
Abstract
Optical imaging methods have been widely used to investigate the function of complex brain tissue. Depending on the approach used, activity can be readily monitored with relatively high temporal resolution throughout large areas of the cortex, or alternatively from individual neurons or sub-cellular compartments. The aim of this chapter is to illustrate the application of optical approaches to provide information about changes in brain metabolism that follow coordinated neuronal firing. The phenomenon of spreading depression (SD) is used to illustrate some of the advantages and challenges of different approaches. SD involves a slowly-propagating coordinated depolarization of neurons and astrocytes and is thought to underlie migraine aura and contribute to enlargement of brain injuries. The stereotyped progression of this event across the cortical surface, coupled with the substantial increase in metabolic demand, has made this response particularly amenable to optical imaging. The discussion will emphasize signals due to changes in mitochondrial redox state, as assessed by changes in levels of NADH autofluorescence. Optical signals associated with changes in blood flow and oxygenation will also be described, since these can significantly impact autofluorescence studies and because the integration of hemodynamic and mitochondrial signals should be valuable for understanding metabolic deficits and neuronal injury that can accompany SD. It is expected that many of the principles discussed here will also apply to less severe metabolic disturbances that accompany reflex activation of discrete neuronal populations.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Autofluorescence
- Intrinsic optical imaging
- Ischemia
- Metabolism
- Mitochondria
- NADH
- Spreading depression
- Stroke
1 Introduction
Optical imaging approaches have a long history of use for the study of brain function and the range of methods employed includes detection of multiple intrinsic optical signals and responses of many different fluorescent indicators. The power and diversity of optical approaches is well-illustrated by studies of firing behavior within large populations of neurons, which is essential to understanding the function of neuronal circuits. Microelectrodes can be used to record activity of single neurons, but the technical complexity of such recordings makes it difficult to simultaneously measure responses from more than a small number of cells. A number of optical approaches allow indirect assessment of activity in very large populations of neurons. It was recognized in the late 1960s that neuronal depolarization changes the light scattering properties of individual neurons, and this permits measurement of neuronal firing in vitro and in vivo (Cohen et al. 1968; Tasaki et al. 1968; Wininger et al. 2009). Although such signals are quite small and averaging of repetitive trials is usually required, these types of intrinsic signals can be very useful. The development of fluorescent probes sensitive to membrane voltage changes provided significant increases in signal-noise characteristics (Cohen et al. 1978) and have been used widely into assess circuit activity and changes in excitability with pathophysiological conditions. While voltage-sensitive indicator signals are directly related to neuronal firing, they can be technically challenging because of the rapid time course of membrane voltage changes and spatial resolution is usually compromised by the high acquisition rates that are needed. Partly because of this problem, complementary optical methods that track longer-lasting intracellular consequences of neuronal depolarization (usually Ca2+ accumulation) have been very widely used, including recent work with two photon excitation in relatively deep brain structures (Cossart et al. 2005). Thus a range of optical approaches are widely used to assess fundamental changes in CNS activity.
The emphasis of this chapter is on optical imaging of different signals that provide more direct information about changes in brain metabolism that follow coordinated neuronal firing. To provide a framework for discussion of the advantages and challenges of different optical approaches, the chapter will focus on the phenomenon of spreading depression (SD). There are a number of reasons why SD is particularly well-suited for this purpose. As discussed below (Sect. 12.4), SD involves a coordinated depolarization of neurons and astrocytes, that propagates slowly in across the brain and is associated with transient suppression of ongoing synaptic activity. The fact that SD can be used experimentally to deliver a dramatic increase in metabolic demand with high temporal precision has made it very attractive as a test stimulus for development and evaluation of optical methods for at least 40 years. In addition, recent work has provided a new impetus to the study of SD in a range of neurological disorders, suggesting that future work with optical methods could be valuable for development of interventions and evaluation of mechanisms underlying deleterious and possibly protective effects of these events.
Optical methods have long been used to assess changes in tissue mitochondrial function that accompany SD, and these will be emphasized here. Because optical signals associated with changes in blood flow and oxygenation can significantly impact autofluorescence studies of mitochondrial redox potential, optical imaging of hemodynamic signals will also be briefly considered. Recent work has advanced mitochondrial imaging studies to include cellular and subcellular origins of autofluorescence signals, and the integration of these signals with assessment of regional cerebral blood flow is likely to be helpful for understanding the metabolic deficits and neuronal injury that can accompany SD. While SD is an unusually strong stimulus that involves activation of large populations of cells, it is expected that many of the principles discussed here will also apply, albeit with different signal/noise characteristics, to less severe metabolic disturbances that accompany reflex activation of discrete neuronal populations.
2 Spreading Depression and Related Events
2.1 Basic Mechanisms
Spreading depression (SD) was originally described by Leao as a wave of profound depression of electrical activity that progressed slowly in a wave-like manner across the rabbit cortex (Leao 1944a, 1947; Leao and Morison 1945). Many excellent reviews have discussed the history of Leao’s initial discoveries and summarized subsequent studies of SD mechanisms (Marshall 1959; Bures et al. 1984; Martins-Ferreira et al. 2000; Gorji 2001; Somjen 2001, 2005; Teive et al. 2005), however some of the key points will be reiterated here to emphasize why optical methods have been used for a number of decades to examine SD in a range of experimental and clinical settings.
Leao’s original discovery came while making recordings from the exposed cortical surface with arrangements of recording electrodes placed on the pia-mater. The original intention of the work was to study mechanisms underlying epileptiform activity, but he found that electrical or mechanical stimulation could result in a “marked, enduring reduction of the spontaneous electrical activity of the cortex”. From recordings made from arrangements of multiple electrodes arranged over the cortical surface, it was shown that the depression of electrocorticographic (ECoG) activity propagated slowly in all directions (Fig. 12.1). Leao coined the term “spreading depression” (SD) and, in addition to blocking spontaneous electrical activity, evoked cortical responses were abolished during the depression phase (Leao 1944a). Early studies showed that SD could be generated by both electrical and mechanical stimuli (such as light touch with a glass rod), and also by K+ or Ca2+ applications. In particular, a piece of filter paper soaked in elevated K+ solutions placed on the brain surface produced a repetitive series of SDs, with a new process commencing even before the surface electrical activity had fully recovered (Leao and Morison 1945). A small plug of filter paper (or similar material) soaked in KCl and placed onto the cortical surface via a small burr hole in the skull is still widely used as a method of generating SD in animal models.

Leao’s spreading depression. This figure from Leao’s original description of SD (Leao 1944) shows electrocorticographic activity recorded between pairs of electrodes arranged across the rabbit cortex. Electrical stimulation (tetanizing frequency for 3-5 s) applied at the most frontal location (S) induced a slowly-propagating suppression of ECoG activity. By approximately 4 min after the stimulus, the entire cortical region appeared electrically silent, but activity subsequently recovered throughout the region as ionic gradients were restored (From Leao 1944a), with permission)
Leao showed that SD can also be described as a slow shift in direct current (DC) potential (Leao 1947), recognized now to be due to sudden opening of ion channels allowing extensive redistribution of ions across the plasma membranes of both neurons and astrocytes. Na+ and Ca2+ accumulation is accompanied by a conspicuous extracellular K+ elevation, as recorded with ion-sensitive electrodes and Ca2+ indicators (Martins-Ferreira et al. 2000; Somjen 2001). Release of glutamate and K+ together are thought to be responsible for feed-forward nature of the response. Extracellular K+ concentration increases above about 12 mM are sufficient to initiate SD, and blockers of NMDA-type glutamate receptors are able to prevent propagation of some forms of SD (Somjen 2001).
2.2 Metabolic Consequences of SD
The extraordinary ionic shifts that accompany SD provide a trigger for substantial metabolic changes that occur with high temporal precision relative to the progression of SD. In early studies, a significant drop in the partial pressure of oxygen (PO2) was reported during the progression of SD (Van Harreveld and Stamm 1952), and decreases in glucose and glycogen content coupled with increased lactate production were also noted (Bures 1956; Krivanek 1958, 1961). Robust increases in glucose utilization (CMRglc) were subsequently recorded throughout the cerebral cortex after SD (Shinohara et al. 1979), and glucose decreases were accompanied by reductions in ATP levels by ∼50% following a single SD event in rat cortex (Mies and Paschen 1984).
Because of the well-appreciated extracellular lactate increase following SD, this event has been used for studies of brain lactate release and uptake mechanisms, including efforts to understand the important question of metabolic coupling between neurons and astrocytes (Kocher 1990; Scheller et al. 1992; Cruz et al. 1999). For example, in 14Cglucose studies of SD in rat, it was concluded that SD results in significant efflux of lactate into venous blood, as well as diffusion throughout other brain regions (Cruz et al. 1999).
While the metabolic demands of SD are large, healthy brain tissue has the capacity to recover quite quickly. This recovery was part of Leao’s original description of the transient nature of ECoG suppression, as spontaneous electrical activity was recovered within a few minutes in vivo (Leao 1944a). Furthermore, when SD is generated by placement of KCl on the brain surface, repetitive events can be generated for hours without apparent injury, despite complete cessation of electrical activity during propagation of each event. Consistent with these observations, large decreases in phosphocreatine levels that occur in the first minutes of a SD event recover to normal levels relatively quickly, together with restoration of extracellular pH (Csiba et al. 1985; Lauritzen et al. 1990; Gault et al. 1994). The lack of neuronal damage produced by repetitive SD events in otherwise healthy brain, has also been demonstrated in detail (Nedergaard and Hansen 1988). As discussed below (Sect. 12.5), significant increases in blood flow accompany SD and the rapid provision of increased oxygen and glucose provides fuel for ATP-dependent pumps involved in restoration of membrane potential. However when SD occurs in conditions where metabolic substrates are limiting, irrecoverable injury can occur (Sect. 12.2.3.2).
2.3 Pathophysiological Consequences
2.3.1 Migraine
Passage of SD across the visual cortex likely underlies visual aura symptoms that accompany some migraines. Migraine aura comprises a region of visual field loss that moves at about 3 mm/min across the visual field. After a description of aura by Lashley in 1941, Leao discussed the similarity between progression of migraine aura and the propagation rate of SD, and proposed that a progressive depression of activity in visual cortex might underlie aura (Leao and Morison 1945). The scintillating border around the scotoma as aura progresses could be explained by a brief period of increased excitability prior to arrival of a SD wave. There has since been much work to support this idea (Lauritzen 1994; Pietrobon and Striessnig 2003; Eikermann-Haerter and Ayata 2010), with supporting evidence including propagating changes in blood flow and fMRI signals during migraine in human subjects (Olesen et al. 1981; Hadjikhani et al. 2001) and signals corresponding to the slow DC shift being recorded with magnetoencephalography during migraine attacks (Bowyer et al. 2001). While there is accumulating evidence that aura is a consequence of SD, whether or not other symptoms of migraine are due to SD is not clear cut. It is possible that SD is one of a set of downstream consequences of some other initiating stimulus, but it is also possible that SD generates migraine headache. These issues are currently being debated (Ayata 2010; Charles 2010).
Recent work has identified multiple mutations that influence susceptibility to migraine in a disorder termed familial hemiplegic migraine (FHM). Mutations that lead to increased conductance of voltage-dependent calcium or sodium channels have been identified, as well as mutations that lead to decreased Na+/K+/ATPase activity. All of these types of mutation can increase cortical excitability and importantly, in cases where these mutations are expressed in mice, there is a significantly decreased seizure threshold (van den Maagdenberg et al. 2004; Eikermann-Haerter et al. 2009). This evidence suggests that increased susceptibility to SD can explain the symptoms of FHM, and raises the possibility that targeting SD could be useful for treatment and prophylaxis.
2.3.2 Brain Injury
While the metabolic demands of SD can be adequately handled by healthy brain, there is considerable evidence to suggest that energy depletion caused by SD can be harmful if tissue metabolism is already partially compromised. A large body of literature has characterized an “anoxic depolarization” that occurs when neurons undergo a large and rapid depolarization following interruption of blood supply, and the injury that rapidly follows if measures are not taken to immediately restore blood supply (Hansen 1985). In clinical situations, it is often difficult to restore perfusion quickly enough to prevent establishment of a core ischemic lesion, but surrounding tissue may still be viable despite a significant perfusion deficit. Such regions have been termed “ischemic penumbra”, and the propagation of SD events through penumbral tissue has been implicated in the enlargement of ischemic injuries.
Events that are very similar to SD are generated following focal ischemia, and since they usually originate from edge of the ischemic core, these have been termed peri-infarct depolarizations (PIDs) (see Hossmann 1996). PIDs have been recorded in the hours following stroke injuries and the number of events correlates with the infarct size in animal models (Strong et al. 1983b; Nedergaard and Astrup 1986; Hartings et al. 2003). These events appear to play a role in the enlargement of injury, rather than simply being a secondary consequence of the pathology. Treatments that decrease PID incidence are neuroprotective (Gill et al. 1992; Iijima et al. 1992; Chen et al. 1993; Mies et al. 1993; Mies et al. 1994; Rawanduzy et al. 1997; Tatlisumak et al. 2000) and experimentally generating SD (which is normally non-injurious) in the context of stroke substantially increases infarct size (Busch et al. 1996; Takano et al. 1996). Depletion of energy substrates by the combination of partial ischemia with the metabolic burden of SD is likely responsible for the increase in injury size (Selman et al. 2004).
An important concept that has emerged is that SD leads to impaired neurovascular coupling. This is based in part on the fact that long-lasting oligemia described following SD is not matched by a long-lasting decrease in regional cortical glucose metabolism following SD (Lauritzen and Diemer 1986) suggesting that the flow decrease is unlikely to be due to decreased tissue metabolic activity. In the vicinity of injured tissue, perfusion decreases (rather than increases) are conspicuous following SD (Back et al. 1994), and this deficiency is likely central to injury produced by SD under a range of conditions.
Results from these and other animal studies provided an additional impetus to test whether SDs are generated in humans following brain injury, and contribute to the enlargement of infarctions. A recent series of reports has appeared from a consortium (Cooperative Study on Brain Injury Depolarisations; COSBID) which has used strip electrodes to make ECoG recordings similar to those originally performed by Leao. Electrodes have been placed after craniotomy, and have led to recordings of SDs for hours and days following large ischemic strokes (Dohmen et al. 2008), subarachnoid hemorrhage (Fabricius et al. 2008; Dreier et al. 2009), and traumatic brain injury (Hartings et al. 2008; Hartings et al. 2009; Sakowitz et al. 2009). In some cases, the passage of SD has been demonstrated to coincide closely with enlargement of the ischemic penumbra, supporting conclusions from animal studies that repetitive SDs are deleterious in the injured brain (Dohmen et al. 2008).
Based on these and other studies, there is strengthened interest in determining the metabolic consequences of SD events in vivo. Identifying the relationship between cellular metabolism and vascular coupling is a central part of this goal, and optical imaging approaches can be of significant utility.
2.3.3 Terminology
The term “cortical spreading depression” (CSD) has been used to refer specifically to the phenomenon originally described by Leao that propagates across normally-perfused cerebral cortex. The mechanisms underlying wave-like depolarizations triggered by ischemia and by a variety of experimental conditions are very similar in many respects (or possibly identical) to classical spreading depression described by Leao. However since the causes and consequences of the events can be very different, a range of different terminologies have been introduced for post-ischemic depolarizations. The term “spreading depolarization” (also abbreviated as SD) has recently been adopted in the clinical literature to emphasize (1) the similar cellular events involved in propagation and (2) the central aspect of neuronal and glial depolarization that occur in both classical cortical spreading depression, and similar events observed following ischemia (Dreier et al. 2006; Dahlem et al. 2008; Dohmen et al. 2008; Petzold et al. 2008; Hartings et al. 2009). In the following discussion, the term “SD” will be used as a general term for cortical spreading depression and related events occurring in ischemia.
3 Autofluorescence Studies
3.1 Introduction
In the 1950s and 1960s, Britton Chance and colleagues were largely responsible for demonstrating that changes in mitochondrial function could be assessed in complex tissues, using relatively straightforward optical methods. The term “autofluorescence” simply refers to fluorescence signals that are derived from endogenous fluorophores, rather than from indicators that are experimentally loaded into tissues. While many tissue components can contribute to autofluorescence, nicotinamide adenine dinucleotide and flavoprotein fluorescence emission changes significantly depending on mitochondrial redox state. In the last five decades, many studies have exploited measures of these endogenous fluorophores to assess metabolic consequences of neuronal activation. The reader is referred to a number of excellent reviews that cover the history and range of applications for both NADH (Anderson and Meyer 2002; Chance 2004; Mayevsky and Rogatsky 2007) and flavoprotein signals (Reinert et al. 2007; Shibuki et al. 2007; Tohmi et al. 2009) in studies of nervous system function and dysfunction.
It is not surprising that there is long history utilizing autofluorescence methods to study SD. SD results in a profound metabolic disturbance that can be quite rigorously controlled and, since the event propagates across the brain surface, the event can be accessible with optical methods. NADH autofluorescence has historically been much more widely used than flavoprotein signals to study SD, and the following discussion will therefore concentrate on the NADH signals.
3.2 NADH Autofluorescence
Mitochondrial ATP generation occurs as protons flux across the inner mitochondrial membrane, via ATP synthetase. A large electrochemical gradient across the mitochondrial inner membrane is required to power this synthesis, and the gradient is maintained by activity of the electron transport chain. NADH is a principal electron donor that supplies this process. NADH generated within mitochondria by the tricarboxylic acid (TCA) cycle is oxidized to NAD+ at complex I, and subsequent electron transfer through the electron transport chain culminates in consumption of molecular oxygen at complex IV (cytochrome oxidase) (Fig. 12.2). Thus NADH oxidation is required to maintain the proton gradient during periods of ATP demand, and cellular O2 consumption is normally tightly coupled to NADH/NAD+ transitions (Nicholls and Ferguson 2002).

Sources of NADH signals associated with metabolism. When excited at UV wavelengths, NADH is fluorescencent but the oxidized form (NAD+) is non-fluorescent. NADH fluorescence is also enhanced in the mitochondrial compartment, likely as a consequence of binding to complex I. Decreases in tissue NADH fluorescence can occur as a consequence of increased demand for mitochondrial ATP synthesis and accelerated electron transport. In contrast, when oxygen levels are limiting, mitochondrial NADH fluorescence increases as inhibition of complex IV activity decreases flux through the electron transport chain. In addition to mitochondrial TCA cycle activity, NADH is generated in the cytosol by the glycolytic enzyme glyceraldehyde phosphate dehydrogenase and oxidized to NAD+ by lactate dehydrogenase. Mitochondrial NADH/NAD+ transitions appear to dominate NADH autofluorescence transients following synaptic stimulation and SD
NADH is fluorescent, with relatively broad excitation and emission spectra (peak excitation ∼340–360 nm and peak emission in the blue range ∼430–450 nm; Chance et al. 1962, 1979; Harbig et al. 1976; Aubin 1979). Importantly, early studies confirmed that pure reduced NADH had nearly identical optical characteristics as autofluorescence generated by the same wavelengths in neuronal tissues (including single neurons, axons and intact brain), with the exception of a small wavelength shift due to enzyme binding in some studies (Chance et al. 1962; Terzuolo et al. 1966; Doane 1967; Galeotti et al. 1970; Harbig et al. 1976). While NADH is strongly fluorescent (particularly in mitochondrial compartments, see below), NAD+ is non-fluorescent and this difference makes it possible to measure changes in the total NADH/NAD+ ratio in brain tissue relatively easily (Chance et al. 1962).
The fluorescence properties of reduced nicotinamide adenine dinucleotide phosphate (NADPH) are very similar to that of NADH, and therefore it is reasonable to expect that NADPH should contribute to total tissue autofluorescence. To acknowledge the possible contribution of NADPH to compound signals, some groups refer to “NAD(P)H” signals in studies of CNS tissues (Schuchmann et al. 2001; Shuttleworth et al. 2003; Kahraman and Fiskum 2007). However as discussed previously, NADPH levels have long been recognized to be much lower than NADH in brain tissue and since NADPH is not involved in electron transfer in the electron transport chain, it is expected to normally make little contribution to mitochondrial autofluorescence transients (see discussion in Mayevsky and Rogatsky 2007; Shuttleworth 2010).
3.3 Technical Considerations for Autofluorescence Imaging of SD
3.3.1 Single Photon Penetration and Toxicity
Because no complex loading procedures are required to deliver a fluorescent reporter into target cells, imaging autofluorescence has a fundamental advantage over most other fluorescence methods. However a tradeoff for this convenience is that it can be difficult to detect signal changes from a small number of active cells, if the target cells are surrounded by tissue with high levels of unchanging fluorescence. For this reason, CNS autofluorescence studies are often most effective when populations of synchronously-firing cells are studied. The highly coordinated waves of neuronal depolarization and metabolic increases that occur during SD make this event very well suited for autofluorescence methods, even with imaging systems with quite low spatial resolution.
Optical approaches can be significantly limited by scattering of excitation photons by brain tissue and overlying structures. Scattering is highly dependent on wavelength, and the short wavelengths required for NADH excitation penetrate the intact skull very poorly. For this reason most in vivo NADH studies have been done by visualization through open cranial windows with the dura removed. This approach usually limits longitudinal studies, but it is possible to create a stable cranial window using a fixed coverglass that can be used for repeated imaging sessions in the same animal (Holtmaat et al. 2009). Such an approach should be suitable for repeated NADH studies of SD in animal models.
NADH imaging via cranial windows is very effective for many types of SD studies, but even with the skull and dura removed, effective NADH excitation by UV light is limited to superficial layers of the cortex. The effective excitation depth in rat cortex was originally speculated to be restricted to superficial cortical layers (Jobsis et al. 1971) and the degree of attenuation with depth has been modeled recently with Monte Carlo simulations (L’Heureux et al. 2009). Increasing the intensity of UV illumination can increase the effective excitation of deeper layers, but it is important to note that if this is done using conventional optics, superficial tissue will be very strongly excited and complicate the analysis of signals from deeper structures. The large proportion of fluorescence emission derived from shallow structures that do not participate in a response will reduce the signal-noise ratio of responses from deeper structures, and the response may be also impaired by phototoxicity caused by excessive UV absorption in superficial tissues.
3.3.2 Two Photon Excitation
Partly because of penetration problems with short wavelength light, multi-photon excitation methods have emerged as useful for NADH imaging studies in vitro and in vivo (Huang et al. 2002; Zipfel et al. 2003; Rocheleau et al. 2004). Multi-photon excitation is based on the principle that a fluorophore requires an appropriate quantum of energy to become excited to a higher energy state. Excitation energy can be delivered either by a single photon, or by multiple photons of lower energy (i.e. longer wavelengths). Multi-photon excitation is most commonly achieved with pairs of photons of approximately double the wavelength used for single photon excitation (two photon excitation). For NADH excitation, pairs of photons (720–740 nm) are often used, and because these wavelengths are much less scattered than UV illumination, penetration into brain is greatly increased. In addition, because pairs of photons must be absorbed by the fluorophore at very nearly exactly the same time, a very high photon density is achieved at the objective focal point by using a high frequency infrared laser source, and effective two photon excitation falls very steeply as a function of distance from the focal point. As a consequence, only a very small focal plane is effectively excited and this limits photobleaching of fluorophore outside the region of interest. In addition to allowing for substantially improved penetration into the brain, two photon excitation results in much higher spatial resolution, and allows NADH signals to be resolved from individual cells within complex brain tissue (Huang et al. 2002; Zipfel et al. 2003; Kasischke et al. 2004).
Because infrared light is much less attenuated by bone than UV wavelengths, effective two photon excitation of NADH can be achieved without complete removal of a section of skull to form a cranial window. A number of investigators have developed approaches to thin a region of the rodent skull down to a few tens of microns (Yang et al. 2010). This needs to be done without excessive heating of the underlying brain, but can provide a region that is nearly transparent with two photon imaging, suitable for repetitive NADH imaging sessions over a period of days or weeks. It should be noted that while two photon approaches give a considerable advantage in excitation depth, the maximally effective depth of NADH imaging is still limited by scattering and absorption of the NADH emission photons (∼450 nm). Thus two photon NADH imaging is still restricted to the order of hundreds of microns below the brain surface, and many interesting deep structures remain out of reach. Surgical approaches to remove overlying tissues, or light guides (fibers or lenses) should be useful to assess NADH dynamics in deeper structures in vivo (Jung et al. 2004; Levene et al. 2004; Barretto et al. 2009).
Confocal microscopy using single photon excitation can improve spatial resolution by excluding emission light that is out of the focal plane and has been very effectively used for NADH imaging in a range of settings (e.g. Combs and Balaban 2001; Duchen et al. 2003). However, the excitation of large volumes of tissue with confocal microscopy can lead to significantly more photobleaching of thick preparations than with two photon methods, and furthermore since confocal methods for NADH still rely on UV excitation, penetration through skull and brain tissue remains very limited. For these reasons, single photon confocal approaches have been largely surpassed by multiphoton methods for imaging of NADH in brain slices and in vivo.
3.3.3 Correction of NADH Signals for Optical Artifacts
3.3.3.1 Interference from Hemodynamic Responses
It was Leao who first described a dramatic change in cerebral blood flow associated with SD (Leao 1944b). He reported that SD was associated with a wave of vasodilation and increased flow in arteries of all sizes, and concluded that the increase in vessel diameter (in the order of 50–100% increase) was significantly larger than that produced by hypercapnia. Leao also noted that flow in veins was increased, and that they rapidly became red. Leao concluded that the vascular response was secondary to local change in activity neuronal tissue and speculated that the increase in flow probably influenced the activity of the neurons (Leao 1944b). The temporal relationship between pial vessel changes and SD was examined in more detail by Van Harreveld and Stamm (1952) who concluded a wave of vasoconstriction occurs together with the slow potential change and decreased PO2, and proposed an asphyxial mechanism for the propagation of suppression of surface electrocorticographic activity. The relationship between blood flow and neuronal activity or injury has since been examined in considerable detail using a range of methods, including laser dopper flow and autoradiographic measures. Although there can be significant species differences in responses, experimental SD is often accompanied by a transient increase in cortical blood flow, and a subsequent longer-lasting flow decrease (Lauritzen et al. 1982; Wahl et al. 1987; Piper et al. 1991; Dreier et al. 1998; Busija et al. 2008). In mice, a significant early hypoperfusion has also been noted that coincides with the duration of the DC shift (Ayata et al. 2004; Brennan et al. 2007b). Prominent vascular responses that occur during the progression of SD have been exploited to track (with fMRI and PET approaches) SD events that occur in human subjects during migraine attacks (Woods et al. 1994; Hadjikhani et al. 2001).
As discussed in more detail below (Sect. 12.4.2), the absorption of light by hemoglobin is dependent on the degree of oxygen binding, and occurs over wavelengths that overlap with the NADH emission fluorescence spectrum. Thus hemodynamic changes have the potential to decrease the amount of autofluorescence emission that is detected, even if there are no changes in NADH redox state.
3.3.3.2 Interference from Cell Swelling
In addition to hemodynamic effects, a substantial tissue swelling response has long been recognized during SD, and this can also be a significant confound to small autofluorescence optical signals.
Van Harreveld first clearly demonstrated a swelling of neurons following SD-like events associated with ischemia in rabbit cortex (Van Harreveld 1957) and subsequently showed that apical dendrites of neurons swelled following SD generated by electrical stimulation in the same preparation (Van Harreveld 1958). The swelling response produces a reliable change in light scattering, and this is of sufficient magnitude that the progression of SD can be seen by the naked eye in some preparations (e.g. Martins-Ferreira and de Castro 1966). The transmission of white (or filtered red light) through thin preparations is also dramatically modified by cellular swelling during SD, and intrinsic optical signals generated by transmitted light have been widely used to track SD in brain slices (Aitken et al. 1999; Jarvis et al. 1999; Anderson and Andrew 2002).
Recent studies have utilized two photon imaging to examine in more detail the optical signals generated by swelling of neurons and astrocytes during SD. In addition to showing a rapid neuronal swelling coincident with SD, a transient loss of dendritic spines was also noted that could contribute to the temporary silencing of electrical activity following SD (Takano et al. 2007). Two photon studies of murine cortical brain slices have demonstrated that astrocytes swell following hyposmotic stress and K+ exposures (Risher et al. 2009), however when the relative contributions of neurons and astrocytes were specifically studied in relation to SD onset in rat hippocampal slices, it was shown that neurons undergo a rapid and transient swelling, while astrocytes undergo a passive swelling in response to K+ elevations (Zhou et al. 2010).
In addition to reversible swelling responses due to water movement, stereotypical changes in light transmission can occur as a consequence of neuronal injury. Ischemia or exposure to high concentrations of glutamate receptor agonists can generate a stereotypical pattern of dendrite swelling punctuated by severely constricted regions. This gives the dendrite the appearance of “beads on a string” and is associated with significant rearrangement of structural proteins and irreversible injury (Swann et al. 2000; Hoskison and Shuttleworth 2006; Greenwood et al. 2007). Dendritic beading is prominent when SD occurs in metabolically compromised conditions, and it has been concluded that the extreme structural rearrangement can underlie persistent increases in light scattering (Obeidat and Andrew 1998; Andrew et al. 1999).
3.3.3.3 Correction from Reflectance Changes
A reflectance subtraction method has been widely used to correct for the impact of swelling and hemodynamic changes on NADH responses. Tissues are illuminated with UV light to excite NADH fluorescence, and the intensity of UV light reflected from the cortical surface is collected together with the NADH fluorescence emission. Subtraction of the reflectance signal generates a corrected NADH fluorescence signal, which was first validated by Jobsis and colleagues for studies of epliptiform activity in perfused cat cortex (Jobsis et al. 1971). A similar correction approach was utilized soon thereafter in cortical slices in vitro by Lipton, to report corrected biphasic changes in NADH fluorescence following sustained electrical stimulation (Lipton 1973a, b). The work of Mayevsky and Chance (1974) provided a detailed justification of the subtraction method for SD studies in vivo, and showed that simple 1:1 subtraction provides appropriate compensation for vascular effects during SD. The initial subtraction method was later extended by methods to include measurement of fluorescence at the isobestic wavelength of hemoglobin-oxygemoglobin transitions (445 nm) in cat cortex (Harbig et al. 1976). These subtraction methods and their limitations have been discussed in detail relatively recently (Bradley and Thorniley 2006; Mayevsky and Rogatsky 2007). While there are some limitations, correction for reflectance changes have been very important for assessment of NADH responses during SD, since the initial oxidation phase of NADH responses during SD in vivo might be completely missed without this correction (see Mayevsky et al. 1974).
The reflectance correction method has been used routinely in NADH studies that utilize fluoremeters to record averaged fluorescence signals from a region of interest on the cortical surface. However, the same approach is not usually applied in CCD-based imaging studies of SD progression across the brain. It has been noted that CCDs lack sensitivity to UV light, making it very difficult to measure meaningful reflectance signals together with the blue light fluorescence emission of NADH (Strong et al. 1996). This limitation requires that hemodynamic and swelling responses are carefully considered in the interpretation of uncorrected NADH signals in vivo.
3.3.3.4 Comparison with Flavoprotein Autofluorescence
Comparison with flavoprotein (FP) autofluorescence signals can be an additional useful test for contamination of NADH autofluorescence signals by changes in tissue swelling or light absorption. Mitochondrial FP fluorescence changes are directly coupled to NADH responses, however the redox sensitivity of the two signals are inverted with respect to each other. Thus mitochondrial oxidation leads to FP fluorescence increases, coincident with NADH fluorescence decreases. Conversely, FP fluorescence decreases and NADH increases occur during transitions to more reduced mitochondrial redox states (see discussion in Shuttleworth 2010). Thus if NADH and FP signals have similar kinetics but are of opposite sign, then it is likely that the responses are both of mitochondrial origin. Conversely, if the fluorescence of both signals change in parallel, it is likely that changes in light absorption and scattering make a major contribution to the signals (Shuttleworth 2010).
The development of a dual-wavelength fluorometer permitted simultaneous NADH/FP measurements to be made (Chance et al. 1971) and scanning measurements of mitochondrial redox ratio have also been achieved (Chance et al. 1979). For CCD-based recording, near-simultaneous imaging can be done by alternating filter sets (Duchen 1992; Mironov and Richter 2001; Shuttleworth et al. 2003; Brennan et al. 2004; Brennan et al. 2007a). However the broad emission spectra of NADH means that it is also possible to using a single filter set (e.g. 505 nm beamsplitter and a single 510/40 nm bandpass filter) to make virtually simultaneous measurements of both fluorphores, if the excitation wavelength is switched quickly with an excitation filter changer of monochromator. This arrangement leads to loss of a great deal of the NADH fluorescence signal, but still allows enough transmission to measure inverted NADH/FP signals (Hepp et al. 2005; Gerich et al. 2006).
In comparison to NADH signals, there have been few reports of FP autofluorescence dynamics during SD in vivo. However FP signals have been utilized together with NADH to characterize mechanisms underlying energy depletion in response to spreading depression in the gerbil (Haselgrove et al. 1990), and to track the propagation of a related propagating depolarization in mouse cerebellar cortex (Chen et al. 2005).
3.4 NADH Autofluorescence Signals During Normoxic SD
3.4.1 Single Site Recordings
In the early 1970s, Jobsis and colleagues utilized a fluorimeter system to make simultaneous fluorescence and reflectance measurements from a region of cat cortex and reported NADH fluorescence dynamics corrected for changes in hemodynamic and/or swelling changes. They reported NADH transients during a range of stimuli, including evoked seizure activity (Jobsis et al. 1971) and graded electrical stimulation (Rosenthal and Jobsis 1971). The same system was soon used by Rosenthal and Somjen for study of mitochondrial activity during SD, elicited by electrical stimulation or elevated K+ in normoxic cat brain (Rosenthal and Somjen 1973). A significant question at the time was whether brain metabolism was deficient prior to SD, and was responsible for the initiation and propagation of the event. However it was clearly found that there was no NADH fluorescence decrease prior to SD, but a substantial decrease occurred following SD. (Fig. 12.3). This early observation was consistent with the accumulating evidence that SD involved a profound metabolic challenge to brain tissue (see Sect. 12.2.2 above), and the authors discussed possible roles for both astrocytes and neurons in the metabolic signals generated (Rosenthal and Somjen 1973).

NADH oxidation during SD. This figure is taken from one of the initial descriptions of NADH decreases in cortex during SD (Rosenthal and Somjen 1973). The top trace (FL) is the fluorescence emission from the cortical surface, generated by UV excitation. This record was corrected for changes in UV absorption during SD by subtracting the reflectance signal (Ref) to generate the characteristic signal attributed to NADH oxidation (F-R). The lower three traces are simultaneous electrical recordings of SD, including slow potential changes at two depths under the corticla surface (0.5 and 2.4 mm) and surface EEG activity (From Rosenthal and Somjen 1973, with permission)
At around the same time (Mayevsky and Chance 1974) reported similar large NADH fluorescence decreases during SD, using a newly-developed dual fluorometer coupled to the recording site by a flexible light pipe. The use of small light guides also allowed positioning of NADH and reflectance measurements close to a double-barreled K+ sensitive microelectrode, and EEG electrodes. From these studies it was confirmed that the durations of NADH decreases were very closely matched with extracellular K+ increases that occurred during the spread of SD (Mayevsky et al. 1974). NADH oxidation signals were interpreted to be triggered by ATP decreases that occurred after strong activation of plasma membrane Na+K+ATPase pumps, and increased O2 consumption that accompanied increased blood flow (Mayevsky et al. 1974). When the relationship between NADH oxidation and K+ elevations was examined further, a linear relationship was observed with moderate electrical stimulation in cat cortex, but the NADH oxidation response was much larger for SD than would be expected from that relationship. It was suggested that this might be due to recruitment of an auxiliary source of ADP (i.e. a reserve ATP pool) that is called upon during SD, or possibly a failure of metabolic regulation following SD (Lothman et al. 1975).
While single-site studies of SD in vivo have often reported monophasic NADH decreases, this is not always the case. Some studies report that initial the oxidation phase is followed by a large and longer-lasting NAD+ reduction phase following passage of SD (e.g. Rex et al. 1999). This may be due to differences in measurement methods, but as discussed below (Sect. 12.7.2) differences in oxygen availability between experimental models can also strongly influence the polarity of NADH signals after SD.
3.4.2 Full Field Imaging
The application of CCD-based imaging and two photon imaging has been very useful to assess propagation of SD events across the brain surface and cellular mechanisms that contribute to recovery. One of the early studies involved measurement of waves of NADH fluorescence during SD triggered by localized K+ application onto the surface of the gerbil cortex (Hashimoto et al. 2000). The CCD was placed over a cranial window (2 × 3 mm), and K+ was applied at a burr hole some distance away to initiate SD events that would spread through the recording field. Images were taken relatively slow frame rate (every 5 s), but it was possible to visualize the propagation of a long-lasting NADH increase across the field of view at a rate consistent with SD. It was curious that an initial NADH oxidation phase was not observed during SD, and it is possible that without adequate correction for reflectance changes (see above), the oxidation phase was obscured by optical interference from hemodynamic changes.
Two photon imaging has recently been employed to examine the spatial distribution of NADH changes following SD evoked by localized injection of KCl under the cortical surface of anesthetized mice (Takano et al. 2007). The high resolution of the images revealed a very interesting heterogeneity of signals that suggested regional differences in oxygen availability (Fig. 12.4). Initial fluorescence decreases were observed throughout the imaging field during SD, but fluorescence increases after SD occurred in an organized pattern that was defined by the vascular architecture. The signals were not corrected for possible heterogeneity in swelling or increased absorption of NADH fluorescence emission, but if these heterogeneity is indeed due to differences in NADH fluorescence, it can be taken to imply that NADH oxidation was limited in tissue where O2 availability was more limited due to longer diffusion distances from vessels (Takano et al. 2007). This high resolution study also directly demonstrated that if responses were averaged over a whole region imaging, regional heterogeneity was lost. It was also concluded that the NADH transitions following SD do not involve a cellular pattern, and are thus unlikely not to involve neurons and/or astrocytes specifically, but may be a metabolic response that is common to all cell types as a consequence of metabolic demands placed by the SD event (Takano et al. 2007).

Regional heterogeneity of NADH signals during SD. The recent application of two photon imaging to SD in vivo revealed a distinct pattern of NADH dynamics during SD that was determined by proximity of tissue to blood vessels. The figure is modified from Takano et al. (2007), and left hand panels show autofluorescence attributed to NADH at the peak of the DC potential shift of SD (top), vascular distribution identified following FITC-dextran injection (middle). The merged image (bottom) shows that autofluorescence decreases are concentrated around vessels, while fluorescence increases are prominent at greater distances from the vessels. The longer diffusion distance for O2 and consequent hypoxia could explain fluorescence increases at sites more distant from vessels during SD. The traces show the DC potential shift (black) and the different fluorescence decreases adjacent to the recording electrode, in regions close (green) and more distant from a vessel (brown) (From Takano et al. 2007)
3.5 Mechanisms Underlying NADH Signals
3.5.1 Change in Mitochondrial Redox Potential
Since early studies of NADH and SD, there has been a strong consensus that the signals are due to changes in mitochondrial redox state. Although NADH is involved in cytosolic as well as mitochondrial metabolism, the fluorescence of NADH in mitochondrial compartments appears to dominate fluorescence recordings under most synaptic stimulation conditions (see discussion in Shuttleworth 2010). Thus in early studies of surface NADH recordings from cat cortex, it was concluded that mitochondrial dehydrogenase activity, rather than cytosolic NADH transitions, was the prominent source of NADH signals following epileptiform activation (Jobsis et al. 1971).
The classical NADH oxidation observed during normoxic SD was described as a “state 4-3-4 metabolic transition at the mitochondrial level” (Mayevsky et al. 1974). These states refer to different mitochondrial redox potentials described by Chance and Williams, where state 4 is a resting state with mainly reduced NADH, and state 3 is an activation state where ADP levels rise and NADH is oxidized as electron transport is accelerated (Chance and Williams 1955a, b, c). Lothman and co-workers reemphasized that “labile” component of NADH signals is derived almost exclusively from intramitochondrial NADH, and that changes in redox level of NADH measured with fluorescence methods report relative rates of oxidative metabolism (Lothman et al. 1975). Much subsequent work was done to characterize the influence of levels of ischemia, anesthesia, drugs on mitochondrial function using NADH signals during SD signals, and has been reviewed previously (Mayevsky and Rogatsky 2007).
In many studies where tissue NADH simply recovered back to pre-stimulus levels after SD, it could be concluded that mitochondria recovered from state 3 back to state 4 as plasma membrane ionic gradients were restored. However in cases where NADH levels significantly increased after SD, additional explanations are required. As noted above, the availability of oxygen to support mitochondrial metabolism is likely a central issue, as limitations will lead to increased NADH fluorescence and this may be easier to detect with higher resolution measurements (e.g. Takano et al. 2007). However it is also possible that cation accumulation itself leads to increased TCA cycle activity and increased mitochondrial NADH.
SD is associated with very large Ca2+ increases (Kunkler and Kraig 2004; Dietz et al. 2008), which are sufficient to cause mitochondrial depolarization (Bahar et al. 2000). Much work has characterized stimulation of mitochondrial dehydrogenases by Ca2+, and led to the suggestion that Ca2+ accumulation couples neuronal activity to metabolism increases. In an important study of mechanisms underlying NADH dynamics in isolated neurons, it was concluded that mitochondrial Ca2+ accumulation and consequent stimulation of dehydrogenases was responsible for the prominent NADH increases that persist after transient depolarization (Duchen 1992). It is not yet known whether the very large increases in neuronal Ca2+ can contribute to NADH increases after SD.
From studies in brain slices, it was also found that NADH increases (“overshoots”) could occur following transient neuronal depolarization in the absence of Ca2+ (Shuttleworth et al. 2003). The mechanism(s) underlying these responses it not yet known, but on the basis of Na+ replacement experiments, it was speculated that metabolic consequences of Na+ accumulation could contribute to increased TCA cycle activity. It has also been suggested that large NADH increases after stimulation could occur partly because of inadequate supply of O2 in the brain slice (Turner et al. 2007). However, this seems unlikely from the responses seen during repetitive challenge with synaptic stimulation at various time points during the “overshoot” phase. If overshoots were due to O2 depletion, then it should be more difficult to generate a state 4 to state 3 transition, when synaptic stimulation is applied at the peak of NADH overshoots, yet initial NADH oxidation responses are virtually unchanged (Brennan et al. 2007a). The relative contributions of localized O2 depletion and metabolic consequences of cation loading to NADH increases after the extraordinary metabolic challenges of SD are yet to be determined.
3.5.2 Possible Contributions from Astrocyte Metabolism
Astrocyte function is likely and important regulator of initiation and propagation of SD (Somjen 2001), presumably because of the central roles of these cells in buffering extracellular glutamate and K+ concentrations (Danbolt 2001; Kofuji and Newman 2004). Consistent with this assumption, selective inhibition of astrocytic mitochondrial metabolism with the toxin fluorocitrate accelerates the onset of, and consequences of SD (Largo et al. 1997; Lian & Stringer 2004) and prolonged exposures to glial toxins alone can initiate SD accompanied by irreversible neuronal injury (Canals et al., 2008). These findings imply that mitochondrial function in astrocytes is important for the regulation of SD, but the extent to which astrocyte mitochondrial function contributes to NADH autofluorescence signals following SD is currently unclear.
Although mitochondrial inhibitors impair astrocyte function in SD, astrocytes are usually considered to rely heavily on glycolysis for ATP synthesis. Glycolysis depends on cytosolic NAD+ as a cofactor for activity of glyceraldehyde phosphate dehydrogenase. Conversion of pyruvate to lactate serves to reoxidize NADH to provide the NAD+ that is required to maintain glycolysis during O2 depletion. Thus strong activation of glycolysis could potentially contribute to NADH increases if O2 is available, but significant cytosolic NADH accumulation would not be expected if there were tight coupling between glycolysis and lactate formation.
Glycolysis has been suggested as a candidate for large NADH increases that occur following prolonged periods of electrical stimulation of guinea-pig cortical slices (Lipton 1973b). A more recent two photon imaging study suggested that NADH increases following synaptic stimulation of rat hippocampal slices were restricted to astrocytes (rather than neurons), and that this was in turn due to stimulation of glycolysis by astrocytic glutamate uptake (Kasischke et al. 2004). The latter suggestion was based on spatial and temporal segregation of NADH signals between neurons and astrocytes, and was suggested to be due to stimulation of glycolysis by astrocytic glutamate uptake. This interesting possibility is difficult to test pharmacologically, since the key experiments require block of glycolysis with procedures that maintain substrate for oxidative metabolism. However from subsequent studies in murine hippocampal slices, it was concluded that either glycolytic inhibition or inhibition of astrocytic glutamate uptake did not reduce overshooting NADH increases, over a range of synaptic stimulation durations (Brennan et al. 2006). These results suggested that glycolysis was not necessary to generate robust NADH overshoots, but it was acknowledged that supply of low pyruave concentrations to support oxidative metabolism could artificially shift metabolism from glycolytic to oxidative processes. For this reason, it was useful to note that comparison of NADH and FP responses under the same stimulation conditions (without supplemental pyruvate) supported the conclusion that both components of NADH responses were unlikely to involve glycolysis in murine brain slices (Brennan et al. 2006, 2007a).
These findings are in agreement with the general assumption that mitochondrial metabolism predominates in optical measures of NADH metabolism following activation of brain tissue, and this topic has been reviewed in more detail recently (Shuttleworth 2010). However it may be useful to emphasize that the ability to measure robust mitochondrial NADH transients with fluorimeter and CCD-based imaging systems does not provide evidence that significant accumulation of NADH in the cytosol due to activation of glycolysis does not occur. What these results do suggest is that if such accumulation occurs, it may be too small to detect in comparison to much larger signals generated by NADH/NAD+ transitions that occur in an environment (mitochondria) that leads to much brighter fluorescence (Shuttleworth 2010). Additional high resolution studies, coupled with pharmacological tests for metabolic pathways involved, are needed to directly test whether glycolytic NADH signals can be clearly demonstrated in the cytosol of astrocytes. Discrimination between mitochondrial and cytosolic signals will be of particular interest during SD, when lactate production accelerates.
3.5.3 Contributions of Neuronal Compartments to NADH Transients
An important early study of mechanisms underlying NADH transients was undertaken using isolated dorsal root ganglion neurons, and showed that brief depolarization resulted in initial NAD(P)H fluorescence decreases, followed by a longer-lasting overshooting NADH. The fact that these signals were generated by direct depolarization of isolated neurons provided clear evidence that neurons alone could be sufficient to explain both phases of biphasic NAD(P)H transients observed in brain slices or in vivo (Duchen 1992). Relatively few studies have dissected the relative contributions of different neuronal compartments to brain autofluorescence transients in complex brain tissue, however it is possible that both presynaptic and postsynaptic neuronal elements could contribute to NADH signals during SD. Mitochondria are densely accumulated in perinuclear regions in nerve cell bodies, but are also found in distributed throughout dendrites and axons (see Kann and Kovacs 2007 for review) and recent work has demonstrated significant trafficking of mitochondria through these processes (Chang and Reynolds 2006; Mironov 2006). Consistent with the idea that dendritic processes can contribute to NADH signals, two photon imaging has shown that localized high K+ produces biphasic NADH transients in single dendritic processes of isolated Purkinje neurons (Hayakawa et al. 2005).
Ionic loading is also expected to place significant metabolic demands on presynaptic compartments following SD presynaptic mitochondrial function may also be expected to contribute to compound NADH transients during these responses. Concentrations of mitochondria have been localized to some presynaptic terminals (Rowland et al. 2000) and pharmacological evidence has implied that mitochondrial Ca2+ uptake can modulate presynaptic Ca2+ transients (Tang and Zucker 1997; Zucker 1999). Consistent with a role in presynaptic metabolism, NADH transients have recently been documented in a preparation of neurosecretory terminals (Kosterin et al. 2005), and NADH fluorescence decreases in nerve terminals at the neuromuscular junction following extended electrical stimulation (Talbot et al. 2007). Within the CNS, parallel fiber stimulation also leads to some pre-synaptic NADH transients (Diez-Garcia et al. 2005), however it does appear that these are relatively small, and signals are dominated by postsynaptic responses.
4 Optical Imaging of Intrinsic Optical and Hemodynamic Signals During SD
4.1 Introduction
As discussed above, vascular changes during SD can interfere significantly with autofluorescence measurements, but it has long been appreciated that intrinsic optical signals attributed to vascular dynamics can provide valuable insights into oxygen delivery and recovery following SD (Rosenthal and Somjen 1973). A range of optical methods has been used to assess blood flow and hemoglobin desaturation during SD in diverse experimental settings, and this large body of work will not be reviewed in detail here. The following section will instead introduce some of the main approaches that can provide high resolution imaging of spreading changes in hemoglobin oxygen saturation and blood flow related to SD.
4.2 Optical Detection of Vascular Changes
The optical spectra of oxygenated and deoxygenated hemoglobin (Hb) that underlie differences in the color of blood are plotted in Fig. 12.5. Light reflected from well-oxygenated vessels has a more intense red appearance because deoxygenated Hb continues to absorb light through red wavelengths (>∼590 nm), while there is almost no absorption in this range by oxygenated Hb. Based on this difference, imaging at wavelengths above 600 nm has commonly been used to assess blood oxygenation supplying brain tissue. From Fig. 12.5, discrimination between oxy and deoxyHb could also theoretically be achieved at shorter wavelengths but longer wavelengths are favored partly because of good penetration into the brain tissue, including through thinned skull windows. Figure 12.5 also shows that the absorption of oxy and deoxy Hb is identical at some wavelengths (e.g. ∼500 nm) and if a range of green wavelengths is assessed (∼530–570 nm) absorption of the two species overlaps to such an extent that changes in reflectance of green light can provide information on changes in local cerebral blood volume, independent of changes in blood oxygen saturation (Fujita et al. 2000; Dunn et al. 2005; Vanzetta et al. 2005). Imaging changes in blood oxygenation and volume using red and green light in vivo is usually termed intrinsic optical imaging, and has had very wide application for monitoring the regional activation and plasticity of cortical structures (Frostig et al. 1990; Nemoto et al. 1999; Hillman 2007; Prakash et al. 2009).

Absorbance spectra of oxyg-hemoglobin and deoxy-hemoglobin. A clear separation in the spectra occurs above 600 nm, so that changes in the amount of reflected red light can be used to assess changes in deoxyhemoglobin levels. Conversely, at green wavelengths (∼530–570 nm), the absorption of the two species overlaps to such an extent that changes in reflectance of green light can provide information on changes in local cerebral blood volume, independent of changes in blood oxygen saturation. Since absorption of both species is very low above ∼800 nm, changes in infrared signals can be used to assess light scattering changes. Data were obtained from http://omlc.ogi.edu/spectra/hemoglobin/, by S. Prahl, Oregon Medical Laser Center. Replotted with permission
High resolution optical imaging of brain hemodynamic signals has usually been achieved by using filter-based systems and CCD cameras, but alternative approaches include laser illumination and near-infrared spectroscopic analysis of oxy and deoxy Hb shifts. A more recent development in optical imaging of SD has been the application of laser speckle contrast analysis (LSCA). LSCA was originally developed for studies of flow in retina (reviewed in Briers 2001) but has since become widely used for studies of cerebral blood flow. The method is an extension of single-point laser Doppler flow (LDF) measurements, and is based on changes in the speckled appearance of brain under laser illumination. The speckling is due to a random interference effect, and the image contrast is highest when particles within the image are stationary, or at least moving slowly. When red blood cells move more quickly in an image field, image contrast is reduced. The interference effect requires laser illumination and, like standard laser dopper flow analysis, there remains difficulty in measuring absolute changes in CBF, although a calibration method has been validated in a study of SDs generated by ischemia in cat cortex (Strong et al. 2006). Many studies have utilized open skull preparations, but recent work has begun to utilize LCSA for imaging through thinned skull windows (Parthasarathy et al. 2008), which should be useful for longitudinal studies of SD in rodents.
4.3 Signals During Normoxic SD
Figure 12.6 provides an example from a study that examined intrinsic optical responses in rat cortex, using a thinned skull preparation (Ba et al. 2002). SD was initiated by a pin prick, using a 32 gauge needle inserted briefly through a burr hole drilled a few millimeters distant from the imaging site. This is a useful example, because the brain surface was illuminated with white light and reflected light were filtered at three wavelength bands centered at 550 (530–570 nm), 610 (605–615 nm) and 850 (845–855 nm), and signals from an intravascular fluorescent dye (Texas Red dextran) are also shown (Ba et al. 2002). As discussed above, 550 nm reflection was used to assess changes in total hemoglobin concentration, 610 nm for oxy-deoxy Hb shifts, and 850 nm signals for light scattering effects that are not significantly contaminated by Hb absorption. In this report, the authors concluded that a decrease in deoxyhemoglobin occurred soon after SD, as blood volume and flow increase to such a degree that O2 supply exceeds demand of the tissue, and conclude that these early signals are similar to the normal neurovascular coupling seen following physiological stimulation of somatosensory cortex (Ba et al. 2002). However at later time points post-SD, an increase in 610 nm signals is interpreted as increased O2 demand being larger than supply (Ba et al. 2002). These and other intrinsic optical imaging studies in a range of preparations are currently being used to investigate regulation of SD propagation and neurovascular coupling (e.g. Chang et al. 2010), and also the propagation of vascular reactivity changes. For example, (Brennan et al. 2007b) recently examined in detail the relationship between the diameter of surface arterioles and intrinsic optical signals recorded from the parenchyma. Interestingly, they found that a dilation response propagated throughout the vascular network significantly faster than the DC shift or parencymal change accompanying SD, and suggested that specific vascular conduction pathways are activated prior to SD (Brennan et al. 2007b).

Intrinsic optical signals during SD. This figure is taken from Ba et al. (2002) and illustrates a number of different optical signals recorded from the rat cortical surface, following initiation of SD with a pinprick As noted in the text, intrinsic signals were centered at 550 (blue trace), 610 (purple trace) and 850 nm (green trace) to assess blood volume, deoxgyhemoglobin and light scattering changes, respectively. The red trace is from intravascular injection of an indicator dye Texas Red dextran and plots from different trials have been aligned to form this composite figure. Soon after SD (∼40 s on trace) a significant increase in 550 and 850 nm absorption (plotted as reflectance decrease) is seen, and the increase in blood flow at this time point is also visualized as an increase in the intravascular dye signal. The decreased deoxyhemablobin signal (610 nm) at this time point suggests a normal neurovascular coupling response, similar to that observed with reflex stimulation. However at later time points (>60 s on trace), decreased blood flow signals and increased deoxyhemoglobin absorption implies inadequate neurovascular coupling (From Ba et al. 2002, with permission)
LSCA of flow changes has emerged as a very useful complementary approach for tracking SD. In an early application, LSCA measurements was used to monitor SD evoked by K+ in mouse cortex, and provided reliable detection of an initial hypoperfusion phase that coincided with the shift in DC potential (Ayata et al. 2004). LSCA has been successfully combined with simultaneous measurements of intrinsic optical signals following SD in rat cortex (Dunn et al. 2003), and a combination of LSCA and glucose microdialysis was used to characterize the temporal relationship between SD progression and the substantial depletion of extracellular glucose concentration that ensues (Hashemi et al. 2008).
5 Optical Imaging of Metabolism During SD in the Context of Brain Ischemia
5.1 Introduction
As part of his early descriptions of SD in rabbit cortex, Leao reported the effects of transient common carotid occlusion. The basilar artery was clipped in these animals, so that bilateral carotid artery occlusion would produce a rapid and profound cerebral ischemia without compensation from vertebral artery supply. After about 2–5 min of ischemia, a slow potential shift underlying SD was recorded. Recordings were made from arrangements of five electrodes placed across the cortical surface, and although a propagation rate was not reported, the asynchrony of the responses was consistent with a spreading response over the brain surface (Leao 1947). Leao also described the effects of brief ischemia (1 min) applied to tissue that was undergoing SD generated by electrical stimulation. Ischemia caused a marked prolongation and enlargement of the voltage shift associated with SD (Leao 1947). These observations first raised the idea that ischemia can prolong ionic disruptions of SD, and that ischemia itself can trigger propagating depolarizing events. As noted above in Sect. 12.2, a significant body of work has concentrated on the contributions of SD to ischemic brain injury in animal models, and recent studies have greatly strengthened the suggestion that SD contributes to the evolution of ischemic infarction in human subjects.
Electrophysiological recordings of DC shifts and ECoG activity have been central to efforts to identify the SD events that accompany ischemia (Strong et al. 1983a; Nedergaard and Astrup 1986). From such studies, a relationship between the number of events and infarct volume has been established, and the effectiveness of non-specific inhibitors of SD on reduction of lesion volume has been demonstrated (see Hossmann 1996) However optical imaging has also contributed significantly to understanding of how repetitive SDs contribute to the expansion of injury, partly because of the ability to examine the relationship between regional changes in metabolism and blood flow. As described in Sect. 12.4.1, a substantial increase in blood flow accompanies normoxic SD, and this helps meet increased energy demands. However the ability to increase flow in peri-infarct areas can be greatly reduced. From a variety of experimental approaches, it has been demonstrated that SD events in peri-infarct regions do not elicit appropriate flow increases, and that PO2 changes are opposite that which is observed in healthy tissue (Back et al. 1994). It is widely presumed that the inappropriate neurovascular coupling that accompanies SD in these regions is a central cause of brain injury (Back 1998). Autofluorescence imaging of NADH has provided a way of assessing regional metabolic status during these events, and when coupled with hemodynamic measures, provides a powerful way of interrogating tissue surrounding infarcts, to predict whether brain tissue will survive or succumb to the repetitive challenges of SD after ischemia.
5.2 Influence on NADH Autofluorescence Signals
The relationship between oxygen tension and NADH responses during SD has been extensively investigated by Mayvesky and others, and has provided important support for the idea that NADH autofluorescence can identify vulnerable brain tissue after SD. A large body of work has been summarized elsewhere (Mayevsky and Rogatsky 2007), but one example (Sonn and Mayevsky 2000) will be used to illustrate some key points (Fig. 12.7). NADH autofluorescence was measured from a small region of interest on the rat cortex, together with a laser Doppler flow measurement of blood flow, and electrode recordings of DC potential and extracellular K+ concentration. These multiple recording probes were embedded in a single rectangular cannula and SD was initiated by KCl delivered from a burr hole a few millimeters away. In normal tissue, the expected NADH decrease was observed during SD (as described in Sect. 12.3.4) which coincided with local flow increases, and this was followed by a longer-lasting fluorescence increase. In contrast, under conditions of hypoxia (inhaled 12% O2, 88% N2; decreasing arterial PO2 from 107 to 57 mmHg), or partial ischemia (permanent bilateral carotid artery occlusion), a quite different NADH response was observed. NADH increases (rather than decreases) were observed, and this switch to a “reduction cycle” coincided with initial local flow decreases (Sonn and Mayevsky 2000). These NADH recordings supported the conclusion that local flow did not compensate for the lack of O2 during hypoxia and the lack of increased flow was considered to arise either due to lack of capacity for further dilation after ischemia, or release of vasoconstrictor factors (including K+) during SD (Sonn and Mayevsky 2000).

Effects of hypoxia on NADH responses to SD. This composite figure is modified from Sonn and Mayevsky (2000), and shows the recording arrangement for simultaneous NADH fluorescence with laser Doppler flow, extracellular K+, and DC potential recordings from the rat cortex (a). Panel b shows the typical responses to SD initiated by topical application of KCl. The raw 366 nm reflectance trace (R) is shown together with the corrected NADH fluorescence signal, and emphasizes the importance of this correction to reveal the initial NADH oxidation during SD. Panel c shows that the initial NADH oxidation was completely lost and replaced by an initial NADH fluorescence increase when the same stimulus is applied to hypoxic brain (arterial PO2 reduced from 107 to 57 mmHg). Note that the DC potential shift and extracellular K+ elevations are also significantly prolonged in the hypoxic condition (From Sonn and Mayevsky 2000, with permission)
5.3 Imaging the Spread of SD Events in Relation to Ischemic Infarcts
The application of CCD-based imaging of brain surface NADH autofluorescence signals has proven very useful for the evaluation of site of origin and pattern of spread of SD events in relation to an ischemic infarct. Neuronal injury at an ischemic core is expected to release large amounts of glutamate and K+ into the extracellular space, and it was proposed that accumulation of these mediators could be sufficient to trigger SDs. Alternatively, SD initiation sites could be determined by a pattern of regional blood flow deficiencies, rather than by neuronal injury. Imaging NADH levels from a relatively large field of view that includes the ischemic region has made it possible to address this question.
After an initial report of NADH dynamics from single locations using a fluorimeter (Strong et al. 1988), Strong and co-workers applied a CCD-based approach to image responses following middle cerebral artery occlusion in cats (Strong et al. 1996) (Fig. 12.8). A large cortical area was imaged at a frame rate high enough to capture the spread of a series of complex spreading events in each animal. NADH decreases and increases were both observed, and since these CCD measurements could not be corrected for UV reflectance measures, it was acknowledged that fluorescence decreases were likely a combination of NADH oxidation as well as increases in blood volume as would be expected for SD propagation through healthy tissue (see Sect. 12.4). In addition, these investigators showed that in some regions, clear NADH increases were observed. Again, uncorrected fluorescence increases could be due to decreased blood volume, as well as conversion of NAD+ to NADH, however in regions where these fluorescence increases were observed, the authors concluded that this represented a conversion to pathologic or “core conditions” (Strong et al. 1996). In addition, the imaging provided evidence that SD events initiated at the edge of ischemic infarcts.

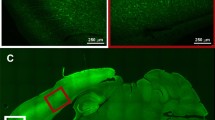
Imaging autofluorescence signals associated with focal ischemic infarction. This figure is taken from a study that demonstrated that waves of autofluorescence increase originated at the site of focal ischemic infarcts and propagated through surrounding tissue. Surface autofluorescence recordings that included NADH (UV excitation, 445–465 nm emission) were made approximately 3 h after generating a middle cerebral artery occlusion in a squirrel monkey. Panel a shows a bright-field image of the recording site (scale bar = 5 mm), and b shows a small central region of sustained fluorescence increase that appears to correspond to a region of irrecoverably injured tissue. Subsequent frames (c–e, 32–34 min from onset of recording) show progression of events around that focal region. The last three frames (f–h, 54–56 min after onset of recording) show a larger wave that invades the entire region, including the initial core site (From Strong et al. 2000, with permission)
These conclusions were extended in studies of squirrel monkey cortex after middle cerebral artery occlusion. It was concluded that the sign of NADH fluorescence responses could be used to distinguish between periinfarct depolarizations (with risk of promoting damage) and responses in normally perfused cortex (Strong et al. 2000). The demonstration that the propagation of such events could be demonstrated in the gyrencephalic cortex of a primate was also important in strengthening the idea that SDs could occur in higher mammals, in addition to the relatively smooth cortical surface of rodents and cats (Strong et al. 2000). Another important advance has been the demonstration that NADH imaging could be used to clearly evaluate the enlargement of areas of ischemic infarction associated with repetitive SD events (Higuchi et al. 2002). In studies of middle cerebral artery occlusion in rats, sustained NADH fluorescence increases identified the ischemic core, and then propagating NADH increases were observed to originate at the edge of the core. Bands of fluorescence increases (∼2–4 mm across) propagated along the margin of the core, and some cases were observed to completely circle the injured region. In about half the cases, areas involved in propagating NADH increases merged with, and could be seen to increase the size of the ischemic core (Higuchi et al. 2002).
The ability to reliably monitor the propagation of periinfarct SDs with NADH is now beginning to be exploited to assess the mechanism of action of drugs and treatments designed to reduce ischemic infarction. For example, CCD-based NADH imaging was recently used suggest that lack of effectiveness of hypothermia against injury produced by permanent vascular occlusion may be a consequence of lack of effect on SD mechanisms (Sasaki et al. 2009).
While the studies above have usually acknowledged the complications of hemodynamic changes for interpretation of NADH signals, it is also possible that pathologic swelling responses modify autofluorescence signals in peri-infarct regions. Recent two photon imaging of neurons has shown dramatic and transient loss of dendritic spines associated with bilateral common carotid artery occlusion in mice (Brown et al. 2008; Murphy et al. 2008) and it is not yet clear whether these changes contribute significantly to NADH signals during ischemia.
Laser speckle contrast imaging (LSCA) has recently been applied to monitor SD events triggered by middle cerebral artery occlusion in cats (Strong et al. 2006). Imaging was performed over a large cortical region, and following the initial drop due to MCAO, flow decreases were observed to propagate in a wave-like manner at a rate (∼2–4 mm/min) consistent with progression of peri-infarct depolarizations. DC potential shifts were recorded simultaneously with surface electrodes, and revealed that LSCA measurements failed to detect all electrical events, but it is likely that the detection limit could be improved with higher image acquisition frequency (Strong et al. 2006). It is also noteworthy that some events detected with LSCA did not propagate to the recording electrode, suggesting that a combination of the electrical and optical methods will be useful for comprehensive detection of SDs associated with focal ischemia (Strong et al. 2006). Because this imaging approach can be stable for hours following ischemia surgery, it is likely that LSCA will be of great value for evaluating therapeutic interventions to reduce SD frequency or propagation after ischemia
6 Conclusions and Perspectives
Much of the important development of autofluorescence imaging for studies of brain metabolism has relied on spreading depression (SD) as an excellent test response. The stimulus evokes a very strong metabolic challenge and NADH signals that can be detected with good signal-noise characteristics in a variety of experimental settings. In addition, the slow and well-organized propagation of SD makes NADH signals quite easy to capture with conventional CCD cameras, and also by high resolution imaging with two photon excitation methods. The validation of autofluorescence methods in SD studies has provided an important foundation for application of similar optical approaches to other questions of brain pathophysiology. Historically, most such applications have involved NADH imaging, but the complementary flavoprotein autofluorescence signals have recently been applied successfully to in vivo studies of plasticity in the rodent cortex, including through thinned skull preparations (Shibuki et al. 2007; Tohmi et al. 2009). It may be useful if more studies can combine NADH and flavoprotein imaging, to assess the relative contributions of mitochondrial and cytosolic metabolism in vivo. Such studies could be helpful for addressing questions of metabolic coupling including, if done at sufficiently high resolution, the important questions of compartmentalization of astrocytic and neuronal signals. A useful complement to such studies could be the application of fluorescent analogues of glucose, which have recently been used in brain slice studies to assess differences in glucose uptake between neurons and astrocytes (Barros et al. 2009). Such methods have not yet been applied to synaptic activation in vivo.
As a consequence of the large number of SD studies associated with development of NADH methods, much has been learned about the temporal and spatial spread of mitochondrial metabolism after SD. This has provided valuable insights about the spread of the events in relation to ischemic infarcts and, when this has been combined with information about regional blood flow, it has been possible to distinguish tissue that is likely to die following repetitive SDs, and tissue which has sufficient coupling to survive. Much of this work has been done in experimental animal models, but recent evidence that SD events are likely a prominent contributor to some types of human brain injury and it is likely that optical approaches to assess tissue viability could be utilized in clinical settings. As noted above, penetration of light through the skull is a limitation for optical studies, but illumination of relatively large brain areas may be possible in a small number of clinical settings where segments of skull have been removed for decompression after injury. Recent work has demonstrated the effectiveness of small optical probes for recording tissue partial pressure of oxygen, together with electrocorticographic and laser dopper flow probes, all arranged together on strip electrodes and placed under the dura in injured human brain (Dreier et al. 2009). It is possible that other optical signals that have been well-validated for distinguishing at-risk tissue in animal models could also be incorporated into subdural strip arrangements, to identify vulnerable regions in human studies.
The relationship of mitochondrial and hemodynamic signals to other cellular consequences of SD in vivo is an important next step. Ca2+ overload has long been appreciated as a major contributor to neuronal injury after SD in brain slices (Somjen 2001), but very little is known about the patterns of Ca2+ deregulation following repetitive SD in vivo. High resolution fluorescence imaging of Ca2+ indicators would be helpful to determine whether dendrites serve as initiation sites for neuronal injury after SD, as has been concluded for excitotoxic injury in brain slices (Shuttleworth and Connor 2001). Pathologic accumulations of Zn2+ have also recently been considered as a contributor to the initiation and pathologic consequences of SD in brain slices (Dietz et al. 2008, 2009), and it will be of interest to determine whether Zn2+ increases are prominent following under similar conditions in vivo. As discussed above for NADH autofluorescence (Sect. 12.5.3), such efforts may be most effective with two photon excitation in via open cranial windows or thinned skull preparations. In contrast to autofluorescence methods (which rely no indicator loading procedures), a challenge for fluorescence measurements of any ion indictor is effective loading of organic indicators into the preparations. The use of mice with sensors genetically encoded into subsets of neurons or astrocytes could be very useful in this context.
Recent work has begun to evaluate optical imaging of voltage-sensitive indicators to assess SD propagation in the rat cortex (Obrenovitch et al. 2009; Farkas et al. 2010). These signals have some advantages over metabolic or vascular signals related to SD, since they can be directly related to the initial wave of depolarization associated with SD, rather than downstream consequences. It will be of significant interest to see whether these signals can be used for relatively long-term recordings during focal ischemia, or other injury models, since they could provide valuable high-resolution information about the progression of electrical silencing that is difficult to achieve with conventional electrical recordings.
It is becoming increasingly appreciated that many neurodegenerative diseases are associated with regional or global metabolic dysfunction, and optical imaging methods in animal models can provide valuable insights into mechanisms underlying injury progression. Transfer of these approaches into human subjects is difficult, but may be feasible in some settings, particularly with the development of small optical probes suitable for the types of long-term monitoring that may be needed.
References
Aitken PG, Fayuk D, Somjen GG, Turner DA (1999) Use of intrinsic optical signals to monitor physiological changes in brain tissue slices. Methods 18:91–103
Anderson TR, Andrew RD (2002) Spreading depression: imaging and blockade in the rat neocortical brain slice. J Neurophysiol 88:2713–25
Anderson RE, Meyer FB (2002) In vivo fluorescent imaging of NADH redox state in brain. Methods Enzymol 352:482–94
Andrew RD, Jarvis CR, Obeidat AS (1999) Potential sources of intrinsic optical signals imaged in live brain slices. Methods 18(185–196):179
Aubin JE (1979) Autofluorescence of viable cultured mammalian cells. J Histochem Cytochem 27:36–43
Ayata C (2010) Cortical spreading depression triggers migraine attack: pro. Headache 50:725–730
Ayata C, Shin HK, Salomone S, Ozdemir-Gursoy Y, Boas DA, Dunn AK, Moskowitz MA (2004) Pronounced hypoperfusion during spreading depression in mouse cortex. J Cereb Blood Flow Metab 24:1172–82
Ba AM, Guiou M, Pouratian N, Muthialu A, Rex DE, Cannestra AF, Chen JW, Toga AW (2002) Multiwavelength optical intrinsic signal imaging of cortical spreading depression. J Neurophysiol 88:2726–35
Back T (1998) Pathophysiology of the ischemic penumbra–revision of a concept. Cell Mol Neurobiol 18:621–38
Back T, Kohno K, Hossmann KA (1994) Cortical negative DC deflections following middle cerebral artery occlusion and KCl-induced spreading depression: effect on blood flow, tissue oxygenation, and electroencephalogram. J Cereb Blood Flow Metab 14:12–19
Bahar S, Fayuk D, Somjen GG, Aitken PG, Turner DA (2000) Mitochondrial and intrinsic optical signals imaged during hypoxia and spreading depression in rat hippocampal slices. J Neurophysiol 84:311–24
Barretto RP, Messerschmidt B, Schnitzer MJ (2009) In vivo fluorescence imaging with high-resolution microlenses. Nat Methods 6:511–12
Barros LF, Courjaret R, Jakoby P, Loaiza A, Lohr C, Deitmer JW (2009) Preferential transport and metabolism of glucose in Bergmann glia over Purkinje cells: a multiphoton study of cerebellar slices. Glia 57:962–70
Bowyer SM, Aurora KS, Moran JE, Tepley N, Welch KM (2001) Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann Neurol 50:582–87
Bradley RS, Thorniley MS (2006) A review of attenuation correction techniques for tissue fluorescence. J R Soc Interface 3:1–13
Brennan AM, Connor JA, Shuttleworth CW (2006) NAD(P)H fluorescence transients after synaptic activity in brain slices: predominant role of mitochondrial function. J Cereb Blood Flow Metab 26:1389–1406
Brennan AM, Smith AJ, Connor JA, Shuttleworth CW (2004) Kinetics of intrinsic fluorescence transients in hippocampal slices following synaptic stimulation. Soc Neuroscience Abstracts:197.123
Brennan AM, Connor JA, Shuttleworth CW (2007a) Modulation of the amplitude of NAD(P)H fluorescence transients after synaptic stimulation. J Neurosci Res 85(15):3233–343
Brennan KC, Beltran-Parrazal L, Lopez-Valdes HE, Theriot J, Toga AW, Charles AC (2007b) Distinct vascular conduction with cortical spreading depression. J Neurophysiol 97:4143–51
Briers JD (2001) Laser Doppler, speckle and related techniques for blood perfusion mapping and imaging. Physiol Meas 22:R35–R66
Brown CE, Wong C, Murphy TH (2008) Rapid morphologic plasticity of peri-infarct dendritic spines after focal ischemic stroke. Stroke 39:1286–91
Bures J (1956) Some metabolic aspects of Leao’s spreading depression. J Neurochem 1:153–158
Bures J, Buresova O, Krivanek J (1984) The meaning and significance of Leao’s spreading depression. An Acad Bras Cienc 56:385–400
Busch E, Gyngell ML, Eis M, Hoehn-Berlage M, Hossmann KA (1996) Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging. J Cereb Blood Flow Metab 16:1090–99
Busija DW, Bari F, Domoki F, Horiguchi T, Shimizu K (2008) Mechanisms involved in the cerebrovascular dilator effects of cortical spreading depression. Prog Neurobiol 86:379–395
Canals S, Larrosa B, Pintor J, Mena MA, Herreras O (2008) Metabolic challenge to glia activates an adenosine-mediated safety mechanism that promotes neuronal survival by delaying the onset of spreading depression waves. J Cereb Blood Flow Metab 28:1835–44
Chance B (2004) Mitochondrial NADH redox state, monitoring discovery and deployment in tissue. Methods Enzymol 385:361–70
Chance B, Williams GR (1955a) Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem 217:383–93
Chance B, Williams GR (1955b) Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem 217:409–27
Chance B, Williams GR (1955c) Respiratory enzymes in oxidative phosphorylation. II. Difference spectra. J Biol Chem 217:395–07
Chance B, Cohen P, Jobsis F, Schoener B (1962) Intracellular oxidation-reduction states in vivo. Science 137:499–508
Chance B, Mayer D, Legallais V (1971) A dual-wavelength spectrophotometer and fluorometer using interference filters. Anal Biochem 42:494–504
Chance B, Schoener B, Oshino R, Itshak F, Nakase Y (1979) Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J Biol Chem 254:4764–71
Chang DT, Reynolds IJ (2006) Mitochondrial trafficking and morphology in healthy and injured neurons. Prog Neurobiol 80:241–68
Chang JC, Shook LL, Biag J, Nguyen EN, Toga AW, Charles AC, Brennan KC (2010) Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain 133:996–1012
Charles A (2010) Does cortical spreading depression initiate a migraine attack? Maybe not. Headache 50:731–33
Chen Q, Chopp M, Bodzin G, Chen H (1993) Temperature modulation of cerebral depolarization during focal cerebral ischemia in rats: correlation with ischemic injury. J Cereb Blood Flow Metab 13:389–94
Chen G, Gao W, Reinert KC, Popa LS, Hendrix CM, Ross ME, Ebner TJ (2005) Involvement of kv1 potassium channels in spreading acidification and depression in the cerebellar cortex. J Neurophysiol 94:1287–98
Cohen LB, Keynes RD, Hille B (1968) Light scattering and birefringence changes during nerve activity. Nature 218:438–41
Cohen LB, Salzberg BM, Grinvald A (1978) Optical methods for monitoring neuron activity. Annu Rev Neurosci 1:171–82
Combs CA, Balaban RS (2001) Direct imaging of dehydrogenase activity within living cells using enzyme-dependent fluorescence recovery after photobleaching (ED-FRAP). Biophys J 80: 2018–28
Cossart R, Ikegaya Y, Yuste R (2005) Calcium imaging of cortical networks dynamics. Cell Calcium 37:451–57
Cruz NF, Adachi K, Dienel GA (1999) Rapid efflux of lactate from cerebral cortex during K+-induced spreading cortical depression. J Cereb Blood Flow Metab 19:380–92
Csiba L, Paschen W, Mies G (1985) Regional changes in tissue pH and glucose content during cortical spreading depression in rat brain. Brain Res 336:167–70
Dahlem MA, Schneider FM, Scholl E (2008) Failure of feedback as a putative common mechanism of spreading depolarizations in migraine and stroke. Chaos 18:026110
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105
Dietz RM, Weiss JH, Shuttleworth CW (2008) Zn2+ influx is critical for some forms of spreading depression in brain slices. J Neurosci 28:8014–24
Dietz RM, Weiss JH, Shuttleworth CW (2009) Contributions of Ca2+ and Zn2+ to spreading depression-like events and neuronal injury. J Neurochem 109(Suppl 1):145–52
Diez-Garcia J, Matsushita S, Mutoh H, Nakai J, Ohkura M, Yokoyama J, Dimitrov D, Knopfel T (2005) Activation of cerebellar parallel fibers monitored in transgenic mice expressing a fluorescent Ca2+ indicator protein. Eur J Neurosci 22:627–35
Doane MG (1967) Fluorometric measurement of pyridine nucleotide reduction in the giant axon of the squid. J Gen Physiol 50:2603–32
Dohmen C, Sakowitz OW, Fabricius M, Bosche B, Reithmeier T, Ernestus RI, Brinker G, Dreier JP, Woitzik J, Strong AJ, Graf R (2008) Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol 63:720–28
Dreier JP, Korner K, Ebert N, Gorner A, Rubin I, Back T, Lindauer U, Wolf T, Villringer A, Einhaupl KM, Lauritzen M, Dirnagl U (1998) Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J Cereb Blood Flow Metab 18:978–90
Dreier JP, Woitzik J, Fabricius M, Bhatia R, Major S, Drenckhahn C, Lehmann TN, Sarrafzadeh A, Willumsen L, Hartings JA, Sakowitz OW, Seemann JH, Thieme A, Lauritzen M, Strong AJ (2006) Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain 129:3224–37
Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, Tolias C, Oliveira-Ferreira AI, Fabricius M, Hartings JA, Vajkoczy P, Lauritzen M, Dirnagl U, Bohner G, Strong AJ (2009) Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 132:1866–81
Duchen MR (1992) Ca(2+)-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J 283(Pt 1):41–50
Duchen MR, Surin A, Jacobson J (2003) Imaging mitochondrial function in intact cells. Methods Enzymol 361:353–89
Dunn AK, Devor A, Bolay H, Andermann ML, Moskowitz MA, Dale AM, Boas DA (2003) Simultaneous imaging of total cerebral hemoglobin concentration, oxygenation, and blood flow during functional activation. Opt Lett 28:28–30
Dunn AK, Devor A, Dale AM, Boas DA (2005) Spatial extent of oxygen metabolism and hemodynamic changes during functional activation of the rat somatosensory cortex. Neuroimage 27:279–90
Eikermann-Haerter K, Ayata C (2010) Cortical spreading depression and migraine. Curr Neurol Neurosci Rep 10:167–73
Eikermann-Haerter K, Dilekoz E, Kudo C, Savitz SI, Waeber C, Baum MJ, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, Ayata C (2009) Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest 119:99–109
Fabricius M, Fuhr S, Willumsen L, Dreier JP, Bhatia R, Boutelle MG, Hartings JA, Bullock R, Strong AJ, Lauritzen M (2008) Association of seizures with cortical spreading depression and peri-infarct depolarisations in the acutely injured human brain. Clin Neurophysiol 119:1973–84
Farkas E, Bari F, Obrenovitch TP (2010) Multi-modal imaging of anoxic depolarization and hemodynamic changes induced by cardiac arrest in the rat cerebral cortex. Neuroimage 51: 734–42
Frostig RD, Lieke EE, Ts’o DY, Grinvald A (1990) Cortical functional architecture and local coupling between neuronal activity and the microcirculation revealed by in vivo high-resolution optical imaging of intrinsic signals. Proc Natl Acad Sci USA 87:6082–86
Fujita H, Matsuura T, Yamada K, Inagaki N, Kanno I (2000) A sealed cranial window system for simultaneous recording of blood flow, and electrical and optical signals in the rat barrel cortex. J Neurosci Methods 99:71–78
Galeotti T, van Rossum GD, Mayer DH, Chance B (1970) On the fluorescence of NAD(P)H in whole-cell preparations of tumours and normal tissues. Eur J Biochem 17:485–96
Gault LM, Lin CW, LaManna JC, Lust WD (1994) Changes in energy metabolites, cGMP and intracellular pH during cortical spreading depression. Brain Res 641:176–80
Gerich FJ, Hepp S, Probst I, Muller M (2006) Mitochondrial inhibition prior to oxygen-withdrawal facilitates the occurrence of hypoxia-induced spreading depression in rat hippocampal slices. J Neurophysiol 96:492–504
Gill R, Andine P, Hillered L, Persson L, Hagberg H (1992) The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischaemia in the rat. J Cereb Blood Flow Metab 12:371–79
Gorji A (2001) Spreading depression: a review of the clinical relevance. Brain Res Brain Res Rev 38:33–60
Greenwood SM, Mizielinska SM, Frenguelli BG, Harvey J, Connolly CN (2007) Mitochondrial dysfunction and dendritic beading during neuronal toxicity. J Biol Chem 282:26235–44
Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA (2001) Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci USA 98: 4687–92
Hansen AJ (1985) Effect of anoxia on ion distribution in the brain. Physiol Rev 65:101–148
Harbig K, Chance B, Kovach AG, Reivich M (1976) In vivo measurement of pyridine nucleotide fluorescence from cat brain cortex. J Appl Physiol 41:480–88
Hartings JA, Rolli ML, Lu XC, Tortella FC (2003) Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: relation to infarct growth and neuroprotection. J Neurosci 23:11602–10
Hartings JA, Gugliotta M, Gilman C, Strong AJ, Tortella FC, Bullock MR (2008) Repetitive cortical spreading depolarizations in a case of severe brain trauma. Neurol Res 30:876–82
Hartings J, Strong AJ, Fabricius M, Manning A, Bhatia R, Dreier J, Mazzeo AT, Tortella FC, Bullock R (2009) Spreading depolarizations and late secondary insults after traumatic brain injury. J Neurotrauma 26:1857–66
Haselgrove JC, Bashford CL, Barlow CH, Quistorff B, Chance B, Mayevsky A (1990) Time resolved 3-dimensional recording of redox ratio during spreading depression in gerbil brain. Brain Res 506:109–14
Hashemi P, Bhatia R, Nakamura H, Dreier JP, Graf R, Strong AJ, Boutelle MG (2008) Persisting depletion of brain glucose following cortical spreading depression, despite apparent hyperaemia: evidence for risk of an adverse effect of Leao’s spreading depression. J Cereb Blood Flow Metab 29:166–175
Hashimoto M, Takeda Y, Sato T, Kawahara H, Nagano O, Hirakawa M (2000) Dynamic changes of NADH fluorescence images and NADH content during spreading depression in the cerebral cortex of gerbils. Brain Res 872:294–300
Hayakawa Y, Nemoto T, Iino M, Kasai H (2005) Rapid Ca2+-dependent increase in oxygen consumption by mitochondria in single mammalian central neurons. Cell Calcium 37:359–70
Hepp S, Gerich FJ, Muller M (2005) Sulfhydryl oxidation reduces hippocampal susceptibility to hypoxia-induced spreading depression by activating BK channels. J Neurophysiol 94: 1091–103
Higuchi T, Takeda Y, Hashimoto M, Nagano O, Hirakawa M (2002) Dynamic changes in cortical NADH fluorescence and direct current potential in rat focal ischemia: relationship between propagation of recurrent depolarization and growth of the ischemic core. J Cereb Blood Flow Metab 22:71–79
Hillman EM (2007) Optical brain imaging in vivo: techniques and applications from animal to man. J Biomed Opt 12:051402
Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hubener M, Keck T, Knott G, Lee WC, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L (2009) Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc 4:1128–44
Hoskison MM, Shuttleworth CW (2006) Microtubule disruption, not calpain-dependent loss of MAP2, contributes to enduring NMDA-induced dendritic dysfunction in acute hippocampal slices. Exp Neurol 202:302–12
Hossmann KA (1996) Periinfarct depolarizations. Cerebrovasc Brain Metab Rev 8:195–208
Huang S, Heikal AA, Webb WW (2002) Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys J 82:2811–25
Iijima T, Mies G, Hossmann KA (1992) Repeated negative DC deflections in rat cortex following middle cerebral artery occlusion are abolished by MK-801: effect on volume of ischemic injury. J Cereb Blood Flow Metab 12:727–33
Jarvis CR, Lilge L, Vipond GJ, Andrew RD (1999) Interpretation of intrinsic optical signals and calcein fluorescence during acute excitotoxic insult in the hippocampal slice. Neuroimage 10:357–72
Jobsis FF, O’Connor M, Vitale A, Vreman H (1971) Intracellular redox changes in functioning cerebral cortex. I. Metabolic effects of epileptiform activity. J Neurophysiol 34:735–49
Jung JC, Mehta AD, Aksay E, Stepnoski R, Schnitzer MJ (2004) In vivo mammalian brain imaging using one- and two-photon fluorescence microendoscopy. J Neurophysiol 92:3121–33
Kahraman S, Fiskum G (2007) Anoxia-induced changes in pyridine nucleotide redox state in cortical neurons and astrocytes. Neurochem Res 32:799–806
Kann O, Kovacs R (2007) Mitochondria and neuronal activity. Am J Physiol Cell Physiol 292: C641–C657
Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW (2004) Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 305:99–103
Kocher M (1990) Metabolic and hemodynamic activation of postischemic rat brain by cortical spreading depression. J Cereb Blood Flow Metab 10:564–71
Kofuji P, Newman EA (2004) Potassium buffering in the central nervous system. Neuroscience 129:1045–56
Kosterin P, Kim GH, Muschol M, Obaid AL, Salzberg BM (2005) Changes in FAD and NADH fluorescence in neurosecretory terminals are triggered by calcium entry and by ADP production. J Membr Biol 208:113–24
Krivanek J (1958) Changes of brain glycogen in the spreading EEG-depression of Leao. J Neurochem 2:337–43
Krivanek J (1961) Some metabolic changes accompanying Leao’s spreading cortical depression in the rat. J Neurochem 6:183–89
Kunkler PE, Kraig RP (2004) P/Q Ca2+ channel blockade stops spreading depression and related pyramidal neuronal Ca2+ rise in hippocampal organ culture. Hippocampus 14:356–67
Largo C, Ibarz JM, Herreras O (1997) Effects of the gliotoxin fluorocitrate on spreading depression and glial membrane potential in rat brain in situ. J Neurophysiol 78:295–307
Lauritzen M (1994) Pathophysiology of the migraine aura. The spreading depression theory. Brain 117(Pt 1):199–210
Lauritzen M, Diemer NH (1986) Uncoupling of cerebral blood flow and metabolism after single episode of cortical spreading depression in the rat brain. Brain Res 370:405–08
Lauritzen M, Jorgensen MB, Diemer NH, Gjedde A, Hansen AJ (1982) Persistent oligemia of rat cerebral cortex in the wake of spreading depression. Ann Neurol 12:469–74
Lauritzen M, Hansen AJ, Kronborg D, Wieloch T (1990) Cortical spreading depression is associated with arachidonic acid accumulation and preservation of energy charge. J Cereb Blood Flow Metab 10:115–22
Leao AAP (1944a) Spreading depression of activity in the cerebral cortex. J Neurophysiol 7:359–90
Leao AAP (1944b) Pial circulation and spreading depression of activity in the cerebral cortex. J Neurophysiol 7:359–90
Leao AAP (1947) Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol 10:409–14
Leao AAP, Morison RS (1945) Propagation of spreading cortical depression. J Neurophysiol 8:33–45
Levene MJ, Dombeck DA, Kasischke KA, Molloy RP, Webb WW (2004) In vivo multiphoton microscopy of deep brain tissue. J Neurophysiol 91:1908–12
L’Heureux B, Gurden H, Pain F (2009) Autofluorescence imaging of NADH and flavoproteins in the rat brain: insights from Monte Carlo simulations. Opt Express 17:9477–90
Lian XY, Stringer JL (2004) Energy failure in astrocytes increases the vulnerability of neurons to spreading depression. Eur J Neurosci 19:2446–54
Lipton P (1973a) Effects of membrane depolarization on light scattering by cerebral cortical slices. J Physiol 231:365–83
Lipton P (1973b) Effects of membrane depolarization on nicotinamide nucleotide fluorescence in brain slices. Biochem J 136:999–1009
Lothman E, Lamanna J, Cordingley G, Rosenthal M, Somjen G (1975) Responses of electrical potential, potassium levels, and oxidative metabolic activity of the cerebral neocortex of cats. Brain Res 88:15–36
Marshall WH (1959) Spreading cortical depression of Leao. Physiol Rev 39:239–79
Martins-Ferreira H, de Castro GO (1966) Light-scattering changes accompanying spreading depression in isolated retina. J Neurophysiol 29:715–26
Martins-Ferreira H, Nedergaard M, Nicholson C (2000) Perspectives on spreading depression. Brain Res Brain Res Rev 32:215–34
Mayevsky A, Chance B (1974) Repetitive patterns of metabolic changes during cortical spreading depression of the awake rat. Brain Res 65:529–33
Mayevsky A, Rogatsky GG (2007) Mitochondrial function in vivo evaluated by NADH fluorescence: from animal models to human studies. Am J Physiol Cell Physiol 292:C615–C640
Mayevsky A, Zeuthen T, Chance B (1974) Measurements of extracellular potassium, ECoG and pyridine nucleotide levels during cortical spreading depression in rats. Brain Res 76:347–49
Mies G, Paschen W (1984) Regional changes of blood flow, glucose, and ATP content determined on brain sections during a single passage of spreading depression in rat brain cortex. Exp Neurol 84:249–58
Mies G, Iijima T, Hossmann KA (1993) Correlation between peri-infarct DC shifts and ischaemic neuronal damage in rat. Neuroreport 4:709–11
Mies G, Kohno K, Hossmann KA (1994) Prevention of periinfarct direct current shifts with glutamate antagonist NBQX following occlusion of the middle cerebral artery in the rat. J Cereb Blood Flow Metab 14:802–07
Mironov SL (2006) Spontaneous and evoked neuronal activities regulate movements of single neuronal mitochondria. Synapse 59:403–11
Mironov SL, Richter DW (2001) Oscillations and hypoxic changes of mitochondrial variables in neurons of the brainstem respiratory centre of mice. J Physiol 533:227–36
Murphy TH, Li P, Betts K, Liu R (2008) Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J Neurosci 28:1756–72
Nedergaard M, Astrup J (1986) Infarct rim: effect of hyperglycemia on direct current potential and [14C]2-deoxyglucose phosphorylation. J Cereb Blood Flow Metab 6:607–15
Nedergaard M, Hansen AJ (1988) Spreading depression is not associated with neuronal injury in the normal brain. Brain Res 449:395–98
Nemoto M, Nomura Y, Sato C, Tamura M, Houkin K, Koyanagi I, Abe H (1999) Analysis of optical signals evoked by peripheral nerve stimulation in rat somatosensory cortex: dynamic changes in hemoglobin concentration and oxygenation. J Cereb Blood Flow Metab 19:246–59
Nicholls DG, Ferguson SJ (2002) Bioenergetics. Elsevier, London
Obeidat AS, Andrew RD (1998) Spreading depression determines acute cellular damage in the hippocampal slice during oxygen/glucose deprivation. Eur J Neurosci 10:3451–61
Obrenovitch TP, Chen S, Farkas E (2009) Simultaneous, live imaging of cortical spreading depression and associated cerebral blood flow changes, by combining voltage-sensitive dye and laser speckle contrast methods. Neuroimage 45:68–74
Olesen J, Larsen B, Lauritzen M (1981) Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann Neurol 9:344–52
Parthasarathy AB, Tom WJ, Gopal A, Zhang X, Dunn AK (2008) Robust flow measurement with multi-exposure speckle imaging. Opt Express 16:1975–1989
Petzold GC, Haack S, von Bohlen Und Halbach O, Priller J, Lehmann TN, Heinemann U, Dirnagl U, Dreier JP (2008) Nitric oxide modulates spreading depolarization threshold in the human and rodent cortex. Stroke 39:1292–99
Pietrobon D, Striessnig J (2003) Neurobiology of migraine. Nat Rev Neurosci 4:386–98
Piper RD, Lambert GA, Duckworth JW (1991) Cortical blood flow changes during spreading depression in cats. Am J Physiol 261:H96–H102
Prakash N, Uhlemann F, Sheth SA, Bookheimer S, Martin N, Toga AW (2009) Current trends in intraoperative optical imaging for functional brain mapping and delineation of lesions of language cortex. Neuroimage 47(Suppl 2):T116–T126
Rawanduzy A, Hansen A, Hansen TW, Nedergaard M (1997) Effective reduction of infarct volume by gap junction blockade in a rodent model of stroke. J Neurosurg 87:916–20
Reinert KC, Gao W, Chen G, Ebner TJ (2007) Flavoprotein autofluorescence imaging in the cerebellar cortex in vivo. J Neurosci Res 85:3221–32
Rex A, Pfeifer L, Fink F, Fink H (1999) Cortical NADH during pharmacological manipulations of the respiratory chain and spreading depression in vivo. J Neurosci Res 57:359–70
Risher WC, Andrew RD, Kirov SA (2009) Real-time passive volume responses of astrocytes to acute osmotic and ischemic stress in cortical slices and in vivo revealed by two-photon microscopy. Glia 57:207–21
Rocheleau JV, Head WS, Piston DW (2004) Quantitative NAD(P)H/flavoprotein autofluorescence imaging reveals metabolic mechanisms of pancreatic islet pyruvate response. J Biol Chem 279:31780–87
Rosenthal M, Jobsis FF (1971) Intracellular redox changes in functioning cerebral cortex. II. Effects of direct cortical stimulation. J Neurophysiol 34:750–62
Rosenthal M, Somjen G (1973) Spreading depression, sustained potential shifts, and metabolic activity of cerebral cortex of cats. J Neurophysiol 36:739–49
Rowland KC, Irby NK, Spirou GA (2000) Specialized synapse-associated structures within the calyx of Held. J Neurosci 20:9135–44
Sakowitz OW, Kiening KL, Krajewski KL, Sarrafzadeh AS, Fabricius M, Strong AJ, Unterberg AW, Dreier JP (2009) Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke 40:e519–e522
Sasaki T, Takeda Y, Taninishi H, Arai M, Shiraishi K, Morita K (2009) Dynamic changes in cortical NADH fluorescence in rat focal ischemia: evaluation of the effects of hypothermia on propagation of peri-infarct depolarization by temporal and spatial analysis. Neurosci Lett 449:61–5
Scheller D, Kolb J, Tegtmeier F (1992) Lactate and pH change in close correlation in the extracellular space of the rat brain during cortical spreading depression. Neurosci Lett 135:83–86
Schuchmann S, Kovacs R, Kann O, Heinemann U, Buchheim K (2001) Monitoring NAD(P)H autofluorescence to assess mitochondrial metabolic functions in rat hippocampal-entorhinal cortex slices. Brain Res Brain Res Protoc 7:267–6
Selman WR, Lust WD, Pundik S, Zhou Y, Ratcheson RA (2004) Compromised metabolic recovery following spontaneous spreading depression in the penumbra. Brain Res 999:167–74
Shibuki K, Hishida R, Kitaura H, Takahashi K, Tohmi M (2007) Coupling of brain function and metabolism: Endogenous flavoprotein fluorescence imaging of neural activities by local changes in energy metabolism. In: Handbook of Neurochemistry & Molecular Neurobiology. 3rd Edition, Vol. 5, Neural Energy Utilization (Gibson G and Dienel G, ed). Springer, New York, pp. 322–42
Shinohara M, Dollinger B, Brown G, Rapoport S, Sokoloff L (1979) Cerebral glucose utilization: local changes during and after recovery from spreading cortical depression. Science 203:188–90
Shuttleworth CW (2010) Use of NAD(P)H and flavoprotein autofluorescence transients to probe neuron and astrocyte responses to synaptic activation. Neurochem Int 56:379–86
Shuttleworth CW, Connor JA (2001) Strain-dependent differences in calcium signaling predict excitotoxicity in murine hippocampal neurons. J Neurosci 21:4225–36
Shuttleworth CW, Brennan AM, Connor JA (2003) NAD(P)H fluorescence imaging of postsynaptic neuronal activation in murine hippocampal slices. J Neurosci 23:3196–08
Somjen GG (2001) Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 81:1065–96
Somjen GG (2005) Aristides Leao’s discovery of cortical spreading depression. J Neurophysiol 94:2–4
Sonn J, Mayevsky A (2000) Effects of brain oxygenation on metabolic, hemodynamic, ionic and electrical responses to spreading depression in the rat. Brain Res 882:212–16
Strong AJ, Venables GS, Gibson G (1983a) The cortical ischaemic penumbra associated with occlusion of the middle cerebral artery in the cat: 1. Topography of changes in blood flow, potassium ion activity, and EEG. J Cereb Blood Flow Metab 3:86–96
Strong AJ, Tomlinson BE, Venables GS, Gibson G, Hardy JA (1983b) The cortical ischaemic penumbra associated with occlusion of the middle cerebral artery in the cat: 2. Studies of histopathology, water content, and in vitro neurotransmitter uptake. J Cereb Blood Flow Metab 3:97–108
Strong AJ, Gibson G, Miller SA, Venables GS (1988) Changes in vascular and metabolic reactivity as indices of ischaemia in the penumbra. J Cereb Blood Flow Metab 8:79–88
Strong AJ, Harland SP, Meldrum BS, Whittington DJ (1996) The use of in vivo fluorescence image sequences to indicate the occurrence and propagation of transient focal depolarizations in cerebral ischemia. J Cereb Blood Flow Metab 16:367–77
Strong AJ, Smith SE, Whittington DJ, Meldrum BS, Parsons AA, Krupinski J, Hunter AJ, Patel S, Robertson C (2000) Factors influencing the frequency of fluorescence transients as markers of peri-infarct depolarizations in focal cerebral ischemia. Stroke 31:214–22
Strong AJ, Bezzina EL, Anderson PJ, Boutelle MG, Hopwood SE, Dunn AK (2006) Evaluation of laser speckle flowmetry for imaging cortical perfusion in experimental stroke studies: quantitation of perfusion and detection of peri-infarct depolarisations. J Cereb Blood Flow Metab 26:645–53
Swann JW, Al-Noori S, Jiang M, Lee CL (2000) Spine loss and other dendritic abnormalities in epilepsy. Hippocampus 10:617–25
Takano K, Latour LL, Formato JE, Carano RA, Helmer KG, Hasegawa Y, Sotak CH, Fisher M (1996) The role of spreading depression in focal ischemia evaluated by diffusion mapping. Ann Neurol 39:308–18
Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M (2007) Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci 10:754–62
Talbot J, Barrett JN, Barrett EF, David G (2007) Stimulation-induced changes in NADH fluorescence and mitochondrial membrane potential in lizard motor nerve terminals. J Physiol 579:783–98
Tang Y, Zucker RS (1997) Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron 18:483–91
Tasaki I, Watanabe A, Sandlin R, Carnay L (1968) Changes in fluorescence, turbidity, and birefringence associated with nerve excitation. Proc Natl Acad Sci USA 61:883–88
Tatlisumak T, Takano K, Meiler MR, Fisher M (2000) A glycine site antagonist ZD9379 reduces number of spreading depressions and infarct size in rats with permanent middle cerebral artery occlusion. Acta Neurochir Suppl 76:331–33
Teive HA, Kowacs PA, Maranhao Filho P, Piovesan EJ, Werneck LC (2005) Leao’s cortical spreading depression: from experimental “artifact” to physiological principle. Neurology 65:1455–59
Terzuolo CA, Chance B, Handelman E, Rossini L, Schmelzer P (1966) Measurements of reduced pyridine nucleotides in a single neuron. Biochim Biophys Acta 126:361–72
Tohmi M, Takahashi K, Kubota Y, Hishida R, Shibuki K (2009) Transcranial flavoprotein fluorescence imaging of mouse cortical activity and plasticity. J Neurochem 109(Suppl 1):3–9
Turner DA, Foster KA, Galeffi F, Somjen GG (2007) Differences in O2 availability resolve the apparent discrepancies in metabolic intrinsic optical signals in vivo and in vitro. Trends Neurosci 30:390–98
van den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T, van de Ven RC, Tottene A, van der Kaa J, Plomp JJ, Frants RR, Ferrari MD (2004) A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41:701–10
Van Harreveld A (1957) Changes in volume of cortical neuronal elements during asphyxiation. Am J Physiol 191:233–42
Van Harreveld A (1958) Changes in the diameter of apical dendrites during spreading depression. Am J Physiol 192:457–63
Van Harreveld A, Stamm JS (1952) Vascular concomitants of spreading cortical depression. J Neurophysiol 15:487–96
Vanzetta I, Hildesheim R, Grinvald A (2005) Compartment-resolved imaging of activity-dependent dynamics of cortical blood volume and oximetry. J Neurosci 25:2233–44
Wahl M, Lauritzen M, Schilling L (1987) Change of cerebrovascular reactivity after cortical spreading depression in cats and rats. Brain Res 411:72–80
Wininger FA, Schei JL, Rector DM (2009) Complete optical neurophysiology: toward optical stimulation and recording of neural tissue. Appl Opt 48:D218–D224
Woods RP, Iacoboni M, Mazziotta JC (1994) Brief report: bilateral spreading cerebral hypoperfusion during spontaneous migraine headache. N Engl J Med 331:1689–92
Yang G, Pan F, Parkhurst CN, Grutzendler J, Gan WB (2010) Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat Protoc 5:201–08
Zhou N, Gordon GR, Feighan D, Macvicar BA (2010) Transient swelling, acidification, and mitochondrial depolarization occurs in neurons but not astrocytes during spreading depression. Cereb Cortex 20:2614–24
Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW (2003) Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc Natl Acad Sci USA 100:7075–80
Zucker RS (1999) Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol 9:305–13
Acknowledgement
Supported by NIH grant NS51288
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Shuttleworth, C.W. (2012). Optical Approaches to Study Metabolic Consequences of Spreading Depression In Vivo . In: Choi, IY., Gruetter, R. (eds) Neural Metabolism In Vivo. Advances in Neurobiology, vol 4. Springer, Boston, MA. https://doi.org/10.1007/978-1-4614-1788-0_12
Download citation
DOI: https://doi.org/10.1007/978-1-4614-1788-0_12
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4614-1787-3
Online ISBN: 978-1-4614-1788-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)