion
-
t(7;22)(p22;q12) with EWSR1–ETV1 fusion
-
t(17;22)(q12;q12) with EWSR1–E1AF fusion
-
t(2;22)(q33;q12) with EWSR1–FEV fusion
-
inv(22)(q12q12) with EWSR1–ZSG fusion
-
These EWSR1 gene fusions contain the N terminal portion of EWSR1 and the C terminal DNA-binding domain of an ETS transcriptional family member (e.g., ERG, ETV1, E1AF, FEV) and are, thus, analogous to the canonical EWSR1–FLI1 fusions
-
-
t(20;22)(q13;q12) with EWSR1–NFATC2 fusion
-
The NFATC2 gene involved in the t(20;22), is a transcription factor that is not a member of the ETS family. The inframe fusion gene contain the C terminal of EWSR1, encoded by the first 8 exons, and the N terminal of NFATc2, encoded by exons 3–10
-
-
t(16;21)(p11;q22) with a FUS–ERG fusion
-
The FUS gene, involved in the t(16;21), belongs to the TET family of RNA-binding proteins, and shows considerable homology with EWSR1
-
FUS is the most frequent gene replacing EWSR1 in other sarcoma translocations
-
A variant t(2;16) (q35;p11) associated with a FUS–FEV fusion gene has been recently reported, suggesting that the FUS rearrangement could be underreported
-
-
-
-
Molecular diagnostics
-
Test indications
-
Establish a diagnosis: the differential diagnosis of small round blue cell tumors is broad and requires ancillary methods. This is especially useful when the tumor presents in an unusual site
-
Recent prospective randomized multi-institute trials described the absence of prognostic importance of type 1 EWS–FLI1 fusion
-
Minimal disease monitoring: RT-PCR may be used for staging patients for bone marrow involvement in the absence of radiologic or morphologic evidence. Moreover, some studies have looked at detecting circulating tumor cells with RT-PCR
-
-
Additional technical considerations
-
FISH: a commercial break apart probe is available for EWSR1 and can detect all molecular variants, but cannot distinguish among chromosomal variants, and cannot distinguish Ewing sarcoma from other sarcomas with EWSR1 translocation (e.g., clear cell sarcoma, intraabdominal desmoplastic round cell tumor, extraskeletal myxoid chondrosarcoma, myxoid liposarcoma)
-
-
RT-PCR: variability of breakpoints and diversity of molecular variants provides an assay design challenge
-
A single primer set to EWSR1 exon 7 and FLI1 exon 6 will detect approximately 80% of cases and enable size-based distinction of the type I and type II transcripts, but will not detect fusion transcripts with more distal FLI1 breakpoints or, potentially, large fusion transcripts with more distal EWSR1 breakpoints
-
A single primer set to EWSR1 exon 7 and FLI1 exon 9 could detect all fusion types, but transcripts with distal EWSR1 breakpoints and/or proximal FLI1 breakpoints may be too large for reliable detection by RT-PCR
-
Chromosomal variants may also be detected due to conservation of EWSR1 breakpoints and high homology between genes partnered with EWSR1: consensus primers have been designed that anneal to FLI1/ERG/FEV and to TEV1/E1AF
-
Southern blot: EWSR1 breakpoints occur over a relatively small area (∼7 kb), enabling Southern blot detection of all fusion genes, but Alu repeat polymorphism in intron 6 in African Americans can complicate the analysis
-
-
Additional interpretive considerations
-
The same translocations are seen in primary osseous and in extraosseous Ewing sarcomas, but are characteristically absent in olfactory neuroblastomas (esthesioneuroblastomas), suggesting that these tumors may be unrelated to Ewing sarcomas
-
Therefore, we need to be aware of these cytogenetic variant Ewing sarcomas in order to modify their detection in clinical practice. A negative results generated by RT-PCR using specific fusion transcript or FISH for EWSR1, should no preclude the diagnosis of Ewing sarcoma in the contest of typical morphology and immunophenotype features. Moreover, cytogenetic analysis is still a gold standard to detect these rare translocations
-
A similar t(16;21) associated with FUS–ERG has been previously reported in a subgroup of AML
-
-


Gastrointestinal Stromal Tumor
-
Basic pathology
-
Mesenchymal tumor of GI viscera
-
Composed most often of pure spindle cells, but epithelioid and mixed variants occur
-
Tumors are now believed to arise from the interstitial cells of Cajal present within the gut wall
-
Contain features of smooth muscle and neural tissues
-
-
Clinical features
-
Most gastrointestinal stromal tumors (GISTs) arise in the stomach (60%) and small intestine (25%), but they can occur anywhere in the GI tract as well as in omentum, retroperitoneum, and mesentery
-
GIST patients vary widely in age, but peak around 60 years
-
Prognosis is largely dependent on size and mitotic activity; patients with gastric GISTs tend to fare better than those with intestinal tumors
-
Surgery is the primary treatment, but recurrence and dissemination are inevitable for high risk lesions (>5 cm, >5 mitoses/50 high power fields). Some GISTs have been treated successfully with imatinib (Gleevec)
-
-
Molecular genetic pathology
-
Unlike the other sarcomas in this chapter, GISTs are not characterized by a chromosomal pathology (i.e., translocations, inversions, etc.), but rather by molecular genetic (i.e., point mutations, insertions, deletions) pathology
-
KIT gene (at 4q12) is mutated in 80–85% of cases
-
Activating mutations include small inframe deletions and insertions, and point mutations
-
Most mutations are in exons 11 and 9, but mutations in exons 13 and 17 have also been described
-
-
A subset of GISTs has mutations in the KIT-related PDGF receptor-α (PDGFRA) gene (also at 4q12), in exons 18 and 12
-
-
Molecular diagnostics
-
Test indications
-
Establish diagnosis
-
Prognosis: exon 9 mutations are associated with malignant GISTs
-
Theranosis: GISTs with exon 11 mutations are most likely to respond to imatinib (Gleevec)
-
Acquired mutations, especially in exons 13 and 17, may confer resistance to imatinib
-
-
Additional technical considerations
-
IHC is a simple, sensitive, and specific means of assessing overexpression of KIT (CD117), but variability between commercial antibody preparations and between different laboratory protocols has led to some inconsistency in published results, particularly with regard to specificity
-
IHC cannot distinguish between mutations that have different implications for therapy response, and is most useful as a screening tool to select cases for molecular analysis
-
-
PCR: DHPLC and high resolution melt curve analysis are quick and sensitive screening methods for detection of both deletions and point mutations that is free of some of the inconsistencies that affect IHC
-
Sanger sequencing is typically used for specific identification of each mutation detected by a screening method
-
-
Additional interpretive considerations
-
GISTs with PDGFRA mutations are negative by IHC, as are some GISTs with KIT mutations and others with acquired imatinib resistance
-
-
Infantile Fibrosarcoma
-
Basic pathology
-
Spindle cell neoplasm of early childhood
-
Bundles of interdigitating cells that are immunoreactive for vimentin, but not smooth muscle, desmin, or S100
-
-
Clinical features
-
One of the more common soft tissue sarcomas of childhood
-
Soft tissue mass, usually noted at, or soon after, birth
-
70% in extremities, followed by head/neck, trunk
-
Excellent prognosis
-
Treatment is complete surgical excision
-
Sensitive to chemotherapy
-
-
Molecular genetic pathology
-
t(12;15)(p13;q26) in approximately 95%
-
ETV6 gene (at 12p13)
-
NTRK3 gene (at 15q26)
-
Fusion protein contains the N terminal helix-loop-helix protein dimerization domain of ETV6 and the C terminal tyrosine kinase domain of NTRK3, presumably leading to oncogenesis through increased activation of NTRK3 kinase and downstream signal transduction; the fusion protein has oncogenic in vitro activity
-
-
Molecular diagnostics
-
Test indications
-
Establish diagnosis
-
Therapy selection: other spindle cell lesions of childhood may lack sensitivity to chemotherapy
-
-
Additional technical considerations
-
Karyotype: the t(12;15) is cryptic, and difficult to detect by standard GTG-banding
-
Polysomies of chromosomes 8, 11, 17, and/or 20 are common
-
-
FISH: a commercial break apart probe is available for ETV6
-
-
Additional interpretive considerations
-
The same t(12;15) has been reported in congenital mesoblastic nephroma
-
A renal lesion with similar histopathology and clinical features
-
In secretory carcinomas of the breast and of the salivary glands
-
-
Inflammatory Myofibroblastic Tumors
-
Basic pathology
-
Proliferation of myofibroblastic spindle cells, usually with a prominent mixed inflammatory component
-
Lesion has many pseudonyms, reflective of its controversial nature
-
Inflammatory pseudotumor
-
Plasma cell granuloma
-
Pseudosarcomatous myofibroblastic proliferation
-
Postoperative spindle cell nodule
-
Atypical fibromyxoid tumor
-
-
Epithelioid inflammatory myofibroblastic sarcoma is a distinctive variant tumor
-
Epithelioid and round cell morphology
-
Nuclear membrane or perinuclear ALK immunostaining
-
Arising in intraabdominal locations
-
Aggressive clinical course and predilection for male patients
-
-
-
Clinical features
-
Most often in children and young adults
-
Can involve both soft tissues and viscera
-
Patients often present with systemic symptoms (fever, weight loss) and anemia
-
Usually indolent course, especially in the lung; more aggressive course in the abdomen (e.g., epithelioid inflammatory myofibroblastic sarcoma)
-
Tumor-related mortality, approximately 10%, usually due to local destruction rather than metastasis
-
Aggressive behavior, with metastasis, has been described, often associated with morphologic change (round cell transformation)
-
Usual management is surgical excision
-
-
Molecular genetic pathology
-
2p23 rearrangement in approximately 50% of cases
-
ALK gene (at 2p23) is invariably fused by chromosomal translocation to a wide variety of partner genes
-
t(1;2)(q25;p23) with TPM3–ALK fusion
-
t(2;19)(p23;p13.1) with TPM4–ALK fusion
-
t(2;17)(p23;q23) with CLTC–ALK fusion
-
t(2;2)(p23;q13) with RANBP2–ALK fusion
-
inv(2)(p12q35) with ATIC–ALK fusion
-
t(2;11)(p23;p15) with CARS–ALK fusion
-
t(2;4)(p23;q21) with SEC31L1–ALK fusion
-
-
-
ALK gene encodes a membrane-associated protein with a cytoplasmic tyrosine kinase domain, and fusion proteins are believed to undergo homodimerization, which is predicted to trigger stimulus-independent activation of ALK tyrosine kinase domain
-
-
Molecular diagnostics
-
Indications for molecular genetic testing
-
Establish a definitive diagnosis: the differential diagnosis is very broad, and ranges from self-limited benign reactive proliferations to aggressive malignancies
-
-
Additional technical considerations
-
Karyotype: 50% of these tumors do not have an ALK rearrangement
-
FISH: a commercial break apart probe is available for ALK
-
RT-PCR is very difficult, given the wide range of possible partner genes
-
IHC: staining for ALK is simplest, quickest, and most sensitive methodology
-
Diffuse cytoplasmic staining when the fusion involves the cytoplasmic proteins TMP3, TPM4, CARS, ATIC, and SEC31L1
-
Granular cytoplasmic staining with CLTC and nuclear membrane staining with RANBP2
-
A correlation between the nuclear membrane pattern and RANBP2–ALK fusion seems to be consistent in epithelioid inflammatory myofibroblastic sarcoma
-
-
-
Additional interpretive consideration
-
ALK rearrangements are also seen in anaplastic large cell lymphoma and in ∼5% of lung adenocarcinomas, where they have been shown to confer a favorable response to treatment with crizotinib, a tyrosine kinase inhibitor
-
In 2011, the U.S. Food and Drug Administration approved a commercial ALK split apart FISH assay (Abbott Molecular) to be used to select lung cancer patients for treatment with crizotinib (Xalkori™, Pfizer)
-
-
Low Grade Fibromyxoid Sarcoma
-
Basic pathology
-
Rare soft tissue neoplasm of low malignant potential with uncertain histogenesis
-
Contains a mixture of hypocellular areas with collagenous stroma and more cellular areas with myxoid stroma
-
Focal collagen rosettes are seen in a subset of cases
-
-
Clinical features
-
Incidence is presumed to be low, but it is likely that these lesions have been underrecognized
-
Painless mass, typically in the lower extremities, especially thigh in young male adults
-
Treatment is wide local resection
-
Local recurrences in approximately 10%, and metastasis is seen in 5%
-
-
Molecular genetic pathology
-
t(7;16)(q33–34;p11) in 95% (see Fig. 15.2G)
-
FUS gene (at 16p11) with breakpoints in exon 5, 6, and 7
-
CREB3L2 (BBF2H7) gene (at 7q33–34) with breakpoints in exon 5 and 6
-
Rarely, ring chromosome assumed derived from chromosomes 7 and 16 has been also reported
-
Fusion protein contains carboxy terminal portion of CREB3L2, including B-ZIP DNA-binding domain and amino terminus of FUS, containing transactivation domain, under regulation of ubiquitously expressed FUS promoter. Mechanism of oncogenesis is likely through dysregulation of CREB3L2 transcriptional targets
-
Molecular variants are related to the different FUS and CREB3L2 breakpoints
-
-
Chromosomal variant; t(11;16)(p11;p11) in 5% of the cases
-
FUS gene (at 16p11) with breakpoint in exon 5
-
CREB3L1 gene at (11p11) with breakpoint in exon 6
-
-
-
Molecular diagnostics
-
Indications for molecular genetic testing
-
Establish a definitive diagnosis
-
-
Additional technical considerations
-
Karyotype: t(7;16) is cryptic and can easily be missed by G-banded chromosome analysis
-
FISH (see Fig. 15.3): a commercial break apart probe is available for FUS
-
RT-PCR: relative proximity of breakpoints facilitates RT-PCR analysis
-
-
Additional interpretive consideration
-
The t(7;16)/FUS–CREB3L2 has been reported in hyalinizing spindle cell tumors with giant rosettes (so-called unusual variant of low grade fibromyxoid sarcoma) and sclerosing epithelioid fibrosarcoma
-
-
Mesenchymal Chondrosarcoma
-
Basic pathology
-
Biphasic sarcoma, with small round/slightly spindled cells and islands of chondroid matrix
-
Differential diagnosis can be challenging in samples with minimal chondroid matrix, and includes other “small round blue cell tumors”
-
-
Clinical features
-
Affects young patients, between 10 and 20 of age
-
Late distal recurrences
-
Poor outcome
-
-
Molecular genetic pathology
-
HEY1–NCOA2 fusion by genome-wide screen of expression data
-
HEY1 gene (at 8q21.1) with breakpoint in exon 4
-
NCOA2 gene (at 8q13.3)/with breakpoint in exon 13
-
The HEY1–NCOA2 fusion replaces the C terminal portion of the HEY1 by the NOAC2 AD1/CIS and AD2 domain, while retaining the Hey1 bHLH DNA-binding/dimerizations domain
-
These genes are only ∼10 Mb apart, and this fusion can be the results of a cryptic interstitial deletion or paracentric inversion between the 8q13.3 and 8q21.1 region
-
-
-
Molecular diagnostics
-
Indications for molecular genetic testing
-
Diagnosis
-
The noncartilage components of this sarcoma are often predominant, and such a lesion can be confused with other small cell neoplasms
-
The 8q rearrangement is cryptic and can easily be missed by G-banded chromosome analysis
-
FISH: no commercial probes are available
-
-
-
Myoepithelioma, Soft Tissue
-
Basic pathology
-
Myoepithelial tumors represent a family of lesions with variable terminology, based on anatomical location
-
Pleomorphic adenoma of salivary glands
-
Benign mixed tumor in the skin, and myoepithelial tumor/parachordoma in the soft tissue
-
Often have uniform rounded cell morphology and clear cytoplasm in deep-seated soft tissue
-
Criteria for confirming the diagnosis include coreactivity for EMA ± cytokeratin AE1/AE3 and S100 ± GFAP
-
-
Clinical features
-
>50% of cases occur in children or young adults
-
Most common in the extremities, followed by head and neck
-
-
Molecular genetic pathology
-
EWSR1(22q12) rearrangement in approximately 50% of cases
-
t(6;22)(p21;q12)
-
EWSR1 gene (at 22q12)
-
POUF5F1 gene (at 6p21)
-
This translocation is identified in a subset of deep-seated tumors of extremities, in children or young adults with distinct clear cell morphology
-
-
t(1;22)(q23;q12)
-
EWSR1 gene (at 22q12)
-
PBX1 gene (at 1q23)
-
This translocation is identified in a subset of tumors with a deceptively bland appearance, composed mainly of spindle cells embedded in a fibrotic stroma, resembling, in areas, desmoid-type fibromatosis
-
-
t(19;22)(q13;q12)
-
EWSR1 gene (at 22q12)
-
ZNF444 gene (at 19q13)
-
Very rare translocation, less 2% of the cases
-
-
EWSR1 negative myoepithelial tumors are more often benign, superficially located, and show ductal differentiation, suggesting the possibility of a distinct subgroup
-
-
Molecular diagnostics
-
Indications for molecular genetic testing
-
EWSR1 rearrangement is a common event in myoepithelial tumors arising outside the salivary glands
-
-
Additional technical considerations
-
FISH: a commercial break apart probe is available for EWSR1 and can detect all molecular variants, but cannot distinguish among chromosomal variants, and cannot distinguish soft tissue myoepitheliomas from other cancers with EWSR1 translocation
-
RT-PCR: variability of breakpoints and diversity of molecular variants provides an assay design challenge
-
-
Additional interpretive considerations
-
t(6;22)/EWSR1–POU5F1 has been reported in three cases of hidradenoma of the skin and one case of mucoepidermoid carcinoma of salivary gland
-
-
Myxoid Chondrosarcoma, Extraskeletal
-
Basic pathology (see Fig. 15.14)
Fig. 15.14 
Extraskeletal myxoid chondrosarcoma. Low-power image on the left shows ribbons and cords of tumor cells in a myxoid stroma. High-power image on the right shows cytologic features of the cancer cells, including small, dark nuclei vacuolated cytoplasm, and bubbly myxoid stroma
-
Rare soft tissue tumor of characteristic histology and disputed histogenesis
-
The name may be a misnomer, as it is clearly a different lesion from skeletal chondrosarcoma
-
Well-circumscribed, lobular mass with gelatinous or mucoid cut surface
-
Microscopically, it is composed of multiple lobules with myxoid stroma and polygonal, stellate, or spindled tumor cells with cytoplasm that may be vacuolated, mimicking signet ring cells or physaliferous cells
-
Myxoid areas characteristically have columns, cords, or strands of tumor cells, while hypercellular areas can have many different patterns of growth, including solid sheets with minimal or no myxoid matrix
-
Cells are immunoreactive for vimentin, S100, and epithelial membrane antigen
-
Differentiated chondrocytes are rare
-
Electron microscopy shows evidence of cartilaginous differentiation
-
-
-
Clinical features
-
This rare tumor is most common in middle age adults, and is very rare in children (5% of patients are younger than age 20); males are more frequently affected than females
-
The lesion is most common in extremities (85%), particularly lower (75%), and is usually located in deep soft tissues, where it presents as a slow growing mass
-
Usual treatment is wide surgical excision, with adjuvant chemotherapy and/or radiotherapy in case of lymph nodes or metastasis
-
Prognosis is poor, with mean survival of 20 months, and a high rate of metastasis, especially to lungs
-
-
Molecular genetic pathology
-
t(9;22)(q22;q12) in approximately one-third of cases (see Fig. 15.2h)
-
EWSR1 gene (at 22q12) breakpoints in introns 7, 11, 12
-
NR4A3 (CHN, TEC, NOR1) gene (at 9q22)
-
Fusion contains nearly entire NR4A3 steroid/thyroid hormone nuclear receptor protein juxtaposed to amino terminal portion of EWSR1, under regulatory control of the constitutively expressed EWS promoter
-
The exact mechanism of oncogenesis is unclear
-
-
-
Molecular variants
-
Type 1: EWSR1 exon 12—NR4A3 exon 3
-
Type 2: EWSR1 exon 7—NR4A3 exon 2
-
EWSR1 exon 11—NR4A3 exon 1
-
-
Chromosomal variants
-
t(9;17)(q22;q11) with TAF15 (TAF2N, TAFI168, RBP56)–NR4A3 fusion
-
t(9;15)(q22;q21) with TCF12 (HTF4)–NR4A3 fusion
-
t(3;9)(q11;q22) with TFG–NR4A3 fusion
-
-
-
Molecular diagnostics
-
Test indications: establish diagnosis, as morphology in hypercellular lesions is not distinctive
-
Additional technical considerations
-
FISH: commercial break apart probe is available for EWSR1, can detect t(9;22) but not other chromosomal variants
-
RT-PCR: different primers for type 1 and type 2 fusion transcripts may be indicated due to large distance between EWSR1 breakpoints in these two molecular variants
-
-

Myxoid Liposarcoma
-
Basic pathology (see Fig. 15.15)
Fig. 15.15 
Myxoid liposarcoma. Low-power image on the left shows characteristic pattern of delicate arborizing capillaries that vaguely resemble chicken wire. High-power image on the right shows characteristic lipoblasts with scalloped nuclei indented by circular globules of optically clear cytoplasmic fat
-
Most common malignant soft tissue tumor in adults
-
Arising from adipose tissue
-
Tumor consists of hypocellular myxoid tissue with a rich capillary network in a classic “chicken wire” pattern
-
Characteristic lipoblasts, mononuclear cells, or multinuclear cells with cytoplasmic lipid vacuoles that push aside and indent the nucleus
-
May be difficult to identify
-
-
Clinical features
-
Primarily affects adults (median age, 55–60)
-
Lower extremities most often affected
-
Usual treatment is surgical excision
-
Five-year survival is good for pure myxoid liposarcoma (70%), but not for round cell liposarcoma (18%)
-
Metastasis is usually to lung
-
-
Molecular genetic pathology
-
t(12;16)(q13;p11) in 90% of cases (see Fig. 15.2i)
-
FUS gene (at 16p11) breakpoints in introns 5, 7, and 8
-
DDIT3 (CHOP, GADD153) gene (at 12q13) breakpoint in intron 1 or exon 2
-
Oncogenicity of FUS–CHOP fusion protein has been shown in vitro and in animal models
-
Molecular variants
-
Type I: FUS exon 7—DDIT3 exon 2 (20%)
-
Type II: FUS exon 5—DDIT3 exon 2 (70%)
-
Type III: FUS exon 8—DDIT3 exon 2 (10%)
-
-
-
Chromosomal variant: t(12;22)(q13;q12) with EWSR1–DDIT3 fusion
-
-
Molecular diagnostics
-
Indication for testing
-
Establish diagnosis
-
-
Additional technical considerations
-
FISH: commercial break apart probe is available for DDIT3, FUS, and EWS. The CHOP (DDIT3) probe is most useful for myxoid liposarcoma, as it enables detection of both t(12;16) and t(12;22)
-
RT-PCR: a single primer set (FUS exon 5 and DDIT3 exon 3) can amplify all three molecular variants, which can then be distinguished by size
-
-
Additional interpretive considerations
-
Round cell liposarcoma, a high grade liposarcoma, also contains the same t(12;16), suggesting that these lesions may be high grade variants of myxoid liposarcoma, rather than a separate category
-
Some areas in myxoid liposarcoma may resemble lipoblastoma, a benign lesion of childhood
-
In very rare cases, myxoid liposarcoma may occur in children, and in these cases molecular diagnosis may be particularly useful
-
Lipoblastomas carry a rearrangement of 8q12, involving the PLAG1 gene
-
-
Balanced translocations involving 12q13–q15 are also seen in benign lipomas, but the gene involved is usually HGMA2 (HMGIC), not DDIT3
-
-

Myxoinflammatory Fibroblastic Sarcoma
-
Basic pathology
-
Multinodular architecture with alternating myxoid and cellular areas
-
Prominent inflammatory infiltrate containing a variable number of mononucleated and multinucleated Reed–Sternberg-like tumor cells
-
Prominent viral inclusion-like nucleoli and mucin-containing pseudolipoblasts
-
-
Clinical features
-
Rare low grade sarcoma
-
Subcutaneous tissues of digital extremities
-
Repeated local recurrences, sometimes requiring amputation, but rare metastases
-
-
Molecular genetic pathology
-
der(10)t(1;10)(p22;q24), typically unbalanced
-
TGFBR3 gene (at 1p22)
-
MGEA5 gene (at 10q24)
-
Both candidate genes for the gene fusion are transcribed in opposite directions and, thus, are unable to form a fusion transcript
-
TGFBR3 and MGEA5 have losses of 5′ sequence
-
The der(10) chromosome contains the residual 3′ sequences from TGFBR3 and MGEA5
-
-
Additional chromosome aberrations involving chromosome 3 (e.g., ring or marker chromosome), with amplification of 3p11.1–12.1, containing VGLL3 gene
-
Overexpression of FGF8 on 10q was identified by microarray analysis, suggesting that this translocation may alter gene transcription away from the breakpoint
-
-
Molecular diagnostics
-
Indications for molecular genetic testing: Currently unclear
-
Additional technical considerations
-
Cytogenetic analysis is best to detect der(10)t(1;10)
-
FISH: no commercial probes are available
-
RT-PCR: Expression levels of both genes do not appear altered
-
-
Additional interpretive considerations
-
The same genetic alteration, der(10)t(1;10), has been reported in hemosiderotic fibrolipomatous tumor, a predominantly fatty lesion arising with predilection in the subcutaneous tissues of the ankle of middle-aged women
-
The coexistence of mixed tumors, with both pathologic features, either synchronously or metachronously in primary lesions or a subsequent recurrence, suggests either different morphologic variants or different level of progression of a single entity
-
-
Synovial Sarcoma
-
Basic pathology (see Fig. 15.16)
Fig. 15.16 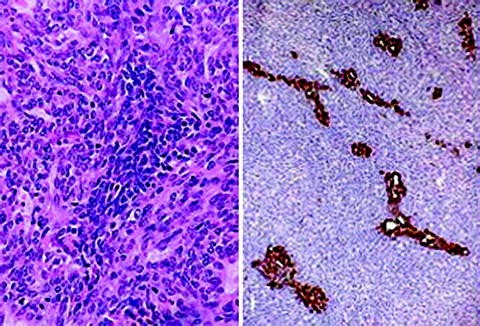
Synovial sarcoma. High-power view on the left shows predominantly spindle-shaped tumor cells with focal epithelial patterns, including a rudimentary glandular structure in the lower right corner. The image on the right shows a low-power view of a biphasic synovial sarcoma stained with polyclonal antibodies directed against a mixture of cytokeratins, demonstrating epithelial elements (brown) growing within the spindle cell population (blue)
-
Malignant soft tissue tumor of uncertain histogenesis
-
Features of both mesenchymal and epithelial differentiation
-
The name is a misnomer, as the tumor is unrelated to synovium
-
Although classic synovial sarcoma is a biphasic tumor with both spindled and epithelial morphology, monophasic variants are equally common
-
Most monophasic synovial sarcomas are spindle cell type
-
Both cell types express both epithelial (e.g., keratin, epithelial membrane antigen, carcinoembryonic antigen [CEA]) and mesenchymal (e.g., vimentin) markers by IHC
-
-
Clinical features
-
Fourth most common sarcoma, with overall incidence of 2.75/100,000
-
Primarily affects adolescents and young adults, and males are more often affected than females
-
Usually presents in para-articular regions of extremities, especially knees and ankles
-
Disease course ranges from very aggressive to very indolent, with 5-year survival 45–60%
-
Favorable prognostic factors include young age, small tumor, low mitotic activity
-
Lymph node metastasis more common than in other sarcomas (10–15% of cases)
-
Complete surgical excision is usual treatment; postoperative radiotherapy and adjuvant chemotherapy may enable limb-sparing surgery and limit metastasis
-
-
Molecular genetic pathology
-
t(X;18)(p11.2;q11.2) in approximately 90% (see Fig. 15.2j)
-
SS18 (SYT) gene (at 18q11.2)
-
SSX1 or SSX2 genes (both at Xp11.2)
-
Fusion protein contains carboxy terminal portion of SSX1 or SSX2, both containing Kruppel-type zinc finger DNA-binding transcriptional repression domains, and amino terminal portion of SYT, containing a novel QPGY transactivation domain, under control of the TATA-less CpG rich SS18 promoter
-
Mechanism of oncogenesis remains under investigation
-
-
Molecular variants involve the fusion of either SSX1 or SSX2 to SS18
-
These genes have extreme (>90%) homology to one another
-
Rare cases of SSX4–SYT fusions have also been reported
-
-
-
Chromosomal variant: a single case of t(X;20)(p11;q13), with an SSX1–SS19L1 fusion being reported
-
-
Molecular diagnostics
-
Indications for molecular genetic testing
-
Establish diagnosis: classic biphasic lesions are generally straightforward to diagnose, but monophasic and poorly differentiated lesions may require cytogenetic and/or molecular tests
-
Prognosis: this is controversial, as early studies suggested that SS18–SSX1 fusions have significantly worse prognosis (survival ∼40% vs. ∼80% for SS18–SSX2), but more recent data has shown that there is no association between fusion type and outcome when a three-part histologic grading scheme is used
-
-
Additional technical considerations
-
Karyotype: rare variant translocation or marker chromosomes can occur
-
FISH: commercial break apart probe for SS18 is available, but cannot distinguish SS18–SSX1 from SS18–SSX2
-
RT-PCR is required to distinguish SS18–SSX1 from SS18–SSX2 transcripts (see Fig. 15.17)
Fig. 15.17 
RT-PCR of synovial sarcomas. The two gels show RT-PCR analysis of two cases (1 and 2), using paired reactions for the SSX1-SS18 and SSX2-SS18 fusion transcripts, from both frozen and formalin-fixed paraffin-embedded tissues. All samples were tested in duplicate. The frozen samples each showed RT-PCR amplification of fusion transcripts with one set of primers, indicating which variant of translocation was present, but amplification failed for paraffin-embedded tissues from the same cases. It is not uncommon for fixed tissue sections to show poor amplification in RT-PCR assays, presumably due to degradation of RNA
-
-
Additional interpretive considerations
-
Biphasic tumors have, almost exclusively, SS18–SSX1 fusions
-
Monophasic tumors can have either fusion, but SS18–SSX2 fusions are more likely
-
Any synovial tumor with abnormal karyotype without a t(X;18) needs to be further investigated by FISH and molecular techniques
-
Cases have been reported of tumors with coexisting SS18–SSX1 and SS18–SSX2 transcripts detected by RT-PCR
-
-


Suggested Readings
Antonescu CR. The role of genetic testing in soft tissue sarcoma. Histopathology. 2006;48:13–21.
Antonescu CR, Nafa K, Segal NH, Dal Cin P, Ladanyi M. EWS-CREB1: a recurrent variant fusion in clear cell sarcoma association with gastrointestinal location and absence of melanocytic differentiation. Clin Cancer Res. 2006;12:5356–62.
Antonescu CR, Dal Cin P, Nafa K, Teot LA, Surti U, Fletcher CD, et al. EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer. 2007;46:1051–60.
Antonescu CR, Zhang L, Chang N, Pawel BR, Travis W, Rosenberg AE, et al. Novel EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of 66 cases, including soft tissue, bone and visceral lesions, showing common involvement of EWSR1 gene rearrangement. Genes Chromosomes Cancer. 2010;49:1114–24.
Antonescu CR, Zhang L, Nielsen P, Rosenberg A, Dal Cin P, Fletcher CD. Consistent t(1;10) with rearrangements of TGFBR3 and MGEA5 in both myxoinflammatory fibroblastic sarcoma and hemosiderotic fibrolipomatous tumor. Genes Chromosomes Cancer. 2011;50:757–64.
Argani P, Lal P, Hutchinson B, Lui MY, Reuter VE, Ladanyi M. Aberrant nuclear immunoreactivity for TFE3 in neoplasms with TFE3 gene fusions: a sensitive and specific immunohistochemical assay. Am J Surg Pathol. 2003;27:750–61.
Atlas of Genetics and Cytogenetics in Oncology and Haematology, http://www.infobiogen.fr/services/chromcancer/index.html. Barr FG, Ladanyi M. Sarcomas. In: Leonard DGB, editor. Diagnostic molecular pathology. Philadelphia: Saunders; 2003, p. 53–76.
Barr FG, Womer RB. Molecular diagnosis of ewing family tumors: too many fusions… ? J Mol Diagn. 2007;9:437–40.
Bovée JV, Hogendoorn PC. Molecular pathology of sarcomas: concepts and clinical implications. Virchows Arch. 2010;456:193–9.
Breitfield PP, Meyer WH. Rhabdomyosarcoma: new windows of opportunity. Oncologist. 2005;10:518–27.
Chang CC, Shidham VB. Molecular genetics of pediatric soft tissue tumors. J Mol Diagn. 2003;5:143–54.
Fisher C. Soft tissue sarcomas with non-EWS translocations: molecular genetic features and pathologic and clinical correlations. Virchows Arch. 2010;456:153–66.
Guillou L, Benhattar J, Bonichon F, et al. Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol. 2004;22:4040–50.
Jain S, Xu R, Prieto VG, Lee P. Molecular classification of soft tissue sarcomas and its clinical applications. Int J Clin Exp Pathol. 2010;3:416–28.
Lee CH, Ou W, Marino-Enriquez A, Zhu M, Myeda M, Wang YX, et al. 14-3-3 fusion oncogenes in high-grade endometrial stromal sarcoma. Proc Natl Acad Sci USA. 2012;109:929–34.
Lemm D, Mügge LO, Mentzel T, Höffken K. Current treatment options in dermatofibrosarcoma protuberans. J Cancer Res Clin Oncol. 2009;135:653–65.
Lessnick SL, Kovar H, Houghton P. The molecular basis of sarcoma. Sarcoma. 2011;2011:864130.
Mariño-Enríquez A, Wang WL, Roy A, Lopez-Terrada D, Lazar AJ, Fletcher CD, et al. Epithelioid inflammatory myofibroblastic sarcoma: an aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol. 2011;35:135–44.
Mertens F, Fletcher CD, Antonescu CR, et al. Clinicopathologic and molecular genetic characterization of low-grade fibromyxoid sarcoma, and cloning of a novel FUS/CREB3L1 fusion gene. Lab Invest. 2005;85:408–15.
O’Leary TJ, Frisman DM. Soft tissue and bones. In: O’Leary TJ, editor. Advanced diagnostic methods in pathology. Saunders: Philadelphia; 2003. p. 421–58.
Ordóñez JL, Osuna D, García-Domínguez DJ, Amaral AT, Otero-Motta AP, Mackintosh C, et al. The clinical relevance of molecular genetics in soft tissue sarcomas. Adv Anat Pathol. 2010;17:162–81.
Romeo S, Dei Tos AP. Soft tissue tumors associated with EWSR1 translocation. Virchows Arch. 2010;456:219–34.
Romeo S, Dei Tos AP. Clinical application of molecular pathology in sarcomas. Curr Opin Oncol. 2011;23:379–84.
Sankar S, Lessnick SL. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet. 2011;204:351–65.
Sorensen PHB, Lynch JC, Qualman SJ, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol. 2002;20:2672–9.
Storlazzi CT, Mertens F, Nascimento A, et al. Fusion of the FUS and BBF2H7 genes in low grade fibromyxoid sarcoma. Hum Mol Genet. 2003;12:2349–58.
Szuhai K, Ijszenga M, de Jong D, Karseladze A, Tanke HJ. Hogendoorn PC The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res. 2009;15(7):2259–68.
Sumegi J, Streblow R, Frayer RW, Dal Cin P, Rosenberg A, Meloni-Ehrig A, et al. Recurrent (2;2) and (2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator (NCOA) family. Genes Chromosomes Cancer. 2010;49:224–36.
Tanas MR, Sboner A, Oliveira AM, Erickson-Johnson MR, Hespelt J, Hanwright PJ, et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med. 2011;3(98):98ra82.
Taylor BS, Barretina J, Maki RG, Antonescu CR, Singer S, Ladanyi M. Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer. 2011;11(8):541–57.
Wang L, Motoi T, Khanin R, Socci N, Olshen A, Mertens F, et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer. 2012;51:127–39.
Williamson D, Lu YJ, Gordon T, et al. Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J Clin Oncol. 2005;23:880–8.
Xia SJ, Barr FG. Chromosome translocations in sarcomas and the emergence of oncogenic transcription factors. Eur J Cancer. 2005;41:2513–27.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Lindeman, N.I., Cin, P.D. (2013). Molecular Pathology of Soft Tissue and Bone Tumors. In: Cheng, L., Eble, J. (eds) Molecular Surgical Pathology. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-4900-3_15
Download citation
DOI: https://doi.org/10.1007/978-1-4614-4900-3_15
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-4899-0
Online ISBN: 978-1-4614-4900-3
eBook Packages: MedicineMedicine (R0)