
Abstract
All cell types from the testis can give rise to neoplasms. It is of relevance to distinguish germ cell tumors and non-germ cell tumors. Understanding the existence of the various types of neoplasms of the testis, and getting insight into their pathogenesis, requires knowledge about the normal anatomy and physiology of both the developing and mature testis. The germ cell tumors represent the most frequent neoplasm, followed by the sex cord–stromal tumors (Leydig cell and Sertoli cell tumors), and others (lymphoma, etc.). Based on morphological criteria, germ cell tumors are subdivided into (classic) seminoma, spermatocytic seminoma, embryonal carcinoma, yolk sac tumor, teratoma, and choriocarcinoma. Alternatively, a classification system has been proposed, in which age at presentation, pattern of genomic imprinting, cell of origin, chromosomal constitution, as well as representative animal model(s) are included. Within the testis, three of these subtypes can be found, each with their own clinical and pathological characteristics. Based on this recognition, an informative set of diagnostic markers has been developed, proven to be of value in a clinical setting. These can even be applied for early diagnosis, i.e., in the pre-invasive stage, in specific groups of individuals known to be at risk. This will possibly prevent the need for systemic treatment, irradiation, or chemotherapy at a relatively young age, known to have significant side effects.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction to Normal Testis
The testis is a complex organ with multiple functions, including generation of [mature] germ cells (spermatogenesis) and hormone production (i.e., testosterone). The first is dependent on the second, although visa versa is not the case. In other words, testis might be completely functional regarding formation of hormones, in spite of a complete lack of germ cell formation and production, resulting in infertility. To allow these processes to occur at the proper time and place, various cell types and structures are required. Most of them are initiated during early embryogenesis, while they further develop at different time points, even up to adult life. The most obvious structures within the testis are the seminiferous tubules and the interstitial space. These compartments contain specific types of cells, dependent on age in various stages of maturation.
-
Testicular functions, i.e., germ cell formation and hormone production, are regulated by a highly sophisticated network of specific cell types in distinct compartments (see Fig. 12.1a). In the interstitial compartment, i.e., the stromal space in between the seminiferous tubules, different cell types and (microscopic) structures are present, from which only the Leydig cells are testis specific. The Leydig cells produce androgens (testosterone) when stimulated by luteinizing hormone (LH) produced by the pituitary gland (see Fig. 12.1b). In addition, INSL3 is formed, required for the first phase of testicular descent. Other cells and microscopic structures of the interstitial compartment include vascular structures, fibroblasts, macrophages, and lymphocytes
Fig. 12.1 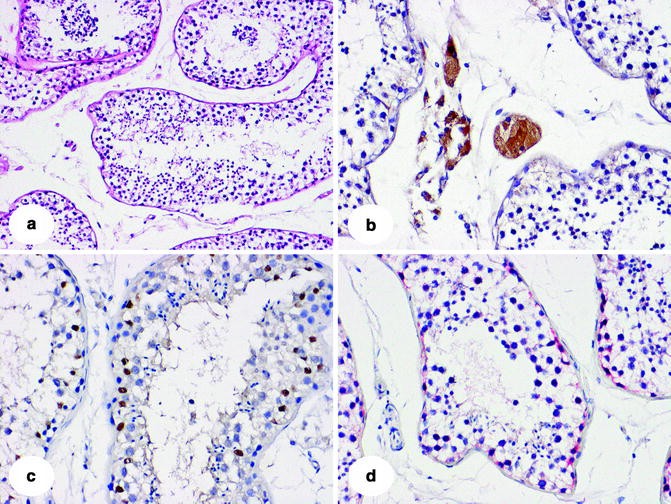
Representative examples of normal adult testis histology, including: (a) hematoxylin and eosin (H&E), 100×; (b) LH-R, (c) SOX9, (d) TSPY, all 200×. The immunohistochemical markers used are informative to identify the germ cells in the stage of spermatogonia, sertoli cells, and Leydig cells, respectively
-
The intratubular compartment is separated from the interstitial space by a highly organized barrier composed of both cells (e.g., peritubular cells) and extracellular matrix components (basal lamina). Within the seminiferous tubule in principle two types of cells are present under physiological conditions. These are the Sertoli cells and the germ cells (see Fig. 12.1c, d). The Sertoli cells are nursing the germ cells, from the initial embryonic phase to the mature spermatozoa. In the adult (postpubertal) testis, the germ cells present, i.e., spermatogonia, undergo a process of both mitosis and meiosis, including defined steps of further maturation. The spermatogonia are situated at the inner side of the basal lamina, under the tight junctions formed by the Sertoli cells. The developmental stages that follow are spermatocytes (undergoing meiosis) and spermatids, and finally spermatozoa. This process of mitosis and meiosis, followed by spermiogenesis, is highly dependent on production of androgens. Because of the epidemiological characteristics, this chapter will focus on germ cell tumors (GCTs), although at the end some characteristics of sex cord–stromal tumors will be mentioned
-
The majority of GCTs do not arise from adult germ cells, as found after puberty, but from early (embryonic) germ cells blocked in their normal maturation during fetal development (see Fig. 12.2). The only exception is the rare type III GCT (spermatocytic seminoma) (see below). The prerequisite for development of a type I or type II GCTs is thus the escape from the strictly regulated maturation process from an embryonic germ cell to a (pre-)spermatogonium
Fig. 12.2 
Schematic representation of normal germ cell development (in black) and malignant germ cell development (in red). Embryogenesis starts with fertilization, and generation of pluripotent stem cells, referred to as embryonic stem cells (ES), which are responsible for formation of the various differentiation lineages (both somatic and extra-embryonic). All these cells have a biparental pattern of genomic imprinting (GI) (represented in orange), resulting from a pure male- (blue) and female- (red) mature germ cell. The primordial germ cell (PGC) erases this biparental pattern, which in the male differentiation lineage in fully becoming paternal, via the stages pre-spermatogonia (A,B), primary and secondary spermatocyte and spermatid leading to fully matured sperm. The types of germ cell tumors originate from different stages of stem cell/germ cell development, representing their developmental potential. Type I are the teratomas and yolk sac tumors found in neonates and infants, Type II are the seminomas and nonseminomas diagnosed in adolescents and young adults, and the Type III are the spermatocytic seminomas, predominantly occurring in elderly males. The timing of birth, start of meiosis, and puberty are indicated
-
Primordial germ cell (PGCs) arise from the proximal epiblast
-
PGCs retain an intrinsic, although suppressed capacity to pluripotency
-
PGCs move along the hindgut to the developing genital ridges (to develop into testes in an XY chromosomal constitution)
-
Migration of PGCs is regulated by the stem cell factor (SCF/KITLG)–c-KIT pathway
-
PGCs lose their biparental pattern of genomic imprinting (erasement)
-
Germ cells entering the genital ridge are called gonocytes
-
During the second and third trimester of pregnancy, gonocytes mature into prespermatogonia
-
Prespermatogonia lose expression of embryonic germ cell markers
-
Proper maturation is required for spermatogenesis
-
Spermatogenesis is activated by androgens at puberty
-
Gonocytes can be delayed or blocked in the maturation process in a suboptimal microenvironment (i.e., cryptorchidism)
-
Presence of germ cells with embryonic (PGC/gonocyte-like) characteristics after the first year of life indicates a maturation defect
-
Delayed/blocked gonocytes can survive in postnatal testis
-
Blocked gonocytes can transform and progress to neoplasm(s)
-


Introduction to Germ Cell Tumors
Germ Cell Tumors: Classification of Germ Cell Tumors (Types I–III)
In principle, all cells of the testis can give rise to neoplasms. This results in the fact that in the testis an enormous variety of histological variants of tumors can be observed, significantly influenced by age, amongst others. Overall, it is of relevance to distinguish two main subgroups: GCTs and non-GCTs. Again within these categories, a large numbers of histological subgroups can be distinguished. Although officially incorrect, the GCTs of the testis are often referred to as testicular cancer, mainly based on epidemiological criteria; they are the most frequent type of neoplasm of this organ. Understanding the existence of the various types of neoplasms of the testis, and to get insight into their pathogenesis, requires knowledge about the normal anatomy and physiology of both the developing and mature testis. This resulted in novel information about the origin of especially the various types of GCTs, and identification of informative diagnostic markers. The GCTs represent the most frequent neoplasm, followed by the sex cord–stromal tumors (Leydig cell and Sertoli cell tumors), and others (lymphoma, etc.). Based on morphological criteria, GCTs are subdivided into (classic) seminoma, spermatocytic seminoma, embryonal carcinoma, yolk sac tumor, teratoma, and choriocarcinoma. These can be either pure or (inter)mixed in composition.
-
In contrast to the histological description of GCTs, on which all pathological classification systems are based, an alternative is presented. This has been appreciated by the World Health Organization as well as specialized pathologists in the field. According to developmental potential, cell of origin, age at clinical presentation, pattern of genomic imprinting, and molecular characteristics, GCTs can be classified into five entities (type I–V) (see Fig. 12.2), each with their own pathogenesis and pattern of (identified) risk factors. In this chapter, only the type I–III GCTs will be discussed, as they can be found in the testis. The type IV GCTs (dermoid cyst of the ovary) and type V GCTs (hydatidiform mole of placenta) are discussed elsewhere
Type I (Pediatric) Germ Cell Tumor
-
Clinical features
-
Tumor of predominantly neonates and infants (incidence of testicular GCT is 1–2 cases per million person-years)
-
First peak of incidence in first 2 years (mean age, 20 months for teratoma), second peak at age of 10
-
Can be found in sacrococcygeal region, retroperitoneum, intracranial, and mediastinum
-
Mostly benign (in contrast to type II teratomas, see below)
-
Malignant progression can occur, leading to yolk sac tumor (malignant)
-
No risk factors have been identified so far and no increase in incidence in the general population have been reported
-
No familial predisposition seems to be significant
-
-
Gross and microscopic features
-
Teratoma
-
Most common GCT in pediatric population
-
Heterogeneous, mixed cystic and solid mass with gray or brown cut surface
-
Mixture of differentiated (mature) somatic tissue, possibly containing (immature) neuroepithelial structures
-
Derivates of all three germinal (somatic) layers might be present (pluripotent)
-
Cartilage, and fetal mesenchymal tissue is often observed
-
Immature neural tissue might be intermixed in various quantities and include nests glands and tubules lined by immature embryonal-like cells with high mitotic activity
-
Malignant transformation to yolk sac tumor is possible
-
Microscopic foci of yolk sac tumor (<3 mm) do not affect prognosis
-
Patients with multiple large foci of yolk sac tumor might require additional chemotherapy (depending of the tumor stage)
-
Can histologically not be distinguished from type II teratoma (see below)
-
-
Yolk sac tumor
-
Can be primary malignancy in the testis
-
Malignant progression to yolk sac tumor can occur in teratoma (might be mixed)
-
Can histologically not be distinguished from type II yolk sac tumor (see below)
-
-
-
Precursor lesions and cell of origin
-
Type I GCT, both teratoma and yolk sac tumor, arise from early immature embryonic stem or germ cells. This is supported by:
-
Pattern of genomic imprinting—similarly to embryonic stem cells or early PGCs (biparental or partially erased)
-
Mouse teratoma models histologically represent type I GCTs: Pten-knockout mouse, Ap2gamma-knockout mouse, Fhit-knockout mouse, p53-knockout mouse, Kit-ligand deficient mouse in the 129/Sv background
-
-
No precursor lesions of type I GCT are histologically identified yet
-
-
Molecular features
-
Type I teratomas show normal chromosomal content (diploidy). In contrast, type I yolk sac tumors are aneuploid, with recurrent chromosomal changes including:
-
Loss of part(s) of 1p, 4, 6q
-
Gain of part(s) of 1q, 12p13, 20q, and 22
-
No genetic mutations have been found so far (teratoma and yolk sac tumor)
-
-
Type II (Adult) Germ Cell Tumors
-
Clinical features
-
Incidence
-
Account for 60% of all malignancies diagnosed in Caucasian males between 20 and 40 years of age
-
Incidence of 6–11 per 100,000, although dependent of Ethnic background
-
Asian and Blacks significant lower incidence, not influenced by migration
-
Plateau in rise related to World War II
-
Highest incidence in the northern European countries (Denmark, Germany, Norway, Sweden)
-
Significant rise in incidence during last decades (3–6%)
-
Family predisposition involved
-
No high penetrance cancer susceptibility gene identified
-
Nonseminomas develop earlier than seminomas (median age, 25 vs. 35 years)
-
In immunocompromised patients (HIV) seminoma present clinically at the age of nonseminoma
-
Bilateral tumors occur in up to 5% of the patients (synchronous or metachronous)
-
Can also be found in retroperitoneal region, intracranial site and mediastinum
-
-
Known risk factors
-
Are associated with the aberrant germ cell maturation during the fetal development
-
Clinical predisposition is related to the testicular dysgenesis syndrome (TDS)
-
TDS includes a spectrum of disorders of the male reproductive system including cryptorchid testis, hypospadias, microlithiasis, sub- or infertility; widely accepted view on its pathogenesis is that environmental endocrine disrupting chemicals act on Leydig cells and/or testicular Sertoli cells, resulting in abnormal development of the testis
-
-
Specific types of disorder of sex development (DSD)
-
DSD (previously intersex) is defined as a congenital condition in which development of a chromosomal, gonadal, or anatomical sex is atypical. The risk for type II GCTs is specifically related to hypovirilization and gonadal dysgenesis, related to presence of part of the Y chromosome (likely TSPY as candidate)
-
-
Genome-wide association studies have implicated single nucleotide polymorphism (SNPs) related to SCF (KITLG) DMRT1, SPRY4, HTERT/CLPM1L, ATF7IP, and BAK1 genes as risk modifiers
-
Low and high birth weight suggested to be associated with increased risk
-
Most likely a combined action between genetic and environmental factors is the most important determinant in risk determination, referred to as GENVIRONMENT
-
-
Prognosis
-
Type II GCTs are malignant neoplasms, with a high tendency to metastasize to retroperitoneal lymph nodes and different other organs, influenced by histological composition. In spite of this, they overall show a good prognosis. In patients with metastasized disease, three prognostic groups are identified: good, intermediate, and poor
-
Overall 10-year survival rate over 90%
-
Radiotherapy is effective for metastatic seminoma
-
Worse prognosis in patients with disseminated choriocarcinoma
-
Late relapses (after 2 years) can occur in nonseminomas, often with worse prognosis
-
Most patients with metastasized disease cured using cisplatin-based chemotherapy
-
Sensitivity to DNA-damaging agents (including irradiation and chemotherapy) supposed to be multifactorial related to the embryonic germ cell origin. Embryonal stem cells are sensitive due to lack of DNA repair mechanisms combined with no G1 arrest checkpoint. In addition, low level for apoptosis induction of germ cells is involved in preventing transmission of mutated DNA to the next generation
-
-
Retroperitoneal lymph node dissection might be indicated
-
Long-term side effects of the chemo(radio)therapeutic treatment are common, including subfertility, fatigue, cardiovascular complications, metabolic syndrome, and, less frequently, secondary cancer
-
-
Serum markers
-
Three principal tumor serum markers for type II GCTs are available and used according to the guidelines for primary diagnosis, staging, monitoring of therapeutic response and followup
-
Alpha-fetoprotein (AFP) (half-life of 4.5 days) is elevated in up to 70% of patients. Predominantly generated by the yolk sac component
-
Beta subunit of the human choriogonadotropin (HCG) (half-life of 24–36 h) elevated in 50% of patients. Predominantly generated by the choriocarcinoma component
-
Marijuana usage can result in false positive hCG finding
-
Lactate dehydrogenase (LDH1), being less specific. Elevated in 40–60% of patients
-
If these tumor markers do not decline expected based on half-life after treatment, residual disease is likely
-
Normal level of the markers does not prove absence of disease (only 40–50 and 30% of relapses in patients under active surveillance for clinical stage I disease and after systemic chemotherapy are associated with marker increases)
-
-
-
-
Gross, microscopic, and immunohistochemical features by specific histological type
-
Histologically, different variants of type II GCT can be identified. These are subdivided into seminoma and nonseminoma. According to the British Classification, a combined tumor is composed of both a seminoma and nonseminoma component, referred to as nonseminoma to the other classification systems. Nonseminoma defines a cancer with the following possible histological types: embryonal carcinoma, teratoma, yolk sac tumor, and choriocarcinoma (with or without a seminoma component). Overall, about 40% of type I GCTs are seminomas and 60% nonseminomas. Nonseminoma show mostly a mixture of different histological components
-
The different histological components are described below in more detail
-
Seminoma (see Fig. 12.3)
Fig. 12.3 
Representative examples of an invasive seminoma, stained with (a) H&E, and the immunohistochemical markers (b) OCT3/4, and (c) SOX17
-
Solid tumors with gray, white, or pink surface
-
Microscopically, the tumor consists of sheets or lobules separated by fibrous septa with lymphoid infiltrate
-
Round to polygonal tumor cells with clear or eosinophilic cytoplasm
-
Nuclei are central and contain prominent nucleoli
-
Epithelioid cells, Langhans giant cells, or sarcoid-like granulomas can be present
-
Up to 25% contain syncytiotrophoblastic giant cells
-
Atypical seminoma show a greater degree of polymorphism and a higher mitotic rate; however the clinical significance of this subtype is doubtful
-
There is nuclear staining for markers of undifferentiated germ cells, including OCT3/4, SALL4, NANOG, AP2gamma (>90% of tumor cells are positive)
-
OCT3/4 is nowadays the most sensitive and specific marker for seminoma (as well as for CIS and embryonal carcinoma), always nuclear in localization
-
Variable membranous staining for markers of germ cell differentiation, including CD117 (c-KIT), D2-40, as well as for placental alkaline phosphatase (PLAP)
-
In contrast to embryonal carcinoma, (see below) no expression of EMA in seminoma
-
Immunohistochemistry for cytokeratins can be positive without clinical impact
-
SOX17 is positive in seminoma and can differentiate seminoma from embryonal carcinoma, which is SOX17 negative. However, normal PGCs and gonocytes as well as spermatogonia are positive as well
-
-
Embryonal carcinoma (see Fig. 12.4)
Fig. 12.4 
Representative example of a mixed nonseminoma, containing an embryonal carcinoma and yolk sac tumor component, stained with (a) H&E, and the immunohistochemical markers (b) OCT3/4, and (c) SOX2
-
Solid tumor with gray to pink appearance and foci of hemorrhage and necrosis
-
Growth pattern varies from solid to papillary and syncytial
-
Typical epithelial-like cells with large irregular nuclei
-
Mitotic figures are frequent
-
Syncytiotrophoblastic giant cells might be scattered
-
There is a nuclear (and cytoplasmic) staining for markers of undifferentiated germ cells, including OCT3/4, SALL4, NANOG (>90% of tumor cells are positive)
-
OCT3/4 also shows both a nuclear and cytoplasmic localization
-
Membranous staining for CD30, EMA, and PLAP
-
Markers differentially expressed in embryonal carcinoma vs. seminoma are SOX2, CD30 (exclusively positive in embryonal carcinoma), and EMA of SOX17 (positive in seminoma)
-
Vascular invasion is often the result of embryonal carcinoma
-
-
Yolk sac tumor (see Fig. 12.4)
-
Solid soft tumors, gray-white to yellow surface
-
Necrosis may be present
-
There are numerous pattern of differentiation: microcystic, macrocystic, endodermal sinus, papillary, glandular, solid, polyvesicular, vitelline, hepatoid, myxoid, parietal pattern
-
Various pattern are usually admixed in one tumor
-
Foci of yolk sac tumor are frequently seen in nonseminomas
-
Pure yolk sac tumors are rare
-
AFP and glypican 3 are variably expressed in yolk sac tumors and can be informative to discriminate from other components and cancers
-
Combination of these markers can increase sensitivity for the detection
-
Low molecular weight cytokeratins are positive in yolk sac tumors
-
-
Choriocarcinoma
-
Tumor represents as nodules with hemorrhage
-
Composed of trophoblast-like cells and syncytial large cells
-
Frequently present in nonseminomatous
-
Rare in a pure form (<0,1% of all TGCT)
-
Pure choriocarcinoma is likely to present as a highly aggressive disease with hematogenous metastases
-
Syncytiotrophoblasts are positive for beta-HCG, inhibin (alpha subunit), and EMA
-
Cytokeratin is expressed in trophoblasts and syncytiotrophoblasts
-
-
Teratoma
-
Malignant tumor showing somatic differentiation with endoderm, ectoderm, and endoderm derivates
-
Teratomatous component is often present in nonseminomas
-
Various somatic malignancies might arise in the background of teratoma including sarcoma, PNET, carcinoma, and nephroblastoma
-
No specific immunoprofile, the differentiated areas of teratoma show immunophenotype which is in accordance with the underlying cell type
-
AFP can be expressed in intestinal or hepatoid areas
-
Intratesticular epidermoid cyst represents a rare benign teratoma in the adult and should not be mixed up with the type II malignant teratoma
-
This benign teratoma shows in contrast to the malignant type II teratoma no CIS (see below) in the adjacent testis. Careful examination of the seminiferous tubules should be done under usage of the immunohistochemistry for OCT3/4 (or another marker for CIS). Detection of CIS supports a malignant type II teratoma
-
-
-
-
Precursor lesions and cell of origin (see Fig. 12.5)
Fig. 12.5 
Representative examples of a seminiferous tubules containing carcinoma in situ (CIS) cells and intratubular seminoma cells, stained with (a) H&E, and the immunohistochemical markers (b) OCT3/4, and (c) stem cell factor (KITLG)
-
The precursor of all type II GCTs of the testis is the so-called carcinoma in situ (CIS) of the testis, also referred to as intratubular germ cell neoplasia unclassified (IGCNU) or testicular intraepithelial neoplasia (TIN). It is expected that all patients with CIS will eventually develop an invasive cancer (in the prospective study, 70% of the patients with CIS developed an invasive GCT within 7 years)
-
CIS cells are located at the inner side of the basal lamina of the seminiferous tubule, most frequently in a single row in close connection with Sertoli cells, under their interconnecting tight junctions
-
CIS is often present in the adjacent parenchyma of invasive type II GCTs, especially nonseminomas
-
Activated immune system as found in seminomas can also eradicate CIS
-
PGCs and CIS cells share the same pattern of genomic imprinting (erased), telomerase activity, and gene and protein expression profile
-
CIS shows homogeneous expression of markers of PGCs/gonocytes, including c-KIT, BLIMP1, AP2gamma, OCT3/4, NANOG, LIN28 (and many more)
-
OCT3/4 is the most specific and sensitive marker for CIS, strongly staining the nucleus of all CIS cells, but not normal spermatogonia
-
The CIS counterpart in dysgenetic gonads with a low level of virilization (i.e., no or limited testicular differentiation) is known as gonadoblastoma
-
Gonadoblastoma is composed on CIS-like cells intermixed with stromal cells expressing FOXL2 (Granulosa differentiation), whereas Sertoli cells (SOX9 positive) are associated with CIS. Gonadoblastoma can mimic CIS, to be differentiated by SOX9 and FOXL2. Presence of gonadoblastoma is proof for the presence of DSD
-
-
Overdiagnosis of CIS is possible due to germ cell maturation delay. This can be avoided using immunohistochemical detection of SCF (KITLG)
-
In a testicular biopsy taken during the first year of life in an individual with possible germ cell maturation delay (e.g., cryptorchidism), overdiagnosis is possible. Distinguishing morphological criteria are not strictly informative; immunohistochemistry with OCT3/4 is also discriminatory. It can be solved using immunohistochemistry for SCF, specifically present in the premalignant cells
-
-
No informative animal model has been identified yet
-
-
Molecular features
-
Chromosomal constitution
-
Seminomas and CIS are hypertriploid and the nonseminomas hypotriploid type II GCT show various losses and gains of (parts of) chromosomes: loss of chromosomes 4, 5, 11, 13, 18, and Y and gain of chromosomes 7, 8, X, and 12
-
Yolk sac tumors show recurrent chromosomal imbalances, including loss of 1p, 4, and 6q, and gain of 1q and 20q
-
All invasive tumors show gain of 12p, mostly due to formation of isochromosomes (i12p). Regional high level amplification can also be observed. No obvious candidate gene(s) has been identified so far, although various have been suggested (CCND2, NANOG KRAS2, etc.)
-
Studies indicate that gain of 12p is progression related (occurs when CIS cells become independent of interaction with Sertoli cells). Cyclin D2 (CNND2) is expressed in all type II GCTs, as well as in CIS, while NANOG is expressed in CIS as well as seminoma and embryonal carcinoma. It is most likely that multiple genes located on 12p are relevant in the pathogenesis. It matches with the observation that gain of 12p can be found in extended in vitro cultures of human embryonic stem cells
-
-
X chromosome is gained in the majority of TGCT
-
Familial predisposition of type II GCT has been linked to the X chromosome. Additional X chromosome is also relevant in the context of Klinefelter syndrome patients, although these patients only develop mediastinal type II GCTs (not testicular). A role of the X chromosome might also be suggested from data of patients with specific forms of DSD. Supernumerical X chromosomes are inactivated in nonseminomas by methylation. This, in parallel to normal embryogenesis, result from function of the non-(protein)-coding XIST gene. This phenomenon is correlated with hypomethylation of the promoter region, reported to be useful as molecular target for this type of cancer
-
-
-
Epigenetic modifications
-
CIS and seminomas show a hypomethylated DNA status, in contrast to the various histological types of nonseminomas, this parallels normal embryogenesis
-
Histone modification proteins BLIMP1 and PRMT5
-
The complex of the transcription factor BLIMP1 and protein arginine methyltransferase-5 PRMT5 protein is expressed in CIS and seminoma. Proposed function of BLIMP1/PRMT5 complex is suppression of premature differentiation and maintenance of pluripotency in PGCs/gonocytes by dimethylation of H2A/H4 at arginine 3 and repression of gene expression (see above). It suggests that histone H2A and H4 arginine 3 dimethylation suppress differentiation of CIS and seminoma, while loss of these histone modifications might induce reprogramming and differentiation to embryonal carcinomas and the various subtypes of differentiated nonseminomas
-
-
-
Embryonal stem cell genes
-
Expression pattern of mRNA in seminoma and embryonal carcinoma shows high levels of mRNA of genes related to the pluripotency (OCT3/4, NANOG, LIN28), in close similarity to the PGCs/gonocytes and embryonal stem cells
-
In embryonal stem cells, pluripotency is regulated by interaction of POU5F1 (OCT3/4) with a member of the SOX family. Studies in cell lines derived from seminoma and embryonal carcinoma provide evidence that the functional partner of POU5F1 in seminomas is SOX17, and in embryonal carcinomas SOX2. The role of POU5F1/SOX17 complex is likely not in regulation of pluripotency but in prevention from apoptosis
-
-
Two specific variants of the protein encoding OCT3/4 are recognized, of which the A (or I) type is a nuclear protein and is related to pluripotency
-
The B (or II) variant is localized in the cytoplasm and is not related to regulation of pluripotency. Detection of OCT3/4 mRNA is hampered by existence of two variants as well as the presence of pseudogenes. This may result in false positive RT-PCR observations in variety of non-GCTs
-
-
Expression pattern of NANOG is similar to OCT3/4
-
-
Involvement c-KIT (CD117)
-
Receptor tyrosine kinase
-
c-KIT is expressed mainly in CIS and also, but less, in seminoma, but not in embryonal carcinoma
-
c-KIT is downregulated during the progression of CIS to seminomas
-
Amplification of c-KIT is found in a selected number of seminomas
-
Activating gene mutations in exon 17 are detected mostly in (bilateral) seminomas
-
It is supposed that activation of c-KIT in early germ cells during fetal phase of germ cell differentiation can potentially lead to survival of immature germ cells in the niche of spermatogonia. It has also been proposed that c-KIT plays a role in the initiation of the germ cell malignancy but might not be relevant for the further steps of progression
-
-
-
Mutational status
-
GCTs show overall an exceptional low mutation rate of genomic DNA
-
This is supported by mutation analysis of individual genes as well as by high-throughput investigation on the mutation status of the kinome. Various deep sequencing projects are currently undertaken. The uniqueness of this low mutation rate is likely to be (again) related to the embryonic cell of origin. Embryonic stem cells keep one of the two DNA strands protected against any form of mutations, the so-called immortal DNA strand. This reduces the probability of transmitting of the DNA anomalies to the next generation
-
-
BRAF and microsatellite instability
-
Overall, type II GCTs, with the exception of teratomas, show an exceptional sensitivity to DNA damaging agents. However, not all nonteratomatous elements are sensitive to cisplatin-based chemotherapy. Various putative mechanisms for resistance are proposed based on limited studies and number of cases. Intriguing findings are the role of microsatellite instability (MSI), BRAF mutations, disturbed apoptotic, etc. MSI is found in about 30% of the refractory cancers, in a significant number related to hypermethylation of the promoter region of hMLH1. Interestingly, this seems to be partially overlapping with activating mutations within the BRAF oncogene (V600E) in all patient groups
-
-
-



Type III Germ Cell Tumors (Spermatocytic Seminoma)
-
Clinical features
-
Rare tumor, up to 4% of all GCT of the testis
-
Incidence 0.4 per 1,000,000
-
Occur in older male compared to type II GCT, average age 52 years
-
Most tumors are unilateral
-
Bilateral tumors can occur
-
Serum markers (AFP, HCG, LDH) are negative
-
Metastases from a pure spermatocytic seminoma are very rare
-
Sarcomatoid dedifferentiation rarely occurs within a spermatocytic seminoma and is associated with a progressive disease
-
No association with cryptorchidism
-
Excellent prognosis with surgery alone
-
-
Gross, microscopic, and immunohistochemical features (see Fig. 12.6)
Fig. 12.6 
Representative example of a spermatocytic seminoma, stained with (a) H&E and (b) DMRT1
-
Soft tumor with mucoid grayish-white cut surface, and friable texture
-
Background of edematous stroma
-
Tumor cells are of various size: large eosinophilic cells, small dark cells, and mono- or multinucleated giant cells
-
High mitotic activity
-
CIS/IGCNU/TIN is not present
-
Specific precursor lesions might be present (so-called intratubular spermatocytic seminoma in situ)
-
Immunohistochemical markers of classic seminoma (OCT3/4, PLAP, c-KIT) are negative
-
Spermatogonial markers DMRT1, OCT2, SSX2–4, and SAGE1 are positive in spermatocytic seminoma (the last suggesting a heterogeneous origin)
-
In the histogenetic model, spermatocytic seminoma arise from mature spermatogonia or spermatocytes
-
-
-
Molecular features
-
Gain of chromosome 9 is consistent
-
DMRT1 is an interesting 9p gene
-
Paternal pattern of genomic imprinting
-
Many testis cancer antigens and genes related to spermatogenesis are expressed
-
HRAS and FGF3 genes are frequently mutated, especially in the cases in elderly men
-
The canine seminomas are the animal model for type III GCTs
-

Sex Cord/Gonadal Stromal Tumors
-
Sex cord tumors are rare; these tumors constitute about 4% of all testicular neoplasms. Metastases can occur, mainly in adult patients, but no histological or molecular prognostic markers had been found so far to predict the clinical behavior
Leydig Cell Tumors
-
Clinical features
-
Two age peaks, in children mostly between 5 and 10, and in adults
-
Hormone producing tumors: testosterone, androstenedione
-
Serum estrogen level might be elevated
-
In children, clinical presentation might be pubertas praecox or/and gynecomastia
-
Adults frequently present with testicular mass, 30% develop also gynecomastia
-
Bilaterality is rare
-
10% of Leydig cell tumors are malignant
-
Malignant tumors do not respond to chemotherapy or radiation
-
-
Gross, microscopic, and immunohistochemical features
-
Solid, yellowish tumor
-
Well circumscribed, sometimes nodular
-
Average tumor size 2–5 cm
-
Necrosis and extratesticular extension might occur
-
Typical histologic features include sheets of eosinophilic cells with distinct cell borders
-
Reinke crystals are pathognomonic and seen in up to 40% of the cases
-
Pseudoglandular and microcystic pattern can be seen
-
Lipomatous changes might occur
-
Malignant behavior correlates with the size (>5 cm), higher mitotic rate (>3 mitotic figures per 10 high power fields), necrosis, vascular invasion, invasion in the neighboring structures, and high proliferative activity
-
Tumors are positive for vimentin, inhibin, and LH-R
-
Sertoli Cell Tumors
-
Clinical features
-
Very rare tumors (<1% of all testicular neoplasms)
-
Mean age at the time of the diagnosis 45 years
-
Some tumors are associated with genetic syndromes
-
Large cell calcifying Sertoli cell tumor (LCCST) can be sporadic or associated with Carney and Peutz–Jeghers syndromes
-
Estrogen production might lead to gynecomastia
-
Most Sertoli cell tumors are benign
-
-
Gross, microscopic, and immunohistochemical features
-
Solid tumor, gray-whitish
-
Nests, tubules, sheets, mostly bland cytology
-
Frequently, stromal hyalinization is present
-
LCCST present with nests of large eosinophilic cells embedded with hyaline stroma (with calcifications in some cases)
-
In Peutz–Jeghers syndrome, multifocal bilateral Sertoli cell proliferation occur mostly in young patients
-
Immunohistochemically, Sertoli cell tumors are positive for inhibin, cytokeratin, vimentin, and SOX9
-
Granulosa Cell Tumors
-
Clinical Features
-
In similarity to the ovary, juvenile and adult types are distinguished
-
Adult type is rare, metastases occur in up to 20% of the patients
-
Juvenile tumors occur mostly in maldescended testis, in young children
-
-
Gross, microscopic, and immunohistochemical features
-
Adult type granulosa cell tumors
-
Solid and well circumscribed, with varying size
-
Several histological pattern can occur, including solid, trabecular, insular, microfollicular
-
Typically, microfollicles surround eosinophilic material (Call–Exner bodies)
-
Tumor cells show grooved nuclei
-
Tumor cells are reactive for vimentin, inhibin, smooth muscle actin
-
-
Juvenile granulosa cell tumors
-
Often cystic
-
Solid and follicle-like zones are admixed
-
Follicles lined by stratified epithelium
-
Tumor cells are reactive for vimentin, inhibin, smooth muscle actin, and focally to anti-Müllerian hormone
-
-
-
Molecular features of sex cord/gonadal stromal tumors
-
Activating mutations in LH-receptor had been detected in pediatric Leydig cell tumors
-
Most ovarian adult type granulosa cell tumors harbor a somatic missense mutation in the FOXL2 gene, but no information is available yet for the testicular granulosa cell tumors
-
Summary of Molecular Pathology of Testicular Cancer
-
Neoplasms of the testis comprise a heterogeneous group of tumors, of which the majority is of the germ cell origin. GCTs of the testis can be classified by the cell of origin, as well as the molecular findings into three main groups
-
Type I GCT is a common neoplasm in the neonates and children and arises from very early germ cells, which are blocked in their differentiation. The most common type I GCT is teratoma, which is a benign tumor. Yolk sac tumor is a malignant type I GCT, developing from transformed early germ cells or, rarely, arises in a type I teratoma
-
Type II GCT are malignant GCTs with complex morphology and different histological subtypes, all arising from a common precursor lesions, the carcinoma in situ
-
Seminoma and embryonal carcinoma as well as carcinoma in situ share (partly) the same gene expression pattern and pattern of genomic imprinting as early fetal germ cells
-
Transition from the precursor lesion to an invasive cancer is associated with gain of the short arm of chromosome 12, in which multiple genes might be involved
-
Knowledge of cell of origin of type II GCT has led to the successful employment of OCT3/4 as a highly specific marker of CIS, seminoma, and embryonal carcinoma
-
Type III GCTs represent a benign lesion composed of germ cells in the maturation stage of spermatogonia/spermatocyte. They consistently show gain of chromosome 9, and are positive for a number of immunohistochemical markers, including DMRT1
Suggested Reading
Barksdale Jr EM, Obokhare I. Teratomas in infants and children. Curr Opin Pediatr. 2009;21:344–9.
Cheng L, Sung MT, Cossu-Rocca P, et al. LHJ: OCT4: biological functions and clinical applications as a marker of germ cell neoplasia. J Pathol. 2007;211:1–9.
Gilbert D, Rapley E, Shipley J. Testicular germ cell tumours: predisposition genes and the male germ cell niche. Nat Rev Cancer. 2011;11:278–88.
Goddard NC, McIntyre A, Summersgill B, et al. KIT and RAS signalling pathways in testicular germ cell tumours: new data and a review of the literature. Int J Androl. 2007;30:337–48.
de Jong J, Stoop H, Gillis AJ, et al. Differential expression of SOX17 and SOX2 in germ cells and stem cells has biological and clinical implications. J Pathol. 2008;215:21–30.
Oosterhuis JW, Looijenga LH. Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer. 2005;5:210–22.
Kollmannsberger C, Daneshmand S, So A, et al. Management of disseminated nonseminomatous germ cell tumors with risk-based chemotherapy followed by response-guided postchemotherapy surgery. J Clin Oncol. 2010;28:537–42.
Looijenga LH. Human testicular (non)seminomatous germ cell tumours: the clinical implications of recent pathobiological insights. J Pathol. 2009;218:146–62.
Oosterhuis JW, Stoop H, Dohle G, et al. A pathologist’s view on the testis biopsy. Int J Androl. 2011;34:14–9.
Rajpert-De Meyts E, Skakkebaek NE. Pathogenesis of testicular carcinoma in situ and germ cell cancer: still more questions than answers. Int J Androl. 2011;34:2–6.
Rajpert-De Meyts E. Developmental model for the pathogenesis of testicular carcinoma in situ: genetic and environmental aspects. Hum Reprod Update. 2006;12:303–23.
Stoop H, Honecker F, van de Geijn GJ, et al. Stem cell factor as a novel diagnostic marker for early malignant germ cells. J Pathol. 2008;216:43–54.
Ye H, Ulbright TM. Difficult differential diagnoses in testicular pathology. Arch Pathol Lab Med. 2012;136:435–46.
Woodward PJ, Heidenreich A, Looijenga LHJ, et al. Germ cell tumors. In: Eble JN, Sauter G, Epstein JI, editors. WHO classification of the tumors of the urinary system and male genital organs. Lyon, France: International Agency for Research on Cancer; 2004. p. 221–58.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Biermann, K., Looijenga, L.H.J. (2013). Molecular Pathology of Testicular Cancer. In: Cheng, L., Eble, J. (eds) Molecular Surgical Pathology. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-4900-3_12
Download citation
DOI: https://doi.org/10.1007/978-1-4614-4900-3_12
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-4899-0
Online ISBN: 978-1-4614-4900-3
eBook Packages: MedicineMedicine (R0)