
Abstract
Synthetic organic chemicals are produced by the transformation of carbonaceous feedstocks into functionalized molecules through one or more chemical reactions. Such transformations are accomplished at vast industrial scales and the resulting products permeate every aspect of modern society. The molecules produced find use largely as monomers for polymer synthesis of ubiquitous plastics, or as task-specific ingredients for a myriad of applications as divergent as paint leveling agents to food preservatives. Advances in technology, significant increases in energy efficiency, as well as the utilization of fossil-fuel derived starting materials has resulted in unprecedented economy of scale and relatively stable product costs in spite of large relative increases in the price of oil and natural gas. The section entitled “Chemical Raw Materials and Feedstocks” covers the most important carbonaceous feedstocks currently utilized in the chemical processing industries; all derived from fossil-fuel based raw materials.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Chapter Background
Synthetic organic chemicals are produced by the transformation of carbonaceous feedstocks into functionalized molecules through one or more chemical reactions. Such transformations are accomplished at vast industrial scales and the resulting products permeate every aspect of modern society. The molecules produced find use largely as monomers for polymer synthesis of ubiquitous plastics, or as task-specific ingredients for a myriad of applications as divergent as paint leveling agents to food preservatives. Advances in technology, significant increases in energy efficiency, as well as the utilization of fossil-fuel derived starting materials has resulted in unprecedented economy of scale and relatively stable product costs in spite of large relative increases in the price of oil and natural gas. The section entitled “Chemical Raw Materials and Feedstocks” covers the most important carbonaceous feedstocks currently utilized in the chemical processing industries; all derived from fossil-fuel based raw materials.
The volume of synthetic organic chemicals produced worldwide grew from less than 5 million metric tons in 1950 to more than 400 million metric tons in 2009, with an average annualized growth rate above 7% [1]. The value of all chemicals produced worldwide reached over $3.7 trillion dollars in 2008, before dropping somewhat to $3.4 trillion dollars during the 2009 recession as shown in Fig. 10.1. The average annualized worldwide growth rate in value of chemicals produced for the decade of 1999–2009 was 7.7% [2].

Global chemical output, $ Billions, 1999–2009
The United States has been the country with the largest chemical industry by volume and value for many years, due to inexpensive natural gas and very large consumer markets. However, this leadership position has deteriorated rapidly over the last decade with the dramatic growth in production of chemicals in China [2]. The value of all chemicals produced between 1999 and 2009 is shown in Fig. 10.2 for the traditional major chemical producing countries of the United States, Japan, and Germany in comparison to China. Average annualized growth rates in the value of chemicals produced over the decade for these countries are summarized in Table 10.1. The growth rate in value of chemicals produced in China far outpaces that in the United States. As a result, the US share of global chemical production value has dropped from 26% in 1999, to 20% in 2009, while that of China has rocketed from 5 to 19% in the same timeframe (Figs. 10.3 and 10.4).

Chemical output of selected countries, 1999–2009, $ Billions

Global chemical output, 1999, $ Billions

Global chemical output, 2009, $ Billions
The cost of a raw material can dramatically alter production patterns over a short period of time. Figure 10.5 shows chemical raw material usage of natural gas and annualized cost for natural gas for the years 1998, 2002, 2006 in the United States. After many decades near $2.00/million BTU, natural gas price in the United States began to rise and spiked dramatically in 2006 at above $9.50/MM BTU, leading to the shuttering of many older, small-scale marginal US facilities for the production of the ultra-commodities of ammonia-based fertilizers and methanol. As of 2007 only one US methanol facility based on coal, Eastman Chemical Company’s Kingsport plant, remained operational. Most of the US production moved overseas to the Middle East, Trinidad, and other regions with lower cost natural gas. Natural gas price since has dropped significantly in the US with the advent of substantial shale gas production.

U.S. usage of natural gas as chemical raw material vs. price, 1998–2006
Chapter Organization
The traditional approach to discussion of synthetic organic chemicals in most survey literature is to concentrate on families of derivatives from each specific chemical feedstock. Although many different chemicals can be made from each of the feedstocks, only a limited number of different types of chemistry have found particular favor in large-scale commercial production of synthetic organic chemicals. These include: (1) oxidation, (2) carbonylation, (3) hydroformylation, (4) chlorination, (5) condensation, (6) hydration/hydrolysis, (7) esterification, (8) hydrogenation, (9) dehydrogenation, (10) sulfonation, and (11) ammonation/ammoxidation. The first nine of these are covered in this chapter. Discussion of the last two is found in other chapters of this book, in Chaps. 14, Chaps. 22, and Chaps. 36. Cross references within Chapter 10 are noted by subsection titles in quotation marks.
Organizing our discussion of the significant commercial processes by these chemistry themes allows one to see the commonality between many seemingly diverse production processes with very different raw materials and end products. In some cases a chemical is made commercially by very different chemistries, but the processes begin with the same feedstock. Examples include: (1) the production of alcohols by the direct or indirect hydration of olefins, (2) the various propylene-based routes to propylene oxide, and (3) the production of methyl methacrylate from ethylene by either carbonylation/esterification/condensation or hydroformylation/oxidation/esterification steps. In other cases, the feedstock may be entirely different, but end at the same product, such as the acetylene, propylene, and butane/butene-based processes for the production of 1,4-butanediol. More often, and to the point of the chapter organization, the chemistry is similar in spite of significant feedstock differences, such as hydroformylation of C2-C5 olefins, oxidation of aldehydes to carboxylic acids, or aldol condensations of a wide variety of carbonyls.
Often one basic chemistry route has become commercially dominant, for example ethylene oxide production via silver catalyzed oxidation of ethylene, terephthalic acid (TPA) by Co/Mn/Br catalyzed oxidation of p-xylene, or methanol by hydrogenation of carbon monoxide/carbon dioxide. In these cases, the key differences between commercial processes are more subtle and may amount to alternative catalysts, reactor conditions and designs, or separation sequences. A portion of these differences are highlighted in the discussion below where pertinent, but the roles of reaction engineering and separation process synthesis/design cannot be adequately covered in the space allotted. Many books and articles have been written on these process design subjects, with a few recommended texts given below [3–14].
The capacity, production, and consumption figures presented throughout the section, “Chemical Raw Materials and Feedstocks”, for individual feedstocks and derivatives were derived from data gathered from a variety of literature sources, spanning the years 2006–2010 [15, 16]. Thus, some numbers reflect reduced production and consumption due to the 2008–2010 recession; some do not. The resulting variation in production/consumption numbers is estimated to be 5–10%.
The simplified flowsheets were drawn specifically for this chapter and are based on a compilation of open literature data (referenced in each subsection). The major processing steps of each flowsheet are named in italics and numbered. These designations appear as bolded numerals in parentheses in the corresponding process description in the text. Each process description includes a summary of temperature, pressure, yields, and selectivity, but open literature references can vary widely on the values.
Chemical Raw Materials and Feedstocks
Background
The formation of new a carbon–carbon bond is one of the more difficult chemical transformations to make. So the foundations of the modern chemical industry is very much a story of learning to exploit a small number of reactive skeletal carbon structures which can be derived from natural sources in relatively simple and efficient manner. In the early twentieth century the nascent chemical industry was based on utilization of coal tar liquids (a by-product of metallurgical coke production) and the fermentation of biologically derived feedstocks such as molasses, sugars, and whey. Coal tar liquids now provide a minor fraction of chemical feedstocks. Early fermentation routes, such as the ABE (acetone-butanol-ethanol) process, have all but disappeared due to poor economics. In spite of massive research efforts, newer bio-based processes have yet to compete favorably except in a few niche applications.
Rather, the substantial growth of the global chemical industry over the past 70 years would not have been possible without the concomitant rise in the fossil fuel energy infrastructure encompassing petroleum refining, natural gas production, and to a growing extent coal mining (primarily due to Chinese gasification projects). This close coupling of the chemical industry with the energy infrastructure is exemplified in the United States for the primary chemical raw materials of natural gas, petroleum products, and liquefied petroleum gases/natural gas liquids (LPG/NGL, C2-C4 hydrocarbons derived from crude natural gas and refinery gases). Figures 10.6, 10.7, and 10.8 show the relative magnitude of the total US energy consumption of these three key categories vs. their use as raw materials and energy sources for chemical production during the years 1998, 2002, and 2006. All quantities are expressed in trillions of BTUs of energy equivalent for easy comparison. Only LPG/NGL is used largely as a chemical raw material. Chemical raw material usages of natural gas and petroleum products represent just 1.6% and 4.5% of much larger power/heating and motor fuels markets respectively in the United States [17–19]. The situation is similar worldwide. Total global fossil fuel demand in 2007 was 282,000 trillion BTU’s. About 9%, 25,500 trillion BTUs, was used as chemical raw materials, with 78.9% from petroleum, 16.9% from LPG/NGL, 3.9% from natural gas, and 0.3% from coal [20].

U.S. natural gas usage, 1998, 2002, and 2006

U.S. LPG/NGL usage, 1998, 2002, and 2006

U.S. usage of refined petroleum materials, 1998, 2002, and 2006
An overwhelming majority of the synthetic organic chemicals produced at commercial scale today begin with one of five major types of feedstocks:
-
1.
Light olefins—ethylene and propylene
-
2.
Aromatics—benzene, toluene, xylenes, or BTX
-
3.
C4 hydrocarbons—butanes, butenes, butadiene
-
4.
Kerosene derived C9-C17 paraffins
-
5.
Synthesis gas—a mixture of carbon monoxide and hydrogen
Figure 10.9 shows a simplified overview of the raw materials, feedstocks, and derivatives of the synthetic organic chemical industry. Major chemical feedstock capacity, production, and consumption for the world, United States, and China are summarized in Table 10.2.

Chemical raw material-feedstock-derivatives overview
As will become apparent below, the production of chemical feedstocks from fossil fuel raw materials typically involve highly endothermic processes. A large amount of energy must be expended for both driving endothermic reactions as well as for separation and purification. The resulting high energy, reactive feedstocks allow subsequent chemistry to proceeds favorably downhill to lower energy products, with mostly exothermic reactions and relatively high yields.
C2-C3 Light Olefins [21–25]
Ethylene and propylene are by far the most important building blocks of the petrochemical industry. The primary derivatives produced from ethylene and propylene feedstocks, the capacity/production/consumption of those primary derivatives, and their major applications are summarized in Tables 10.3, 10.4, 10.5, 10.6, 10.7, and 10.8. The family of derivatives produced from ethylene and propylene are presented in Figs. 10.10 and 10.11, respectively. Light olefins can be produced from a variety of raw materials and methods:

Ethylene derivative tree

Propylene derivative tree
-
1.
Steam cracking (thermal pyrolysis) of hydrocarbon raw materials ranging from LPG/NGL to naphthas and gas oils
-
2.
Methanol to olefins
-
3.
Recovery from refinery gases and FCC (fluid catalytic cracking) gases
-
4.
Interconversion of butenes, ethylene, and propylene
-
5.
Dehydrogenation of propane (propylene only)
-
6.
Dehydration of bio-derived ethanol (ethylene only)
Steam cracking accounts for almost all of the ethylene and about 60% of the propylene produced worldwide. A simplified diagram of a naphtha-based cracker is shown in Fig. 10.12. In a thermal cracking unit, steam and the hydrocarbon raw material are heated in a short residence time tubular reactor/furnace (1) to 775–950°C, 0.17–0.24 MPa pressure, to initiate free radical cision, decomposition, isomerization, and aromatization reactions. The reactions are highly endothermic, requiring +1,600 to +2,800 kJ/kg of hydrocarbon fed, or roughly +55 to +219 kJ/gmole of ethylene/propylene produced. The yield of ethylene vs. propylene is highly dependent on the hydrocarbon raw material used as well as the severity of conditions. Ethylene production is favored by use of light hydrocarbons and higher temperature conditions. Typical yields for several common hydrocarbon feeds are given in Table 10.9. Major by-products are methane, hydrogen, butanes/butenes/butadiene, pyrolysis gasoline (benzene, toluene, C8 aromatics), and heavy oils. Steam feed helps mitigate coking, with increasing amounts of water required for heavier feedstocks, typically 0.3/1 to 0.75/1 kg water/kg hydrocarbon.

Flowsheet for naphtha cracker
The cracked gases are rapidly quenched to halt further reaction, first by indirect cooling against water, then by direct contact cooling with recycled heavy ends (2). The effluent is fractionated to separate the light gases (containing ethylene, propylene, C4−C5+ fractions) from pyrolysis gasoline, heavy oil fractions, and to remove water (3). The light gas is compressed to about 3.5 MPa, caustic washed to remove acid gases (CO2, H2S, COS, mercaptans), further cooled, and partially liquefied (4). The demethanizer (5) removes methane and hydrogen overhead, followed by the distillation of ethane/ethylene/(acetylenes) in the deethanizer (7). If desired, methane and hydrogen are separated by Joule–Thomson expansion/cooling to give about 85–90% hydrogen (6). The ethylene-rich fraction from the deethanizer is first hydrogenated over Pd or Ni fixed bed catalysts (8) to convert alkynes to alkenes to prevent fouling. Propane/propylene are taken overhead in the depropanizer (10), and C4 components split from remaining heavy materials in the debutanizer (11). A rather complex ethylene/propylene cycle is used to provide refrigeration needs in the plant.
Ethane/ethylene are split in a column comprising over 100 stages (9), with ethane underflow recycled to cracking. The propane/propylene separation requires several 100 stages to achieve polymer-grade purity (12). Propane underflow is also recycled to cracking. The distillative separation of ethane/ethylene (relative volatility ≈ 1.4) and propane/propylene (relative volatility ≈ 1.2) to produce polymer grade ethylene and propylene (typically 99.5% or greater purity) requires significant staging and reflux. The production of ethylene and propylene are the largest single users of energy in the chemical industries, equating to (not an insignificant) 1% of total energy demand globally.
About a third of propylene is recovered from refinery operations, such as FCC of heavy oils. FCC produces primarily motor gasoline components, but also 5–9% propylene, which can be recovered by fractionation [26]. See Chap. 18. Less than 10% of propylene currently comes from on-purpose propane dehydrogenation, ethylene–butene disproportionation/metathesis, and other sources [27]. During the next decade in the United States in particular, the significant increase in availability of NGL from shale gas is expected to move to higher usage of light feedstocks for cracking. This will exacerbate the relative shortage of propylene and may lead to more serious investment in metathesis and dehydrogenation processes.
It has been known for many years that methanol could be reacted over acidic zeolites at high temperatures to produce primarily an aromatic-heavy gasoline as well as some C2-C4 olefins. This type of process was commercialized briefly in New Zealand in the 1980s for gasoline production and later shut down due to poor economics [28]. The catalyst was the ubiquitous ZSM-5 zeolite. In all process variations, methanol is first converted into dimethyl ether with release of water. DME then further generates a “pool” of CH2 equivalents on the catalyst surface.


These CH2 equivalents then participate in the formation of larger molecules of many possible types, such as n-olefins, aromatics, naphthenes, relatively small amounts of n-paraffins, CO2, hydrogen, methane, and trace amounts of lower carboxylic acids [29–31]. The product distribution is heavily dependent on process conditions as well as zeolite geometry. High temperatures favor aromatic formation, while the pore size of the zeolite, if properly chosen, can significantly reduce or virtually eliminate aromatic formation. For example, SAPO-34, a template-based silica aluminophosphate molecular sieve zeolite with a pore opening of 3.8 Ǻ, controls the size of the olefin produced to mostly C2-C3, as well as minimizing aromatics formation. ZSM-5 and MFI zeolites, with pore openings of 5.1–5.6 Ǻ, lead to much higher aromatics production.
Modern methanol-to-olefins processes capable of producing relatively high yields of ethylene and propylene, are just beginning to be commercialized, in particular in China, where the methanol is largely derived from coal [32–34]. In the UOP/Hydro Methanol-to-Olefins (MTO) process methanol is reacted at 350–550°C, 0.2–0.4 MPa, over SAPO-34 zeolite to produce predominantly ethylene and propylene. Approximately 80% of the input carbon is converted into ethylene and propylene, and as high as about 90% with recycle of the butanes fraction. The ethylene/propylene product mass ratio can be varied between 0.75 and 1.05, dependent on process conditions. For 100 kg of methanol input, typical mass yields are 17 kg ethylene, 17 kg propylene, 5 kg mixed C4’s, 1.9 kg C5+, 1.7 kg fuel gases, 57 kg water, 1 kg and minor materials (CO2, coke, carboxylic acids). This process uses a fluid bed reactor with a separate regenerator vessel (using air), much like an FCC unit. The reactor effluent is separated from the water by-product and unreacted DME is recovered. The hydrocarbon fraction is washed with caustic to remove acids, dried, and compressed. Recovery of pure ethylene and propylene occurs in a distillation train very similar to that used in steam cracking for olefin production.
In the Lurgi MTP® process, methanol is converted to DME over γ-alumina in a separate reactor, followed by zeolite-based conversion to hydrocarbons in a six-stage adiabatic reactor system. Propylene yield is about 70% on input carbon, with relatively minor amounts of ethylene. A gasoline fraction of relatively modest octane is the major by-product.
As of 2011, at least one plant has been built in Brazil for dehydration of sugarcane-derived ethanol to ethylene (200 KMTA of ethylene, <0.2% of world capacity), and additional facilities are planned in Brazil and India [35–37].
C6-C8 Aromatics: BTX [38–42]
In the early twentieth century, the primary source of the C6−C8 aromatics (benzene, toluene, o-, m-, p-xylenes, BTX) was coal tar liquids (the aromatics-rich fraction is also called benzole) from the carbonization of coal to produce metallurgical coke. The rise of the petroleum refining industry has significantly diminished the contribution of this source of aromatics, less than 2% of aromatics are still derived from coal tars. Globally BTX is produced primarily by catalytic reformer operations (55–60%), and by recovery from pyrolysis gasoline (40–45%). Because of the large gasoline demand (as well as the tradtional use of LPG/NGL for steam cracking), about 70–75% of aromatics produced in the United States comes from reformates. The situation is reversed in Europe, Japan, and China. The predominance of naphtha steam cracking in these locations leads to larger amounts of aromatics from pyrolysis gasoline.
In catalytic reformers, dehydroisomerization and cyclization/dehydrogenation reactions convert cycloparaffins and alkylcyclopentanes (naphthenes), along with paraffins into predominately aromatic liquid products at high temperature over platinum-based catalysts (with H2 and lighter hydrocarbon as by-products). Reforming significantly improves the octane, or gasoline blending quality of the stream. A typical reformate contains 50–60% aromatics, mostly toluene and xylenes. In the United States about 90% of this reformate is used for gasoline production, the other 10% becomes chemical feedstocks.
The amount and composition of the BTX fraction recovered from pyrolysis gasoline is highly dependent on the steam cracking feedstock and severity of conditions (see Table 10.9). Due to its high content of polymerizable components (not good for internal combustion engines), virtually all of the pyrolysis gasoline is used for chemical feedstock production rather than gasoline production.
The most important aromatics for chemical production are benzene and p-xylene. Reformer and pyrolysis gasoline (pygas) aromatic fractions as produced typically do not meet the demand patterns for these aromatics. Four families of processes are widely used to adjust the natural distribution between benzene, toluene, xylene isomers, and lesser amounts of polyalkylbenzenes and ethylbenzene [43–47]. See also the discussion of alkylation in Section 10.3 below.
-
1.
Hydrodealkylation involves the thermal or catalytic reaction of alkylated aromatics (normally methyl aromatics) with hydrogen to produce light alkanes (normally methane) and benzene.
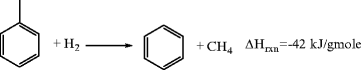
Toluene is the most common feed substrate, but higher alkylated benzenes (e.g., xylenes and trimethyl benzene) are also used. In the catalytic process, temperatures stay below 500°C to prevent metal sintering, with pressures from 2.5 to 7.0 MPa. Common catalysts are Group VIII metals and metal oxides, e.g., chromium oxides, Pt, Pt oxide, on alumina. Thermal dealkylation requires a temperature of 600–660°C, pressures of 3.5–7.0 MPa. The heat of reaction is controlled by recycling cold hydrogen at high (6:1 to 8:1) molar ratio to substrate. Because of the formation of the highly stable by-product of methane, the reaction goes very far to completion. Selectivity to benzene approaches 99% in the thermal process and 98–99% in the catalytic processes. Many versions of hydrodealkylation have been commercialized and are available for license.
-
2.
Transalkylation/disporportionation involves the migration of methyl groups among aromatic rings in the presence of hydrogen. For example two toluene molecules produce a mixture of xylene isomers.
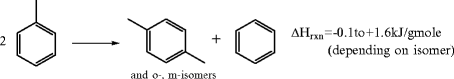
The reaction proceeds over aluminosilicate or silicoaluminophosphate zeolites (often containing noble metal (Pt) or rare earths) at 400–470°C, pressures of 1.4–3 MPa, with a hydrogen:aromatic molar ratio of 5:1 to 12:1. Yield is well above 90%. Under normal reaction conditions the xylene isomer composition approaches the equilibrium value. At 400°C the distribution is about 21.6% ortho-, 24.4% para-, and 54% meta-xylene. Thus, if p-xylene is the desired product (usually), conversion to para-xylene is low per pass. Separation and recycle are critical (and expensive).
A fairly recent innovation has been the use of shape selective zeolite catalysts that allow toluene into the zeolite cage, but only allow para-xylene, with its narrow cross sectional area, to leave. Ortho- and meta-isomers are trapped and isomerized until para-xylene is formed. Significantly greater than equilibrium levels of para-xylene can be produced with much less separation and recycle requirements.
The other significant C8 isomer, ethylbenzene (EB), can be present in appreciable amounts, depending on the origin of the stream. Difficult to separate cleanly (requiring superfractionation with over 200 stages), it can be converted via several reaction pathways: (1) de-ethylation to benzene (similar to HDA); (2) exhaustive hydrogenation to ethylcyclohexane, rearrangement to dimethylcyclohexanes, and dehydrogenation to xylene isomers; (3) disproportionation with xylenes to give benzene and ethyl xylenes. All three are operational on most transalkylation and disproportionation catalysts, typically giving at least 30% up to almost 100% EB conversion per pass.
-
3.
Isomerization is a related process using much the same catalysts and reaction conditions as transalkylation. Some versions do not require hydrogen co-feed and can operate at 200–260°C. Shape selective catalysts that enhance para-xylene formation are also employed for isomerization.
-
4.
Toluene methanation is a fourth process to convert toluene to xylenes that has been commercialized in the last decade to a limited extent. Methanol is reacted with excess toluene at high temperature over acidic or zeolite catalysts to produce xylene isomers and water.
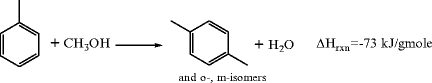
Toluene is distilled and recycled, while the product mixture of xylene isomers must be separated and isomerized to maximize p-xylene. Shape selective, para-enhancing zeolites are also used here. Note that this reaction cannot be accomplished with methane as the alkylating agent.



Recovery and purification of individual BTX components are challenging separations problems. Separation of paraffins and olefins from aromatics is typically done by some combination of simple distillation, extraction, and extractive distillation. Benzene and toluene (normal boiling points of 80.1°C and 110.6°C respectively) can be separated from C8 and higher aromatics by simple distillation. However, the C8 isomers all boil within about 8°C of each other (ethylbenzene at 136.2°C, p-xylene at 138.3°C, m-xylene at 139.1°C, and o-xylene at 144.4°C), rendering simple distillation impractical. Crystallization and shape-selective molecular sieve adsorption have become standard techniques for xylene isomer purification. A rich literature on these separations is readily available [48–53]. An aromatics complex often has many of these reaction and separation processes working in concert. Further information on aromatics production is given in Chap. 18.
The primary derivatives produced from BTX feedstocks, the capacity/production/consumption of those primary derivatives, and their major applications are summarized in Tables 10.10, 10.11, and 10.12. The family of derivatives produced from BTX is presented in Fig. 10.13.

BTX derivative tree
C4 Hydrocarbons: Butanes, Butenes, Butadiene [54–58]
The two saturated C4 hydrocarbons, n-butane and isobutane (2-methylpropane), are obtained directly as components of NGL or from a variety of petroleum refining operations. Crude natural gas typically contains 1–2 mole% butanes, with a normal to iso ratio between 1:2 and 2:1. Much of these butanes become raw material for stream cracking to light olefins. Refinery processes giving appreciable amounts of butanes include crude distillation, catalytic cracking, catalytic reforming, delayed coking, fluid coking, visbreaking, and hydrocracking. Further information on these processes is given in Chap. 18. In the United States, refinery operations account for close to 80% of the butanes produced. A significant portion of the refinery butanes is used for fuel, gasoline blending, and other gasoline-producing operations. Only about 10% is diverted as a raw material for chemical synthesis.
Butenes comprise C4 mono-olefin isomers, 1-butene, cis-2-butene, trans-2-butene, and isobutylene. These isomers are co-produced with butanes in refinery and steam cracking operations. Typical yields of butanes, butenes, and butadiene for several processes are given in Table 10.13. These C4 fractions cannot be separated economically into pure components by simple distillation due to close boiling points. Rather, separation and reaction methods that exploit property differences due to molecular structure or between functional groups are employed, such as shape-selective adsorption, extraction, extractive distillation, isomerization, and dehydrogenation. Separation sequences typically begin with removal of 1,3-butadiene via extraction or extractive distillation (see below). The remaining butanes/butenes can be treated by a variety of methods including adsorption (to separate branched and normal species); reactions that exploit the reactivity of the olefins, especially isobutylene, over paraffins; extraction or extractive distillation (to separate butenes from butanes), or isomerization (to convert n-butenes to isobutylene or visa versa, n-butane to isobutane or visa versa, or 2-butene to 1-butene). A large body of information on these technologies is available in the open literature [55, 58–60]. Dehydrogenation of C4 species is covered briefly in the section, “Butenes and 1,3-Butadiene.”
During the 1990s in the United States, a significant fraction of the butenes pool was used for production of ether oxygenates, such as MTBE (methyl tert-butyl ether) for gasoline blending to meet specifications for reformulated gasoline. MTBE use in gasoline was later discontinued and replaced with corn-based ethanol. A large portion of the butenes is now used for alkylation (isobutane reacted with propylene and/or butenes) and for the production of higher octane butene dimer blending components. Further information on these processes is given in Chap. 18.
Over 95% of the 1,3-butadiene (1,3-BD) produced globally is recovered as a by-product of light olefins production from steam cracking. A number of extractive distillation processes, such as the n-methylpyrrolidone (NMP)-based process illustrated in Fig. 10.14, have been commercialized for recovery of 1,3-BD from such mixed C4 streams [61–64]. The mixed C4 stream, containing n/i-butanes, n/-i-butenes, 1,3-BD, 1,2-butadiene, and alkynes, is fed to a predistillation column (1), where propyne, propadiene, and other lights are removed overhead. The underflow of this column is fed to the bottom of the stripping section of the extractive distillation column using NMP as the extractive distillation solvent (2). The distillate is a butadiene-free butanes/butenes mixture (commonly referred to as raffinate-1). The bottom butadiene-rich stream is sent to the rectifying section of the extractive distillation, a divided wall column (3), where butenes are stripped out of the column feed mixture in one compartment (3a), with the stripped materials returned to the extractive stripping column. In the second compartment (3b), the more soluble C4 alkynes are removed from the crude 1,3-BD by extraction into NMP. The bottoms of the rectifying section are distilled in the stripping column (4) to remove light hydrocarbons from the recycle solvent. Acetylenes banding in this column are removed as a sidedraw, scrubbed of NMP (6), and added to the raffinate-1 stream if desired. The stipped hydrocarbons are recompressed (5) and recycled to the rectifying section of extractive column. The overhead from the rectifying section (3b) is distilled in the final butadiene column (7), with water removed overhead, 1,3-BD taken as a liquid sidedraw product, and C4-C5 hydrocarbons underflowed. Recovery of 1,3-BD is above 98%.

Recovery of 1,3-butadiene from mixed C4’s by extractive distillation
A number of older processes for on-purpose synthesis of 1,3-BD, such aldol condensation of acetaldehyde, followed by hydrogenation/dehydration, ethanol to acetaldehyde to butadiene(via aldol), or acetylene plus formaldehyde [65], are largely obsolete. A small amount of butadiene is produced by dehydrogenation or oxidative dehydrogenation of butane/butenes, as discussed in section, “Butenes and 1,3-Butadiene.”
The primary derivatives produced from C4 hydrocarbon feedstocks, the capacity/production/consumption of those primary derivatives, and their major applications are summarized in Tables 10.14, 10.15, and 10.16. The family of derivatives produced from C4 hydrocarbon feedstocks is presented in Fig. 10.15.

C4’s derivative tree
C9-C17 Paraffins [66–68]
Higher C9-C17 alkanes derived from petroleum, find limited but important uses as raw materials for the production of higher C9-C17 n-olefins. These higher n-olefins are intermediates in the manufacture of detergents and anionic surfactants. Because of their wide availability and generally low cost, heavy naphthas (boiling range of 150–200°C) and kerosene (boiling range of 190–250°C) are the raw materials for production these olefins. The naphtha and kerosene fractions typically contain 20% or more n-paraffin content. After hydrotreating to remove sulfur which poisons downstream catalysts, the naphtha and kerosene cuts are separated by shape-selective adsorption that takes advantage of the larger cross-sectional area of the branched isomers. The n-alkanes are preferentially adsorbed, while the branched materials largely pass through. The n-alkanes are desorbed by a light hydrocarbon, and distilled to recover the desorbent. Both vapor and liquid phase adsorption processes are practiced. The high n-alkane product is then dehydrogenated to give most internal olefins. Further details on the dehydrogenation process are given in the section, “Internal Olefins from Higher n-Alkanes.” The primary derivatives produced from C9-C17 hydrocarbon feedstocks, the capacity/production/consumption of those primary derivatives, and their major applications are summarized in Tables 10.17, 10.18, and 10.19. The family of derivatives produced from C9-C17 hydrocarbon feedstocks is presented in Fig. 10.16.

C9-C17 normal paraffins derivative tree
Synthesis Gas [69–77]
Synthesis gas, or “syngas,” is a mixture of hydrogen and carbon monoxide produced by the partial oxidation of carbonaceous feedstocks. The ratio of hydrogen to carbon monoxide in the crude syngas (as well as carbon dioxide content and impurities) is highly dependent on the carbonaceous raw material as well as the process used to generate the syngas. Natural gas is the most important raw material for syngas generation worldwide, with limited coal-based production in the United States, South Africa, India, and significantly growing production in China. Liquid hydrocarbons (naphtha, heavy fuel oils, and vacuum resid) are also used to some extent.
Syngas production involves the breaking of C–C and C–H bonds of the raw material molecules at high temperature via reaction with water (steam reforming), oxygen (partial oxidation), or carbon dioxide (carbon dioxide reforming), or combinations therein (autothermal reforming or gasification). The primary reactions in steam methane reforming (SMR), stream hydrocarbon reforming (SHR), and carbon dioxide reforming (CMR—not yet commercial) are endothermic, with only the water-gas shift reaction providing heat. Heat must be added externally, normally by superheating steam and raw materials in a fired furnace.
Syngas can also be produced by partial oxidation, typically with high purity oxygen (to avoid dilution with large quantities of hard to separate nitrogen). In partial oxidation, a fraction of the raw material is simultaneously combusted to completion to provide in situ heat input. Roughly 70–85% of the energy in the raw material is preserved in the syngas produced. For methane, pertinent reactions are given below, with reforming and water-gas shift reactions also occurring to some extent.
Gasification of solid carbonaceous raw materials adds additional endothermic reactions of solid carbon with steam and exothermic reactions with oxygen. Methanation may also occur under appropriate conditions.
Typical H2/CO ratios for the most important syngas-generating processes are summarized in Table 10.20. Further information on syngas production from natural gas and via coal gasification is given in Chaps. 19, Chaps. 20, and Chaps. 22.
It is not much of an exaggeration to say that coal contains every element on the periodic table, many of which end up in the crude syngas, such as mercury, arsenic, and sulfur as H2S and COS. To a lesser extent natural gas often contains H2S, COS, as well as mercaptans. Many of the downstream catalysts used in syngas derivative processes are particularly susceptible to poisoning by these trace elements. A wide variety of techniques, mostly involving absorption and adsorption, have been developed and implemented for removal of such trace elements.
Most syngas derivatives are produced optimally at a specific H2/CO ratio, with a limited acceptable CO2 content [78]. See Fig. 10.17 for the syngas family of derivatives produced from different H2/CO ratios. If not produced directly from the syngas generator, a number of reaction and separation techniques are used to adjust the composition of the gas.

Syngas and methane derivative tree
-
1.
The water-gas shift reaction is the primary mechanism for reactive adjustments of the H2/CO ratio. Both sweet (very low amounts of H2S and other sulfur moieties present) and sour (high sulfur levels) shifts are practiced. The feed gas enters the shift reactor at 200°C or higher.
$$\begin{array}{lll} {\text{CO}} + {{\text{H}}_{{2}}}{\text{O}} \leftrightarrow {\text{C}}{{\text{O}}_{{2}}} + {{\text{H}}_{{2}}}\cr \Delta {{H}_{\rm{r}}} = - {42}\;{\text{kJ/gmole}}({\text{water gas reaction}}) \end{array}$$The reaction is normally run adiabatically with a significant temperature, often of 200°C or more. As temperature is increased, the equilibrium shifts more toward the left. Multiple stages, with heat interstage steam generation for cooling, is required for high levels of conversion, i.e., for hydrogen production.
-
2.
Carbon dioxide is removed by absorption with either physical or chemical absorbents, the choice depending on gas pressure, CO2 concentrations, and desired level of removal.
-
3.
Small adjustments of H2/CO may be accomplished by membrane permeation (hydrogen permeates primarily), decreasing the H2/CO ratio of the retentate. Membrane permeation is particularly common for production of 1/1 H2/CO syngas for hydroformylation.
-
4.
High purity hydrogen is produced by water-gas shift, followed by CO2 absorption, and pressure swing adsorption.
-
5.
CO may be produced at up to about 95% purity by membrane permeation (more extreme case of 3). High purity CO (>99.9%) is made by cryogenic distillation of syngas, or less commonly by chemical absorption processes (CO absorbed).
Estimated global production of syngas, at common H2/CO ratios, is given in Table 10.21. The capacity/production/consumption of primary derivatives produced from syngas feedstocks and their major applications are summarized in Tables 10.22 and 10.23.
Minor Feedstocks: Methane and Acetylene
Methane itself is relatively unreactive. Much effort has been expended to synthesize functionalized chemicals from methane directly. Examples of large-scale industrial syntheses are carbon disulfide, hydrogen cyanide, and to some extent chloromethanes. However, most other direct methane processes have been ultimately uneconomical and have fallen by the wayside or never been commercialized.
Until the rise of the massive global petrochemical infrastructure in the late 1940s and 1950s, acetylene was one of the most important feedstocks for the production synthetic organic chemicals. Although highly reactive, high conversion and generally very good selectivities can be achieved in acetylene-based chemistries, Such routes thus were favored for many years. Acetylene was once a major feedstock for the production of vinyl chloride monomer (VCM), acrylic acid, acrylonitrile, and many chlorinated solvents. Olefin-based processes have largely supplanted those starting with acetylene. Between 1965 (near its peak demand) and 2007, acetylene usage for chemical synthesis has dropped at least 70% in all areas of the world except China. Acetylene demand, especially for VCM, has enjoyed somewhat of a renaissance in China, with capacity growth above 15% per year over the last decade. The fact that acetylene can be made from a wide variety of feedstocks, including coal, is a particular advantage in oil-poor China. Currently about 75% of global acetylene feedstock usage occurs in China. Capacity/consumption statistics for acetylene are given in Table 10.24 for the year 2007. Remaining large scale applications of acetylene are summarized in Table 10.25.
Acetylene can be produced by a wide variety of processes [79, 80]. These include:
-
1.
Calcium carbide processes, in which calcium oxide and coke are electrothermally reacted at 2,250°C to produce calcium carbide. The reaction is highly endothermic at +465 kJ/gmole. Calcium carbide is reacted with water in an exothermic reaction acetylene and calcium hydroxide. Yield of acetylene is about 76% on calcium carbide.
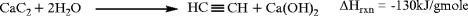
-
2.
Electric arc processes, in which a carbonaceous raw material is cracked at very high temperatures up to 20,000°C in an electric arc-generated plasma with very short residence times of 0.001–0.01 s. Rapid quenching to 200–300°C freezes the composition and prevents decomposition of acetylene to thermodynamically favorable soot and hydrogen. Typical yields are around 45 wt% of feed carbon to acetylene and 25% to ethylene.
-
3.
Autothermal partial oxidation processes, in which light hydrocarbons (predominantly methane in practice) are reacted with oxygen in a controlled fashion. In one version methane and oxygen at a 2:1 M ratio are preheated separately to 500–600°C, mixed, and reacted in proprietary burner configurations at 1,500°C, millisecond residence times to produce syngas and acetylene. The endothermic acetylene formation reaction is coupled with the highly exothermic partial combustion of methane to provide in situ heat integration:
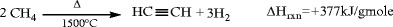
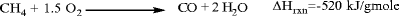
The reaction gases are rapidly quenched with water or oil to prevent decomposition to soot and hydrogen. The cooled gas, comprising 8% acetylene, 57% H2, and 26% CO, is contacted with N-methylpyrrolidone (NMP) in a countercurrent absorber to recover acetylene (see the section, “C4 Hydrocarbons: Butanes, Butenes, Butadiene,” for a similar use of NMP). About 25–30% of the carbon in the raw material is converted into acetylene, with about 3–5% going to coke. Most of the rest ends up as CO.



Downstream Derivatives
Many of the primary derivatives of the major petrochemical feedstocks are intermediates for the synthesis of additional downstream derivatives The raw materials and chemistries of these downstream derivatives are summarized in Table 10.26. Capacity/production/consumption figures and major uses of the downstream derivatives are presented in Tables 10.27 and 10.28, respectively.
Manipulation of Hydrocarbons: Oligomerization, Metathesis, Alkylation
Oligomerization
Linear α-Olefins [81–84]
Linear α-olefins can be produced from higher n-paraffins by dehydrogenation, as described in the section, “Internal Olefins from Higher n-Alkanes.” Linear terminal olefins with an even number of carbon atoms can be produced by a family of related processes based on oligomerization/metathesis of ethylene using trialkyl aluminum or titanium (Ziegler) catalysts or liganded Ni catalysts. The reaction takes place in two steps:
-
1.
In the oligomerization, or chain growth step, ethylene is inserted progressively into the carbon-catalyst metal bond, leading to a longer linear alkyl chain.
-
2.
To terminate the chain growth, ethylene displaces the longer chain alkyl unit from the C-metal bond, releasing a longer chain α-olefin, and starting the oligomerization cycle over again.
The product composition follows the Shulz–Flory distribution of chain lengths, with substantial amounts of undesirable high and low molecular weight tails. Both one-reactor and two-reactor processes are practiced commercially. The one-reactor process leads to a somewhat broader distribution of molecular weights, with typical conditions of 170–290°C, 14–28 MPa. For a two-reactor process with a Ziegler catalyst, typical conditions are 100–120°C, 7–20 MPa for oligomerization and termination at 260–300°C, 2–5 MPa. With a nickel catalyst, more mild oligomerization conditions are possible, 80–120°C, 7–14 MPa. A typical product distribution is 5–14 wt% C4, 25–50 wt% C6-C10, 15–20 wt% C12-C14, 11–15 wt% C16-C18, and 3–15 wt% C20+ One process can produce a very narrow distribution of closer to 75% C6-C10 olefins. Depending on the process conditions, catalyst, and molecular weight of the olefin in question, the product comprises 88–98% linear α-olefins. Distillation is used to separate the crude olefin product into molecular weight ranges required for various applications (e.g., oxo for detergent alcohols, LAB production).
Two methods are commonly used for narrowing the molecular weight range of the ultimate product. Low molecular weight olefins may be recycled to participate in further oligomerization (see above). Secondly, light and heavy fractions may be metathesized to produce two smaller olefinic molecules (see section “Metathesis”, directly below).
Metathesis [85]
An intriguing reaction in which the carbon–carbon double bonds of two olefins are broken and remade, resulting in the switching of their alkylidene groups, is referred to as olefin metathesis.

The analogous reaction for carbon–carbon triple bonds, alkyne metathesis, was also discovered at about the same time, in the early 1960s.

One of the earliest applications of olefin metathesis was developed at Phillips Petroleum and is known as the Triolefin Process [86]. It illustrates the simplest case of olefin metathesis in which propylene is converted to ethylene and both cis and trans isomers of 2-butene.

The process was practiced as described (propylene-to-ethylene/butene) by Shawinigan Chemical from 1964 to 1972 but the demand for propylene vs. ethylene shifted. Now processes have been developed by ARCO (now Lummus OCT: olefin conversion technology) and Axen (Meta-4) to execute variations of the opposite in which C4 streams, derived from hydrocarbon cracking, are reacted with ethylene to produce propylene. This technology thus provides the opportunity to enrich the ethylene/propylene product ratio of a cracker to favor propylene. In OCT, ethylene is dimerized to butenes and then cracked with additional ethylene (“ethenolysis”) to yield propylene, allowing total conversion of such a stream to propylene. Several different metal oxides, such as CoO-MoO3/Al2O3 oxide (120–210°C), WO3/SiO2 (450–500°C) and Re2O7/Al2O3 (450–500°C), are employed as catalysts.

Another example of ethenolysis to produce valuable products is the cracking of unsaturated fatty acid esters for production of fuels from renewable materials [87].
Olefin metathesis is an equilibrium controlled reaction, and the equilibria of a related set of olefins are typically close to a statistical mixture, thus limiting the conversion of reactants. However, if one olefin of the equilibrating mixture can be selectively removed then a metathesis reaction can be driven to high conversion. A simple strategy which exploits this concept is the reactions of a-olefins which produce low boiling ethylene (the oppositie of ethenolysis). As the volatile alkene escapes from solution the reaction proceeds to completion. This approach is exploited in polymer synthesis by reacting various α-, ω-dienes to generate ethylene and a growing polymer chain (referred to as “Acyclic Diene Metathesis” ADMET, polymerization). Likewise, dienes which can produce a favored ring size yield “Ring Closing Metathesis.”

A second strategy for driving a metathesis reaction toward complete conversion is demonstrated by polymerization of cyclic monomers, such as norbornene, which release enough energy upon ring-opening to drive the polymerization. This method is known as ring-opening metathesis polymerization (ROMP) [88].

The reaction above gives an early example of the homogeneous catalysis of metathesis reactions by metals such as ruthenium or iridium [89, 90]. The development of single-component metal-carbene metathesis catalysts, in particular those of ruthenium, and their applications in organic synthesis have been the subject of an enormous body of research in the last 20 years and the 2005 Nobel prize in chemistry was awarded to three of the pioneers in this area: Yves Chauvin, Robert Grubbs and Richard Schrock. ROMP is one of the most promising techniques to emerge in this work with ready applications in polymer and material science. The structures of some of the most widely utilized catalysts are given in Fig. 10.18.

Flowsheet alkylation of benzene to ethylbenzene
In 1977, Shell commercialized a process known as the SHOP Process (Shell Higher Olefin Process) for the production of detergent alcohols at their plant in Geismar, LA. The 2010 capacity of this plant for olefin production is over 900,000 M-tons/year. Detergent alcohols are produced in four steps from ethylene. First, the ethylene is oligomerzed using a homogeneous nickel-based catalyst with a chelating P-O ligand ([Ni] ~50 ppm). Oligomerization is carried out at 80–140oC and 7–14 MPa in a polar organic solvent (1,4-butanediol) which phase separates from C4-C30 α-olefin products [91]. The nickel catalyst solution phase is separated in a high-pressure separator and the C4-C20 α-olefins are distilled from the mixture and are purified and sold as Shell’s Neodene linear α-olefins.
In the next step the mixture of α-olefins are isomerized in the liquid phase using a heterogeneous catalyst such as MgO granules. This produces all possible isomers of the even-numbered linear olefins. Then, in the third step, olefin metathesis of this mixture over a molybdenum/alumina catalyst (100–125°C, 10 MPa) further complicates this mixture by doubling the number of olefins within the range to include not only the even (C4, C6, C8, etc.) but also all of the odd-numbered (C4, C5, C6, C7, etc.) olefins in a thermodynamically controlled equilibrium. The yield of desired C11-C14 olefins is 10–15% per pass. These olefins are treated in the final step with syngas (CO:H2 = 1:2) using a phosphine-modified cobalt catalyst (170°C, 10 MPa). In this process internal double bonds are isomerized rapidly, resulting in a highly linear alcohol product (75–90% linear).
Alkylation [92, 93]
Chemistry
Catalytic alkylation of aromatics is a substitution reaction in which a hydrogen atom on the ring or side chain of the aromatic is replaced by an alkyl group derived from an alkylating agent. Alkylation may occur on both unsubstituted as well as substituted aromatics. The reaction is shown below for ring substitution with an olefinic alkylating agent:
Acid-catalyzed electrophilic substitution (by way of a carbonium ion) gives replacement on the ring. If the alkylation is to occur on the side chain, then nucleophilic (base-catalyzed), or free radical reactions are involved. Most large-scale commercial alkylations are electrophilic in nature, e.g., cumene, ethylbenzene, and linear alkylbenzenes (LAB’s) syntheses, but a few are nucleophilic, e.g., isobutylbenzene, dimethylnaphthalene syntheses.
Alkylating agents can be olefins, alkynes, dienes, alcohols, and ethers. For electrophilic substitution, any substrate capable of forming a carbonium ion may be used, although the use of olefins predominates for all applications except introduction of a methyl group. Methanol is a common methylating agent as it can give a carbonium ion equivalent, whereas methane itself is unreactive.
Acid catalysts useful for aromatic alkylations are Brønsted acids containing acidic protons. Examples include protonic acids such as sulfuric, HF, phosphoric, solid phosphoric acid (SPA), polysulfonate cation-exchange resins; acidic aluminas, silicas, alumina-silicas, zeolites (aluminosilicates and silicoaluminophosphates); and acidic halides, such as AlCl3 and BF3. The latter are relatively inactive in their pure states, but become catalytically active when coupled with trace concentrations of water, alcohols, or hydrogen halides (giving the needed Brønsted acidity). These Lewis acids were the original highly effective Friedel-Crafts catalysts, with high activity at low temperature (<100°C), but have largely been replaced commercially with more selective, less corrosive, and environmentally friendly zeolites.
With strong Brønsted acid catalysis, the olefin is protonated to form an activated electrophile (carbonium ion), which attacks the electron-rich π-complex of the aromatic ring. A new C–C-bond is formed, followed by regeneration of an acidic proton. Acid-catalyzed skeletonal rearrangement of the alkylating agent leads to formation of the most substituted and stable ion (tert > iso > normal). Relative rates of alkylation of the ring and isomerization of the alkylating agent dictate the product distribution. Typically for small alkylating agents like butenes, (especially isobutene) rearrangement is fast, giving almost exclusively tert-butyl aromatics. Thus synthesis of isopropylbenzene (cumene) and diisopropylbenzene from propylene and benzene is highly selective while isobutylbenzene formation must be base catalyzed. Alklyation rate also roughly follows carbonium ion stability, with isopropylation roughly 1,500 times faster than ethylation and 20,000 times faster than methylation.
The rate of alkylation of benzene is slower than alkylation of alkylaromatics, so polyalkylation is kinetically favored. However, many of the most important alkylation products (e.g., cumene and ethylbenzene) are monosubstituted. The traditional method of limiting polyalkylation is to use high ratios of aromatic to alkylating agent, leading to low per pass aromatics conversion, and high separation/recycle costs. One of the most intriguing breakthroughs in alkylation technology in the last two decades has been the commercialization of shape-selective zeolite catalysts, which can largely eliminate polyalkylation, as well as allow much higher than equilibrium production of para-isomers (see the section, “C6-C8 Aromatics: BTX,” above).
Ethylbenzene [94–98]
Well over 90% of the ethylbenzene consumed worldwide is formed by the alkylation of benzene with ethylene.

Early processes employed AlCl3 in liquid phase processes operating below 130°C. Significant polyalklyation occurred, but was controlled by high benzene to ethylene feed ratios and recycle of polyethylbenzene (PEB) materials for equilibration in the alkylation reactor. High temperature (>300°C) vapor phase zeolite (ZSM-5) processes made significant inroads starting in the 1970s, but reactors coked quickly and required frequent regenerations.
Greenfield plants predominantly rely on next generation large pore zeolite catalysts to allow liquid-phase operation at 170–240°C (more than 100°C lower than the older vapor phase processes), at 3.5 MPa, with a much smaller excess of benzene to ethylene required. Low temperature operation decreases formation of by-product xylenes, polyaromatics, and PEB. In a typical commercial process (Fig. 10.19), benzene at less than 7:1 B:E molar ratio is fed to zeolite-filled alkylation beds in series (1), with cooled ethylene added between stages. Older vapor phase processes required a B:E of 18:1. Interstage cooling provides additional temperature control. Purification is accomplished in a three column distillation sequence. Unreacted benzene (for recycle) is taken overhead in the benzene column (2), EB overhead in the product column (3), and PEB for recycle in the third column (4), with polyaromatics and other heavies as underflow. PEB are converted to EB by reaction with benzene in a separate transalkylation unit (5). Ethylene and benzene consumption are respectively 0.265 and 0.738 kg/kg EB. Ethylene containing up to about 30% ethane can be used with the addition of a column for recovery of ethylene from off-gas ethane by absorption into benzene feedstock.

Single-component olefin metathesis catalysts
Cumene and Diisopropylbenzene [99–103]
Cumene production via alkylation of benzene with propylene, began in World War II as a high octane aviation fuel component. This early vapor phase process operated at 200–250°C, 1.5–3.5 MPa, over a silicophosphoric acid (SPA), with a 7:1 benzene:propylene ratio, with benzene-based yields above 95%.

The SPA process was widely used for decades with little change, competing well against even zeolite based processes. Although successful for ethylbenzene synthesis, similar early generation zeolites were found to be unsuitable for cumene production. These narrow pore zeolites promote excessive n-propylbenzene formation due to (unwanted) shape selectivity, as well as propylene oligomerization (propylene is more reactive to oligomerization than ethylene). In the mid-1990s, a proprietary zeolite was invented with unique crystal structure that largely eliminated these former disadvantages, even at benzene:propylene ratios less than 2:1. Commercialized liquid phase processes using this zeolite are capable of greater than 99.7% yield of cumene, at 99.97% purity. The process flowsheet for cumene production looks very similar to that of ethylbenzene, with a mult-stage series alkylation system, a separate transalkylation step, and a three-stage distillation sequence for purification. Benzene, cumene, polyisopropylbenzene are taken overhead in successive columns, and heavies as underflow in the final column. Para-DIPB can be made in essentially an identical process, but with a higher propylene to benzene feed ratio.
Higher Alkylbenzenes [104–107]
Linear alkylbenzenes (LAB’s) with a C10-C14 side chain are important intermediates for the production of alkylbenzenesulfonate anionic detergents (see Chap. 36). The name is somewhat of a misnomer, as the alkylation reaction produces predominately branched 2-phenylalkane derivatives, rather than true linear 1-phenylalkanes.
Alkylation with the propylene tetramer has largely become obsolete, as the resulting branched alkylbenzene sulfonates are not readily biodegradable. The higher alkylbenzenes are synthesized by the liquid-phase reaction of C10-C14 α-olefins and internal olefins with benzene, using typical Friedel-Crafts catalysts such as HF, AlCl3, or zeolites. The higher chain olefins are sourced from either: (1) catalytic dehydration of the n-paraffin fraction recovered from the kerosene crude distillation cut to give largely internal olefins (see the section, “Internal Olefins from Higher n-Alkanes”), or (2) ethylene oligomerization processes (see above, the section, “Oligomerization”), which give largely α-olefins. Newer zeolite catalysts favor the more desirable 2-phenylalkane isomers with both internal and α-olefin feedstocks. Most greenfield LAB plants are now zeolite-based, with their clear advantage in safety, catalyst and capital cost, with elimination of catalyst neutralization stepsand corrosion concerns.
Oxidation
Chemistry [108–115]
Roughly two-thirds of high value synthetic organic chemicals are oxygenated. These include aldehydes, ketones, alcohols, organic acids, anhydrides, esters, and epoxides. Many of these functional groups ultimately are derived from some form of selective oxidation. In its broadest sense, oxidation is the process whereby the oxidation state of a substrate (typically carbon in this chapter) is increased. Although conceptually simple the controlled addition of oxygen to a hydrocarbon feedstock, i.e., partial oxidation, is a tricky feat to master. The principle technical challenges in partial oxidation of organic substrates are (1) finding a catalyst or conditions that give high selectivity toward the desired oxidation state and low selectivity for over or complete oxidation; and (2) carefully controlling reaction conditions, especially temperature and composition, to prevent loss of selectivity, runaway exothermicity, or explosive conditions.
Selective oxidations can be classified into two broad categories:
-
1.
Electrophilic oxidations involve activation of dioxygen into electron-seeking forms such as O −2 or O− and formation of free radical intermediates.
-
2.
Nucleophilic oxidations involve activation of the organic substrate followed by stepwise nucleophilic addition of O −2 and hydrogen abstraction.
In order of increasing severity, electrophilic oxidations include; (1) addition of atomic oxygen to a high electron density double bond to form an epoxide, (2) formation of an alkyl hydroperoxide via hemolytic cleavage of a C–H bond by an oxygen radical to form an alkyl radical followed by reaction with dioxygen, (3) fission of C–C bonds to form two aldehydic groups, (4) rapid stepwise fission of C–H and C–C bonds to form CO2 and water—i.e., total combustion. Nucleophilic oxidations in increasing severity include; (1) abstraction of hydrogen from an aldehyde followed by nucleophilic addition of oxygen to form an acid, (2) abstraction of hydrogen from an alcohol substrate to form an aldehyde or ketone, (3) abstraction of hydrogen from a substrate to form olefins, diolefins, aromatics from hydrocarbons, (4) abstraction of hydrogen from an olefin followed by successive nucleophilic additions of heteroatoms such as O, Cl, S, and N.
Electrophilic oxidations alter the carbon skeleton and π electron systems, while nucleophilic oxidations do not. Nucleophilic oxidations often involve a redox mechanism, in which oxygen is supplied indirectly to the substrate from the oxide lattice of the catalyst, and the catalyst lattice is then reoxidized by dissolved or gaseous dioxygen. Important industrial oxidation reactions described in this chapter are classified and summarized in Table 10.29.
The design and operation of reactors for oxidation service are demanding tasks. The desired products of selective partial oxidation are invariably not the thermodynamically stable products. Thus kinetic controls must be exploited to avoid overoxidation or complete combustion. Moreover, complete combustion releases typically four to ten times the energy of selective partial oxidations. Heat management is of paramount importance as non-selective reactions often have higher activation energies than the selective pathways, and are thus accelerated disproportionally by poor heat control. A large body of literature has been built up on heat and mass transfer, as well as reactor design. Some representative references are given below [116–118].
Responsible industrial implementation of oxidation chemistry requires understanding the implications of generating potentially flammable or explosive mixtures. The literature on safety issues, data and discussions of explosive limits is quite extensive. A few key references are cited here [119–123].
Homogeneous Aromatic Oxidations
Terephthalic, Isophthalic, Trimellitic Acids [124–126]
A number of aromatic carboxylic acids such as TPA, isophthalic acid (IPA), and trimellitic anhydride (TMA), are produced by the liquid phase air oxidation of the parent aromatic (p-xylene, m-xylene, and 1,2,4-trimethyl benzene respectively) with a homogenous catalyst comprising cobalt, manganese, and bromine salts. The key breakthrough in the development of this family of processes was the addition of the bromine promoter which resulted in a remarkably higher reaction rate and selectivity than with Co/Mn alone [127]. TPA is by far the largest volume product of this group and is produced via the stepwise oxidation of p-xylene:
The oxidation sequence begins by with hydrogen abstraction from one of the methyl groups of p-xylene by a bromine radical to form a benzyl radical. Dioxygen reacts with this species to form a hydroperoxide intermediate that passes through tolualdehyde on the way to toluic acid. Toluic acid is somewhat deactivated toward oxidation compared to p-xylene, but conversion of the second methyl group proceeds via a similar pathway, albeit at a slower rate.
A simplified flowsheet for one commercial process is given in Fig. 10.20. Air is compressed (1) and fed along with p-xylene, acetic acid solvent, and catalyst salts into a bubble column reactor or CSTR (2). The reaction occurs in the liquid phase at 175–230°C, 1.5–3.0 MPa. The exothermic heat of reaction is removed by evaporation of the reaction mixture. These vapors are carried into the water column (3), where the water of reaction is removed overhead, and off-gases are vented to a scrubber to prevent emission of bromine species. The underflow of the water column, comprising acetic acid is returned to the process. Conversion of p-xylene is typically above 99%, with greater than 98% selectivity to TPA. Small amounts of acetic acid (about 36 kg/metric ton of TPA) and p-xylene are lost by complete oxidation to carbon dioxide and water. The inlet air rate is controlled so that the off-gases contain less than the minimum oxygen composition needed to form explosive mixtures.

Flowsheet for oxidation of p-xylene to terephthalic acid
TPA is highly insoluble at reaction conditions and largely precipitates as it forms. Efficient absorption and dispersion of oxygen is critical for the reduction of partially oxidized intermediates which can cause purity problems in the final TPA. 4-Carboxybenzaldehyde (4-CBA), generated at up 5,000 ppm in the oxidizer, tends to occlude in the TPA crystals and must be removed to give high quality TPA monomer. A class of compounds generated in ppm levels, known as florenones, are highly colored and result in yellowed polymer if not removed or destroyed in the TPA purification process.
Upon exiting the oxidizer, the crude TPA slurry is centrifuged and washed with acetic acid (4). Recycle of the Co/Mn/Br catalyst and purging of by-products in the filtrate and wash streams typically involve proprietary processes (5) that are extremely important for good economics. In order to reduce 4-CBA and toluic acid to acceptable levels, the crude TPA is subjected to a series of long residence time post oxidations (6) with air at high temperature and pressure. Although still a slurry in the post oxidizer, the crystals continually remelt and recrystallize in a steady state fashion, allowing occluded impurities to be further oxidized in solution. The TPA slurry effluent from the post oxidizers is fed to the flash crystallizer (7) for concentration and to build crystal size. Further solvent is removed in the vacuum flash drum (8). Crystals are collected on the rotary vacuum filter (9) and the dryer (10) produces the final polymer grade TPA product with less than 200 ppm CBA and less than 10 ppm toluic acid. The high corrosivity of process streams necessitates the use of expensive titanium reactors and hastelloy in most other locations.
In another widely practiced process, the crude TPA from the centrifuge is dissolved in water at around 260°C. The aqueous TPA solution is hydrogenated over a Pd/carbon catalyst at high temperature and pressure to convert the 4-CBA to p-toluic acid and for color body destruction. The hydrogenated effluent is cooled, crystallized, centrifuged, washed, and dried to produce polymer grade TPA.
For many years the best method for purifying TPA to remove color bodies and monofunctional by-products was via esterification with methanol to produce dimethyl terephthalate (DMT), with subsequent distillative purification of DMT. However, the hydrogenation and post oxidation steps have demonstrated superior economics, although giving not quite as pure a product as the DMT route. DMT still finds use in a number of niche applications.
The production of IPA is very similar to TPA. However, IPA is 8–12 times more soluble (depending on temperature) in the reaction media than TPA, does not precipitate as readily, and has less issues with 3-CBA occlusion. On the negative side, IPA’s higher solubility requires more stringent crystallization and purging protocols for high recovery.
Benzoic Acid [128, 129]
In a common industrial process, benzoic acid is produced via the liquid-phase air oxidation of toluene with a homogenous cobalt catalyst at 130–170°C, 0.3–1.0 MPa.
The reaction is carried out normally in a bubble column or CSTR. The exothermic heat of reaction is removed by evaporation of the reaction mixture, or via a circulating cooler. Partial conversion of toluene is typical, with selectivity on toluene of about 90% to benzoic acid, 7–9% to benzaldehyde, and the remainder a mix of benzyl alcohol, benzyl formate, methyl biphenylsm and other minor by-products. Unreacted toluene is flashed off and benzoic acid is purified by distillation. The heavy ends are extracted to recover cobalt. High purity benzoic acid may also be produced by recrystallization from water. As with TPA, the reaction proceeds through a free-radical chain process, with a hydroperoxide intermediate. Cobalt shortens the induction period, and retards by-product formation. Oxygen in the off-gas is kept below the critical explosive concentration to ensure safe operation.
Benzoic acid may be produced with the same catalyst system (Co/Mn/Br) at similar conditions to TPA and IPA, with essentially complete conversion and high yield. However, the use of Br requires expensive titanium and hastelloy metallurgy, whereas the cobalt system can be practiced in cheaper stainless steels.
Phthalic Anhydride [130–133]
In the early 1960s naphthalene from coke oven gases was the predominant raw material for phthalic anhydride production. Since the mid-1990s, over 90% of phthalic anhydride has been made from o-xylene over TiO2-supported V2O5 catalsysts with promoters such as K, Cs, P, Sb, or Nb. The formation of phthalic anhydride from o-xylene is highly exothermic, with a heat of reaction of −1,109 kJ/gmole. Nonselective complete combustion is about four times more energetic per mole of xylene than the desired reaction. The reaction takes place at 360–390°C, less than 0.1 MPa.
Conversion of o-xylene is 92–96% per pass, with the molar yield on o-xylene is around 75–85%. Major by-products are CO2, benzoic acid, phthalide, o-toualdehyde, and maleic anhydride. Formation of potentially explosive feed and product mixtures dictates a very low o-xylene to air mass ratio of 1/60 to 1/120 (or about 1.2 mole% in air) [134].
A simplified flow sheet for the high temperature partial oxidation of ortho-xylene in air is given in Fig. 10.21. The o-xylene feed is vaporized into air and fed into the fixed bed tubular reactor (1) where the heat of reaction is controlled by shell-side generation of high pressure steam or exchange against molten salt. The reactor effluent, containing around 1% phthalic anhydride and other by-products, is cooled by steam generation (2), and desublimated in a device called a switch condenser (3) (a cyclically operated finned heat exchanger in which the anhydride is solidified and periodically melted). The crude anhydride is thermally treated (4) at 230–300°C to decompose or polymerize some of the by-products, followed by distillation. The first column removes light ends such as benzoic acid and maleic anhydride (5), and the product column (6) takes phthalic anhydride overhead from heavies. Off-gases are scrubbed with water (7), to recover maleic and fumaric acids.

Flowsheet for oxidation of o-xylene to phthalic anhydride
Peroxidations of Secondary or Tert-Alkyl Benzene Derivatives
Phenol/Acetone or MEK and Hydroquinone/Acetone [135–140]
Greater than 90% of phenol and acetone are produced worldwide by the air oxidation of cumene via a free radical chain mechanism in which cumene hydroperoxide (CHP) is the chain initiator. Footnote 1 The reaction is autocatalytic and reasonably exothermic. As shown in Fig. 10.22, cumene and air are reacted in two to four bubble columns in series (1) at 85–110°C, 0.5–0.8 MPa, to produce CHP at 25–30 wt% concentration in excess cumene.

Flowsheet for cumene hydroperoxidation to phenol/acetone
Temperature is controlled by evaporation of cumene and water as well as by cooling in external heat exchangers. Conversion of cumene is essentially complete with a selectivity of greater than 95% to CHP. Phenol is an inhibitor to oxidation, so cleavage to phenol is undesirable at this point. In some processes a small amount of sodium carbonate or sodium hydroxide is added to maintain a neutral solution and prevent the acid-catalyzed decomposition of CHP. Thermal decomposition of CHP (rapid above 130°C) leads to the by-products α,α-dimethylphenyl methanol (DMPM), acetophenone, methanol, formic acid, and acetic acid. Alkaline earth bismuthates, stannates, antimonates, and metal phthalocyanines are known to catalyze the reaction, but are not used industrially as they tend to also catalyze the decomposition of CHP to by-products. As both cumene and CHP are potentially explosive, the oxygen in the reactor off-gas is maintained at 1–6 volume %, below its critical concentration of 8.5–9.5% under reaction conditions.
The crude CHP from the bubble columns is vacuum-distilled (2) to remove cumene and concentrated to 65–85 wt% CHP. The concentrated solution is cleaved at 70–90°C in the presence of a strong acid catalyst such as sulfuric acid, to produce phenol and acetone in an exothermic reaction.
Temperature is controlled by boiling of acetone. Under these conditions DMPM dehydrates to α-methylstyrene (AMS), while diacetone alcohol and mesityl oxide form via aldol/dehydration of acetone. The cleavage mixture is neutralized with caustic or sodium phenolate, phase separated (4) and the organic layer is refined in a series of distillations. Many distillation sequences are possible and are practiced. One variation is shown in Fig. 10.22. The neutralized organic layer is fed to the crude acetone column (5) to distill acetone, mesityl oxide, water, lights, cumene, and AMS overhead from crude phenol and heavies. This distillate is further fractionated under vacuum in the acetone product column (6) to remove lights (aldehydes) overhead, acetone as a sidedraw near the top, and heavy water/hydrocarbon azeotropes out the bottom. Traces of caustic are sometimes added to this column to catalyze aldol condensations of aldehyde impurities into higher boiling, easily separable materials. The bottoms stream separates into two liquid phases (7). The organic phase is distilled to remove low-boiling AMS-cumene-phenol azeotropes overhead (8). After removal of heavies in the AMS column underflow (9), the AMS is subjected to hydrogenation (10) over Ni, Cu, or Pd catalysts and converted back into cumene for recycle.
In the crude phenol column (11), the bulk of the phenol is removed overhead with light organics. The crude phenol bottoms further distilled (12) for additional phenol recovery, with the residue used as fuel. Hydrocarbons are azeotroped out of the distilled phenol (13) with water and the bottoms phenol product is passed through a resin bed (14) to convert any carbonyls present to heavy ends. This treated material is subjected to a final distillation (15), with trace hydrocarbon/water removal overhead, and phenol product recovered as a sidedraw.
Decanter water streams are acidified, if needed, to convert the sodium phenolate contained therein back into phenol. Phenol is then recovered by extraction into cumene or other suitable solvents, distilled, and recycled. Overall selectivity on cumene is about 97.5%. Many other process variations are practiced.
Hydroperoxide Co-product Processes for Propylene Oxide
Although differing from ethylene oxide by only one extra carbon in the backbone, propylene oxide (PO) has not been made successfully at commercial scale by the direct oxidation of propylene. The presence of allylic hydrogens dramatically and adversely affects the efficacy of silver-based catalysts for the selective oxidation of propylene to PO with oxygen [141]. In spite of significant research over the last several decades, no catalysts have been found that show commercially viable yields or rates. Instead, all world scale PO plants rely on indirect oxidation of propylene by either the chlorohydrin route (50% of total, see the section, “Propylene Oxide via Propylene Chlorohydrin”) or co-product hydroperoxide routes (50% of total, described herein).
Styrene/Propylene Oxide Co-production [142]
About 15% of commercial styrene is produced by the rather complex air oxidation of ethylbenzene with coproduction of propylene oxide. The remaining 85% is produced via dehydrogenation of ethylbenzene (see the section, “Styrene from Ethylbenzene”). The process consists of four steps:
-
1.
Ethylbenzene is oxidized directly by air to produce ethylbenzene hydroperoxide at around 130°C, 0.2 MPa, and 13% conversion per pass. Yields are about 90% to the hydroperoxide, 5–7% to acetophenone and α-methylbenzyl alcohol.
-
2.
The hydroperoxide is reacted with propylene (molybdenum or titanium catalysts) at about 110°C, 4.0 MPa, with essentially complete peroxide conversion to give α-methylbenzyl alcohol and propylene oxide. Propylene oxide and ethylbenzene for recycle are separated by distillation from the acetophenone and α-methylbenzyl alcohol.
-
3.
α-Methylbenzyl alcohol is dehydrated in the vapor phase at about 250°C and low pressure over an acidic oxide such as alumina, to produce styrene. Styrene is purified by distillation.
-
4.
Recovered acetophenone is hydrogenated in the liquid phase (Zn-Cu oxide catalyst) at 90–150°C, 8.0 MPa, to give α-methylbenzyl alcohol at 92% selectivity.
This route offers a pathway to propylene oxide without chlorohydrin production, but produces a weight ratio of styrene to propylene oxide of 2.5 to 1, which does not match the relative market demands of the two products and is quite capital intensive.
Tert-Butanol/Propylene Oxide Co-production [143]
This route is similar to the styrene co-product process:
-
1.
Isobutene is oxidized in the liquid phase with pure oxygen to produce tert-butyl hydroperoxide at around 130°C, 2.5–3.5 MPa, and 15–40% conversion per pass. Yields are about 50% to the hydroperoxide, 46% to tert-butanol, and a minor amount of aldehydes and ketones.
-
2.
The hydroperoxide is reacted with propylene at a 1:10 M ratio (molybdenum catalyst in toluene) in a two-stage process. The first stage is held at about 110°C, 4.0 MPa, with the second stage elevated to 120°C to ensure essentially complete peroxide conversion. Propylene conversion is about 9% per pass at greater than 90% selectivity. Propylene and other lights are removed overhead by distillation for recycle, followed recovery of crude propylene oxide overhead and tert-butanol, catalyst, and other higher boiling by-products as underflow.
-
3.
Production of high purity PO requires further distillation to remove lights such as ethylene oxide, and extractive distillation with a heavy hydrocarbon to remove close-boiling or azeotrope-forming hydrocarbons or carbonyls.
This route also offers a pathway to propylene oxide without chlorohydrin production, but produces a weight ratio of tert-butanol to propylene oxide of about 3 to 1. Economic utilization of the tert-butanol relies on demand for methyl-tert-butyl ether, or other secondary isobutylene markets.
Ethylene Oxide [144–149]
Ethylene oxide (EO) is one of the most versatile and reactive chemicals produced commercially at large scale. Formerly, EO was produced via the chlorohydrin of ethylene, similar to propylene oxide (see below). This route has been abandoned commercially in favor of the direct oxidation of ethylene. The use of high purity oxygen (>99%) instead of air as the source of the oxidant has become standard. A simplified flowsheet of a typical oxygen-based EO plant is shown in Fig. 10.23. Ethylene is reacted with compressed (1) oxygen over a silver-based catalyst in a multi-tubular fixed bed reactor (2), with a hot spot temperature of 250–275°C, 1–2.2 MPa pressure.

Flowsheet for oxidation of ethylene to ethylene oxide
The complete combustion of ethylene to CO2 and water releases −1,688 kJ/gmole. Commercial selectivities range from 80 to 90% on ethylene over the lifetime of the catalyst, resulting in a net heat load of 230–450 kJ/gmole. Ethylene conversion is 7–15% per pass. The off-gas from the reactor typically comprises 1–2 vol% EO, 5–6 vol% CO2, 5–9 vol% O2, 15–40 vol% ethylene, 5–15% inerts (Ar, N2, ethane), and 1–60 vol% methane. The methane ballast gas acts as a heat sink and keeps the recycle gas above the upper explosive limit at the oxygen-recycle gas mix point.
The reactor effluent is cooled and EO is absorbed into water (3), or in some specialized plants into ethylene carbonate. The EO is recovered by distillation (4, 5, 6) from the water, if desired in pure form. Alternatively, the EO is reacted with water to produce a mixture of mono-, di-, and tri-ethylene glycols directly, or with CO2, then water, to produce higher purity monoethylene glycol (see Section 10.8 below). The safe handling of concentrated EO requires an appreciation of its volatility (normal boiling point of 10.8°C), and ability to form explosive mixtures without an additional oxidant (the upper explosive limit goes up to 100% EO) [150, 151].
Carbon dioxide is a modest reversible retardant to the epoxidation reaction; a slipstream of EO-free recycle gas is treated with hot potassium carbonate to absorb out the CO2 (7). The CO2-rich absorbent is regenerated by vacuum steam stripping (8) [152]. A small purge is also removed to prevent the build-up of inerts from the ethylene and oxygen feeds, before recompression and recycle (9).
The only effective catalyst discovered thus far for this epoxidation reaction comprises silver on ultrapure γ-alumina, typically with a cesium promoter. Catalyst selectivity has dramatically improved over the past 40 years, from about 65% in 1966 to a maximum of about 90% today. A small continuous feed of a chlorinated hydrocarbon (1–10 ppmv), such as 1,2-dichloroethane, vinyl chloride, or ethyl chloride helps to suppress the combustion of ethylene, significantly improving yield and controllability. The beneficial effect of the chlorinated hydrocarbon was found serendipitously in the early days of commercial silver-catalyzed ethylene oxidation. The air intake for an EO plant was located near the off-gas outlet of a vinyl chloride plant. Unexpected improved performance of the EO reactor was eventually traced back to small quantities of chlorinated hydrocarbons present in the inlet air.
By adjustment of the Cs/Ag ratio on the catalyst, the analogous epoxide of butadiene, 1,2-epoxybutene, can be made by a similar vapor phase oxidation process [153].
Wacker Oxidation: Acetaldehyde [154–158]
Acetaldehyde, a very reactive and versatile molecule, historically was an important intermediate for the production of a wide variety of chemicals. Large volume applications included:
-
(a)
n-Butanol, via aldol condensation to crotonaldehyde, followed by hydrogenation—now supplanted by hydroformylation of propylene (see section “Hydroformylation, Lower C2-C5 Olefins”).
-
(b)
Terephthalic acid, added as a peroxidant rate accelerator for the oxidation of p-xylene to TPA—now supplanted by more effective Co/Mn/Br catalyst systems.
-
(c)
Vinyl acetate, via reaction with acetic anhydride or ketene to form ethylidene diacetate (EDA), with subsequent cracking to VAM—now supplanted by oxidative esterification of ethylene with acetic acid (see section “Esters via Oxidative Acetylation of Olefins, VAM”).
-
(d)
Acetic acid, via oxidation (see section “Oxidation of Aldehydes to Acids”)—largely supplanted by carbonylation of methanol (see “Acetic Acid”).
-
(e)
Ethyl acetate, via Tishchenko reaction (see section “Tishchenko Reactions: Ethyl Acetate and 2,2,4-Trimethyl-1,3-Pentanediol Derivatives”)—still practiced, competes against acetic acid-ethanol esterification (see section “Low-Boiling Esters”).
-
(f)
Pyridine derivatives, via reaction of paraldehyde (acetaldehyde cyclic trimer) with aqueous ammonia and formaldehyde [159]—still a significant outlet for acetaldehyde. Current applications are dominated by the latter three, while the first three, once the biggest uses, are essentially gone.
Acetaldehyde has been made by dehydrogenation of ethanol over Ag (similar to formaldehyde, See section, “Formaldehyde”). However, much of the remaining world capacity is based on the homogeneous PdCl2/CuCl2-catalyzed direct oxidation of ethylene with air developed in the late 1950s. The catalytic cycle begins with the complexation of Pd with ethylene. Water addition leads to hydride transfer and reductive elimination to produce acetaldehyde, reduced Pd, and hydrochloric acid. Cupric chloride reoxidizes the reduced Pd, regenerating the active Pd species. Finally, cuprous chloride is re-oxidized by oxygen back to Cu(II). The net reaction is the addition of 0.5 moles of oxygen to ethylene.

The palladium catalyzed oxidation of an olefin is highly efficient and only a small concentration of expensive Pd is required for commercially viable rates if sufficient (and comparatively inexpensive) reoxidant is present.
Both one-stage (ethylene reaction and entire redox cycle occur in one vessel) and two-stage (ethylene reaction and reoxidation of cuprous chloride with air in separate vessels) processes have been commercialized, although the two-step process was and is more prevalent. In a typical commercial two-step process, as illustrated in Fig. 10.24, ethylene gas is mixed with freshly reoxidized aqueous catalyst solution and passed through a tubular serpentine reactor (1) at 105–110°C, 0.9–1.0 MPa, where acetaldehyde is formed with close to 100% conversion of ethylene. The catalyst solution contains a substantially higher molar concentration of Cu(II) than Pd (typically 150 to 200 to 1), allowing multiple Pd turnovers per reactor pass. The reactor effluent is then flashed (2) to remove most of the acetaldehyde as vapor, along with water, and by-products, prior to the lean reduced catalyst solution entering the second tubular reactor (3). Air is compressed to about 1.0 MPa and allowed to react with Cu(I) ions at about 100–110°C, to regenerate Cu(II). Recycled and make-up water is added to maintain the catalyst concentration before reentering the ethylene reactor. Conversion of ethylene is close to 99%, with a selectivity of greater than 94%. By-products include chloroacetaldehydes (1–2 mole%), ethyl chloride, chloromethane (0.5–1.0%), carbon dioxide (~0.8%), and acetic acid (2–4%) resulting from over-oxidation of acetaldehyde.

Flowsheet for Wacker oxidation of ethylene to acetaldehyde
The crude acetaldehyde is purified by a series of distillations. Acetaldehyde is concentrated in the crude column (4), with underflow returned as reflux to the flash tower. In the light ends column (5), low-boiling substances (chloromethanes, chloroethane, carbon dioxide) are removed overhead. Aqueous acetaldehyde and high boilers are distilled further in the product column (6), with high purity acetaldehyde collected as distillate, water/acetic acid as underflow. A fraction of the underflow is purged to prevent buildup of acetic acid, the rest recycled. Mid-boiling chloroaldehydes are removed as azeotropes with water in a sidedraw. This stream may be further concentrated if desired in a sidedraw stripper. The catalyst solution contains appreciable free hydrochloric acid and is quite corrosive. Significant parts of the plant must be constructed of corrosion-resistant materials such as titanium. Small amounts of chloride are lost in the chlorinated by-products. A constant makeup of chloride is fed as hydrochloric acid.
This process may also be adapted for higher olefins, with production of the corresponding internal carbonyl (i.e., ketone) predominating. Thus, propylene gives acetone and 1-/2-butenes give methyl ethyl ketone (MEK), typically 88–92% yield on the olefin. Acetone was produced commercially in Japan for a number of years via the Wacker oxidation of propylene, but the plant has subsequently shut down.
Considerable literature has been published in the past two decades on the use of vanadium-molybdenum-phosphorus homogeneous heteropolyacids (HPA) as replacements for the copper-based reoxidant. The required PdCl2 concentration is considerably reduced, HCl concentration is essentially zero, and formation of chlororinated by-product species is claimed to be virtually eliminated [160, 161]. No commercialized applications of HPA systems are known.
Acrolein/Acrylic Acid and Methacrolein/Methacrylic Acid [162–165]
Acrylic and methacrylic acids are produced commercially by similar two-step oxidations of propylene and isobutene/isobutene. Propylene oxidation is now the dominant route for acrylic acid, while several methacrylic acid processes are competitive including acetone cyanohydrin (see Chaps. 14 and Chaps. 22), propionaldehyde/formaldehyde aldol/oxidation (see section “Mannich Base Condensation: Methacrolein”), ethylene carbonylation, and isobutene oxidation. For acrylic acid, propylene/air/steam at a ratio of 1:8:4 (plus recycled gas) is fed over a bismuth molybdate oxide catalyst in a multi-tubular fixed bed reactor at 300–400°C, 0.15–0.25 MPa.
Conversion of propylene is greater than 95% per pass, with a yield of 80% acrolein, 5–10% acrylic acid. By-products are CO, CO2, acetic acid, acetaldehyde, and polyacrolein. The reactor is cooled by molten salt circulation and steam generation. A world scale reactor with 40,000 tubes in a single shell can produce 100,000 metric tons/year, with a catalyst lifetime up to 10 years.
Pure acrolein can be obtained by water washing of the reactor gases to knock out acids, followed by quench absorption in water. The crude, wet acrolein is distilled overhead, with additional steps for light/heavy ends removal. Acrolein is prone to oligomerization; hydroquinone or a derivative is added as a stabilizer.
More commonly acrylic acid is the desired end product. The crude acrolein gaseous effluent is sent directly to a second salt/steam cooled multi-tubular fixed bed reactor operating at around 260°C to complete the oxidation to acrylic acid.
Conversion of acrolein is essentially 100% at greater than 90% yield. Modern catalysts comprise Molybdenum–vanadium oxides with copper, tungsten, or cerium promoters. The reactor effluent is cooled further by steam generation.
Many different approaches are practiced for the recovery and purification of the acrylic acid. In one variation, the crude acrylic acid is quenched and absorbed into water to produce a 20–70 wt% solution in water. The acrylic acid is concentrated by extraction into an organic solvent (examples are ethyl acetate, MEK, butyl acetate, ethyl acrylate), followed by azeotropic distillation of the extract phase. Water is removed overhead as the solvent-water azeotrope, and the dry acrylic acid bottoms product is distilled overhead in a second column, with oligomers, dimers, and polymers taken as underflow. Additional acrylic acid is recovered from the heavy ends by decomposition of acrylate dimers in an evaporator.
In a second recovery scheme, the cooled acrylic acid reactor effluent is absorbed into a high boiling ester rather than water. This eliminates the extractor, but azeotropic distillation is required for drying, and the acrylic must be distilled from the ester. In both processes, an inhibitor (phenothiazone, hydroquinone, or hydroquinone monomethyl ether) is added to prevent polymerization of the acrylic acid.
In an analogous process for methacrylic acid, isobutene/air/steam at a ratio of 1:11.5:7.5 (plus recycled gas) is fed over a bismuth molybdate oxide catalyst at about 350°C, followed by second stage oxidation over Mo–V oxides at about 280°C
The multi-tubular fixed bed reactors are cooled by molten salt circulation and steam generation. Conversion of methacrolein is around 90% per pass to keep selectivity to 80–90% methacrylic acid. Product recovery by distillation is similar.
KA Oil: Cyclohexanone/Cyclohexanol [166–168]
A mixture of cyclohexanone and cyclohexanol known as KA (Ketone-Alcohol) oil is made predominately by the liquid phase cobalt-catalyzed air oxidation of cyclohexane at 140–160°C, 0.8–2.0 MPa. A typical catalyst is the soluble Co(II) naphthenate, which gives a ketone to alcohol ratio of 1:1 to 1:3.5. Conversion must be kept low (about 5–7% per pass) in order to achieve selectivities as high 75–77%. The addition of chromium increases the ketone to alcohol ratio close to 2:1, while promotion with boron compounds such as boric anhydride (B2O3), boric acid (H3BO3), or metaboric acid (HBO2) results in a ketone to alcohol ratio as low as 1:10 and increased selectivity to 85–90%. Stoicheometric amounts of slurried borate are required and must be recycled. In an alternative approach, the cyclohexyl hydroperoxide intermediate is formed in an initial uncatalyzed air oxidation step, followed by decomposition in the presence of cobalt or other transition metal catalyst. Yields as high as 84% are achievable with the high hydroperoxide route. In all process variations, the crude KA oil is purified by water extraction for removal of dibasic acids such as adipic, glutaric, and succinic, as well as C4-C6 carboxylic and hydroxycarboxylate acids, followed by distillation.
KA oil is an important intermediate for the production of both adipic acid and caprolactam for nylon-6,6 production (see Chap. 22). Purified cyclohexanol and cyclohexanone both find limited uses as solvents.
Maleic Anhydride [169–172]
Prior to 1975, benzene was the predominant raw material for the production of maleic anhydride. Currently worldwide, over 80% of maleic anhydride is made from butane or butane/butene mixtures of high alkane content. About 2% is recovered as a by-product of phthalic anhydride production, and the remainder is derived from benzene. The formation of maleic anhydride from either benzene or butane is highly exothermic.
Nonselective complete combustion of benzene (−3,136 kJ/gmole) and butane (−2,655 kJ/gmole) are much more energetic than the desired reaction.
Benzene is oxidized in air over V2O5 or MoO3 catalysts in a multi-tubular fixed bed reactor at 340–500°C, 0.15–0.25 MPa, with molten salt-steam generation for heat control. The benzene content is kept below the lower explosive limit (LEL). Typically more steam is generated than is needed in the process. Selectivity to maleic anhydride is around 73 mole% at 96% conversion per pass.
Typical conditions for the air oxidation of n-butane are a temperature of about 400°C, pressure of 0.15–0.25 MPa, with 80% conversion per pass, at about 70% selectivity to maleic anhydride. Vanadyl pyrophosphate, (VO)2P2O7, often bound with TiO2, SiO2, or Al2O3, (with the latter two supports used for abrasive service in fluid beds) is the only material found thus far with high enough selectivity to be used commercially for butane oxidation. Multi-tubular fixed bed, fluid bed, and transport reactors have all been used for this service. The inlet gas to a multi-tubular reactor is kept below the LEL, about 1.8% butane, while the excellent heat control and thermal mass of the circulating catalyst in the fluid bed design allows for operation in the explosive regime at close to the stoichiometric O2 to butane ratio (about 5.5% butane). As a result, fluid bed processes have much smaller equipment in the reactor loop, and are favored for new installations.
Two alternative recovery and purification are practiced commercially. The reactor effluent is further cooled by generating lower pressure steam, and then is absorbed into either an organic solvent or water. If water is chosen as the absorbent, a substantial amount of the maleic anhydride may be hydrolyzed to maleic acid. The maleic acid/anhydride mixture must then be converted back. A common method for water removal is heterogeneous azeotropic distillation with an entrainer such as xylene. Maleic acid is prone to thermal isomerization to fumaric acid, a high melting (286°C) by-product. Thus, residence time at the high temperatures in the dehydrating column base must be kept to a minimum. The resulting concentrated maleic anhydride solution is then distilled to remove lights and traces of solvent, and then taken overhead to remove heavies.
When a high-boiling, water-immiscible solvent, such as dibutyl hexahydrophthalate, is used for absorption, much less maleic anhydride is converted into the acid. No separate dehydration step is required, resulting in much lower energy requirements. Maleic anhydride is stripped overhead from the rich solvent stream from the absorber. This crude anhydride is distilled to separate light and heavy impurities. A slipstream of lean recycle solvent is treated to eliminate any heavy by-product that may build up.
The transport process, commercialized in 1996, was revolutionary in its use of separate butane oxidation and catalyst reoxidation zones. The butane was oxidized by the oxygen lattice of the catalyst, which was then regenerated with air in a separate vessel. Air and butane were never mixed. However, an unexpectedly high make rate of fumaric acid in the transport reactor system caused severe operational difficulties and poor plant availability. These problems led to the permanent shut down and dismantling of the one and only commercialized transport reactor process in 2004 [173].
Formaldehyde [174–179]
The production of formaldehyde from methanol illustrates that many different process alternatives, such as choice of catalyst, conversion per pass, separation selection, and safety considerations, can lead to a commercially practiced, economically viable process. With the mixed metal oxide catalyst (Fe/Mo, often with Cr promoter), formaldehyde is produced via the endothermic oxidative dehydrogenation of methanol:

The additional pathway of equilibrium-limited, endothermic dehydrogenation occurs simultaneously in a ratio of 40% dehydrogenation, 60% oxidative dehydrogenation on silver catalysts:

In a typical silver catalyst process, a mixture of air–steam–methanol is reacted adiabatically over a thin layer (2.5–3.0 cm thick) of silver gauze or crystals at slightly above atmospheric pressure and high temperature. The formaldehyde-rich effluent is cooled initially by steam generation, and formaldehyde is recovered from the gas in a water-fed circulating quench absorber. The hydrogen-rich absorber tail gas is burned for steam generation. Both complete and partial methanol conversion processes are practiced commercially. In the complete conversion process, the feed gas comprises sufficient methanol to be above the upper explosive limit (25–27 mole% methanol), along with 46–54 mole% air, and 20–30 mole% steam. Conversion is greater than 99% at reaction temperature of 600–720°C, at a selectivity of 89–91%. A product of 40–55 wt% formaldehyde with <1 wt% methanol can be produced without further distillation. In the partial conversion process a mixture of 40–45% methanol, 20–25% air, and 20–35% steam is adiabatically converted to formaldehyde at 550–650°C. Methanol conversion is 70–80%, with a selectivity of 90–92%. Distillation is required to concentrate the product formaldehyde and remove unreacted methanol for recycle. Some vendors offer as high as 68 wt% formaldehyde with less than 1 wt% methanol.
The mixed metal oxide process operates below the LEL with 9 mole% methanol in air diluted with recycled off-gas and steam. The methanol is converted in a non-isothermal, multi-tubular fixed bed reactor at 350–390°C, where the heat of reaction is controlled by shell-side generation of high pressure steam. Formaldehyde is recovered by absorption into sufficient water to produce a solution of 50 wt% formaldehyde, with less than 1 wt% methanol without further distillation. Conversion is greater than 99%, at a selectivity of 93–96%. Purged gas does not contain sufficient combustibles and must be incinerated or catalytically oxidized with supplemental fuel. Formaldehyde is susceptible to oligomerization in the liquid phase and must be kept warm if concentrated, and is often stabilized with the addition of a small amount of methanol [180, 181].
Esters via Oxidative Acetylation of Olefins, VAM [182–186]
Many routes for the production of the important monomer vinyl acetate (VAM), the unsaturated two-carbon ester of acetic, have been developed. These include:
-
1.
Addition of acetic acid to acetylene form VAM directly (once a significant commercial process)
-
2.
Reaction of acetic anhydride with acetaldehyde to form EDA, with subsequent cleavage to VAM and acetic acid (also once a significant commercial approach)
-
3.
Carbonylation of methyl acetate with CO and H2 to form EDA, with subsequent cleavage (never commercialized fully)
-
4.
A vapor or liquid phase reaction of acetic acid, ethylene, and oxygen to form VAM and water
The latter route, as a gas phase reaction dominates commercial practice. In the typical industrial process acetic acid is vaporized and mixed with a fresh and recycle stream of ethylene. The combined gas is then mixed with high purity oxygen in a highly reinforced and shielded mixing unit. The feed gas to the reactor typically comprises 10–20 mol% acetic acid, 10–30 mol% CO2, 10 mol% N2, Ar or other inerts, 50 mol% ethylene, and less than 8 mol% oxygen. This composition is above the upper explosive limit.
The exothermic reaction occurs in a multitubular fixed bed reactor over a silica supported Pd/Ag/K catalyst at 140–180°C, 0.5–1.2 MPa. Steam is generated on the reactor shell side for heat removal and control of the hot spot temperature. By-products are water, carbon dioxide, and small amounts of EDA, ethyl acetate, acetaldehyde, methyl acetate, and glycol acetates.
The reactor effluent is interchanged with the incoming feed gases and fed to a predehydration column, where about 50% of the water contained therein is removed by azeotropic distillation using the hot incoming gases for boil-up. The water/VAM azeotrope is taken overhead, decanted and VAM refluxed to the column. The crude VAM in the bottoms stream (20–40% VAM, 6–10 wt% water, the rest acetic acid and by-products) is dehydrated further by a similar water/VAM heterogeneous azeotropic distillation, with the VAM phase refluxed. The aqueous phase is further steam stripped to recover VAM for recycle to the first column and wastewater discharged as underflow. Final purification occurs in a third column, with the product VAM taken overhead, ethyl acetate as a sidedraw, and acetic acid and heavies (EDA, glycol acetates) as the bottoms. The acetic acid is recycled, with a heavy purge taken from the sludge of the acetic acid evaporator.
The ethylene-rich off-gas from the predehydrator is largely recycled, but a fraction is purged to prevent build-up of argon and nitrogen, and another portion is scrubbed with water to remove acetic acid prior to contacting with hot potassium carbonate system for carbon dioxide removal. Per pass conversion of ethylene, acetic acid, and oxygen range from 8–15%, 15–35%, and up to 90%, respectively. Selectivities of 94–96% on ethylene and greater than 98% on acetic acid are achievable.
Oxidation of Aldehydes to Acids [187]
The principle industrial route to C3-C10 carboxylic acids is by the homogeneous liquid phase oxidation of the corresponding aldehyde, due to their wide availability from hydroformylation.
Examples of acids produced at large scale by aldehyde oxidation are propionic, n-butyric, isobutyric, n-valeric, isovaleric, and 2-ethylhexanoic acid. This chemistry typically is not sufficiently selective for use with unsaturated aldehydes (e.g., acrolein, methacrolein). Other methods are used for these substrates (see above, this section).
The oxidation may be carried out with oxygen or more typically with air, usually without an additional solvent, and with or without a catalyst present. Oxidation of a branched-chain aldehyde, exemplified by 2-ethylhexanal, is extremely sluggish and non-selective without a catalyst, while straight chain moieties can be oxidized successfully without a catalyst. The most effective catalysts are primarily transition metal ions that can transfer only one electron, and have two valence states of equal stability to allow efficient oxidation/reduction, such as CuI/CuII, CoII/CoIII, and MnII/MnIII. The metal is most conveniently introduced into the reaction system as the carboxylic acid salt of either acetic acid (commonly available) or as the salt of the acid to be produced. Although these oxidations typically give greater than 90–95% selectivity, common types of by-products include formate esters, ketones, and n−1 hydrocarbons and alcohols.
A transition metal-catalyzed oxidation reaction proceeds through a number of identifiable steps: (1) a hydrogen atom is abstracted from an aldehyde molecule by a metal ion to form a carbonyl radical, with reduction of the oxidation state of the metal; (2) dioxygen adds to the carbonyl radical to form a percarbonyl radical; (3) the percarbonyl reacts with another aldehyde molecule to form a peracid and regenerates a carbonyl radical (chain propagation); (4) the peracid reacts with another aldehyde molecule to form two acid molecules; (5) the metal is returned to its original oxidation state via reaction with a peracid, with formation of an acid molecule. The catalyst acts as an initiator for rapid generation of a high flux of carbonyl radicals, but also can inhibit the reaction if at too high a concentration (too many radicals, not enough free aldehyde to accomplish step 3). Typically there is an optimal catalyst concentration that balances radical chain formation and decomposition. Without a catalyst, there is often a lengthy induction period, before sufficient radials are generated to kick off the chain mechanism.
In a generic industrial process illustrated in Fig. 10.25, air, the aldehyde substrate, and the catalyst are fed to a bubble column reactor or CSTR (1) at 50–150°C, close to atmospheric pressure. Conversion of aldehyde is 80–95%, depending on the substrate. The off-gas from the reactor is cooled to remove condensable species, separated into vapor and liquid phases (2), and off-gases are scrubbed to remove traces of organics before emission to the atmosphere. Temperature, air, and aldehyde flow are adjusted to ensure that the exit gas is outside of the explosive region.

Flowsheet for oxidation of aldehydes to carboxylic acids
Liquid product from the reactor, containing unreacted aldehyde, product acid, catalyst, and a range of high, medium, and low boiling by-products is distilled (3), with materials boiling lighter than the product acid taken overhead and acid, catalyst, heavies as bottoms product. Depending on the by-product slate (some oxidations produce formate esters and ketones boiling closely to the aldehyde), the distillate of the crude column may be further treated in one or two columns to remove lights (4), medium boilers (5), and to recycle unreacted aldehyde. The crude acid is taken overhead in the product column (6), with concentrated catalyst underflow for recycle, less a purge for heavy ends.
Carbonylation
Chemistry
Carbonylation is a catalytic reaction between carbon monoxide (CO) and an organic substrate to form a new carbon–carbon bond with the introduction of an oxygen-containing functional group into the product. Technically any reaction involving CO alone or in tandem with another molecule, e.g., H2 or H2O is a carbonylation. However, historically reactions involving the insertion of CO and H2 commonly have been known as hydroformylation or oxo reactions. This important class of carbonylations is treated separately in the section, “Hydroformylation.” In this section, the types of carbonylation reactions highlighted are:
-
1.
Liquid phase reaction of CO with methanol or methyl acetate catalyzed by homogeneous Rh or Ir complexes to form acetic acid or anhydride
-
2.
Liquid phase reaction of CO with methanol catalyzed by a strong base to form methyl formate
-
3.
Liquid phase reaction of CO and ethylene with methanol over a strong base catalyst to form methyl propionate
-
4.
Liquid phase reaction of CO, water, and olefins catalyzed by a strong acid to form neo acids, also known as Koch carbonylation or hydrocarboxylation
Acetic Acid [188–193]
The carbonylation of methanol has largely displaced earlier synthetic routes to acetic acid such as partial oxidation of hydrocarbons, fermentation of carbohydrates, and oxidation of acetaldehyde derived from ethylene. Methanol carbonylation, a net insertion of carbon monoxide into methanol, has been known since 1913 and practiced commercially since the 1950s, initially as a high temperature, high pressure process (250°C, 70 MPa) using cobalt iodide as a catalyst. The breakthrough for methanol carbonylation occurred in the late 1960s with the discovery of rhodium catalysts promoted with methyl iodide, allowing operation at much lower temperatures and pressures and much higher productivity and yield than the original cobalt systems.

Under reaction conditions, the active catalyst is the [RhI2(CO)2]− anion. Oxidative addition of methyl iodide (formed from HI and methanol) [RhI2(CO)2]− followed by insertion of CO into the Rh-CH3 bond and coordination of CO yields [CH3C(O)RhI3(CO)2]−. Reductive elimination of acetyl iodide (CH3COI) liberates the original [RhI2(CO)2]− complex, and rapid hydration of acetyl iodide produces acetic acid and regenerates HI. The reaction is zero order in methanol and CO and first order in methyl iodide and rhodium. The concentration of methyl iodide in the reactor is substantial, around 20–25 wt%. With the original Rh—methyl iodide system, high water concentration (14–15 wt%) is required for high activity and for Rh stability. This high water level also promotes the water-gas shift reaction, resulting in byproduct CO2 and H2. Propionic acid is the major heavy byproduct, formed primarily via carbonylation of ethanol resulting from small quantities of acetaldehyde formed by acetic acid reduction or homologation of methanol.
Many catalytic advances have been made since the 1970s, enhancing selectivity, productivity, and reducing energy usage. The four basic catalytic systems used in practice today are summarized in Table 10.30. The latter three allow high selectivity and productivity at much lower water content in the reactor loop than the original Rh-methyl iodide system. The water-rich end of acetic acid–water vapor–liquid equilibrium is fairly pinched, the heat of vaporization of water is high, and all the water present in the reactor loop must be distilled overhead, so low water content translates into substantial energy and capital savings.
The addition of LiI greatly enhances rates by assisting in formation of methyl iodide from methanol and stabilizes the rhodium complex under low water conditions. Use of a heterogeneous rhodium catalyst, addresses many of the drawbacks of the original homogeneous Rh system. The rhodium is immobilized in a highly stable state on a polyvinyl pyridine resin. The bound, catalytically active [RhI2(CO)2]− remains stable at low water levels, and without LiI addition. The reaction is accomplished in a three phase gas lift reactor. By-product propionic acid, CO2 and H2 production are reduced.
Iridium-based carbonylation has been commercialized fairly recently by BP (the Cativa process), and is yet a third method to reduce water content. The rate of methyl iodide addition to the active [IrI2(CO)2]− complex is about 150 times faster than to [RhI2(CO)2]−. Insertion of CO to form the iridium acetyl species is the slow step, but is accelerated by addition of Re or Ru promoters. Substantial Ir concentrations are required for good rates, but low water operation is possible with lower methyl iodide concentrations and without LiI addition. With Ir, production of propionic acid, CO2 and H2 is reduced, but methane formation is higher.
A flow sheet for a typical modern acetic acid plant is shown in Fig. 10.26. High purity carbon monoxide, methanol, catalyst, and promoters (methyl iodide and others), are introduced into a CSTR or gas lift (thermosyphon) reactor system (1) operated at (150–200°C, 2–4 MPa). The exothermic heat of reaction is controlled by flashing of the reactor effluent to produce a crude acetic acid vapor product and a catalyst residue for recycle to the reactor (2). Noncondensable by-products, (methane, hydrogen, carbon dioxide) are vented from the reactor loop to control the CO partial pressure. These off-gases must be thoroughly scrubbed, typically by a staged absorption system of methanol, acetic acid, water to ensure that no iodine containing species are released (7).

Flowsheet for carbonylation of methanol to acetic acid
The crude acetic acid from the flash unit is sent to the light ends column (3) where methyl iodide, water, and methyl acetate are taken overhead as a two phase mixture (4) for recycle to the reactor. Bottoms from this column are recycled to the reactor and wet acetic acid is taken as a vapor side draw above the feed. The wet acid is dehydrated in another column (5), with dry acid as underflow, and water/acetic acid distillate for recycle. The product column (6) produces glacial acetic acid as a sidedraw high in the column. Overhead material is recycled and by-product propionic acid and heavies are purged as underflow. Overall selectivity on methanol is typically 99% and greater than 90% on carbon monoxide.
Although highly selective and productive, Rh and Ir are very expensive, requiring millions of dollars for catalyst inventory. Moreover, the processes require high metallurgy (many parts Hastelloy) and appropriate systems to handle volatile iodide species.
Acetic Anhydride [194–196]
Acetic anhydride is an important acetylating agent for the production of such materials as cellulose acetate esters, ibuprofen and other pharmaceuticals, and hard-to-esterify esters. The predominant route to acetic anhydride involves the thermal cracking of acetic acid to ketene and water, followed by the reactive absorption of ketene into acetic acid (see the section, “Ketene”). However since 1983, a significant fraction of acetic anhydride (about 20% worldwide, over 50% in the USA) is produced via the carbonylation of methyl acetate:
The carbonylation of methyl acetate is similar in many respects to the carbonylation of methanol described above, but differs in some key aspects:
-
1.
Methanol carbonylation is done in an aqueous medium. Anhydride production necessitates anhydrous conditions, with significantly reduced Rh stability/solubility at reaction conditions.
-
2.
Hydrogen addition is required to generate and maintain the active catalyst form. Unlike in the aqueous methanol system, no water is available for in situ hydrogen generation via water-gas shift.
-
3.
LiI promoter is absolutely required to generate the active Rh catalyst species and to activate the alkyl methyl group of methyl acetate under anhydrous conditions.
-
4.
Methyl acetate carbonylation is equilibrium-limited, with conversion of methyl acetate between 50 and 75% vs. essentially 100% in methanol carbonylation.
-
5.
The by-product slate is different. EDA, “tars,” acetone, and CO2 are the primary by-products. Propionic acid is not formed to any extent, nor is methane.
-
6.
Production of “tars,” primarily derived from by-product EDA, is significantly higher under methyl acetate carbonylation conditions. These tars must be purged and tend to bind Rh more effectively than acetic acid residue. A much more complex tar purge/Rh recovery system is required.
In the anhydride production process, methyl acetate, recycle methyl iodide, catalyst, CO, a small amount of H2, and LiI promoter are fed to a back-mixed reactor system at 160–190°C, 2–5 MPa to produce crude anhydride. Unreacted CO and other purge gases are scrubbed after leaving the reactor to recover acetyl and iodide moieties. The liquid draw off from the reactor is expanded and flash distilled under low CO/H2 pressure to prevent catalyst decomposition. The flash bottoms are largely recycled, but a purge is taken for tar removal and Rhodium recovery. The crude anhydride flash distillate is purified in a three column sequence. In the first, methyl iodide and methyl acetate are distilled for recycle to the reactor. In the second, acetic acid id distilled overhead for recycle to the methyl acetate production process. The methyl acetate feedstock can be produced via reactive distillation, as shown in the section, “Methyl Acetate.” The final column produces 99% acetic anhydride product as a distillate, with tars and heavies as the bottoms stream.
Methyl Formate [197]
The equilibrium for the direct synthesis of formic acid by the addition of water to carbon monoxide is quite unfavorable and has not been exploited to produce commercial quantities of formic acid. Instead, formic acid is produced by acidification of formate salts (30% of global production) or hydrolysis of methyl formate (70% of global production). The acidification route involves reacting sodium or calcium formate (largely generated as a by-product of aldol/cannizzaro reactions, see the section, “Neopentyl Polyhydric Alcohols”) with a strong acid, usually sulfuric acid, to yield dilute free formic acid and sodium or calcium sulfate. The second route begins with carbonylation of methanol to give methyl formate in a liquid phase reaction using a dilute strong base catalyst (e.g., sodium methoxide) at a temperature around 80°C and pressure of 4–5 MPa:

This carbonylation is equilibrium-limited, resulting in 30–50% methanol and 95% CO conversion per pass. Sodium methoxide can react with methyl formate to give sodium sulfate and dimethyl ether. Dry conditions are required to limit hydrolysis of methyl formate to sodium formate and methanol, and to prevent leveling of the strong alkoxide base to the catalytically ineffective weaker hydroxide ion. The crude methyl formate is flashed to atmospheric pressure and distilled. The low-boiling methyl formate/methanol azeotrope is taken overhead for hydrolysis and the underflow is recycled to the carbonylation step, with purge for removal of sodium formate by crystallization.
The equilibrium-limited hydrolysis of methyl formate is accomplished in both two-stage (1-1 water-methyl formate molar ratio, followed by addition of excess methyl formate—30% conversion/pass) and one-stage processes (5-1 water-methyl formate molar ratio, 60% conversion/pass):

Unreacted methyl formate and methanol are distilled from the crude hydrolysis product, leaving a dilute water-formic acid mixture as underflow. Production of concentrated formic acid from this underflow product presents particular problems due to the high-boiling water/formic acid azeotrope. A number of separation schemes are practiced commercially; the most common approach is pressure-swing distillation. Excess water is distilled overhead in a first higher pressure column to produce azeotropic formic acid/water as a bottoms product. This azeotrope composition is distilled in a second vacuum column to produce concentrated formic acid (90–98% purity) as distillate, and the low pressure azeotrope composition is recycled to the first column [198]. Energy usage is improved with the one-stage hydrolysis process if excess water is removed by extraction (with a secondary amide or other solvents), prior to pressure-swing distillation.
Methyl Propionate [199–201]
When ethylene and carbon monoxide are reacted in methanol, in the presence of a palladium diphosphine catalyst, either polyketone or methyl propionate may result depending on the identity of the diphosphine ligand. Lucite, formerly a subsidiary of ICI, has developed this carbomethoxylation as a step of their “Alpha Process” for the production of methyl methacrylate. In the Alpha Process, methyl propionate is reacted with formaldehyde to ultimately produce MMA. The rate and selectivity of this carbonylation catalysis are extraordinarily high under relatively mild conditions.

Mechanistic studies have established the catalytic cycle depicted below with the key intermediates being a palladium hydride, which reacts with ethylene to produce a palladium ethyl species that rapidly inserts a carbon monoxide yielding the palladium propionyl complex. This complex is methanolyzed to produce methyl propionate product and regenerate the palladium hydride intermediate.

Shell developed very similar catalysis for the production of their Carilon polyketone in which the diphosphine ligand in bis(diphenylphosphino)propane rather than the 1,2-bis(di-t-butylphosphinomethyl)benzene employed by Lucite, illustrating the exquisite sensitivity of this catalysis to the structural details of the palladium diphosphine catalyst. A commercial scale MMA plant (Jurong Island, Singapore) based on the Alpha process has operated at design capacity since 2008.
Koch Acids
Koch carbonylation (also called hydrocarboxylation or the Koch-Haaf reaction) is the synthesis of predominantly tertiary (neo) carboxylic acids from olefins, CO, and water using a strong acid catalyst, typically H3PO4/BF3 or an acidic zeolite. In the initial step of the reaction, the olefin is protonated, generally with isomerization and rearrangement, to form the most stable (i.e., tertiary) carbonium ion. Carbon monoxide then adds to the carbonium ion to form an acylium cation. Further reaction with water and regeneration of the proton results in the formation of the tertiary carboxylic acid.
An example of this chemistry is conversion of mixed butenes, especially isobutylene to pivalic (neopentanoic) acid.

In the typical industrial process, CO is contacted with the olefin and H3PO4/BF3 catalyst in a CSTR at 20–80°C, 2–10 MPa. Conversion of the olefin is generally very high with 80–100% selectivity to the neo acid. Main by-products are neo acids of the oligomerized feedstock olefins. Product neo-acids are washed with water and decanted for removal of the catalyst acids. C5 to C11 neo acids are made industrially form the corresponding C4-C10 olefins. The higher olefinic feedstocks are typically derived from ethylene oligomerization or dehydration of paraffinic fractions.
Hydroformylation [202–207]
Chemistry
Hydroformylation, commonly known as oxo synthesis, is a C–C bond-forming reaction of an olefin with syngas (~1:1 M ratio of CO:H2) to produce aldehydes. When the olefin is a terminal, or α-olefin, two products may be formed and are referred to as the normal (straight chain) and iso (branched chain) products.

The reaction is employed in practice with C2 to C14 olefins, but hydroformylation of propylene is the primary application. The oxo aldehydes are valuable and reactive intermediates for a number of secondary transformations, including hydrogenation to alcohols, oxidation to acids, and aldol condensation to higher aldehydes and ketones, with subsequent further transformations of these products to esters, anhydrides, ketenes, glycol ethers, and sulfonates to name a few.
Process conditions vary significantly with olefin substrate and catalyst employed, but typically fall in the range of 1–20 MPa and 40–200°C. All current commercial catalysts are based on either Co or Rh transition metal complexes. The first commercial catalyst, HCo(CO)4, discovered by Otto Roelen in Germany in 1938, required high pressures (20–35 MPa) for acceptable operation. Subsequent work demonstrated that phosphine ligands could, allowed practical operation at much lower pressures 1–10 MPa and could further hydrogenate the product aldehydes to alcohols (Shell process). Another quantum leap in productivity and selectivity occurred with the introduction of Rh-based catalyst complexes in the early-1970s, with operating pressures as low as 1–2 MPa. Although Rh is roughly 1,000 times more expensive than Co, it is also several orders of magnitude more active than Co, for a substantial net benefit. Low pressure Rh operation reduces syngas compression energy over fivefold and significantly lowers capital costs over that of the original cobalt hydrido carbonyl catalyst system. The formation of an aldehyde via Rh-catalyzed oxo synthesis involves the following catalytic steps:
-
1.
An olefin coordinates to a coordinatively unsaturated metal hydrido carbonyl complex, exemplified by HCo(CO)3 and HRh(CO)(PPh3)2, followed by insertion of the olefin into the metal hydride bond to give an alkyl complex.
-
2.
A second migratory insertion of CO converts the alkyl into an acyl group.
-
3.
Oxidative addition of H2 yielding a metal dihydride.
-
4.
Reductive elimination of the aldehyde product regenerates the coordinatively unsaturated metal hydrido carbonyl complex (back to step 1).
A simplified expression for the rate of aldehyde formation illustrates some of the kinetic tradeoffs: [208, 209]
For lower olefins, especially ethylene and propylene, the concentration of the olefin in the liquid reaction phase is proportional to pressure, higher pressure gives higher solubility. Likewise, high hydrogen partial pressures favor aldehyde formation, as does increasing catalyst concentration (for Rh, this approach is expensive). At some point hydrogenation of the olefin to paraffin or loss of selectivity by hydrogenation of the product aldehyde may also occur. However, reducing CO partial pressure enhances the rate, but a minimum CO pressure is needed to maintain stability of the metal catalyst complexes, as well as for product formation. In practice, a complex tradeoff of these effects is required, with low pressure, low catalyst concentration, and about 1:1 H2/CO molar ratio tending toward optimal.
A key consideration in hydroformylation of α-olefins is the n:i ratio of products for C3 and higher olefin substrates. For example, with propylene, the C–C bond may form to give a linear (n), or a branched product (i):
Linear aldehydes are more valuable intermediates for production of plasticizers and detergents (linear molecules are more biodegradable). Significant research effort has been expended over the last 80 years developing high n:i ratio catalysts. Formation of straight chain olefins is favored by bulky, electron-rich ligands. The vast majority of companies practicing oxo synthesis use catalyst systems that preferentially produce normal as opposed to iso aldehyde products. A company uses proprietary rhodium—halophosphite catalysts that produce an unusually low n:i product mix.



























The chain-length of the feedstock olefin and desired n:i ratio of the aldehyde product largely dictate which metal–ligand catalyst combination is more desirable. Table 10.31 summarizes operating conditions and results for a number of commercial systems, with highlights about each given below. Corresponding catalyst structures are presented in Fig. 10.27.

Hydroformylation catalyst structures
Catalyst I. HCo(CO) 4 : The first generation cobalt carbonyl/hydrocarbonyl catalyst, originally developed in the 1930s, required a high pressure, 20–35 MPa, for good activity. It is not used commercially for lower olefins any longer, but finds some applications with higher olefins.
Catalyst II. Cobalt, phospine ligand: The second-generation phosphine-modified cobalt catalyst, developed in the 1960s, significantly reduced the required operating pressure, improved the n:i ratio to between 6:1 to 9:1, and gives mostly terminal hydroformylation, even when fed internal olefins (by promoting double bond migration). This process is used mostly for production of detergent range alcohols, where the greater propensity of cobalt to hydrogenate the oxo aldehyde is not a disadvantage. Trialkyl phosphines are preferred and derivatives of the phobanes depicted in Fig. 10.27 are used commercially by Shell.
Catalyst III. HRh(CO)4: A rhodium carbonyl hydride catalyst without a phosphine or phosphite is currently used for hydroformylation of C6 to C14 linear and branched olefins. Without expensive ligands to recover, a unique adsorption/ion exchange method is used for efficient catalyst recovery. Isononanol and isodecanol are important products.
Catalyst IV. Rhodium, triphenyl phosphine ligand: The phosphine-modified rhodium catalyst was developed in the 1970s and has been successfully employed since, largely for propylene and butene hydroformylation. This system dramatically reduced required operating pressures, while giving several orders of magnitude rate improvement and higher n:i ratios. Several variations are practiced; (1) vapor stripped reactor for product removal and catalyst separation (product taken as vapor directly from reactor, (2) liquid take-off with flash separation of aldehyde and catalyst.
Catalyst V. Rhodium, water soluble phosphine: The phosphine-modified rhodium catalyst was made water-soluble by adding sulfonate groups to the phenyl rings. In this way a simple separation of catalyst for recycle is facilitated by decantation of the catalyst-bearing aqueous phase from the product-laden organic phase. This low pressure process is used commercially for hydroformylation of propylene to high-normal butyraldehyde n;i ratio of about 10:1 to 20:1.
Catalyst VI. Rhodium, bisphosphite ligand: The family of bisphosphite ligands dramatically increases the n:i ratio to about 30:1, the highest level yet achievable. Both propylene and butene feedstocks are usable.
Catalyst VII. Rhodium, tridentate halophosphite ligands: Unlike the other six catalyst systems, tridentate halophosphite ligands produce the lowest commercialized n/i ratio, typically less than 3:1. Ethylene and propylene are common feedstocks. With these halophosphite ligand, the n:i butyraldehyde ratio dependson the ligand to rhodium molar ratios, the reaction temperature, and the carbon monoxide partial pressure [210].
More detailed descriptions of typical oxo processes for short and long chain olefins are given below.
Lower C2-C5 Olefins
In a typical commercial process for catalyst types V, VI, and VII, a mixture of 1:1 syngas and lower olefins (can be a mixture) are fed to a stirred tank or bubble column reactor along with fresh and recycle homogeneous catalyst solution, where hydroformylation takes place. Temperature and pressure conditions depend on the metal-ligand catalyst chosen, as well as the chain length of the olefin. Heat is removed by the reactor jacket, an external cooling loop, or alternatively, boiling of the product aldehyde. If a vapor-stripped reactor system is used, the vaporous effluent and unreacted gases from the reactor are cooled, allowed to condense, and separated into gas and liquid streams. If conversion of olefin is low, the off-gases are compressed and recycled, with some amount purged to prevent buildup of inerts (i.e., alkanes in the olefin feed). If the aldehyde is the final end product, the crude aldehyde is distilled to remove water and high boilers (usually alcohols and esters), and possibly further fractionated to separate iso and normal isomers. The phosphine or phosphite ligands tend to degrade at a low, but finite rate, so it is necessary to purge some amount of the catalyst solution. Subsequent recovery of Rh, typically involves distillation, evaporation, decantation, or extraction. Decantation is used, for example, with the water soluble ligand system described in Table 10.31.
Higher Oxo: Detergent Alcohols [211]
In one commercial process, linear and branched C6-C14 olefins derived from light olefin oligomerization or paraffin dehydrogenation, are reacted in the liquid phase with 1:1 syngas at 95–180°C, about 25 MPa, using a homogenous rhodium carbonyl catalyst (without additional ligands) to form isomeric oxo aldehydes. Depending on the reactivity of the olefin feedstock (branched < internal olefin < α-olefin), conversion can reach 95%, but is typically limited to 80% for a first pass. The less reactive olefins are removed and sent to a second pass with more severe conditions and higher catalyst loading, with 75% second pass conversion (95% overall conversion for two pass). Mild conditions favor linear, terminal olefin synthesis, while more forcing conditions tend to lead to isomerization of double bonds, yielding higher levels of internal olefins and thus branched aldehydes products.
The pressure of the reactor effluent is let down to remove dissolved CO and hydrogen. This off-gas is recycled, with a purge to prevent build-up of inerts. Since there is no expensive ligand to recover, Rh ions are adsorbed from the degassed olefin/aldehyde liquid product by passing through a series of ion exchange resin beds. The ion exchange bed is periodically incinerated, converting the Rh metal to Rh oxide, which is readily recycled to the Rh salt catalyst precursor. The crude catalyst-free aldehyde is typically sent directly to hydrogenation for conversion into alcohols before distillation. See the section, “Aldehydes and Ketones to Alcohols,” for information on the hydrogenation of the higher aldehydes to alcohols.
Chlorination [212, 213]
Chemistry
As the name implies, chlorination is a family of reactions in which one or more chlorine atoms are incorporated into a molecule. For large scale commercial synthesis the most common chlorine sources are Cl2, HCl, and HOCl (hypochlorous acid). Chlorinated molecules are extremely important to many industries, including construction, automotive, toys (vinyl chloride, PVC), silicon products (methyl chloride), dry cleaning and solvent applications, and air conditioning (HFC’s). Some rank among the largest volume chemicals produced. Several basic types of chlorination reactions are employed industrially:
-
1.
Substitution is the abstraction and replacement of a hydrogen atom by a chlorine atom (from Cl2 gas), with HCl as the co-product. The reaction involves a free radical chain mechanism initiated by thermal dissociation of Cl2, with a chloride radical as the chain carrier. Selectivity is generally poor, as all hydrogen atoms on a molecule are more or less available for substitution in the order tertiary > secondary > primary, saturate > unsaturated > aromatic. Further information on relative reactivity of various substrates is available in standard texts [214]. The substitution reaction can be extremely fast and exothermic, with a heat of reaction on the order of −90 to −135 kJ/gmole of Cl added. Less reactive substrates require either high temperature or a Lewis acid catalyst. Alkane and aromatic substrates are used:
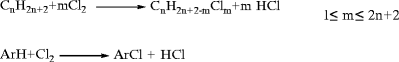
-
2.
Addition involves the insertion of Cl2 across an unsaturated (double or triple) bond in a hydrocarbon molecule. Mild temperatures, 70–120°C, are required. The addition is highly specific; chlorine atoms are symmetrically distributed, and selectivities above 90% are common. The substitution reaction is exothermic, with a heat of reaction on the order of −180 to −230 kJ/gmole of Cl2 added. Lewic acids such as FeCl3, ZnCl2, and PCl3 are common catalysts.
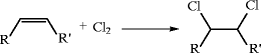
-
3.
Oxychlorination is the reaction of HCl and oxygen with a substrate to produce a chlorinated species and water. Reaction temperature can range from 80 to over 200°C, depending on substrate. The reaction is generally selective (>90%). The heat of reaction is very high, around −250 to −300 kJ/gmole. Common catalysts are CuCl2 and ZnCl2.
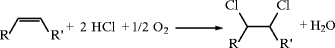
-
4.
Hydrochlorination is the reaction of HCl with a substrate to produce a mono-chlorinated species. If the substrate is an alcohol, then the by-product is water:

The reaction with an alcohol is generally very selective (>90%), with the diether as the major by-product. The heat of reaction, around −30 to −50 kJ/gmole, is relatively low compared to other types of chlorinations. HCl may also be added across a double bond:
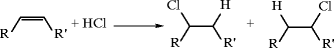
-
5.
Perchlorination (sometimes called chlorinolysis) is the high temperature (400–700°C) exhaustive chlorination of a multi-carbon substrate with concomitant breaking of at least one C–C bond to form smaller fully chlorinated fragments. Both hydrocarbon and partially substituted chlorohydrocarbons are potential feedstocks. At full conversion and complete fragmentation to C1 pieces, the reaction stoichiometry is:
At high temperature (>600°C), an additional equilibrium exists between carbon tetrachloride and perchloroethylene.
Large excesses of Cl2 or high pressure are needed to drive to mostly single carbon fragments. Perchlorination is an important component of an integrated chlorination facility, taking in low value chlorinated by-products from other processes, to produce exhaustively chlorinated, lower carbon number materials. Any aliphatic, aromatic, or oxygenated hydrocarbon may be reacted via perchlorination. Oxygenates produce phosgene as a by-product.
-
6.
Chlorohydrination is the reaction of an olefin with Cl2 and water to form a vicinal hydroxy chloride moiety (chlorohydrin), with HCl as a co-product.
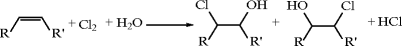
A large excess of water and low olefin concentration is required to prevent by-product formation by addition and substitution reactions. When R ≠ R′, isomers differing in the position of the hydroxyl and Cl may be formed. Chlorohydrins are important intermediates in the synthesis of epoxides, especially propylene oxide.








Hydrochlorination and oxychlorination reactions are often more economical uses of HCl generated by substitution and addition reactions than disposal of excess HCl as a gaseous or dilute aqueous co-product. Examples of some industrially important chlorination reactions are summarized in Table 10.32. The types of reactions discussed above may also be applied to other common halogens, with reactivity in the order F > Cl > Br > I. Fluorine is so reactive and exothermic that fluorinations are not normally done directly, but by indirect methods exemplified by halogen substitution [215].
Chloromethanes [216–218]
The chloromethanes (one-carbon molecules containing one to four chlorines), are primarily used as intermediates for other chemistries: methyl chloride for silicones production; methylene chloride for solvents; chloroform for hydrochlorofluorocarbon production; and carbon tetrachloride for chlorofluorocarbons. The more substituted chloromethanes are made predominately by successive substitutions of methyl chloride or perchlorination of C1-C3 chlorinated streams. The use of carbon disulfide as a feedstock for methyl chloride synthesis is essentially obsolete, and thermal substitution of methane is falling out of favor. The flowsheet for a typical integrated chloromethanes process is shown in Fig. 10.28. Methyl chloride is now largely produced via gas phase hydrochlorination of methanol with excess HCl at 280–350°C, 0.3–0.6 MPa, in a multitubular steam-generating fixed bed reactor filled with γ-alumina catalyst (1).

Flowsheet for production of chloromethanes

An older process using 70% aqueous zinc chloride/HCl at 130–150°C is not used much anymore. The reactor effluent is cooled by direct contact quench with recycle ~30 wt% aqueous HCl (2). Diluted ~20 wt% aqueous HCl exits as underflow from the quench tower, and gaseous crude methyl chloride is scrubbed further with caustic (3), and then concentrated (96 wt%) H2SO4 to remove dimethyl ether (which reacts to form methyl sulfate) and water (4). The diluted H2SO4 (~80 wt%) is a by-product. Dry methyl chloride is compressed (5) to around 2 MPa, condensed, and distilled overhead as a pure product, or used along with the higher chlorinated by-products as feedstock for further substitution chlorination.
In order to produce a slate of chloromethanes, methyl chloride and by-products from the hydrochlorination step are further thermally substituted with gaseous chlorine at 350–400°C, 0.8–1.5 MPa, in an adiabatic reactor (6), usually without catalyst. If the reaction mixture is allowed to reach about 450°C or above, rapid pyrolysis may occur, with the very undesirable formation of carbon soot. The reactor effluent is quenched (7), separated from co-product HCl by multistage condensation (8), stripped of residual HCl (9), and distilled to take mono-, di-, tri- and tetra-chloromethanes overhead in successive columns (10), (11), (12), (13) respectively. Depending on the desired product distribution, any partially substituted product may be recycled to the substitution reactor for further chlorination. Overall yield on methanol is at least 95% and greater than 98% on chlorine.
The heavy chlorinated residue from the last column and other chlorination by-products (e.g., 1,2-dichloropropane from propylene oxide production, vinyl chloride residues) may be fed to a perchlorination unit for production of smaller chain fully substituted materials. A typical high pressure tubular perchlorination unit operates adiabatically at up to about 620°C, 10–20 MPa, with a Cl2/substrate ratio dependent on the desired perchloroethylene/carbon tetrachloride product mix. The effluent, comprising carbon tetrachloride, HCl, Cl2, perchloroethylene, and small amounts of hexachlorobenzene (recyclable to extinction), is quenched with cold carbon tetrachloride, flashed to remove Cl2 and HCl, and distilled to produce carbon tetrachloride and perchloroethylene as pure streams. The reactor must be Ni clad to prevent corrosion. With the decline in applications for carbon tetrachloride, the production of most perchlorination units are slated heavily to perchloroethylene by a ratio of roughly 3-1.
Chloroaromatics [219–221]
A wide variety of chlorinated aromatics, with and without additional functional groups on the aromatic ring, may be produced by substitution chlorination, with co-production of HCl. These include mono- and di-chlorobenzenes, chlorotoluene, benzyl chloride. The product mix ultimately obtained is highly dependent on Cl2/aromatic molar ratio, temperature, catalyst composition, co-catalyst (if any), location of other groups already on the benzene ring, and contacting mode. High temperature and high Cl2/aromatic ratio favor multiple chlorinations. Backmixed (CSTR) operation leads to increased levels of multi-chlorinated products; batch or reactive distillation approaches tend to increase monochloride content. The heats of reaction for mono- and dichlorobenzenes are summarized in Table 10.32. A typical continuous process for mono and di-chlorination of benzene operates at 20–80°C, with dissolved ferric chloride catalyst/organic sulfide co-catalyst, and with very low water content (25–35 ppm) to prevent catalyst deactivation. The resulting effluent is neutralized with caustic, or in more modern plants distilled directly to separate HCl and Cl2 from aromatics. Unreacted benzene is recovered overhead first, with monochlorobenzene taken overhead in a second column. Dichlorobenzenes are close boiling and generally must be separated by crystallization to obtain high purity single isomers. Product distribution between mono- and dichlorobenzenes is highly dependent on the ratio of benzene to chlorine fed to the reactor.
,2-Dichloroethane (Ethylene Dichloride) and Vinyl Chloride Monomer (VCM) [222–225]
Ethylene dichloride (EDC), and its primary end product vinyl chloride monomer (VCM), are produced at truly immense scale, constituting about 8% of global output of chemicals. EDC and VCM were once produced primarily by chlorination of acetylene. Limited application for the by-product HCl posed a major restriction to the growth of EDC and VCM. However, major producers now use a combination of direct chlorination and oxychlorination of ethylene to produce EDC and VCM without significant coproduction of HCl. Figure 10.29 shows such an integrated plant for producing EDC and vinyl chloride from ethylene, chlorine, and air. The chemical reactions are as follows:

Flowsheet for production of vinyl chloride monomer
The oxychlorination step (1), catalyzed by Cu(II)Cl2-impregnated alumina at 200°C and 0.2–0.5 MPa pressure, uses recycled HCl from the EDC pyrolysis step, with oxygen and ethylene to produce EDC and water. This reaction is even more exothermic (−290 kJ/gmole) than direct chlorination. Both multi-tubular fixed bed and fluidized bed reactor designs with steam generation are used commercially to control the heat release. The fluid bed design, although suffering from lower selectivity than a PFR due to back mixing, can be run advantageously in the explosive region. Conversion is typically 93–97% per pass, with a selectivity on ethylene around 95%. Major by products are heavy tri-chlorinated species, trichloroacetaldehyde, and oxygenated lights such as CO, carbon dioxide, formic acid.
The addition chlorination of ethylene is done usually in a liquid-phase reactor (3), catalyzed by the Lewis acid, iron(III) chloride at 40–100°C, 0.4–0.5 MPa. Ethylene absorption is the rate limiting step. The heat of reaction (−180 kJ/gmole) can be removed either by heat exchange in the reactor, or more economically by running the reaction above 100°C and using the generated heat to distill the EDC. High-purity ethylene, with low propylene content, is preferred to avoid formation of chlorinated C3 by-products. Ethylene conversion approaches 100%, with a selectivity of 98% on ethylene and 99% on chlorine.
The effluents from reactors (1) and (3) are disengaged from vapors and scrubbed with caustic for additional removal of HCl (2),(4). The crude EDC is azeotropically distilled to remove residual water (5), and subjected to distillation to separate light (6) and heavy (7) chlorinated by products. These chlorinated species may be used in chlorinolysis or production of other halogenated species.
The cracking (pyrolysis) of EDC to VCM typically is carried out at temperatures of 500–550°C without a catalyst (8). Selectivity to VCM is greater than 98%. The hot gases are quenched (9) and distilled to strip out HCl (10) and then VCM (11). The unconverted EDC is returned to the EDC purification train.
About 35% of the chlorine produced globally goes to the manufacture of VCM. More than 95% of all VCM is used to produce polyvinyl chloride (PVC), an important polymer for the housing and automotive industries (See Chap. 15). The rest of the VCM goes into the production of chlorinated solvents and ethylenediamine.
Chloroethanes [226–228]
The commercial importance of the chlorinated ethane derivatives, perchloroethylene, 1,1,1-trichloroethane, and trichloroethylene, has declined significantly since the 1980s due to t the declining use of chlorofluorocarbons. Major uses of all three are as precursors for fluorocarbon production, dry cleaning agents, and solvents. With declining use of carbon tetrachloride, perchlorination (see Section 10.7.1) is becoming a more significant route to perchloroethylene. 1,1,1-trichloroethane is produced predominantly by a two-step process starting with vinyl chloride monomer (VCM): (1) hydrochlorination of VCM with HCl produce predominantly 1,1-dichloroethane; (2) thermal or photochemical (more selective) substitution chlorination of 1,1-dichloroethane to 1,1,1-trichloroethane. Perchloroethylene and trichloroethylene are also made by the oxychlorination of 1,2-dichloroethane (VCM intermediate, see above):
Propylene Oxide via Propylene Chlorohydrin [229, 230]
Although differing from ethylene oxide by only one extra carbon in the backbone, propylene oxide (PO) has not been made successfully at commercial scale by the direct oxidation of propylene. The presence of allylic hydrogens dramatically and adversely affects the efficacy of silver-based catalysts for the selective oxidation of propylene to PO with oxygen [231]. Instead all world scale PO plants are based either on indirect oxidation of propylene by either the co-product hydroperoxide routes (50% of total, see the section, “Hydroperoxide Co-product Processes for Propylene Oxide”) or the chlorohydrin route described herein.
The chlorohydrin route entails two main steps: (1) synthesis of propylene chlorohydrins (PCH) by the reaction of propylene, chlorine, and water (chlorohydrination) and (2) synthesis of PO via the reaction of PCH with an alkali or alkaline earth hydroxide (dehydrochlorination).
In the first step chemical or polymer grade propylene, gaseous chlorine reacts in equimolar amounts in the presence of excess water at 45–90°C, 0.11–0.19 MPa in bubble column or CSTR, to produce a solution of about 4 wt% PCH isomers, roughly a mole of HCl per mole of PCH, and other chlorinated by-products. Excess water (around a 50:1 mole ratio of water to propylene) is critical to suppress undesired substitution and addition reactions, but 1,2-dichloropropane (DCP), 1,3-dichloropropan-2-ol or 2,3-dichloropropan-1-ol (DCH), 2,2′-dichlorodiisopropyl ether (DICPE) are major by-products. Yields are typically 88–96% PCH isomers, 3–10% DCP, 0.3–1.2% DCH, 0.2–0.8% DCIPE, and 1% of others including allyl chloride. The PCH solution is disengaged from the vent gases (subsequently scrubbed with caustic and thermally oxidized), and conveyed to the dehydrochlorination reactor.
The PCH solution is reacted with slightly more than two equivalents of base (either NaOH or Ca(OH)2) to neutralize the HCl produced in the first step and to saponify PCH to propylene oxide and salt. The reactions are rapid and exothermic. The crude PO is distilled in a two column sequence, with lights taken overhead in the first column and PO distilled overhead in the second column. DCP, DCIPE, and epichlorohydrin are underflowed in the second column. Almost all streams in the chlorhydrin process are extremely corrosive, requiring expensive materials of construction such as Teflon coatings, fiber reinforced plastics, graphite, rubber, brick-linings, Inconel, Hastelloy, or Monel alloys.
This process produces roughly 2 kg of either NaCl or CaCl2 per kg of PO. Economics dictate that the PO unit be associated with a world-scale chloralkali, mercury, or membrane chlorine processes for regeneration of the chlorine and base. 1,2-Butylene oxide (and 1,2-butanediol upon hydrolysis) is made from 1-butene at a substantially smaller scale by a similar chlorohydration/dehydrochlorination route.
Epichlorohydrin [232, 233]
Epichlorhydrin, an important intermediate for coatings and urethane applications, is produced in 90% yield via dehydrochlorination of 1,3-dichloropropan-2-ol or 2,3-dichloropropan-1-ol (DCH).

The DCH may be a by-product of propylene oxide production (see above), or formed on purpose in a two-step process from propylene. In 1936, it was discovered that when propylene is reacted with chlorine at high temperature (500–510°C in practice), free radical substitution of an allyl hydrogen predominates over double bond addition, giving allyl chloride in 80–85% yield.



Excess propylene (4:1 M ratio) is required to ensure essentially complete conversion of chlorine, and for good selectivity. In the second step, allyl chloride is reacted with aqueous hypochlorous acid at 50–60°C to yield a mixture of DCH isomers.
Condensation
In its broadest sense, a condensation reaction involves the combination of two molecules or functional groups of the same molecule to form a new species, typically with the elimination of a small molecule such as water or an alcohol. In the present context, condensation reactions are limited to the coupling of two molecules via the formation of a new carbon–carbon bond, with elimination of water. The discussion to follow covers aldol condensations and the acid-catalyzed condensation of phenols and ketones, as exemplified by the synthesis of bisphenol A (BPA).
Aldol Condensation [234–236]
Chemistry
The aldol reaction is one of the most important industrial means of forming carbon–carbon bonds to directly synthesize complex functionalized molecules. A large number of commercially significant molecules are produced via aldol chemistry as shown in Table 10.33. Many of these compounds are intermediates for further transformations such as hydrogenation or oxidation.
In the aldol addition reaction, an enolizable ketone or aldehyde, i.e., one that has at least one acidic proton alpha to the carbonyl, reacts with another aldehyde or ketone to form a β-hydroxycarbonyl compound. The enolizable carbonyl acts as a nucleophile and other carbonyl as an accepting electrophile. The reaction is called a crossed aldol addition when the nucleophile and electrophile are different and a self aldol dimerization when they are the same.
Aldol reactions are catalyzed most commonly by bases such as alkali earth and alkaline earth hydroxides (e.g., NaOH or Ca(OH)2), and amines (e.g., triethylamine, dimethylamine). Acid catalysis also occurs (and is often a source of by-product formation in acid catalyzed reaction where ketones or aldehydes are present), although is less effective for on-purpose production. The aldol addition reaction may proceed via two fundamentally different mechanisms, depending on whether an acid or base catalyst is used.
With an acid catalyst, the initial step in the reaction mechanism involves acid-catalyzed tautomerization of the carbonyl compound to the enol. The acid also serves to activate the carbonyl group of another molecule by protonation, rendering it highly electrophilic. The enol is nucleophilic at the α-carbon, allowing it to attack the protonated carbonyl compound, leading to the aldol after deprotonation. The secondary or tertiary hydroxyl thus formed is highly favored to dehydrate to give the α,β-unsaturated carbonyl compound under acidic conditions. The combination of the aldol addition and dehydration steps is sometimes called an aldol condensation reaction.
Base-catalyzed aldol reactions begin as acid–base reactions and are thus extremely fast. An α-proton next to a carbonyl group is weakly acidic (pKa of 12–14). In the presence of a strong base, the α-carbon is deprotonated to a small extent to form an enolate ion. The coupling reaction occurs via nucleophilic attack by the resonance-stabilized enolate on the electrophilic carbonyl group of another molecule, which form a β-hydroxy carbonyl derivative (the aldol product) upon regeneration of the base catalyst. The aldol product may then undergo based-catalyzed dehydration to give the α,β-unsaturated carbonyl compound.

Ketones generally have a slightly lower pKa than aldehydes. Thus in a mixed ketone-aldehyde system, the ketone is favored to form the enolate and the aldehyde to act as the acceptor. Both steps are equilibrium limited, but the extent of the formation of addition and aldol dehydration products dictated by the structure of the reactants. For the following nucleophile-electrophile pairs the order of greatest shift to aldol products is:
Aldehyde-Aldehyde > Ketone-Aldehyde
> Ketone-Ketone
Reactivity also diminishes with steric hindrance and with reduced solubility in the catalyst-containing phase (if the reaction is performed with a separate caustic-rich phase). A selection of industrially significant aldol-based processes is described in more detail below.
MIBK, Diacetone Alcohol (DAA), and Mesityl Oxide (MO) [237–239]
MIBK (4-methyl-2-pentanone), related intermediates, and its derivatives are the third largest end use for acetone behind methyl methacrylate and BPA. The synthesis of MIBK is a classic example of an aldol-based reaction pathway comprising self-aldol addition, dehydration, and hydrogenation:

This chemistry is practiced in both three-step and one-step process implementations. In the three-step process, acetone, dilute caustic is fed to a CSTR controlled at 10–20°C to enhance the equilibrium amount of DAA, but acetone conversion is typically only 8–10%. The reactor effluent is neutralized with phosphoric acid and stripped of unreacted acetone. Additional phosphoric acid is added to acidify the mixture to catalyze dehydration of DAA during distillation. Back-reacted acetone and the Mesityl oxide/water heterogeneous azeotrope are taken overhead and by-product salty water is underflowed. The organic layer from the overhead decanter is dehydrated and stripped of residual acetone in the next column. The dry underflow mesityl oxide (98–99 wt%) is hydrogenated over a Pd or Group VIII metal (e.g., Ni, Cu, Cr) in either a liquid phase reactor or in a low pressure hydrogenation/distillation column. One advantage of this process is that it allows for isolation of intermediates DAA and mesityl oxide.
The one-step process is much less capital intensive and generally allows for 30–50% per pass conversion of acetone. Acetone and hydrogen are fed to a packed bed reactor, slurry reactor, or catalytic distillation, typically at 130–140°C, 3–6 MPa, with an appropriate dual function catalyst combination to perform the aldol addition, dehydration, and hydrogenation reactions simultaneously. Many catalyst combinations have been reported in the open literature, including cation exchange resin impregnated with Pd, physically mixed exchange resin and supported Pd, zirconium phosphate-Pd, and niobium-Pd catalysts. Acetone, MIBK, and many of the reaction intermediates are present and active for further aldol and hydrogenation reactions, resulting in a very large reaction network. Typical yields of MIBK are 90–96% on acetone.
Neopentyl Polyhydric Alcohols [240, 241]
An important family of polyhydric alcohols with a neopentyl structure are pentaerythritol, trimethylolethane (TME), trimethylolpropane (TMP), and neopentyl glycol (NPG). These compounds are formed by the successive aldol addition reactions of formaldehyde with C2 to C4 aldehydes followed by reduction of the resulting hydroxy-aldehyde intermediate, as shown in Table 10.34. The aldol addition reactions are rapid, cannot be readily stopped at a lower number of formaldehyde additions, and do not lead to dehydration.
The typical process for pentaerythritol has not changed significantly for several decades. Aqueous formaldehyde, acetaldehyde, and aqueous Ca(OH)2 or NaOH are fed to a CSTR maintained at 15–45°C. Three successive aldol additions of formaldehyde to acetaldehyde is followed by a Cannizzaro reaction (reduction of trimethylol acetlaldehyde by formaldehyde with formation of formic acid as a co-product) to form pentaerythritol. Overall the aldol-Cannizzarro sequence releases about 92 kJ/gmole of heat, necessitating effective reactor cooling. Although stoichiometry dictates the use of 4 moles of formaldehyde per mole of aldehyde, up to 16 moles per mole is added to limit formation of other acetaldehyde aldol and dipentaerythritol by-products.
Following neutralization of excess base with formic acid, unreacted formaldehyde is recovered by pressure distillation. The product polyol is recovered by stepwise vacuum concentration and fractional crystallization. If caustic is used as the base, an additional recrystallization from water is required to produce pentaerythritol free of sodium formate. On the other hand, calcium formate has very low solubility in water and can be precipitated prior to pentaerythritol concentration/crystallization. No effective methods for direct hydrogenation of trimethylol acetaldehyde to pentaerythritol have been commercialized. Yields are typically about 90% on acetaldehyde. Finding industrial uses for the formate salts limits the application of this chemistry.
TME and TMP are produced commercially by very similar aldol-Cannazzaro processes, with the exception that the polyol often is extracted from the reaction mixture with a solvent, such as acetate esters, alcohols, or cyclohexanol, then purified by distillation. Yields are similar to pentaerythritol, reaching about 90%.
A significant portion of the neopentyl glycol produced commercially is synthesized by aldol addition with either sodium hydroxide or calcium hydroxide catalyst. The intermediate, hydroxypivaldehyde, can be finished via a Cannizzaro reaction, or more commonly by direct catalytic hydrogenation with Cu, Cu/Cr,Co, or Ni catalysts at 80–200°C, greater than 3.5 MPa pressure. Because the self-aldol of isobutyraldehyde requires a base stronger than hydroxide ion to occur to a significant extent (see section “Tischenko Reactions: Ethyl Acetate and 2,2,4-Trimethyl-1,3-Propanediol Derivatives” below), a relatively small excess of formaldehyde is sufficient for high conversion and a selectivity greater than 90%. Major by-products are 2,2,4-trimethyl-1,3-propanediol, neopentyl glycol isobutyrate, formic acid, formate salts, and NPG-hydroxypivalic acid esters. Purification involves extraction of the hydroxypivaldehyde intermediate with a solvent, followed by a series of distillations.
State-of-the-art NPG plants now use a tertiary amine catalyst in place of hydroxide catalysts. Complete conversion of formaldehyde occurs with a slight excess of isobutyraldehyde. The excess aldehyde is distilled with the amine and the two are recycled together. The reaction is quite selective and the most of acidic by-products produced by formaldehyde reduction are absent. The synthesis is finished with a similar catalytic hydrogenation. In both processes, hydroxypivaldehyde may be isolated and oxidized to the product hydroxypivalic acid.
-Ethylhexanal [242, 243]
2-ethylhexanal is an extremely important intermediate for solvent and plasticizer applications. It is manufactured from n-butyraldehyde (see section “Hydroformylation”) by aldol condensation in an alkaline medium or with a basic ion exchange resin at 80–130°C, 0.3–1.0 MPa. This is followed by catalytic hydrogenation of the double bond of the α,β-unsaturated aldehyde under mild conditions, 50–100°C, 0.2–2 MPa, to give 2-ethylhexanal. Supported Pd is a typical catalyst.

Conversion of n-butyraldehyde is high. The yield to 2-ethyhexanal are well above 90%, with major by-products of 2-ethylhexanol, and higher aldolized species. Under more severe conditions the α,β-unsaturated aldehyde intermediate can be converted directly to 2-ethylhexanol (see the section, “Hydrogenation”). Purification of 2-ethylhexanal occurs by distillation.
Tishchenko Reactions: Ethyl Acetate and 2,2,4-Trimethyl-1,3-Pentanediol Derivatives [244–246]
A closely related cousin of the aldol addition is the Tishchenko reaction occurring with aldehydes. In the presence of a base such as an aluminum alkoxide, one aldehyde molecule is reduced and the other is oxidized, with the direct formation of an ester. The largest industrial use of this reaction is in the formation of ethyl acetate from acetaldehyde:

With a stronger alkali metal alkoxide base (e.g., sodium alkoxide), aldol addition is a competitive parallel reaction. An important application of the tandem Aldol-Tishchenko pathway is in the synthesis of 3-hydroxy-2,2,4-trimethylpentyl isobutyrate from isobutyraldehyde in the liquid phase using an alkali metal alkoxide salt as the catalyst.

A parallel Tishchenko reaction pathway produces isobutyl isobutyrate while equilibrium-limited transesterifications lead to 2,2,4-trimethyl-1,3-pentanediol and 2,2,4-trimethyl-1,3-pentanediol isobutyrate co-products. The products are separated by distillation.
Mannich Base Condensation: Methacrolein [247, 248]
The industrial use of a Mannich base (carboxylic or inorganic acid salt of a secondary amine) for aldol-type condensations is illustrated by the synthesis of methacrolein, the initial step in an alternate synthesis of methylacrylic acid. Propionaldehyde is condensed with aqueous formaldehyde in the liquid-phase in the presence of the Mannich base to form methacrolein and water:

The best-performing base has been found to be a 40% aqueous solution of the dimethylamine-acetic acid salt. The synthesis methacrolein is remarkably selective (greater than 95%) and conversion of both formaldehyde and aldehyde are very high. Methacrolein is recovered by distillation from the reactor effluent as the methacrolein/water heterogeneous azeotrope, and further purified by distillation. The dimethylamine-acetic acid base is recycled to the reactor. The methacrolein thus produced can be oxidized in the same fashion as described in the section, “Acrolein/Acrylic Acid and Methacrolein/Methacrylic Acid.”
Condensation of Formaldehyde with Acetylene: 1,4-Butanediol [249, 250]
One of the major remaining applications of acetylene as a feedstock is in the synthesis of 2-butyne-1,4-diol, a precursor of 1,4-butanediol. This route still commands about 40% of the capacity for 1,4-butanediol. Acetylene is first reacted with 30–55 wt% formaldehyde in a series of three to five trickle bed columns at 80–110°C, 0.2–2.0 MPa over a silica supported Cu acetylide/Bi-promoted catalyst to produce 2-butyne-1,4-diol. Selectivity is about 90% on acetylene and greater than 90% on formaldehyde.
The CuO is converted to an acetylide under acetylene partial pressure. The reaction is first order in formaldehyde and zero order in acetylene. Back-mixed reactors helps to keep the unconverted formaldehyde level low in order to prevent catalyst inhibition and formation of formals. Conversion of formaldehyde is typically 97–99%. Dilute alkali is added to keep the pH in the 5–8 range. The Bi2O3 inhibits the formation of water-insoluble Cu-acetylene oligomers. The oligomers are quite shock sensitive, especially when dry. Deposition of these complexes in the reactor head spaces may lead to the formation of potentially explosive conditions and are to be avoided. Major by-products are propagyl alcohol, and sodium formate, along with unreacted formaldehyde. The butyne-diol is purified by vacuum distillation of the reactor effluent. Propagyl alcohol is removed as an azeotrope.
In the second step, the butyne is hydrogenated to 1,4-butanediol in the liquid phase at 70–170°C, 14–30 MPa, if Raney Ni is used, or 180–200°C, 20 MPa with Ni/Cu/Cr catalysts.
Both one- and two-stage fixed bed hydrogenation processes are used. Major by-products are methanol, n-propanol, n-butanol, hydroxyl-buyraldehyde, 2-methyl-1,4-butanediol, and formals Selectivity to butanediol is about 95%. The product is purified by distillation.

Bis-Hydroxyaryl Alkanes: Bisphenol A [251–253]
The bis-hydroxyaryl alkane structure, comprising two phenol molecules linked by a hydrocarbon chain, is synthesized by the acid-catalyzed liquid phase condensation of phenol with an aldehyde or ketone. Cyclohexanone, formaldehyde, and acetone adducts are produced commercially by similar processes. The acetone adduct, known as BPA, is by far the most important. BPA is produced from acetone and phenol in a fixed liquid-circulating bed or CSTR at 50–90°C, 0.11–0.40 MPa:

A high molar ratio of phenol to acetone, up to 15:1, helps suppress aldol reactions of acetone and multiple acetone/phenol condensations. The new carbon–carbon bonds occur almost exclusively in the 4-position of the phenol molecules. Acetone conversion is essentially complete, with yields greater than 90%. Sulfonated acid ion exchange resins are favored in newer installations over mineral acids, such as HCl or H2SO4, due to lower corrosion and ease of separation from the crude BPA.

Many flowsheet variations are practiced, but in one licensed process the reactor effluent is distilled to remove water and any unreacted acetone overhead. The bottoms of this column is distilled further to remove phenol for recycle and to produce a concentrated BPA bottoms stream. Solvent is added (alkane or aromatic) and the 1-1 phenol:BPA adduct is crystallized from the concentrate. Distilled to recover solvent, the remaining mother liquor is recycled to the reactor. A purge of the recycle, along with phenol-laden process water, is treated for removal of heavies and recovery of phenol. The adduct is melted and separated by distillation. Phenol is taken overhead, and the molten BPA underflow is prilled in a final processing step.
Hydration/Hydrolysis/Dehydration/Alcoholysis
Hydration of Olefins: Ethanol, Isopropanol, sec-Butanol, tert-Butanol [254–258]
Strong acid catalyzed hydration of olefins is an important industrial method for the production of alcohols. Ethanol, isopropanol, sec-butanol, and tert-butanol are produced in this fashion from ethylene, propylene, 1-butene, and isobutylene respectively. Both indirect and direct methods continue to be used. For unsymmetical olefins (anything greater than C2), the addition of water follows Markovnikov’s rule. Initial protonation of the olefin occurs so as to give the more stable, most substituted carbocation [259].
In the indirect method the olefin is absorbed into concentrated (about 60 wt%) sulfuric acid resulting in protonation of the olefin and formation of a carbocation. The HSO −4 ion then adds to the carbocation to form mono- and di-alkyl sulfate intermediates. The alkyl sulfates are hydrolyzed to the corresponding alcohol by sparging with steam, which also serves to azeotropically distill the alcohol overhead and reconcentrate the sulfuric acid solution for recycle. Reactor conditions of 75–80°C, 0.6–3.0 MPa are typical, with selectivities of greater than 90%. The concentrated sulfuric acid solutions employed in the indirect process require the use of expensive corrosion-resistant materials of construction.
In direct processes, the carbocation intermediate is formed by a heterogeneous strong acid catalyst, such as a sulfonated polystyrene ion exchange resin, tungsten oxide, or supported phosphoric acid catalyst. Water then adds to form the corresponding alcohol in one step. Direct hydrolysis is an equilibrium-limited reaction, favored by low temperature, high pressure, and high water to olefin molar ratios. Both high (260–300°C, 7–20 MPa) and low (130–160°C, 8–10 MPa) temperature fixed bed processes are employed, with 20–75% conversion of the olefin per pass and selectivities of greater than 94%. Due to more mild conditions and lower corrosivity, direct hydrolysis has become the favored method.
In both direct and indirect methods, higher alcohols resulting from olefin oligomerization/hydrolysis, as well as the corresponding dialkyl ether of the product alcohol are typical by-products. All of the C2-C4 alkanols form low-boiling azeotropes with water. Thus, the recovery of an anhydrous product cannot be accomplished in a simple single-feed distillation step. Rather, dehydration of the alcohol is accomplished by a two or three column azeotropic distillation/decantation sequence. If dilute in the reactor effluent, the crude alcohol is first concentrated to close to its azeotropic concentration with water. An entrainer is added that forms an appropriate low-boiling heterogeneous azeotrope with water. Water is removed overhead as the heterogeneous water-entrainer azeotrope in a second distillation. This overhead mixture is decanted, with the water phase steam stripped to recover the entrainer and any alcohol, and the organic phase refluxed to the column. Anhydrous alcohol is recovered as the bottoms of the azeo column.
A convenient entrainer for isopropanol is diisopropylether made as a by-product in the hydration reaction. No additional entrainer is required for sec-butanol, nor for tert-butanol dehydration, as these form heterogeneous azeotropes with water. Diethyl ether can be used for ethanol, as well as other compounds such as n-hexane. A hybrid distillation/adsorption process, utilizing small pore molecular sieves for final removal of water is becoming more common, especially for dehydration of ethanol.
Hydrolysis of Epoxides and Carbonates: Ethylene Glycol, Propylene Glycol, and Higher Analogues [260, 261]
The ring opening of epoxide rings by addition of water is an extremely important industrial reaction, resulting in the formation of glycols and oligomeric ether glycols. Hydrolysis of ethylene oxide is the major industrial route to the production of ethylene glycol. In the typical industrial process, ethylene oxide is reacted thermally at 200°C with a high excess of water (20-1 mole ratio) to produce mono-, di-, and tri-ethylene glycols at a molar selectivity of 90-9-1 respectively.

The hydrolyzed mixture is purified in a series of distillation columns with increasing vacuum level, to successively remove excess water, monethylene glycol, diethylene glycol, triethylene glycol overhead, and higher glycol oligomers as the bottoms of the final column. The purification train is quite energy intensive.
A recently commercialized process involves the hydrolysis of ethylene carbonate, with very high selectivity (>98%) to monoethylene glycol. In this process, ethylene oxide is reacted with carbon dioxide to form ethylene carbonate. Base-catalyzed hydrolysis of ethylene carbonate lead to predominantly monoethylene glycol.




Alcoholysis of Epoxides: Glymes and Ether Alcohols [262–266]
Another important use of epoxides is the formation of glycol ethers by alcoholysis:
When R´ is not hydrogen, such as with propylene oxide, two isomers may be formed. One isomer has a primary hydroxyl group and secondary ether group, the other has the opposite.
The reaction may be catalyzed by both acids and bases, exhibiting a catalyst-dependent product isomer mixture. The epoxide ring may open at either the primary or secondary C–O bond. With anionic (basic) catalysts, the epoxide ring opens preferentially at the least sterically hindered position, typically resulting in 95% or more secondary alcohol product (i.e., 1-alkoxy-2-alkanol). Acid catalysis affords a mixture of the 1-alkoxy-2-alkanol and 2-alkoxy-1-alkanol, with the relative ratio affected by the particular acid catalyst chosen and steric effects related to the size and structure of the reactant alcohol.
Large quantities of glycol ethers derived from C1-C4, C6, and C8 alcohols and ethylene oxide or propylene oxide are produced worldwide, mostly for solvent applications. In order to minimize the further reaction of the formed glycol ether with additional epoxide, a large excess of alcohol is used, at least 5:1 moles of alcohol per mole of epoxide. Alcoholysis of an epoxide is exothermic, with an average heat of reaction of about −80 to −100 kJ/gmole. Reaction conditions depend on the alcohol and epoxide, but are typically 170–220°C, 1.0–1.5 MPa. Conversion of the epoxide is essentially complete, with 80–90% molar selectivity to the mono glycol ether. The product is distilled to recover unreacted alcohol overhead for recycle, and the product glycol ether is distilled under vacuum in a second column, with higher glycol ethers as underflow. If markets dictate, diglycol and higher ethers may be recovered by further distillation of the bottoms.
The reaction of ethylene and propylene oxides with C10-C14 detergent range alcohols affords ethoxylates and propoxylates, which are commonly employed as nonionic surfactants. The solubility of the product alkoxylate can be varied according to the number and type of epoxide molecules incorporated in to the molecule, as well as the chain length of the fatty alcohol. Longer chain groups reduce the solubility in water. Alcohols may be derived from natural fatty acids or higher oxo alcohols (either via ethylene oligomers or kerosene-based olefins).
Dehydration of Acetic Acid: Ketene [267–271]
Ketene, a highly reactive and useful intermediate, has been produced commercially since the 1920s by the high temperature, low pressure, equilibrium-limited, thermal decomposition of acetic acid, with concomitant generation of water (700–750°C, 0.005–0.02 MPa):
The equilibrium is shifted toward ketene production as the temperature is increased. The reaction occurs without a catalyst, but the higher temperatures needed to achieve reasonable rates and high equilibrium conversion without a catalyst lead to excessive decomposition to methane, CO, and CO2. Commercial processes universally are catalyzed, typically with a derivative of phosphoric acid. Phosphoric acid itself, trialkyl phosphates, especially triethyl phosphate, and many other similar compounds are reported in the literature. Heterogeneous catalysis has been extensively studied, but has not been successfully commercialized [272].
In order to efficiently provide the large endothermic heat of reaction, the synthesis is carried out in a plug flow tubular reactor inside a fired furnace. Residence time is a matter of seconds, with achievable conversion of acetic acid of 70–80% per pass at the exit of the tubes. The temperature must be reduced quickly, with effective and rapid separation of liquid water and acetic acid from ketene vapor to prevent extensive back reaction of ketene with water. This quenching is usually accomplished by a series of low residence time, low pressure drop condensers. Overall conversion ends up at 40–70% per pass, depending on condenser design. Ammonia is added, either to the feed or to the furnace effluent to neutralize the phosphoric moieties and help prevent catalysis of the back reaction.
The condensed liquids, with concentration of 30–45% acetic acid in water, are processed, typically by extraction and/or azeotropic distillation to recycle low-water acetic acid to the furnace feed. The ketene thus produced is an efficient acylating agent, and is employed in a variety of downstream processes, such as direct production of esters via reaction with alcohols, synthesis of diketene, and predominantly for the formation of acetic anhydride. In the 1950s and early 1960s the major route to acrylic acid involved the pyrolysis of β-propriolactone, formed by the reaction of formaldehyde with ketene. This route has been completely supplanted by propylene oxidation.
Ketene may also be produced by thermal decomposition of acetone above about 550–600°C (ketene + methane), or from acetic anhydride above about 600–650°C (ketene + acetic acid). The anhydride route is used to some extent to make ketene derivatives in India.
For acetic anhydride production, crude ketene effluent from the condensing train is scrubbed from the vapor phase by acetic acid in an absorber at 45–55°C. Rapid reaction with acetic acid produces acetic anhydride at essentially complete conversion and greater than 95% selectivity.
Heat is removed by a cooled circulation loop at the bottom of the absorber. The crude acetic anhydride, typically 80–90% purity, containing acetic acid and high boiling tars, is distilled in a two column sequence. Acetic acid and lights are recovered as the first distillate and largely recycled to the furnace, while acetic anhydride is recovered overhead in the second column, with tars removed as underflow.
Diketene is produced in a similar fashion by absorption of ketene into crude diketene in an absorber. The [2+2] cycloaddition product of ketene with itself is the four-member lactone, diketene:

Diketene is an extremely reactive and energetic molecule, but can be purified by distillation at reduced pressure and temperature. Diketene is used to produce a variety of specialty acetoacetates and arylides.
Esterification and Related Reactions [273–277]
Chemistry
Esterification is the reaction of a carboxylic acid with an alcohol to produce the corresponding ester and water:

In general, esterifications follow an AAc2 reaction pathway. A strong acid catalyst normally is necessary to achieve industrially viable rates. Typical catalysts are sulfuric acid, sulfonic acids, such as toluene sulfonic and methanesulfonic acids, or sulfonated polystyrene ion exchange resins in the hydrogen form. Although less expensive than sulfonic acids, sulfuric acid tends to promote dehydration of the alcohols to ethers and olefins more so than sulfonic acids. This is especially true for secondary and tertiary alcohols, which also suffer from significantly lower rates of reaction and equilibrium amounts of ester than primary alcohols.
The equilibrium constant for esterification, K eq, can be expressed in terms of liquid concentrations or mole fractions and activity coefficients,

The equilibrium constant is usually on the order of magnitude of unity and the heat of reaction is close to neutral, as shown in Table 10.35 for a selection of esters. Thus, high conversion would require large excesses of alcohol or acid and corresponding large recycles of unreacted materials. To overcome these limitations, a common commercial approach is to use reactive distillation with the aid of azeotropes and two-phase liquid-liquid formation to allow for high conversion at or near stoichiometric feeds. Ester formation is an excellent illustration of LeChatlier’s principle; an equilibrium reaction can be driven to completion by removal of one or more products of the reaction. Several flowsheets are possible, depending on the relative boiling points and azeotropes formed by the acid-water-ester-alcohol system.
Low Boiling Esters
When the ester forms a heterogeneous binary low-boiling azeotrope with water or a heterogeneous tertiary water-ester-alcohol azeotrope, the continuous flow sheet illustrated in Fig. 10.30 can be used. Large volume esters made by this process include ethyl acetate, n-propyl acetate, isopropyl acetate, and n-butyl acetate. In this process, the acid and alcohol are continuously fed to the reboiler-reactor (1) of the esterification column (2), along with a homogeneous strong acid catalyst, such as sulfuric acid, methanesulfonic acid, or toluene sulfonic acid. The low-boiling water-laden azeotrope is taken as the distillate product, which decants into two phases (3). The water layer is steam-stripped (4), with organic distillates returned to the ester column and underflow water discarded. If the water content of the ester-water azeotrope is more than the amount of water created in the esterification reaction, then some of this water may be recycled to the ester column. A portion of the organic layer from the esterification column decanter is refluxed to the ester column, and the rest is distilled in a low boiler column (5). The low boiler column distillate, containing alcohol, water, and ester are returned to the reactor. The dry, alcohol-free ester underflow from the low boiler column is fed to a refining column (6) for final purification from heavies. Heavies, sludge, and spent catalyst is removed as the bottoms of the esterification column and discarded. Sometimes extra water is added near the top of the esterification column to aid in azeotroping the ester overhead.

Flowsheet for production of low-boiling esters
High Boiling Esters
With high boiling esters that cannot be conveniently taken overhead, or those that boil too closely or form azeotropes with their raw material acids and alcohols to allow for easy separation, the reaction still can be driven to completion by using an azeotropic distillation-sidedraw scheme as shown in Fig. 10.31. The acid and alcohol are continuously fed to the reboiler-reactor (1) of the esterification column (2), along with a homogeneous strong acid catalyst, such as sulfuric acid, methanesulfonic acid, or toluene sulfonic acid. Water is removed overhead by azeotroping out as any convenient heterogenous acid-water-ester-alcohol azeotrope or by adding an inert species such as cyclohexane or toluene that forms a low-boiling heterogeneous azeotrope with water. The azeotrope is decanted (3), with the organic layer refluxed to the ester column. The water layer is steam-stripped (4), with organic distillates returned to the ester column and underflow water discarded. The crude product ester is removed low down in the ester column as a sidedraw product with further purification by a sidedraw stripper (5). The ester is taken overhead in the product column (6), with heavy residue as underflow. Many glycol ether esters and higher boiling esters such as butyl butyrate can be made with such a flow sheet.

Flowsheet for production of high-boiling esters
Methyl Acetate [278]
Methyl acetate cannot be produced in high purity using the simple esterification schemes outlined above due to unfavorable vapor–liquid and liquid-liquid equilibrium behavior:
-
1.
The methyl acetate-methanol–water system does not form a ternary azeotrope.
-
2.
The methyl acetate-methanol azeotrope is lower boiling than the water-methyl acetate azeotrope.
-
3.
There is a distillation boundary between the two azeotropes.
-
4.
The liquid-liquid region does not include either the methyl acetate-water or the methyl acetate-methanol azeotrope, nor does it cross the distillation boundary.
Consequently the water generated by esterification cannot be removed effectively, nor can the methyl acetate azeotropes be broken readily. The reactive distillation process (see Fig. 10.32) overcomes both unfavorable reaction and poor phase equilibriums to produce high purity methyl acetate and water in one column from near stoichiometric acetic acid-methanol feed ratios. Conceptually this column can be thought of as four heat-integrated distillation columns and a reactor stacked on top of each other. Reaction occurs below the sulfuric acid feed in a series of countercurrent high hold-up trays. This countercurrent separation, along with the low feed point of methanol results in high local excesses of at each end of the reactive section (3) in spite of the overall stoichiometric feed. The acetic acid feed acts as an extractive distillation agent above the reaction zone (2) to break the methyl acetate-methanol azeotrope. The upper rectification section (1) knocks back any acetic acid from the methyl acetate product, and the lowermost stripping section (4) removes methanol from the water bottoms. High purity methyl acetate is the distillate product, and clean water underflows.

Flowsheet for production of methyl acetate by reactive distillation
Plasticizer Esters
One of the primary uses of higher boiling and low volatility esters is as plasticizers. A plasticizer increases the flexibility and ductility of a brittle thermoplastic polymer by reducing the glass transition of the polymer. PVC is the most commonly plasticized polymer. A large number of plasticizers based on phthalic anhydride, TPA, adipic acid, TMA, benzoic acid with 2-ethylhexanol, or C9 and C10 iso-alcohols are made commercially, by anhydride reactions with alcohols (e.g., di-2-ethylhexylphthalate) or high boiling acid-alcohol reactions driven to completion by introduction of excess alcohol and removal of water generated [279].
Esterifications with Anhydrides
For hindered, unreactive alcohols, those susceptible to dehydration, and those producing very high boiling esters, it is often more favorable to produce the ester via reaction with an anhydride. The basic reaction of a symmetric anhydride with an alcohol is given by:

The equilibrium is quite favorable and lies essentially completely toward the ester. Since the reaction is first order in both alcohol and anhydride, a small excess (less than 5 mole%) of either the alcohol or anhydride is added in practice to ensure complete conversion in reasonable reaction times. Typically a catalyst is not necessary, but strong acids or pyridine derivatives can be used to accelerate the rate.
Many carboxylic acid anhydrides can be made via reactive distillation in which a low boiling anhydride is reacted with a higher boiling carboxylic acid. This reaction, occurring stepwise, is equilibrium-limited and can be driven to complete formation of the symmetric anhydride of the high boiling acid by distillative removal of the generated low-boiling acid. The most common low boiling anhydride used in such a process is acetic anhydride.
Hydrogenation [280–283]
Chemistry
Hydrogenation involves the addition of molecular hydrogen (H2) to a functional group to reduce its oxidation state. Typical substrates and products of hydrogenation reactions include: alkynes, alkenes, and aromatic rings to alkanes, aldehydes to primary alcohols, ketones to secondary alcohols, esters and carboxylic acid to alcohols, nitriles to imines and amines, and amides to amines. A closely related type of reaction, hydrogenolysis, involves hydrogen addition with concomitant breaking of other carbon–carbon or carbon-heteroatom (oxygen, nitrogen or halogen) bonds. Hydrogenation differs from protonation or hydride addition; in a hydrogenation the products have the same charge as the reactants. The ease of hydrogenation of various functional groups generally follows the decreasing order: [284]
Steric factors and substitution patterns further dictate the ease of hydrogenation, decreasing in order of straight chain > branched far from the functional group to be hydrogenated > branched adjacent to the functional group to be hydrogenated. Bulky substituents also tend decrease reactivity. Selective hydrogenation of one functional group in the presence of another functional group (e.g., hydrogenation of the double bond of an α β-unsaturated ketone, while preserving the ketone group) is a fairly common issue, often solved by judicious selection of catalyst metal as well as reaction conditions.
A hydrogenation reactor must be designed for effective heat and mass transfer. Most hydrogenation reactions are highly exothermic, with heats of reaction at least above −50 kJ/gmole and high adiabatic temperature rises. Typical heats of reaction for various functional group hydrogenations are given in Table 10.36. Moreover, hydrogen gas must get to the surface of the catalyst through vapor or liquid films. Common reactor formats include CSTR’s with internal cooling coils or external exchanger loops, Buss loop reactors, and trickle beds with cooling loops. These formats provide good mixing for mass and heat transfer, but as backmixed systems require large reactor volumes for high, i.e., greater than 95%, conversion. Often a small plug flow polishing fixed bed is included after the main reactor to get the conversion up to above 99%. See for example, the hydrogenation of benzene to cyclohexane. Examples of commercially significant hydrogenation reactions are given in the subsections which follow.
Methanol [285–289]
Methanol is produced by the hydrogenation of carbon monoxide and carbon dioxide. The first industrial production of methanol from CO, CO2, and H2, i.e., synthesis gas, was carried out over a zinc oxide/chromium oxide catalyst at high pressure (25–35 MPa) and temperature (300–450°C). Developments during the late 1960s and early 1970s resulted in the first modern Cu/ZnO/catalysts capable of operation at much lower temperatures (200–310°C) and pressures (4–10 MPa). The low pressure process results in lower investment and production costs, higher yields, improved reliability, and much larger potential plant size. Plant capacity has increased dramatically from less than 1,000 metric tons per day for old plants, to more typically greater than 2,500 tons per day, with single train capacities of 5,000 metric tons per day or more now in operation.
The synthesis of methanol can be thought of as the hydrogenation of carbon monoxide and carbon dioxide. A typical feed gas contains somewhat higher than a 2–1 M ratio of H2 to CO, and 2–12 mole% CO2. The feed gas composition often is characterized by the stoichiometric number, SN, defined as:
The value of SN should be about 2.05 for optimal performance. Syngas for methanol production can (and does) originate from a variety of sources and processes including natural gas via SMR, partial oxidation (POX), or autothermal reforming (ATR), as well as gasification of coal. ATR generally produces the appropriate SN for methanol directly. SMR needs CO2 addition; coal gasification and POX require additional water-gas shift and subsequent CO2 separation to enhance hydrogen sufficiently to give the desired SN of 2.05. See Section “Synthesis Gas”, and Chaps. 19, Chaps. 20, and Chaps. 22 for further details on syngas production.
The net stoichiometry of the reaction is:

In reality, the main source of the carbon incorporated into the methanol molecule is actually derived from carbon dioxide via reaction with hydrogen:

As the carbon dioxide in the feed is consumed, the reverse water-gas shift reaction kicks in to supply more:

The overall methanol synthesis network is equilibrium-limited, exothermic, and results in a reduction of volume upon reaction. Low temperatures and high reaction pressures favor conversion to methanol.
The modern low pressure methanol process is relatively simple as shown in Fig. 10.33. A syngas with SN = 2.05, less than 0.05 ppm sulfur (as H2S), is compressed (1), if needed, to the reactor loop pressure of 4–10 MPa,Footnote 2 interchanged with the reactor effluent, combined with recycle gas, and fed to the fixed bead reactor containing the Cu/ZnO/Al2O3 catalyst (2). Both multistage intercooled adiabatic, and multi-tubular steam cooled reactors are used commercially. Some commercial processes use reactors in series at different conditions for higher per pass conversion. The reactor temperature is usually maintained below 260°C. Conversion of CO is about 50% per pass. Major by-products are ethanol (150–200 ppm); higher alcohols (300–400 ppm); acetone, MEK, and other ketones (less than 10 ppm); and depending on the generation of catalyst, 20–100 ppm dimethyl ether. Yield from CO is quite high, usually more than 99%.

Flowsheet for production of methanol
The methanol–rich reactor effluent is cooled, and crude methanol is separated from non-condensables before further purification (3). A small purge is taken from the separator off-gas to prevent build-up of inerts such as argon, nitrogen, methane entering with the inlet syngas. The remainder of the gas (typically at a SN of 3 to about 10) is boosted to loop pressure and recycled (4).
The crude methanol is typically purified in a two or three column distillation sequence. The first column (5) removes light ends overhead (dissolved gases and other species). The product methanol is then distilled overhead, with water taken as underflow, and intermediate boiling-impurities (ethanol, higher alcohols, ketones) as a sidedraw, if high purity is needed. The product distillation is often done as a dual-column pressure swing setup to reduce energy consumption. In this scheme, steam is fed only to the reboiler of first high pressure methanol product column (6). A portion of the methanol is distilled overhead, with methanol–water as bottoms. The vaporous product methanol from the first column is not condensed, but used to drive the reboiler (7) of the second lower pressure column (8). The remainder of the methanol is distilled in the second column, with water taken as bottoms product, and impurities as a sidedraw if needed.
Aromatic Ring Saturation: Cyclohexane [290–292]
Cyclohexane, via the hydrogenation of benzene, is an important intermediate in the production of adipic acid and caprolactam, monomers for nylon 6,6.

Both vapor and liquid phase processes are practiced commercially. In one common liquid phase process, benzene is hydrogenated isothermally over a fine slurry of suspended Raney nickel at 180–200°C, 5 MPa. Soluble catalysts are also used. The heat of reaction is removed by a combination of an external steam generating heat exchanger and by vaporization of part of the reactor contents. Vapors from the first stage reactor are passed to a fixed-bed polishing reactor for final conversion. Conversion is about 95% in the first stage, with essentially complete conversion in the polishing bed. The vapor effluent is flashed, condensed and separated from hydrogen and light gases for recycle and purging. If the hydrogen purity is low, then the cyclohexane is fed to a small stabilizer column to remove light ends. Low temperature operation significantly reduces the equilibrium isomerization of cyclohexane to methylcyclopentane. Selectivity is greater than 99.8% to benzene with less than 100 ppm methylcyclopentane.
α,β-Unsaturated Carbonyls to Saturated Ketones and Aldehydes [293, 294]
An important aspect of the production of ketones and aldehydes via aldolization (see section “Aldol Condensation”) is the selective hydrogenation of the double bond of the α,β-unsaturated carbonyl intermediate without formation of alcohols. Pd on alumina, silica, titania, or carbon have been found to be very effective catalysts for this transformation, with 100/1. Ni, Cu, Cu/Cr, and Pt catalysts normally are not selective for double bond hydrogenation without excessive alcohol formation.
Much research effort has been expended on developing selective catalysts for reduction of the α,β-unsaturated carbonyl to an alcohol, while retaining the double bond functionality.
Aldehydes and Ketones to Alcohols [295, 296]
Aldehydes are readily hydrogenated to their corresponding alcohols under relatively mild conditions. Three alcohols of significant volume produced by the hydrogenation of Oxo-derived aldehydes are n-propanol, n-butanol, and i-butanol:



The hydrogenation may be conducted in either the gas or liquid phase with supported Ni, Cu, or Cu/Cr, or Cu/Zn catalysts at about 115–160°C, 0.2–8 MPa. Yields are typically above 98%. By-products consist of minor amounts of acetals, ethers, and higher boiling condensation products. Detergent range alcohols are also produced in this fashion from higher Oxo aldehydes.
The important plasticizer alcohol, 2-ethyl-hexanol, may be synthesized by a similar hydrogenation of 2-ethylhexaldehyde, or alternatively directly from the aldol-derived α β-unsaturated aldehyde by the simultaneous hydrogenation of the double bond and carbonyl.
Both vapor and liquid phase processes are utilized with supported Ni, Ni/Cu, Cu, or Cu/Cr catalysts. Vapor phase processes are commonly run in two stages, with about 90% conversion in the first stage. The first stage is operated at 100–170 or 200–250°C, 0.5–5.0 MPa. The final polishing stage is operated at 200–250°C, 0.5–5.0 MPa.
Carbinols, the common name for ketone-derived secondary alcohols, can be produced in very similar vapor or liquid hydrogenation processes under slightly more forcing temperature and pressure conditions than aldehyde conversion. The conversion of more hindered ketones, such as MIBK (4-methyl-2-pentanone) to methyl isobutyl carbinol (MIBC) require somewhat longer residence times, temperature, and/or hydrogen pressure, but yields are essentially quantitative. The equilibrium for formation of ketols from ketones and product alcohols is much less favorable than for aldehyde-alcohol acetal formation.
Supported heterogeneous Pt and Ru catalysts are successfully employed for the reduction of highly hindered or sensitive carbonyls, as exemplified by the conversion of the ring-strained 2,2,4,4-tetramethyl-1,3-cyclobutadione to 2,2,4,4-tetramethyl-1,3-cyclobutadiol [297], but are not normally used for more commodity applications.


Esters to Alcohols: Dimethyl Maleate to γ-Butyrolactone, THF, and 1,4-Butanediol [298–301]
In one commercial process, maleic anhydride is esterified with methanol in a reactive distillation column to form the intermediate dimethyl maleate. The reaction is driven to completion by removal of water and methanol from the top of the column. Methanol and water are separated by distillation, with the methanol recycled. The maleate ester underflow from the first column is vaporized in excess hydrogen and is hydrogenated in a fixed bed adiabatic reactor. The reactor effluent is cooled, condensed, and the crude BDO product is separated from hydrogen (which is recompressed and recycled). The crude BDO is separated in a three-column distillation sequence. THF and other lights are removed overhead in the first column. Methanol is distilled overhead in second column for recycle, and high purity product BDO is the distillate of the third column. Unreacted maleate ester and γ–butyrolactone by-product in the underflow of the column are recycled for further conversion. The molar yield of BDO from maleic anhydride is about 97%. Conditions can be modified to produce all THF or to recover GBL. In another commercial process maleic anhydride is hydrogenated directly without first making the diester.
Esters to Alcohols: Dimethyl Terephthalate to 1,4-Cyclohexanedimethanol [302]
An example of the large-scale hydrogenation of an ester to alcohols is the production of 1.4-cyclohexanedimethanol (CHDM) from DMT. DMT in a mixture of methanol and CHDM is hydrogenated in the liquid phase over a supported Pd catalyst at 110–180°C, 30–40 MPa to give a mixture of cis/trans-cyclohexane-1,4-dicarboxylic acid dimethyl ester. Further hydrogenation over copper chromite at more severe conditions yields CHDM.
Co-polyesters of TPA, ethylene glycol, and CHDM have enhanced toughness, better injection molding and sheet extrusion properties than simple polyethyleneterephthalate polymers.
Dehydrogenation [303–305]
Chemistry
Conceptually, direct dehydrogenation, a reaction producing hydrogen as a by-product, is simply the reverse of hydrogenation. Direct dehydrogenation reactions find commercial use primarily in the conversion of saturated alkanes and alkyl aromatics into olefinic and diolefinic compounds (butanes to butenes, butenes to butadiene, ethylbenzene to styrene, long chain n-paraffins to olefins), as well as the conversion of secondary alcohols into ketones (isopropanol to acetone, sec-butanol to 2-butanone, methanol to formaldehyde). Oxidative dehydrogenation, wherein the hydrogen is converted in situ to water, is also practiced for accomplishing the same ends as direct dehydrogenation. See for example the discussion on formaldehyde earlier in this chapter.
Dehydrogenation reactions are endothermic and generally severely equilibrium-limited. Reasonable rates and conversion are favored by high temperature and low pressure, as the number of moles increases with generation of hydrogen. Figure 10.34 presents the effect of temperature on equilibrium conversion for several important dehydrogenation systems (at 0.1 MPa total pressure, with no diluents added). Note that very high temperatures are required with most hydrocarbon systems for a reasonable conversion of 50%. This graph illustrates at least one reason ethane to ethylene dehydrogenation has not been commercialized. To get conversions of ethane comparable to steam cracking, one has to operate at temperatures where thermal cracking is significant anyway. High temperature operation leads to other collateral problems:

Effect of temperature on equilibrium conversion for several dehydrogenation reactions, pressure of 0.1 MPa
-
1.
Significant high temperature (i.e., expensive) energy is needed for heating feeds and reaction mixtures.
-
2.
The thermal cracking of molecules has a high activation energy and tends to compete with dehydrogenation above about 650°C.
-
3.
Consecutive dehydrogenations of the same molecule (often energetically favorable) tend to lead to rapid coke formation and catalyst fouling, with frequent regenerations required.
-
4.
Metal sintering and permanent deactivation is accelerated at high temperatures.
Reactor designs reflect these factors. Common designs include, (a) direct-fired furnace feed heaters with adiabatic fixed beds in series (often with hot shots of feed between beds), (b) isothermal fixed beds (reactor in a furnace), (c) moving beds to allow for regeneration, and (d) fluid beds for heat control, with separate continuous regenerators (much like FCC units).
Manipulation of the partial pressure of products generated is another approach to allow lower temperature operation. Common methods include:
-
1.
Operation at subatmospheric pressure.
-
2.
Addition of steam or other inerts to lower hydrogen and product partial pressures.
-
3.
React away hydrogen as it is formed or form water instead (i.e., oxidative dehydrogenation).
The effect of the magnitude of the equilibrium constant, inert addition, feed hydrogen, and total system pressure on equilibrium conversion can be calculated readily by the relationship given below [306]:
-
where
K p = temperature-dependent equilibrium constant
-
X = molar conversion of reactant
-
H = moles of H2 in feed to moles of reactant
-
P = total system pressure
-
I = moles of inerts in feed to moles of reactant
The process designer has many degrees of freedom, with tradeoff on temperature, partial pressure, and diluents addition, as well as reactor format. Thus, many flowsheet alternatives can accomplish the same end result, and many versions of dehydrogenation processes, particularly for alkanes to olefins, have been successfully commercialized.
Catalysts used for hydrogenations, such as precious metals and Group VIII transition metals, generally also are useful for dehydrogenation reactions. Supported Pt—Sn and Cr-based catalysts are the most common used commercially. Although inactive itself for dehydrogenation, the addition of Sn to Pt catalysts acts to suppress hydrogenolysis, moderates coking rate (promotes migration of coke from the active catalyst metal to the support), and reduces sintering. Table 10.37 summarizes commercial conditions and catalysts for a number of important industrial dehydrogenation reactions. More detailed discussions of a few key processes are given below.
Butenes and 1,3-Butadiene [307–309]
A large number of similar commercial processes have been developed for the dehydrogenation of butanes to butenes and butanes/butenes to 1,3-butadiene, using heterogeneous catalysts (e.g., Pt/Sn on γ-Al2O3; zirconia, or Zn/Ca-aluminate; Chromia on γ-Al2O3; Cromium oxide-Ca-Ni-phosphate; iron oxide) at 530–700°C, 0.03–0.5 MPa.

Butadiene is particularly susceptible to polymerization and coking, so high steam to feed ratios are often used to reduce coke formation and to lower the partial pressure of hydrogen to shift the equilibrium toward butadiene. Even with these steps, coking occurs rapidly. Multiple reactors are used to allow regeneration/operation to occur in cycles lasting on the order of an hour or less.
In an alternative approach, oxidative dehydrogenation methods have also been employed to overcome the unfavorable equilibrium. In one version, butanes and/or butenes, steam, and air are fed to a fixed bed reactor at a relatively low inlet temperature of about 370°C, where combustion of produced hydrogen supplies the necessary heat of reaction for the dehydrogenation. The outlet temperature rises to about 480–600°C due this combustion. Conversion over the ferrite/Zn or ferrite/Mn catalyst is greater than 60%, at up to about 90–93% selectivity.
Internal Olefins from Higher n-Alkanes [310, 311]
The dehydrogenation of C10-C14 n-paraffinic materials derived from kerosene fuels is an important source of internal olefins for linear alkylbenzene production (see Section 10.3 above). In a typical commercial process, a narrow-boiling kerosene cut is first treated by shape-selective adsorption to recover a stream high in n-paraffins. This material is dehydrogenated in the vapor phase over Pt/Sn on alumina (often with In and Li promoters) at 300–550°C, 0.1–0.3 MPa, with H:C ratio of (5–9):1. The higher alkanes are particularly susceptible to thermal cracking reactions, and the hydrogen helps prevent this decomposition pathway. Conversion is 10–15% per pass at selectivities of 90–94%. Unreacted n-paraffins for recycle are separated from olefins by adsorption. This is another example of a separation exploiting functional group differences to effect the separation in a single step, whereas separation by volatility differences (distillation) would be prohibitively complicated.
Styrene from Ethylbenzene [312–315]
Direct dehydrogenation of ethylbenzene to styrene accounts for about 85% of commercial production. The remaining styrene is produced via coproduction with propylene oxide (see section 1 of this chapter). The major reaction is the endothermic (+125 kJ/gmole), high temperature, vapor phase conversion of ethylbenzene to styrene and hydrogen. Side reactions include the thermal degradation of ethylbenzene to benzene and ethylene, and catalyzed formation of toluene and methane from styrene and hydrogen.
In a typical commercial process as shown in Fig. 10.35, superheated steam (1) and ethylbenzene (1:1 weight ratio, 6:1 M ratio) are fed at about 620°C over potassium promoted iron oxide catalyst in a series of adiabatic fixed bed reactors (2), (3), under vacuum (around 0.06 MPa). The effluent from each stage must be reheated by superheated steam injection or indirect fired heating to keep equilibrium conversion and rates sufficiently high. Overall conversion is typically 60–70% per pass at up to 97% selectivity to styrene. Steam plays an important role in the process. It:

Flowsheet for dehydration of ethylbenzene to styrene
-
1.
Brings in heat to get the reaction mixture up to temperature
-
2.
Reduces the partial pressures of products allowing a tradeoff between temperature and conversion
-
3.
Removes coke by steam–carbon reaction (catalyzed by potassium)
-
4.
Minimizes EB cracking
-
5.
Keeps the iron catalyst in the correct oxidation state (H2 will reduce iron to its catalytically inactive elemental state)
-
6.
Redistributes potassium on the working catalyst, enhancing lifetime
The reactor effluent is cooled, separated from off-gases (4) and allowed to phase separate to remove the bulk of the water (5). Purification is accomplished by a straight-forward series of distillations, but time at high temperatures must be limited to minimize losses to styrene polymerization. Toluene and benzene are distilled overhead first (6), followed by distillation of ethylbenzene for recycle (7), and finally the styrene product is taken overhead from heavies (8). Additional styrene is recovered from the tar stream in a flash step (9). Styrene purities range from 99.85 to 99.95%. Ethylbenzene and styrene are close boiling, with a relative volatility of about 1.3. Thus, 70–100 fractionation stages are required, depending on purity requirements. Most modern plants use high efficiency packing to reduce pressure drop, increase stage efficiency for the same height, and to increase throughput for a given column diameter. Styrene is quite prone to polymerization.
An oxidative reheat process is also commercially practiced, typically for retrofits to increase capacity of bottlenecked plants. Air is introduced between the dehydrogenation stages to convert some of the hydrogen into water to overcome the equilibrium limitations and to generate heat. Advantages include increasing the ethylbenzene conversion to about 75% per pass, unloading the costly ethylbenzene/styrene fractionation column, reducing interstage heating requirements, and lowering superheated steam consumption.
Dehydrogenation of Alcohols: MEK, Acetone, and Formaldehyde [316–320]
Aldehydes and ketones can be made by the dehydrogenation of alcohols, liberating hydrogen as a by-product, as illustrated for acetone and MEK:


These reactions are equilibrium-limited, modestly endothermic, and are favored by low pressure and high temperatures. Required temperatures are considerably lower than for alkane dehydrogenation. Compare Figs. 10.34 and 10.36. Both 2-butanone (MEK) and acetone are produced commercially by similar high temperature vapor phase catalytic dehydrogenations, from sec-butanol and isopropanol respectively. Typical conditions are 220–450°C, 0.1–0.6 MPa. A multitubular fixed bed reactor with hot oil circulation provides the endothermic heat of reaction. Copper catalysts are highly active for these dehydrogenation reactions and give high selectivities, typically greater than 90%. The main side products are the alkene (butenes or propylene) formed by dehydration of the parent alcohol, as well as small amounts of aldol condensation products. The high temperatures and the formation of unsaturated by-products lead to relatively rapid coking and deactivation. The catalyst must be burned off roughly every 3–6 months for optimal productivity.

Effect of temperature on equilibrium conversion for alcohol dehydrogenations
Low temperature (150°C) liquid phase processes using Raney nickel or copper chromite are also practiced for the production of MEK, especially in Europe. Selectivity is somewhat better than the vapor phase process. Dehydrogenation of isopropanol to acetone is currently a relatively minor contributor to acetone production worldwide, with most acetone is derived as a by-product of phenol production (see section “Phenol/Acetone or MEK and Hydroquinone/Acetone” for a discussion of the phenol/acetone process).
A similar direct dehydrogenation of methanol to formaldehyde occurs to a limited extent with commercial silver catalysts for the conversion of methanol to formaldehyde, but is combined with oxidative dehydrogenation for more efficient operation (see the section, “Formaldehyde,” for further details).
Notes
- 1.
Most of the remainder of world acetone production is derived from dehydrogenation of isopropanol. See the section, “Dehydrogenation.”
- 2.
Coal gasification-derived gas is usually high enough pressure, SMR, ATR, POX gas needs compression.
References
James JL, Shore D (1993) Chemicals from petroleum. In: McKetta JJ (eds) Chemical processing handbook. Marcel Dekker, New York, p 23 and calculations by the author
American Chemistry Council (2011) Global business of chemistry. http://www.americanchemistry.com/s_acc/sec_directory.asp?CID=292&DID=747
Rase HF (2000) Handbook of commercial catalysts. CRC Press, Boca Raton
Leach BE (1983) Applied industrial catalysis, vol 1–3. Academic, New York
Rase HF (1990) Fixed bed reactor design and diagnostics. Butterworth, Stoneham
Satterfield (1977) Chemical reactor design for process plants, vols 1 & 2. Wiley, New York
Rase HF (1977) Chemical reactor design for process plants, vol 1 & 2. Wiley, New York
Doherty MF, Fidkowski ZT, Malone MF, Taylor R (2008) Distillation (section 13). In: Green DW (ed) Perry’s chemical engineers’ handbook, 8th edn. McGraw Hill, New York
Barnicki SD, Siirola JJ (1997) Enhanced distillation, In: Perry RH, Green DW, Maloney JO (eds) Perry’s chemical engineers’ handbook, 7th edn. McGraw Hill, New York, pp. 13–54, 13–85
Barnicki SD, Hoyme CA, Siirola JJ (2006) Separations process synthesis, In: Seidel A (ed) Kirk-othmer encyclopedia of chemical technology, vol 10, 6th edn. Wiley, Hoboken, pp. 297–339
Dimian AC, Bildea CS (2008) Chemical process design. Wiley-VCH Verlag, Weinheim
Sundmacher K, Kienle A, Seidel-Morgenstern A (2005) Integrated chemical processes: synthesis, operation, analysis, and control. Wiley-VCH Verlag GmbH & Co, Weinheim
Stichlmair JG, Fair JR (1998) Distillation principles and practices. Wiley-VCH Verlag, New York
Rousseau RW (ed) (1987) Handbook of separations process technology. Wiley, New York
Chemical economics handbook. Marketing research reports. SRI Consulting, Menlo Park, CA, (2006–2010)
Chemical profiles. ICIS chemical business, http://www.isis.com, (2006–2010)
Manufacturing energy consumption surveys (MECS). U.S. energy information agency, department of energy, Washington, DC, USA 1998, 2002, 2006. http://www.eia.doe.gov/emeu/mecs/
Annual energy review 2009, Report No. DOE/EIA-0384(2009) U.S. energy information agency, department of energy, Washington, DC, USA. 19 Aug 2010
Petroleum and fuel oil, data for all years including revisions, State Energy Data System (SEDS). U.S. Energy Information Agency, Department of Energy, Washington, DC, USA (2009) http://www.eia.gov/emeu/states/_seds_updates.html
Davis S (2008) Petrochemical industry overview. In: CEH Marketing Research Report, SRI Consulting, Menlo Park, CA, USA, p 7
Sundaham KM, Shreehan MM, Olszewski EF (2005) Ethylene. In: Seidel A (ed) Kirk-Othmer encyclopedia of chemical technology, vol 10, 5th edn. Wiley, Hoboken, pp 598–632
Devanney MT (2009) Ethylene In: CEH Marketing Research Report, SRI Consulting, Menlo Park, CA, USA, pp 6–17
Devanney MT (2008) Propylene. In: CEH Marketing Research Report, SRI Consulting, Menlo Park, CA, USA, pp 6–14
Ethylene. (2010) In: Hydrocarbon processing’s petrochemical processes 2010, Gulf Publishing Company, Houston. p. 121–130
Chauvel A, Lefebvre G (1989) Petrochemical processes, 1. Synthesis-gas derivatives and major hydrocarbons. Gulf Publishing, Houston, pp 117–155
Propylene. (2010) In: Hydrocarbon processing’s petrochemical processes 2010. Gulf Publishing Company, Houston. p. 245–248
Propylene. (2010) In: Hydrocarbon processing’s petrochemical processes 2010. Gulf Publishing Company, Houston, TX. p. 249–250,252
MacDougall LV (1991) Methanol to fuels routes—the achievements and remaining problems. Catal Today 8:337–369
Park T-Y, Froment GF (2001) Kinetic modeling of the methanol to olefins process. 1. model formulation. Ind Eng Chem Res 40:4172–4186
Park T-Y, Froment GF (2004) Analysis of fundamental reaction rates in the methanol-to-olefins process on ZSM-5 as a basis for reactor design and operation. Ind Eng Chem Res 43:682–689
Alwahabi SM, Froment GF (2004) Single event kinetic modeling of the methanol to olefins process on SAPO-34. Ind Eng Chem Res 43:5098–5111
Propylene. (2010) In: Hydrocarbon processing’s petrochemical processes 2010, Gulf Publishing Company, Houston, TX. p. 251,253
Liebner W (2005) Lurgi MTP® technology. In: Meyers RA (ed) Handboook of petrochemicals production processes. McGraw-Hill, New York, pp 10.3–10.14
Pujado PR, Andersen JM (2005) UOP/Hydro MTO process. In: Meyers RA (ed) Handboook of petrochemicals production processes, McGraw-Hill, New York, pp 10.15–10.26
World’s largest ethylene-from-ethanol plant in Brazil. Plastics today, 1 Oct 2010. http://www.plasticstoday.com/articles/worlds-largest-ethylene-ethanol-plant-brazil
Dow chemical explores methods for producing its key feedstocks from renewable resources. ICIS chemical business, 28 Feb 2011. http://www.icis.com/Articles/2011/02/28/9438198/dow-studies-bio-based-propylene-routes.html
Wells GM (1995) Ethylene from ethyl alcohol by dehydration, Handbook of petrochemicals and processes. Gower Publishing Company, Brookfield, pp 173–174
Aromatization, Benzene, Benzene and Toluene, and BTX Aromatics. (2010) In: Hydrocarbon processing’s petrochemical processes 2010, Gulf Publishing Company, Houston, TX, pp. 70, 72–76, 79–83
Franck H.-G, Stadelhofer AW (1998) Industrial aromatic chemistry. Springer, Heidelberg, pp. 27–98, 120–31
Folkins HO (2003) Benzene. In: Ullmann’s encyclopedia of chemical technology, vol 4, 6th edn, Wiley-VCH Verlag GmbH, Weinheim, pp.720–729
Fruscella W (2004) Benzene. In: Seidel A (ed) Kirk-Othmer encyclopedia of chemical technology, vol 5, 3rd edn. Wiley, Hoboken, pp 596–624
Chauvel A, Lefebvre G (1989) Petrochemical processes, 1. Synthesis-gas derivatives and major hydrocarbons. Gulf Publishing, Houston, pp 235–300
Paraxylene and Xylene Isomerization. In: Hydrocarbon processing’s petrochemical processes 2010, Gulf Publishing Company, Houston, TX. 2010, pp. 192–196, 284–288
Benzene/Toluene, PERP 06/07-6, Chemsystems PERP Program, Nexant, White Plains, New York, June, 2007
Aromatics, Transalkylation and Benzene, Ethylbenzene Dealkylation, and BTX Aromatics. In: Hydrocarbon processing’s petrochemical processes 2010, Gulf Publishing Company, Houston, TX. 2010, pp. 71, 77
Stern DL, Brown SH, Beck JS (2008) Isomerization and transalkylation of alkylaromatics. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, pp 3168–3194
Bradley TW (2005) ExxonMobil PxMaxSM p-Xylene from toluene, chapter 13.1 and ExxonMobil XyMaxSM xylene isomerization, chapter 13.2. In: Meyers RA (ed) Handboook of petrochemicals production processes, McGraw-Hill, New York, pp. 13.3–13.21
Aromatics Extraction and Aromatics Extractive Distillation. In: Hydrocarbon processing’s petrochemical processes 2010, Gulf Publishing Company, Houston, TX, 2010, pp. 65–69
Franck H-G, Stadelhofer AW (1998) Industrial aromatic chemistry. Springer, Heidelberg, pp 105–121
Folkins HO (2003) Benzene. In: Ullmann’s encyclopedia of chemical technology. vol 4, 6th edn. Wiley-VCH Verlag GmbH, Weinheim, pp. 730–738
Paraxylene and Paraxylene, Crystallization. In Hydrocarbon processing’s petrochemical processes 2010, Gulf Publishing Company, Houston, TX, 2010, pp. 192, 195–200
Chauvel A, Lefebvre G (1989) Petrochemical processes, 1. Synthesis-gas derivatives and major hydrocarbons. Gulf Publishing, Houston, pp 238–273
Commissaris SE (2005) UOP ParexTM process for p-Xylene production, chapter 13.3. In: Meyers RA (Ed) Handboook of petrochemicals production processes. McGraw-Hill, New York, pp 13.23–3.30
Ormonde E, Yoneyama M (2009) Butanes. In: CEH Marketing Research Report, SRI Consulting, Menlo Park, CA, pp. 10–13
Butadiene/Butylenes. PERP 09/10-5, Chemsystems PERP Program, Nexant, White Plains, New York. (2010)
Davis S, Yoneyama M (2008) Butylenes. In: CEH Marketing Research Report, SRI Consulting, Menlo Park, CA, pp 7–20
Calamur N, Carrera ME, Wilsak RA (2004) Butylenes. In: Seidel A (ed) Kirk-othmer encyclopedia of chemical technology, vol 4, 5th edn. Wiley, Hoboken, pp 402–433
Chauvel A, Lefebvre G (1989) Petrochemical processes, 1. Synthesis-Gas Derivatives and Major Hydrocarbons. Gulf Publishing, Houston, TX, pp 195–226
Butene-1, Butene-1, Polymerization Grade, Butenes (Extraction from Butanes/Butenes), and Isomerization. In: Hydrocarbon processing’s petrochemical processes 2010, Gulf Publishing Company, Houston, TX. 2010, pp 91, 93, 94, 154
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 67–74
Brummer R (2005) BASF butadiene extraction technology, Chapter 3.1. In: Meyers RA (ed) Handboook of petrochemicals production processes. McGraw-Hill, New York, New York, USA, p 3.3–3.9
Butadiene extraction, BASF NMP process. http://www.lurgi.com/website/Butadiene.55.0.html?&L=1
Chauvel A, Lefebvre G (1989) Petrochemical processes, 1. Synthesis-gas derivatives and major hydrocarbons. Gulf Publishing, Houston, TX, USA, p 199–208
Butadiene. In: Hydrocarbon processing’s petrochemical processes 2010, Gulf Publishing Company, Houston, TX. 2010, pp. 87–89
Krupa S, Foley T, McColl S (2005) UOP KLP 1,3-butadiene from acetylene process, Chapter 3.2. In: Meyers RA (ed) Handboook of petrochemicals production processes. McGraw-Hill, New York, New York, USA, p 3.11–3.13
Jeanneret JJ, Mowry JR (1997) UOP sorbex family of technologies, Chapter 10.3. Cusher, NA. UOP IsoSiv Process—Chapter 10.5. Sohn SW. Kerosene IsoSiv process for production of normal paraffins, Chapter 10.6. Sohn SW. UOP molex process for production of normal paraffins—Chapter 10.7. Sohn SW. UOP olex process for olefin recovery in handboook of petrochemicals production processes. 2nd Ed. Meyers RA. Editor-in-chief, McGraw-Hill, New York, NY. p.10.45-10.52, 10.61-10.81
Vora BP, Peterson GA, Sohn, SW, Riley MG (2009) Detergent alkylate and detergent olefins production. In: Zoller U (ed) Handbook of Detergents, Part F: Production. CRC Press, Taylor & Francis, LLC, Boca Raton, pp 45–47
Modler R, Kalin T, Gubler R, Inoguchi Y (2009) Normal paraffins (C9-C17). In: CEH Marketing Research Report. SRI Consulting, Menlo Park, CA, USA, p 5–16
Higman C, van der Burgt M (2003) Gasification. Gulf professional publishing, Elsevier science, USA, pp. 41–146, 298–328
Hiller H, et al. (2003) Gas production. In: Ullmann’s encyclopedia of chemical technology. vol 21, 6th edn. Wiley-VCH Verlag GmbH, Weinheim, pp. 305–470
Shadle LJ, Berry DA, Syamlal M (2004) Coal Gasification. In: Seidel A (ed) Kirk-Othmer Encyclopedia of Chemical Technology, Vol. 4, 5th Edn., John Wiley & Sons, Inc., Hoboken, NJ, p. 771–832
Bartholomew CH, Farrauto RJ (2006) Production of hydrogen and synthesis gas via steam reforming (section 6.2). In: Fundamentals of industrial catalytic processes, 2nd edn. Wiley, Hoboken, pp. 342–371
Aasberg-Petersen K et al (2001) Technologies for large-scale gas. Appl Catal Gen 221:379–387
Jones RM, Shilling NZ (2004) The impact of gas turbine fuel flexibility on IGCC growth. IChemE 6th European Gasification Conference. pp.1–10
Heaven DL (1996) Gasification converts a variety of problematic feedstocks and wastes. Oil Gas J 94:49–54
Chauvel A, Lefebvre G (1989) Petrochemical processes, 1. Synthesis-Gas Derivatives and Major Hydrocarbons. Gulf Publishing, Houston, pp 19–64
Kohl AL (1997) Sulfur recovery processes, chapter 8. Liquid phase oxidation processes for hydrogen sulfide removal, chapter 9. Thermal and catalytic convesion of gas impurities, chapter 13. Physical solvents for acid gas removal. Gas purification, 5th edn. Gulf Publishing Company, Houston, pp. 670–885, 1136–1237
Wender I (1996) Reactions of synthesis gas. Fuel Process Technol 48:189–297
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 91–98
Wells GM (1995) Acetylene, Handbook of petrochemicals and processes. Gower Publishing Company, Brookfield, pp 31–38
Chauvel A, Lefebvre G (1989) Petrochemical processes. 1. Synthesis-Gas Derivatives and Major Hydrocarbons. Gulf Publishing, Houston, pp 180–184
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 75–77
Greiner EOC, Inoguchi Y (2010) Linear alpha-olefins. In: CEH Marketing Research Report, SRI Consulting, Menlo Park, CA, pp. 10–17
Calderazzo F, Catellani M, Chiusoli GP (2006) Carbon-carbon bond formation( chapter 5). In: Chiusoli GP, Maitlis PM (eds) Metal-catalysis in industrial organic processes. RSC Publishing, Cambridge, pp 187–192
Mol JC (2004) Industrial applications of olefin metathesis. J Mol Catal 213:39–45
Banks RL (1984) Olefin metathesis: technology and application. Appl Ind Catal 3:215–239
Newman TH, Rand CL, Burdett KA, Maughon RR, Morrison DL, Wasserman, EP (2006) Dow chemical. US. Patent 7,119,216
Bielawski CW, Grubbs RH (2007) Living ring-opening metathesis polymerization. Prog Polym Sci 32:1–29
Michelotti FW (1967) Interchemical Corp. US. Pat. 3,336,275, 15Aug 1967
Michelotti FW, Keaveney WP (1965) Coordinated polymerization of the bicyclo-[2.2.1]-heptene-2 ring system (Norbornene) in polar media. J Polym Sci A 3:895–905
Vogt D (1998) SHOP process. In: Cornils B, Herrmann W (eds) Aqueous-phase organometallic catalysis. Wiley-VCH Verlag, Weinheim, pp 541–547
Clark MC, Smith CM, Stern DL, Beck JS (2008) Alkylation of aromatics. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis. vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, pp 3153–3168
Stefanidakis G, Gwyn JE (1993) Alkylation. In: McKetta JJ (ed) Chemical processing handbook. Marcel Dekker, New York, pp 110–124
Franck H-G, Stadelhofer AW (1998) Industrial aromatic chemistry. Springer, Heidelberg, pp 133–137
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 335–339
Clark MC, Smith CM, Stern DL, Beck JS (2008) Alkylation of aromatics. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis. vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, pp 3156–3157
Ethylbenzene. In: Hydrocarbon Processing’s Petrochemical Processes 2010, Gulf Publishing Company, Houston, TX, 2010. pp 118–120
Ethylbenzene, Part 5. In Meyers RA(Ed) Handboook of petrochemicals production processes.. McGraw-Hill, New York, NY, 2005. pp 5-1 to 5–35
Clark MC, Smith CM, Stern DL, Beck JS (2008) Alkylation of aromatics. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, Germany, p 3157–3160
Cumene. In: Hydrocarbon Processing’s Petrochemical Processes 2010, Gulf Publishing Company, Houston, TX, 2010. p. 102
Franck H-G, Stadelhofer AW (1998) Industrial aromatic chemistry. Springer, Heidelberg, pp 146–148
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, New York, USA, p 342–343
Cumene, Part 4. In: Meyers RA (ed) Handboook of petrochemicals production processes, McGraw-Hill, New York, New York, USA, 2005, p 4-1–4-19
Clark MC, Smith CM, Stern DL, Beck JS (2008) Alkylation of aromatics. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, Germany, p 3161
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 343–345
Vora BP, M.C, Peterson GA, Sohn SW, Riley MG (2009) Detergent alkylate and detergent olefins production. In: Zoller U (ed) Handbook of detergents, part F: production. CRC Press, Taylor & Francis Group, LLC, Boca Raton, pp. 39–45
Linear Alkylbenzenes (LAB) (2009) PERP 07/08S7, Chemsystems PERP Program, Nexant, White Plains, New York. Jan 2009
Haber J et al (2008) Selective oxidations (section 14.11). In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Weinheim, Wiley-VCH Verlag GmbH, pp 3359–3547
Hodnett BK (2000) Heterogeneous catalytic oxidation. Wiley, Chichester
Bartholomew CH, Farrauto RJ (2006) Catalytic oxidations of inorganic and organic compounds (chapter 8). In: Fundamentals of industrial catalytic processes, 2nd edn. Wiley, Hoboken. pp. 560–562, 578–584
Bielanski A, Haber J (1991) Oxygen in catalysis. Marcel Dekker, New York
Haber J (2003) Selective oxidation—heterogeneous. In: Horvath LT (ed) Encyclopedia of catalysis, vol 6. Wiley, Hoboken, pp 141–189
Sheldon RA, Ten Brink GJ, Arends IWCE (2003) Selective oxidation—homogeneous. In: Horvath LT (ed) Encyclopedia of catalysis, vol 6. Wiley, Hoboken, pp 189–239
Centi G, Perathoner S (2003) Selective oxidation—industrial. In: Horvath LT (ed) Encyclopedia of catalysis, vol 6. Wiley, Hoboken, pp 239–299
Clerici MG, Ricci M, Strukul G (2006) Formation of C–O bonds by oxidation (chapter 2). In: Chiusoli GP, Maitlis PM (eds) Metal-catalysis in industrial organic processes. RSC Publishing, Cambridge, pp 23–78
Doraiswamy LK (1984) Heterogeneous reactions: analysis, examples, and reactor designs, vol 1–2. Wiley, Hoboken
Rase HF (1977) Chemical reactor design for process plants, vol 1–2. Wiley Interscience, New York
Suresh AK, Sharma MM, Sridhar T (2000) Engineering aspects of industrial liquid-phase air oxidation of hydrocarbons. Ind Eng Chem Res 39:3958–3997
Coward HF, Jones GW (1952) Limits of flammability of gases and vapors, bulletin 503 bureau of mines. United States Government Printing Office, Washington, DC, USA
Stull DR (1977) Fundamentals of Fire and Explosion, AIChE Monograph Series, AIChE, Vol 73(10), New York, NY
Medard LA (1989) Ignition temperatures of explosive gaseous mixtures, Chapter 7. Flammability limits of explosive gaseous mixtures, Chapter 8. Flammability limits: safety applications, Chapter 10. In: Accidental explosions, vol 1. Wiley, Hoboken. pp 147–218
Lees FP (1996) Loss prevention in the process industries. vols 1–3, 2nd Edn., Elsevier, New York, NY, 1996.
Bodurtha FT (1980) Industrial explosion prevention and protection. McGraw-Hill, New York
Sheehan RJ (2003) Terephthalic acid, Dimethyl terephthalate, and Isophthalic acid. in Ullmann’s encyclopedia of chemical technology. Vol 35, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p.639–651
Castillo-Walter F (2005) E PTA: The Lurgi/Eastman/SK Process, Chapter 12.1. In: Meyers RA (editor-in-chief) Handboook of petrochemicals production processes. McGraw-Hill, New York, NY. p.12.3–12.12
Rosowski F, Storck S, Zuhlke J (2008) Oxyfunctionalization of alkyl aromatics. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, pp 3425–3433
Partenheimer W (1995) Methodology and scope of metal/bromide autoxidation of hydrocarbons. Catal Today 23:69–158
Maki T, Takeda K (2003) Benzoic acid and derivatives. in Ullmann’s encyclopedia of chemical technolog. Vol 5, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p.59–72
Bizzari SN, Blagoev M, Kumamoto T (2010). Benzoic acid. CEH marketing research report, SRI consulting, Menlo Park, CA. 2010, p. 10–11
Phthalic acid and derivatives. Industrial organic chemicals. Vol 7, Wiley-VCH Verlag GmbH, Weinheim. 1999, p. 3868–3877
Bizzari SN (2010) Phthalic anhydride. CEH Marketing Research Report SRI Consulting, Menlo Park, CA, USA, pp 13–14
Phthalic anhydride. ICIS chemical business. Feb 8–14, 2010, p. 35
Centi G, Perathoner S (2003) Selective oxidation—industrial. In: Horvath LT (ed) Encyclopedia of catalysis, vol 6. John Wiley & Sons, Inc, Hoboken, NJ, pp 294–296
Shu C-M, Wen P-J, Chang R-H (2002) Investigations on flammability models and zones for o-xylene under various initial pressures, temperatures, and oxygen concentrations. Thermochim Acta 392–393:271–287
Ghirardini M, Tampieri, Polimeri Europa Cumene-Phenol Processes, Chapter 9.1. Schmidt, R.J., “Sunoco/UOP Phenol Process, Chapter 9.2”; Moore, A.; Birkhoff, R., “KBR Phenol Process, Chapter 9.3”, in Handboook of Petrochemicals Production Processes, Meyers, R.A., Editor-in-chief, McGraw-Hill, New York, New York, USA, 2005, pp.9.3 to 9.12, 9.13 to 9.29, 9.31 to 9.50.
Phenol. In Hydrocarbon processing’s petrochemical processes 2010. Gulf Publishing Company, Houston, TX. 2010, p. 203–205
Sifniades S, Levy AB (2003) Acetone. in Ullmann’s encyclopedia of chemical technology, Vol 1, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p.197–200
Phenol/Acetone/Cumene. PERP 05/06-4, Chemsystems PERP program, Nexant, White Plains, NY. Mar 2007
Jordan W, van Barneveld H, Gerlich O, Kleine-Boymann M, Ullrich J (2003) Phenol. in Ullmann’s encyclopedia of chemical technology, Vol 25, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p.589–604
Schmidt RJ (2005) Industrial catalytic processes—phenol production. Appl Catal Gen 280:89–103
Clerici MG, Ricci M, Strukul G (2006) Formation of C–O bonds by oxidation, Chapter 2. In: Chiusoli GP, Maitlis PM (eds) Metal-catalysis in industrial organic processes. RSC Publishing, Cambridge, pp 39–40
James DH, Castor WM (2003) Styrene. in Ullmann’s encyclopedia of chemical technology. Vol 34, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p. 390
Kahlich D, Wiechern U, Lindner J (2003) Propylene oxide. in Ullmann’s encyclopedia of chemical technology. Vol 30, 6th Ed. Wiley-VCH Verlag GmbH, Weinheim. p.279–303
Clerici MG, Ricci M, Strukul G (2006) Formation of C–O bonds by oxidation, Chapter 2. In: Chiusoli GP, Maitlis PM (eds) Metal-catalysis in industrial organic processes. RSC Publishing, Cambridge, pp 35–39
Kobe JM, Evans WE, June RL, Lemanski MF (2003) Epoxidation- industrial. In: Horvath LT (ed) Encyclopedia of catalysis, vol 3. Wiley, Hoboken, NJ, USA, p 239–257
Rebsdat S, Mayer D (2003) Ethylene oxide. in Ullmann’s encyclopedia of chemical technology, Vol 12, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p. 609–635
Ethylene oxide/EG. PERP 08/09-2. Chemsystems PERP Program, Nexant, White Plains, New York. Feb 2010.
Bartholomew CH, Farrauto RJ (2006) Ethylene to ethylene oxide, Section 8.4.3. In: Fundamentals of industrial catalytic processes, 2nd edn. Wiley, Hoboken, NJ, USA, p 597–604
Linic S, Barteau MA (2008) Heterogeneous catalysis of alkene epoxidation. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, Germany, p 3449–3456
Schroeder V, Baumeier A, Franzen S, Buettgen F (2007) Explosion limits of decomposition of mixtures of ethylene oxide, propylene oxide, and nitrogen. Chemie Ing Tech 79(8):1241–1245
Pekalski AA, Zevenbergen JF, Braithwaite M, Lemkowitz SM, Pasman HJ (2005) Explosive decomposition of ethylene oxide at elevated conditions: effect of ignition energy, nitrogen dilution, and turbulence. J Haz Mat 118(1–3):19–34
Kohl AL, Nielsen RB (1997) Alkaline salt solutions for acid gas removal. In: Gas purification, 5th edn. Gulf Publishing Company, Houston, TX, USA, p 330–414
Monnier JR, Stavinoha JL, MInga RL (2004) Stability and distribution of cesium in Cs-promoted silver catalysts used for butadiene epoxidation. J Catal 226(2):401–409
Fleischmann G, Jira R, Bolt HM, Golka K (2003) Acetaldehyde. in Ullmann’s encyclopedia of chemical technology, Vol 1, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p. 131–148
Jira R (2002) Oxidation of olefins to carbonyl compounds (Wacker process). Section 2.4.1. In: Cornils B, Herrmann WA (eds) Applied homogenous catalysis with organometallic compounds, vol 1, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, pp 386–412
Miller SA (1969) Ethylene and its industrial derivatives. Ernest Benn Limitied, London, England, pp 480–530
Centi G, Perathoner S (2003) Selective oxidation—industrial. In: Horvath LT (ed) Encyclopedia of catalysis, vol 6. John Wiley & Sons, Inc, Hoboken, NJ, pp 265–269
Wells GM (1995) Acetaldehyde. Handbook of petrochemicals and processes. Gower Publishing Company, Brookfield, Vermont, pp 1–8
Goe, G.L (1982) Pyridine and pyridine derivatives. In: Mark H (editor-in-chief) Kirk-othmer encyclopedia of chemical technology. Vol. 19, 3rd Edn., John Wiley & Sons, Inc., USA, p. 454–483
Grate JH, Hamm DR, Mahajan S (1993) Palladium and phosphomolybdovanadate catalyzed olefin oxidation to carbonyls. Molecular Engineering 3(1–3):205–229
Lambert A, Derouane EG, Kozhevnikov IV (2002) Kinetics of one-stage wacker-type oxidation of C2-C4 olefins by an aqueous PdCl2 heteropoly-anion system. J Catalysis 211(2):445–450
Ohara T, Sato T, Shimizo N, Prescher G, Schwind H, Weiberg O, Marten K (2003) Acrylic acid and derivatives. in Ullmann’s encyclopedia of chemical technology. Vol 1, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p. 289–306
Acrylic Acid. PERP 08/09-3. Chemsystems PERP Program, Nexant, White Plains, New York. Jul 2010, p. 18–33
Bauer W (2003) Methacrylic acid and derivatives. in Ullmann’s encyclopedia of chemical technology. Vol 21, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p. 585–597
Wells GM (1995) Acrolein, Acrylic acid. Handbook of petrochemicals and processes. Gower Publishing Company, Brookfield, Vermont, pp 39–47
Adipic acid. PERP 08/09-2, Chemsystems PERP Program, Nexant, White Plains, New York. Feb 2010
Clerici MG, Ricci M, Strukul G (2006) Formation of C–O bonds by oxidation, Chapter 2. In: Chiusoli GP, Maitlis PM (eds) Metal-catalysis in industrial organic processes. RSC Publishing, Cambridge, pp 29–30
Malveda M.P, Mori H (2009) Cyclohexanol and cyclohexanone. CEH Marketing research report. SRI Consulting, Menlo Park, CA. p. 5, 8
Lohbeck K, Haferkorn H, Fuhrmann W, Fedtke N (2003) Maleic and fumaric acids. in Ullmann’s encyclopedia of chemical technology. Vol 20, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p. 463–473
Greiner EOC, Funada C (2009) Maleic anhydride. CEH marketing research report. SRI Consulting, Menlo Park, CA, pp 7–11
Bartholomew CH, Farrauto RJ (2006) n-Butane to Maleic Anhydride, Section 8.4.5. in Fundamentals of industrial catalytic processes. 2nd Edn. John Wiley & Sons, Hoboken, NJ. p. 610–618
Maleic anhydride. in Hydrocarbon Processing’s Petrochemical Processes 2010. Gulf Publishing Company, Houston, TX. 2010, p. 158–160
Maleic anhydride. PERP 07/08-8, Chemsystems PERP Program, Nexant, White Plains, New York. Feb 2009 p. 12.
Reuss G, Disteldorf W, Gamer AO, Hilt A (2003) Formaldehyde. in Ullmann’s encyclopedia of chemical technology, Vol 15, 6th Ed. Wiley-VCH Verlag GmbH, Weinheim. p.1–34
Bizzari SN (2010) Formaldehyde. CEH Marketing Research Report. SRI Consulting, Menlo Park, CA. p. 5, 14–15
Formaldehyde. ICIS chemical business. March 31-April 6, 2008, p. 56
Formaldehyde and derivatives. PERP 04/05S10, Chemsystems PERP Program, Nexant, White Plains, NY. 2006
Davies P, Donald RT, Harbord NH (1989) Methanol oxidation, Section 10.3. In: Twigg MV (ed) Catalyst Handbook, 2nd edn. Wolfe Publishing, Ltd., England, pp 490–503
Bartholomew CH, Farrauto RJ (2006) Methanol to formaldehyde, Section 8.4.2, in Fundamentals of industrial catalytic processes. 2nd Edn. John Wiley & Sons, Hoboken, NJ. p. 584–597
Maurer G (1986) Vapor–liquid equilibrium of formaldehyde and water containing multicomponent mixtures. AIChE J 32(6):932–948
Ott M, Fischer HH, Maiwald M, Albert K, Hasse H (2005) Kinetics of oligmoerization reactions of formaldehyde solutions. Chem Eng Process 44:653–660
Roscher G (2003) Vinyl esters. in Ullmann’s encyclopedia of chemical technology, Vol 38, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p.59–75
Haber J (2003) Selective oxidation—heterogeneous. In: Horvath LT (ed) Encyclopedia of catalysis, vol 6. John Wiley & Sons, Inc, Hoboken, NJ, pp 283–284
Wells GM (1995) Vinyl acetate. Handbook of petrochemicals and processes. Gower Publishing Company, Brookfield, Vermont, pp 363–367
Clerici MG, Ricci M, Strukul G (2006) Formation of C–O bonds by oxidation, chapter 2. In: Chiusoli GP, Maitlis PM (eds) Metal-catalysis in industrial organic processes. RSC Publishing, Cambridge, pp 70–73
Schunk SA, de Oliveria AL (2008) Acetoxylation of ethylene. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, pp 3464–3478
Koch F (2002) Aliphatic carboxylic acids via aliphatic aldehydes. In: Cornils B, Herrmann WA (eds) Applied homogenous catalysis with organometallic compounds, vol 1, 2nd edn. Weinheim, Wiley-VCH Verlag GmbH, pp 427–432
Cheung H, Tanke RS, Torrence GP (2003) Acetic acid. in Ullmann’s encyclopedia of chemical technology, Vol 1, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p.149–178
Howard MJ, Jones MD, Roberts MS, Taylor SA (1993) C1 to acetyls: catalysis and process. Catal Today 18:325–354
Yoneda N, Kusano S, Yasui M, Pujado P, Wilcher S (2001) Recent advances in processes and catalysts for production of acetic acid. Appl Catal Gen 221:253–265
Zoeller JR (1993) Manufacture via methanol carbonylation. In: Agreda VH, Zoeller JR (eds) Acetic acid and its derivatives, handbook, 7th edn. Marcel Dekker, Inc., New York, NY, pp 35–51
Yoneda N, Hosono Y (2004) Acetic acid process catalyzed by ionically immobilized rhodium complex to solid resin support”. J ChE Japan 37(4):536–545
Acetic acid. in Hydrocarbon processing’s petrochemical processes 2010. Gulf Publishing Company, Houston, TX. 2010, p. 43
Held H, Rengstl A, Mayer D (2003) Acetic anhydrides and mixed fatty acid anhydrides. in Ullmann’s encyclopedia of chemical technology, Vol 1, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p.179–194
Zoeller JR, Agreda VH, Cook SL, Lafferty NL, Polichnowski SW, Pond DM (1992) Eastman chemical company acetic anhydride process. Cataly Today 13:73–91
Malveda MP, Funada C (2010) Acetic anhydride, CEH marketing research report. SRI Consulting, Menlo Park, CA, pp 4–9
Reutemann W, Kieczka H (2003) Formic Acid. in Ullmann’s encyclopedia of chemical technology, Vol 15, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p.49–71
Horsley LH (1973) Azeotrope data for formic acid-water, Azeotrope Data –III, Adv. In Chemistry Sereies 116, ACS, Washington, DC. p.13
Clegg William, Eastham GR, Elsegood MRJ, Tooze RP, Wang XL, Whitson K. J Chem Soc, Chem Commun. 1877 (1999)
Eastham GR, Heaton BT, Iggo JA, Tooze RT, Whyman R, Zacchini S. J Chem Soc, Chem Commun. 609 (2000)
Tooze RP, Eastham GR, Whitson K, Wang XL. J Chem Soc, U.S. Pat. 6,348,621 (Feb. 19, 2002)
Kohlpaintner C (2003) Hydroformylation—industrial. In: Horvath LT (ed) Encyclopedia of catalysis, vol 3. John Wiley & Sons, Inc, Hoboken, NJ, pp 787–808
Bahrmann H, Bach H (2003) Oxo synthesis. in Ullmann’s encyclopedia of chemical technology, Vol 24, 6th Edn.Wiley-VCH Verlag GmbH, Weinheim. p.553–560
Oxo alcohols. PEP Report 21E. SRI Consulting, The Woodlands, TX. Sep 2010
Oxo alcohols. PERP 06/07-8, Chemsystems PERP Program. Nexant, White Plains, NY. 2007
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, NY, pp 129–133
Maitlis PM, Haynes A (2006) Syntheses based on carbon monoxide, chapter 4. In: Chiusoli GP, Maitlis PM (eds) Metal-catalysis in industrial organic processes. RSC Publishing, Cambridge, pp 141–150
Natta G, Ercoli R, Castellano S, Barbieri FH (1954) The influence of hdrogen and carbon monoxide partial pressures on the rate of the hydroformylation reaction. JACS 76:4049–4050
Yang C, Mao Z-S, Wang Y, Chen J (2002) Kinetics of hydroformylation of propylene using RhCl(CO)(TPPTS)2/TPPTS complex catalyst in aqueous system. Catal Today 74:111–119
Puckette TA (2007) Halophosphite ligands for the rhodium-catalyzed low-pressure hydroformylation reaction. In: Schmidt SR (ed) Catalysis of organic reactions. CRC Press, LLC, Boca Raton, FL, pp 31–38
Butcher, J.; Reynolds, G (2005) Johnson matthey oxo alcohols process™. In: Meyers RA (editor-in-chief) Handbook of petrochemicals production processes. McGraw-Hill, New York, NY. p.8.3–8.13
Lee R, Lee AO (1993) Chlorination, liquid phase and vapor phase. In: McKetta JJ (ed) Chemical processing handbook. Marcel Dekker, Inc., New York, NY, pp 295–352
McBee ET, Hass HB, Bordenca C (1943) Chlorinolysis of chloroparaffins. Ind Eng Chem 35(3):317–320
Smith MB, March J (2001) March’s advanced organic chemistry, 5th edn. John Wiley & Sons, Inc, New York, NY, pp 894–911
Smart, B.E (1980) Fluorine Compounds, Organic. In: Mark HF (editor-in-chief). Kirk-Othmer Encyclopedia of Chemical Technology. Vol. 10, 3rd Edn. John Wiley & Sons, Inc, Hoboken, NJ. p. 829–870
DeForest EM (1993) Chloromethanes. In: McKetta JJ (ed) Chemical processing handbook. Marcel Dekker, Inc., New York, NY, pp 427–482
Rossberg M, Lendle W, Pfleiderer G, Togel A, Dreher E.-L, Rassaerts H, Kleinschmidt P, Strack H, Cook R, Beck U, Lipper K.-A, Torkelson TR, Loser E, Beutel KK (2003) Chloronated hydrocarbons, 3.Chloroethanes. in Ullmann’s encyclopedia of chemical technology. Vol 8, 6th Ed. Wiley-VCH Verlag GmbH, Weinheim. p.3–25
Wells, G.M (1995) Carbon Tetrachloride, Chloroform, Methyl Chloride, Methylene Chloride. Handbook of petrochemicals and processes. Gower Publishing Company, Brookfield, Vermont. p. 115–119, 124–128, 246–254.
Kurtz BE, Smalley EW (1993) Chlorobenzene and dichlorobenzene. In: McKetta JJ (ed) Chemical processing handbook. Marcel Dekker, Inc., New York, NY, pp 483–500
Rossberg M, Lendle W, Pfleiderer G, Togel A, Dreher E.-L, Rassaerts H, Kleinschmidt P, Strack H, Cook R, Beck U, Lipper K.-A, Torkelson TR, Loser E, Beutel KK (2003) Chloronated hydrocarbons, 1.Chloroethanes. in Ullmann’s encyclopedia of chemical technology. Vol 8, 6th Ed. Wiley-VCH Verlag GmbH, Weinheim. p.107–116
Wells GM (1995) Chlorobenzene. Handbook of petrochemicals and processes. Gower Publishing Company, Brookfield, Vermont, pp 120–123
Rossberg M, Lendle W, Pfleiderer G, Togel A, Dreher E.-L, Rassaerts H, Kleinschmidt P, Strack H, Cook R, Beck U, Lipper K.-A, Torkelson TR, Loser E, Beutel KK (2003) Chloronated hydrocarbons, 3.Chloroethanes. in Ullmann’s encyclopedia of chemical technology. Vol 8, 6th Ed. Wiley-VCH Verlag GmbH, Weinheim. p.26–32
Vinyl Chloride/EDC. PERP 08/09-4, Chemsystems PERP Program. Nexant, White Plains, NY, Sep 2009
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, NY, pp 217–221
Wells GM (1995) Ethylene dichloride. Handbook of petrochemicals and Processes. Gower Publishing Company, Brookfield, Vermont, pp 175–182
Jordan JI (1993) Chloroethanes. In: McKetta JJ (ed) Chemical processing handbook. Marcel Dekker, Inc., New York, NY, pp 356–381
Rossberg M, Lendle W, Pfleiderer G, Togel A, Dreher E.-L, Rassaerts H, Kleinschmidt P, Strack H, Cook R, Beck U, Lipper K.-A, Torkelson TR, Loser E, Beutel KK (2003) Chloronated hydrocarbons, 2.Chloroethanes. in Ullmann’s encyclopedia of chemical technology, Vol 8, 6th Edn. Wiley-VCH Verlag GmbH, Weinheim. p.30–50
Glauser J, Ishikawa Y (2009) C2 Chlorinated Solvents. CEH Marketing Research Report. SRI Consulting, Menlo Park, CA, pp 12–17
Kahlich D, Wiechern U, Lindner J (2003) Propylene oxide. in Ullmann’s encyclopedia of chemical technology. 6th Edn., Vol 30. Wiley-VCH Verlag GmbH, Weinheim. p.279–303
Trent DL (2006) Propylene oxide. In: Seidel A (editor-in-Chief). Kirk-othmer encyclopedia of chemical technology. Vol. 20, 5th Edn. John Wiley & Sons, Inc, Hoboken, NJ. p. 790–822
Clerici MG, Ricci M, Strukul G (2006) Is the epoxidation of olefins other than ethylene feasible on silver catalysts, section 2.4.3., in Formation of C-O bonds by oxidation, Chapter 2. In: Chiusoli GP, Maitlis PM (eds) Metal-catalysis in industrial organic processes. RSC Publishing, Cambridge, pp 39–40
Rowe CE (1993) Chlorohydrins. In: McKetta JJ (ed) Chemical processing handbook. Marcel Dekker, Inc, New York, NY, pp 382–426
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, NY, pp 295–296
Braun M (2003) Fundamentals and transition-state models. Aldol additions of group 1 and 2 enolates. In: Mahrwald R (ed) Modern aldol reaction, vol 1. Weinheim, Wiley-VCH Verlag GmbH, pp 1–4
Parker SP (editor) McGraw-Hill encyclopedia of chemistry. 2nd Edn. Macmillan Publishing Co., Inc, USA. 1993, p.245–246
Streitwieser A, Heathcock CH (1981) Introduction to organic chemistry, 2nd edn. McGraw-Hill, Inc, New York, NY, pp 392–396
Muthusamy D, Fisher I (2006) Methyl isobutyl ketone. In: Seidel A (editor-in-chief) Kirk-othmer encyclopedia of chemical technology. Vol. 16, 5th Edn. John Wiley & Sons, Inc, Hoboken, NJ. p. 329–355
Simons RM (1989) Methyl isobutyl ketone. In: McKetta JJ, Cunningham WA (eds) Encyclopedia of chemical processing and design, vol 34. Marcel Dekker, New York, NY USA, pp 50–82
O’Keefe WK, Ng FTT, Rempel GL (2007) Experimental studies on the syntheses of mesityl oxide and methyl isobutyl ketone via catalytic distillation. Ind Eng Chem Res 46:716–725
Werle P, Morawietz M (2003) Alcohols, Polyhydric. in Ullmann’s encyclopedia of chemical technology. 6th Edn., Vol 2. Wiley-VCH Verlag GmbH, Weinheim. p.47–64
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, NY, pp 210–213
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, NY, 138
Gautreaux MF, Davis WT, Travis ED (1978) Alcohols, higher aliphatic (Synthetic), in Kirk-othmer encyclopedia of chemical technology. Vol. 1, 3rd Edn. John Wiley & Sons, Inc, New York. p.751
Smith MB, March J (2001) The tishchenko reaction, Section 19-61. in March’s advanced organic chemistry. 5th Edn. John Wiley & Sons, Inc, New York. p. 1565–66
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, NY, pp 186–187
Mahrwald R (2003) The aldol-tishchenko reaction. In: Mahrwald R (ed) Modern aldol reactions, vol 1. Wiley-VCH Verlag GmbH, Weinheim Germany, pp 327–329
Merger F, Foerster H-J (1983) Preparation of alpha-alkylacroleins. U.S. Patent 4,408,079, 4 Oct 1983
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, USA, p 285
Grafje H et al (2003) Butanediols, Butenediol, and Butynediol, in Ullmann’s Encyclopedia of Chemical Technology, 6th Ed., Volume 5, Wiley-VCH Verlag GmbH, Weinheim, Germany, p.703-711.
Weissermel K, Arpe H-J (1997) Industrial Organic Chemistry, 3rd edn. VCH Publishers, New York, USA, p 99
Franck H-G, Stadelhofer AW (1998) Industrial aromatic chemistry. Springer, Heidelberg, Germany, pp 158–160
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, USA, pp 358–359
“Phenol Derivatives”, Industrial Organic Chemicals, Volume *, Wiley-VCH Verlag GmbH, Weinheim, Germany, 1999, pp. 3768–377.
Propanols (1999) Industrial organic chemicals, vol 7. Wiley-VCH Verlag GmbH, Weinheim, Germany, pp 4067–4070
Sherman PW, Kavasmaneck PR (1980) Ethanol. In: Kirk-Othmer (ed) Encyclopedia of chemical technology, vol 9, 3rd edn. Wiley, New York, USA, p 342–350
Linak E, Janshekar H, Inoguchi Y (2009) Ethanol. In: CEH Marketing Research Report, SRI Consulting, Menlo Park, CA, USA p 15–16.
Neier W, Strehlke G (2003) 2-Butanone. In: Ullmann’s encyclopedia of chemical technology, vol 5, 6th edn, Wiley-VCH Verlag GmbH, Weinheim, Germany, p 726
Wells GM (1995) Ethyl alcohol, isopropyl alcohol, Handbook of petrochemicals and processes. Gower Publishing Company, Brookfield, Vermont, USA, 1995, p 154–159, 225–229
Streitwieser A, Heathcock CH (1981) Introduction to organic chemistry, 2nd edn. Macmillan, New York, p 306
Rebstat S, Mayer D (2003) Ethylene glycol. In: Ullmann’s encyclopedia of chemical technology, vol 12, 6th edn. Wiley-VCH Verlag GmbH, Weinheim, Germany, pp 593–608
“Ethylene Oxide/Ethylene Glycol”, PERP 08/09-8, Chemsystems PERP Program, Nexant, White Plains, New York, USA, December, 2009.
Chitwood HC, Freure BT (1946) The reaction of propylene oxide with alcohols. JACS 68(4):680–683
Parker RE, Isaacs NS (1959) Mechanisms of epoxide reactions. Chem Rev 4:737–799
Groggins PH (1958) Unit processes in organic synthesis, 5th edn. McGraw Hill, New York, pp 843–844
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 160–161
Levinson MI (2009) Surfactant production: present realities and future perspectives. In: Zoller U (ed) Handbook of detergents, Part F: production, CRC Press, Taylor & Francis Group, LLC, Boca Raton, FL, USA, pp 26–29
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 181–183
Held H, Rengstl A, Mayer D (2003) Acetic anhydride and mixed fatty acid anhydrides. In: Ullmann’s encyclopedia of chemical technology, vol 1, 6th edn. Wiley-VCH Verlag GmbH, Weinheim, Germany, p. 179–194
Raynolds PW (1993) Ketene (chapter 10). In: Agreda VH, Zoeller JR (eds) Acetic acid and its derivatives. Marcel Dekker, New York, pp 161–171
Wells GM (1995) Handbook of petrochemicals and processes. Gower Publishing Company, Vermont, pp 18–19
Clemens RJ, Witzeman JS (1993) Diketene and acetoacetates (chapter 11). In: Agreda VH, Zoeller JR (eds) Acetic acid and its derivatives. Marcel Dekker, New York, pp 173–224
Martinez R, Huff MC, Barteau MA (2004) Ketonization of acetic acid on titania-functionalized silica monoliths. J Catal 222(2):404–409
Bender ML (1960) Mechanisms of catalysis of nucleophilic reactions of carboxylic acid derivatives. Chem Rev 60(1):53–113
Witzeman JS, Agreda VH (1993) Alcohol acetates (chapter 14). In: Agreda VH, Zoeller JR (eds) Acetic acid and its derivatives. Marcel Dekker, New York, pp 257–284
Simons RM (1993) Esterification. In: McKetta JJ (ed) Chemical processing handbook. Marcel Dekker, New York, pp 537–658
Barnicki SD, Siirola JJ (1997) Enhanced distillation. In: Perry RH, Green DW, Maloney JO (eds) Perry’s chemical engineers’ handbook, 7th edn. McGraw Hill, New York, pp. 13–83 to 13–85.
Groggins PH (1958) Esterification. In: Unit processes in organic synthesis, 5th edn. McGraw Hill Book Company, New York, New York, USA, p 694–749
Agreda VH, Partin LR, Heise WH (1990) High-purity methyl acetate via reactive distillation. Chem Eng Prog 86(2):40–46
Cadogan DF (1999) Plasticizers. In: Kirk-Othmer (ed) Concise encyclopedia of chemical technology, 4th Edn. Wiley, New York, pp 1577–1578
Bartholomew CH, Farrauto RJ (2006) Hydrogenation and dehydrogenation of organic compounds, section 7. In: Fundamentals of industrial catalytic processes, 2nd edn, Wiley, Hoboken, pp 487–532
Gallezot P (2003) Hydrogenation—heterogeneous. In: Horvath LT (ed) Encyclopedia of catalysis, vol 4. Wiley, Hoboken, pp 17–55
Rylander PN (2003) Hydrogenation and dehydrogenation. In: Ullmann’s encyclopedia of chemical technology, vol 17, 6th edn. Wiley-VCH Verlag GmbH, Weinheim, pp 241–252
Chen B, Dingerdissen U, Krauter JGE, Lansink Rotgerink HGJ, Mobus K, Ostgard DJ, Panster P, Riermeier TH, Seebald S, Tacke T, Traauthwein H (2005) New developments in hydrogenation catalysis particularly in synthesis of fine and intermediate chemicals. Appl Catal Gen 280:17–46
Barnicki SD (1991) Separation system synthesis: a knowledge-based approach. PhD Dissertation, The University of Texas at Austin, pp. 99–100
Fiedler E, Grossmann G, Kiersebohm DB, Weiss G, Witte C (2003) Methanol. In: Ullmann’s encyclopedia of chemical technology, vol 1, 6th edn. Wiley-VCH Verlag GmbH, Weinheim, pp 611–635
“Methanol Synthesis Catalyst Product Bulletin”, Sud-Chemie, 2007, pp 1–16
Vanden Bussche KM, Froment GF (1996) A steady-state kinetic model for methanol synthesis and the water gas shift reaction on a commercial Cu/Zn/AL2O3 catalyst. J Catal 161:1–10
Bridger GW, Spencer MS (1989) Methanol syntheis (chapter 9). In: Twigg MV (ed) Catalyst handbook, 2nd edn. Wolfe Publishing, England, pp 441–468
“Methanol”, in Hydrocarbon Processing’s petrochemical processes 2010, Gulf Publishing Company, Houston, 2010, pp 163–172
“Cyclohexane”, In: Hydrocarbon processing’s petrochemical processes 2010, Gulf Publishing Company, Houston, TX, USA, 2010, p. 103
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 345–347
Wells GM (1995) Handbook of petrochemicals and processes. Gower Publishing Company, Brookfield, pp 133–136
Rylander PN (1985) Hydrogenation methods. Academic, Orlando, pp 66–71
Claus P, Onal Y (2008) Regioselective hydrogenations. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, pp 3313–3326
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 134–139
Bartholomew CH, Farrauto RJ (2006) Hydrogenation of carbonyl groups (section 7.3.5). In: Fundamentals of industrial catalytic processes, 2nd edn. Wiley, Hoboken, pp. 531–532
McCusker-Orth JE, Stavinoha JL, Messina AD, Perri ST, Liu Z, Heidt PC, Tennant, BA (2008) Process for the preparation of tetraalkylcyclobutane-1,3-diols from the corresponding butanediones, using a Ru-promoted Co-based catalyst. U.S. Patent Appl. Publ. 20080154069, June 26 2008
Haas T, Jaeger B, Weber R, Mitchell SF, King CF (2005) New diol processes: 1,3-propandiol and 1,4-butanediol. Appl Catal Gen 280:83–88
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 370–372
Chen B, Dingerdissen U, Krauter JGE, Lansink Rotgerink HGJ, Mobus K, Ostgard DJ, Panster P, Riermeier TH, Seebald S, Tacke T, Traauthwein H (2005) Section 1.2.6. Esters, anhydrides, and carboxylic acids. In: New developments in hydrogenation catalysis particularly in synthesis of fine and intermediate chemicals, Appl Cat A: Gen., vol 280, pp. 22–23.
“1,4-Butanediol/THF” PERP 06/07-4, Chemsystems PERP Program, Nexant, White Plains, March, 2008.
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 403–404
Voge HH (1993) Dehydrogenation. In: McKetta JJ (ed) Chemical processing handbook. Marcel Dekker, New York, pp 541–555
Caspary KJ, Gehrke H, Heinritz-Adrian M, Schwefer M (2008) Dehydrogenation of alkanes. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, pp 3206–3229
Bartholomew CH, Farrauto RJ (2006) Dehydrogenation: reaction chemistry; Catalyst and reactor technologies (section 7.5.1). In: Fundamentals of industrial catalytic processes, 2nd edn. Wiley, Hoboken, pp. 533–536
Resasco DE (2003) Dehydrogenation—Heterogeneous. In: Horvath LT (ed) Encyclopedia of catalysis. Wiley, Hoboken, pp 49–79
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 109–112
Caspary KJ, Gehrke H, Heinritz-Adrian M, Schwefer M (2008) Dehydrogenation of alkanes. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, Germany, p 3221–3224
Bartholomew CH, Farrauto RJ (2006) Dehydrogenation of C3 and C4 alkanes to alkenes (section 7.5.1). In: Fundamentals of industrial catalytic processes, 2nd edn. Wiley, Hoboken, pp 536–542
Caspary KJ, Gehrke H, Heinritz-Adrian M, Schwefer M (2008) Dehydrogenation of alkanes. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, p. 3225
Bartholomew CH, Farrauto RJ (2006) Dehydrogenation of C6-C15 alkanes to alkenes (section 7.5.1). In: Fundamentals of industrial catalytic processes, 2nd edn. Wiley, Hoboken, pp 542–543
Styrene (2010) In: Hydrocarbon processing’s petrochemical processes 2010, Gulf Publishing Company, Houston, pp. 264–265
James DH, Castor WM (2003) Styrene. In: Ullmann’s encyclopedia of chemical technology, vol 34, 6th edn. Wiley-VCH Verlag GmbH, Weinheim, pp 386–390
Kochloefl K, Muhler M (2008) Dehydrogenation of ethylbenzene. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Wiley-VCH Verlag GmbH, Weinheim, pp 3229–3238
Bartholomew CH, Farrauto RJ (2006) Dehydrogenation of ethylbenzene (section 7.5.2). In: Fundamentals of industrial catalytic processes, 2nd edn. Wiley, Hoboken, pp. 543–545
Neier W, Strehlke G (2003) 2-Butanone. In: Ullmann’s encyclopedia of chemical technology, vol 5, 6th edn. Wiley-VCH Verlag GmbH, Weinheim, pp.725–732
Papa AJ, Sherman PD (1981) Ketones. In: Mark HF (ed) Kirk-Othmer encyclopedia of chemical technology, vol 13, vol 13, 3rd edn. Wiley, Hoboken, pp 894–941
Sifniades S, Levy AB (2003) Acetone. In: Ullmann’s encyclopedia of chemical technology, vol 1, 6th edn. Wiley-VCH Verlag GmbH, Weinheim, pp. 200–201
Weissermel K, Arpe H-J (1997) Industrial organic chemistry, 3rd edn. VCH Publishers, New York, pp 278–279
Wells GM (1995) Methyl ethyl ketone, Handbook of petrochemicals and processes. Gower Publishing Company, Brookfield, pp 255–258
Burridge E (2008) TDI, Chemical profile, ICIS Chemical Business, 21–27 Jan 2008, p. 38.
Simons RM (1983) Esterification. In: McKetta JJ, Cunningham WA (eds) Encyclopedia of chemical processing and design, vol 19. Marcel Dekker, New York, pp 381–402
R. L. Rowley, W. V. Wilding, J. L. Oscarson, Y. Yang, R. J. Rowley, T. E. Daubert, R. P. Danner, DIPPR® Data Compilation of Pure Compound Properties, dippr@BYU.edu, Design Institute for Physical Property Data, AIChE®, New York, NY, USA, 2001.
RL Rowley, W. V. Wilding, J. L. Oscarson, Y. Yang, R. J. Rowley, T. E. Daubert, R. P. Danner, DIPPR® Data Compilation of Pure Compound Properties, dippr@BYU.edu, Design Institute for Physical Property Data, AIChE®, New York, NY, USA, 2001.
Caspary KJ, Gehrke H, Heinritz-Adrian M, Schwefer M (2008) Dehydrogenation of alkanes. In: Ertl G, Knozinger H, Schuth F, Weitkamp J (eds) Handbook of heterogenous catalysis, vol 8, 2nd edn. Weinheim, Wiley-VCH Verlag GmbH, p 3222
Acknowledgements
The author would like to thank Robert T. Hembre and Steven L. Perri, both of Eastman Chemical Co., Kingsport, for taking their time to read and edit the chapter. The author would also like to acknowledge Robert Hembre’s helpful revisions to the sections, “Carbonylation” and “Hydroformylation,” in particular, as well as his addition of the section, “Metathesis.”
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media New York
About this chapter
Cite this chapter
Barnicki, S.D. (2012). Synthetic Organic Chemicals. In: Kent, J. (eds) Handbook of Industrial Chemistry and Biotechnology. Springer, Boston, MA. https://doi.org/10.1007/978-1-4614-4259-2_10
Download citation
DOI: https://doi.org/10.1007/978-1-4614-4259-2_10
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4614-4258-5
Online ISBN: 978-1-4614-4259-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)