
Abstract
Bleeding complications of surgery are amongst the greatest of a surgeon’s concerns threatening not only the patient but also the success of the surgical procedure. This chapter focuses on disorders and processes that enhance the risk of bleeding in the setting of surgery. A practical method for the assessment of such risk prior to surgery will be developed emphasizing an approach utilizing information derived from the patients’ history, deemphasizing a reliance on the laboratory.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
- Systemic Lupus Erythematosus
- Fresh Freeze Plasma
- Idiopathic Thrombocytopenic Purpura
- Lupus Anticoagulant
- Primary Hemostasis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
FormalPara Objectives-
To appreciate the importance of coagulation problems in the perioperative setting.
-
To develop an approach to the assessment of bleeding risk that emphasizes information obtained for the medical history, deemphasizing the role of screening laboratory assessment.
-
To understand the physiological processes underlying hemostasis.
-
To develop an orderly method for the detection of coagulation disorders preoperatively.
-
To review the hematological basis for the disorders of primary and secondary hemostasis.
-
Bleeding problems are amongst the greatest fears of the surgeon.
-
Hemostasis as a physiological process is well understood.
-
Disorders of hemostasis can be the result of primary (platelet related) dysfunction or secondary (clotting factor related).
-
Patients at increased risk can be identified based on approaches that rely on the patient’s history, deemphasizing the reliance on the laboratory.
Introduction
Hematological problems are not uncommon in the perioperative setting. Indeed the bleeding complications of surgery are amongst the greatest of a surgeon’s concerns threatening not only the patient but also the success of the surgical procedure. Conversely, thrombosis is a particular fear to the orthopedic surgeon. As a problem commanding a prodigious literature, the thromboembolic complications of orthopedic surgery are reviewed separately in Chap. 19. This chapter will focus on disorders and processes that enhance the risk of bleeding. A practical method for their detection prior to surgery will be developed emphasizing an approach utilizing information derived from the patients’ history, deemphasizing a reliance on the laboratory. As such the content of this chapter complements the aforementioned discussion concerning thrombosis but does not represent a comprehensive discourse of the full range of hematological issues that may be seen in the surgical setting.
The Preoperative Evaluation
The methodologies currently employed for the preoperative identification of patients at risk for bleeding in the setting of surgery have followed two distinct strategies [1]. The traditional approach has been to rely on the laboratory, utilizing routine testing such as the platelet count (primary hemostasis) and the prothrombin (PT) and activated thromboplastin times (aPTT) for the secondary disorders of hemostasis. Despite the persistence of this practice, its shortcomings in the prediction of postoperative bleeding are well recognized and predictable based on the complex physiology of bleeding, a multifactorial process not well assessed in the laboratory. Indeed these laboratory studies were not developed to determine the risk of postoperative bleeding but rather for the detection of clotting factor deficiencies.
When a blood vessel is injured the vascular, platelet, coagulation, and fibrinolytic systems react in concert to mitigate the loss of blood; simultaneously thrombus forms, localizing to the site of injury. Bleeding may arise from a disruption in any of these hemostatic mechanisms, operating alone or in combination. The physiology is multifaceted and how such physiological processes play out in the clinical setting is not well assessed by the commonly employed in vivo assessments [2]. Indeed a recent extensive review of the literature, from which practical guidelines have been derived, recommended the discontinuation of such indiscriminate testing favoring a more selective approach in which patients are selected for further evaluation based on their bleeding history. Nonetheless, despite these challenges, the laboratory approach to the assessment of bleeding risk remains a remarkably enduring practice.
The second, more recent method for the risk assessment of bleeding involves the identification of patients at risk based on their medical history and physical examination with targeted laboratory testing performed only on those with specific risk factors. Supported by the observations of Girolami et al. stressing the role of the clinical examination [3], Koscielny et al. have enhanced and systematized this view with the development of a bleeding risk questionnaire (Table 20.1) used to identify patients who should undergo further laboratory testing of their coagulation [4]. The questionnaire, developed retrospectively, has been validated in a large prospective study [5]. Using this methodology, 88 % (5,021/5,649) possessed no risk factors for bleeding; contemporaneous laboratory studies revealed a prolonged aPTT in 9 of these patients, all the result of a lupus anticoagulant (which increases the risk of thrombosis, not hemorrhage). No other laboratory test (PT, platelet count, platelet function study, and von Willebrand factor assay) uncovered any bleeding disorder. With respect to the questionnaire the most reliable (sensitive) questions related to bleeding of minor wounds (85 %), frequent bruising (73 %), and use of nonsteroidal anti-inflammatory agents (62 %); if any four of the questions were answered in the affirmative, the positive predictive value for the presence of a bleeding diathesis was 99%.
Further in distinguishing between acquired and hereditary disorders, the clinical history is helpful [1, 3]. Table 20.2 summarizes these considerations emphasizing the presence or absence of a family history of bleeding, age of onset of bleeding, the pattern of bleeding, comorbidities, and a history of transfusion as historically important. Physical findings also provide important clues to diagnosis. Table 20.3 attempts to make this point. Employing a number of bleeding presentations, largely based on the site involved, various etiologies are implied though as Girolami describes there is considerable overlap [3]; further a distinct differential diagnosis could be structured around the organ system involved in the bleeding. Nonetheless while imprecision in terminology and semantics may somewhat cloud the discussion, certain bleeding patterns are generally appreciated by clinicians and are of some importance in diagnosis.
Whether identified by a history and physical examination, or via screening laboratory testing, once concerns have been raised about a patient’s hemostatic capacity a work-up is indicated. Hemostasis is conceptualized as primary (platelet related) or secondary (coagulation factors), and its evaluation can be developed according to these distinctions.
Hemostasis
When a blood vessel is damaged, the process of hemostasis is activated in order to arrest bleeding. The process has three phases:
-
1.
Vascular phase: involves a transient, localized vasoconstrictor response in the damaged blood vessel thus stopping the flow of blood.
-
2.
Platelet phase: damaged endothelial cells release von Willebrand’s factor resulting in “sticky” endothelial cells, a process known as platelet adhesion. Platelets that adhere to the blood vessel wall in this manner secrete adenosine diphosphate (ADP), a chemical that causes nearby free platelets to attach to each other and to those already fixed to the vessel wall, thereby forming a platelet plug. This “clumping” phenomenon served a number of important functions: the plug may seal the defect in the vessel wall; aggregated platelets release Platelet Thromboplastin (Factor III) which activates the clotting process; further the clumped platelets secrete thromboxane, a potent vasoconstrictor.
-
3.
Coagulation Phase (Fig. 20.1): this phase, which begins within minutes of the initiation of the vascular and platelet phases, involves the formation of insoluble protein Fibrin (from Fibrinogen via the action of the enzyme Thrombin). Once formed Fibrin produces a network of fibers that traps blood cells and platelets thereby forming the clot. This process depends on the presence of 11 different clotting factors and calcium (Factor V), factors required to generate the production of Prothrombin Activator (Factor X). Two distinct pathways with different triggers may be activated.
Fig. 20.1 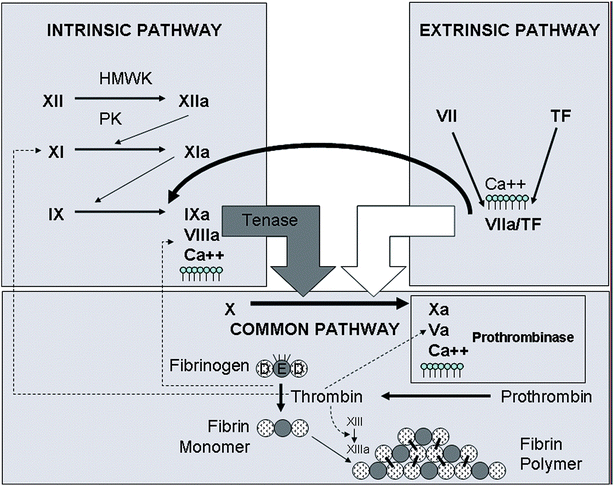
Diagram of the coagulation cascade, depicting the intrinsic and extrinsic pathways of activation. The extrinsic pathway of activation is started with exposure of tissue factor (TF), coupled with factor VIIa that leads to the activation of factor X. The intrinsic pathway is started by the contact activation factors (factor XII, high molecular weight kininogen (HMWK), and prekallikrein (PK)) with eventual activation of factor X by the tenase complex (factors IXa, VIIIa, calcium (Ca2+), and phospholipid). Activated factor X (Xa) participates in the prothrombinase complex (factor Xa, Va, Ca2+, phospholipids) for the conversion of prothrombin to thrombin, which converts fibrinogen to fibrin monomer. Fibrin then polymerizes and is cross-linked by factor XIIIa. Further activation of coagulation is fostered by thrombin’s activation of factors V, VIII, and XI (Used with permission from Kottke-Marchant K. The Role of Coagulation in Arterial and Venous Thrombosis. In Askari AT, Lincoff AM (eds): Antithrombotic Drug Therapy in Cardiovascular Disease. New York: Springer Science; 2010)
The Extrinsic pathway is initiated by the material tissue Thromboplastin (Factor III). Released by the damaged tissue and thus “outside” (extrinsic) the blood, this process is rapid and provides a shortcut to the clotting process. The resultant clot is small and thus considered a “quick patch” phenomenon. Alternatively there is the second Intrinsic pathway which is initiated when the blood itself comes in contact with the exposed collagen of the damaged blood vessel. Although a slower (5–10 min) process, it results in the formation of much larger amounts of thrombin and thus more robust clots. This process involves the sequential activation of multiple clotting factors: Factor XI, activated by contact with the exposed endothelium, activates Factor XI, which in concert with Factor XI activates Factor IX leading to the production of Factor VIII. It is Factor VIII, coupled with calcium and Factor II (derived from platelets), that ultimately activates Factor X (Prothrombin Activator). This is the point in the hemostatic process where the two pathways converge following the same course (Common Pathway) to Fibrin formation: a composite of clotting factors (Factor V, Ca2+, and platelet-derived phospholipids) engage Factor X creating the Factor V Complex which initiates the conversion of Prothrombin to the active enzyme Thrombin. Thrombin completes the cascade by accelerating the formation of Fibrin thread from Fibrinogen (Factor I).
-
4.
Clot Retraction: this process occurs several days later mediated by contractile proteins contained in the platelets pulling the edges of the wound together and assisting in the reparative process.
-
5.
Fibrinolysis: this refers to the dissolution of the clot a process driven by the proteolytic enzyme Plasmin.

Evaluation and Approach of Primary Hemostasis: Platelet Deficiency
The assessment of primary hemostasis focuses on the platelet. While the platelet count is a dependable test it does not provide information concerning platelet function and thus is not a sufficient assessment of the bleeding risk attributable to platelet-mediated problems. The normal platelet count is in the 140–160/mm3 though bleeding problems do not occur until substantial reductions (i.e., <50,000/mm3) are present although there is one important caveat. Anesthesiologists require higher counts (i.e., >70,000/mm3) in order to employ neuraxial blockage, an important consideration in orthopedic surgery where such techniques are often employed. Indeed the American Society of Regional Anesthesia (ASRA) has published recommendations (Table 20.4) concerning such blocks the intent of which is a reduction in the risk of paraspinal hematomas [6]. Nonetheless platelet counts of <100K are likely to represent an underlying platelet disorder and warrant investigation. The underlying mechanisms of thrombocytopenia can be divided into three broad categories: impaired production, peripheral consumption, and redistribution/dilution (Table 20.5). Further there is also pseudo-thrombocytopenia, a relatively common laboratory phenomenon occurring in approximately 1.9 % of hospitalized patients [7, 8], resulting from the ethylenediamine tetra-acetic acid or EDTA, a chelating agent used in blood collection tubes for CBC determinations. The in vitro agglutination of platelets produced by the EDTA results in low platelet counts but no bleeding tendency. Last congenital thrombocytopenia is a rare condition potentially treated with a number of agents including desmopressin (DDAVP), antifibrinolytics, platelet transfusions, and recombinant factor VIIIa.
More commonly asymptomatic thrombocytopenia is caused by the presence of antibodies to circulating platelets. Most often arising as a consequence of autoimmune platelet destruction, a primary form of the disease known as Idiopathic Thrombocytopenic Purpura (ITP) has been long known. In contrast, secondary ITP may arise as autoimmune sequelae of infection (HIV), systemic lupus erythematosus (SLE), antiphospholipid syndrome, or B-cell malignancies [3]. Heparin-induced thrombocytopenia (HIT), another important platelet-related disorder, arises as a consequence of prolonged heparin administration (≥5 days). Although not often seen after orthopedic surgery, HIT is a serious problem resulting in severe thrombocytopenia coupled with a prothrombotic state (arterial and venous thrombosis, pulmonary embolism, cerebral sinus thrombosis). It is a very serious condition that should be suspected in patients who experience a drop of >50 % in their platelet count in the setting of heparin therapy, usually 5–10 days after the initiation of such therapy [9].
Due to their widespread use in the primary and secondary prevention of cardiac and cerebrovascular, the most common cause of platelet dysfunction in the perioperative setting is the use of antiplatelet agents; aspirin and thienopyridine agents (primarily clopidogrel) are amongst the most prevalent. The former, ASA, irreversibly acetylates platelet cyclooxygenase-1 (COX-1) inhibiting thromboxane A2 thus inhibiting platelet function (for the life of the platelet, 5–7 days), while the more potent clopidogrel inhibits (also permanently) platelet aggregation; the recovery time for its effect is approximately 7 days. The impact of ASA on bleeding risk in association with surgery appears mild and outweighed by its influence on graft patency in patients with known coronary artery and cerebrovascular disease. Further, the continuation of ASA in patients undergoing neuraxial block is not associated with bleeding risk [10]. The perioperative management of these agents is fully discussed in Chap. 11. Algorithms for the perioperative management of patients receiving antiplatelet agents have been published. One useful decision tree is shown in Fig. 20.2.

Algorithm for patients receiving antiplatelet agents. ASC acute coronary syndrome, ASA acetyl-salicylic acid, IC intracranial, MI myocardial infarction, PAD peripheral arterial disease, PCI percutaneous coronary intervention (Used with permission from Marcucci C, Chassot PG, Asmis LM et al. Hematologic Risk Assessment. In: Perioperative Medicine: Managing for Outcome. Eds: Newman MF, Fleisher LA, Fink MP. Philadelphia: Saunders Elsevier, 2008)
Last another important disorder of the platelet, occasionally encountered in the perioperative setting, is von Willebrand’s disease (vWD). The most common of the inherited bleeding disorders, its prevalence is approximately 1 % in the general population [11]. The hallmark of this condition is a deficiency of von Willebrand factor; while not an intrinsic platelet defect, this deficiency reduces platelet adhesion and aggregation, thereby producing a bleeding tendency. Given the usual absence of laboratory markers, vWD is diagnosed from the history of abnormal bleeding in the setting of surgical and dental procedures. Several subtypes exist, an important consideration as therapy varies according to the form of the disease. An array of agents is employed perioperatively and includes desmopressin (DDAVP), cryoprecipitate, or purified plasma factor concentrates under the guidance of a hematologist.
Evaluation and Approach to Secondary Hemostasis: Clotting Factor Deficiency
Amongst the various tests of secondary hemostasis, the prothrombin (PT) and partial thromboplastin times (aPTT) are the most commonly employed preoperatively in spite of a prodigious literature demonstrating their poor predictive value for the development of postoperative bleeding [3]. Indeed as implied by the Koscielny algorithm, an unsuspected bleeding diathesis is very unlikely to be uncovered by such testing preoperatively in patients with a negative bleeding history [4, 5]. Indeed in this extensive study, none of the commonly employed screening studies (platelet count, PT, aPTT) identified a single patient without a suspicion of a preexisting bleeding problem based on their history. Further, with respect to the aPTT specifically, the prolongation of this parameter is a fairly common circumstance resulting from either a mild Factor XII deficiency or the presence of a lupus anticoagulant; indeed in the Koscielny study, all patients with this laboratory abnormality (and negative bleeding history) were found to have of a lupus inhibitor. The lupus anticoagulant, a phenomenon known to increase the risk of thrombosis not the risk of bleeding, is itself relatively common being found in 1.2–3.8 % of healthy individuals, though its incidence increases with age and chronic disease, most significantly in patients with systemic lupus erythematosus [12].
Of course, specific clotting factor deficiencies do exist and in those rare patients with severe deficiencies their risk of surgery-related bleeding may be significantly increased. Deficiencies of factors II, V, VII, XI, and XII are well known to hematologists. Clinical clues to the presence of such disorders include that Factor VII deficiency is the only hereditary clotting factor deficiency with a prolonged PT and normal aPTT; Factor XI deficiency occurs in its highest frequency among Ashkenazi Jews (its incidence outside that population is about 1 per 1 million) and is a condition that may be unrecognized until excessive bleeding occurs in the setting of surgery; and Factor XII (Hageman) deficiency results in an elevated aPTT but not a bleeding diathesis. Rather such patients experience thromboembolic phenomenon.
Although generally not an occult disease process, the hemophilias should be mentioned as important hereditary disorders of coagulation. There are two forms: Hemophilia A (Factor VIII deficiency) and Hemophilia B (Factor IX deficiency). Owing to their propensity for intra-articular hemorrhage and ultimately joint destruction, these have been important conditions to the orthopedist. Nonetheless modern clotting factor replacement therapy has significantly mitigated the chronic joint destruction formerly experienced by these patients. Treatment paradigms for the chronic as well as perioperative management of these conditions are well established.
Specific Chronic Diseases
Perhaps most relevant to a discussion of coagulation in the perioperative setting is the contribution made by kidney and liver disease.
In patients with chronic renal failure the most common clotting related abnormality is platelet dysfunction, a problem unrelated to the platelet itself but rather a consequence of the uremic state. Thus, in the patient with renal insufficiency accompanied by low platelet counts, platelet transfusions are generally not indicated though dialysis may be appropriate in certain settings.
In contrast, patients with chronic hepatic failure are particularly worrisome in the perioperative setting where clotting factor deficiency and portal venous insufficiency contribute to an increased risk of bleeding. Deficiencies of the liver-dependent factors (II, VII, IX, X) result in a coagulopathy identified by a prolonged prothrombin time (PT). In such individuals, particularly those who are poorly nourished, vitamin K may be indicated preoperatively. However, in this clinical setting the primary problem may be insufficient hepatic synthesis, a state likely to be unresponsive to vitamin K and require repletion of clotting factors with fresh frozen plasma (FFP) or specific factor concentrates. Large dosages of FFP are often required; near normalization of the PT is the desired outcome.
Summary
In conclusion, problems of coagulation are important considerations to the orthopedic surgeon, anesthesiologist, and particularly to the medical consultant who serves as the first line of defense in the recognition of such conditions. Suspicions concerning their presence can generally be gleaned from the patient’s history though a general understanding of the processes underlying normal hemostasis is also necessary and forms the basis upon which a logical approach to the detection, characterization, and treatment of these conditions is formulated.
Summary Bullet Points
-
Coagulation problems are amongst the surgeons’ most feared complication of surgery.
-
The presence of disordered coagulation can be determined with a targeted medical history.
-
Available preoperative laboratory assessments of coagulation do not predict postoperative bleeding complications and should be deemphasized in practice.
-
Disordered coagulation can be divided into primary and secondary forms.
-
Diseases of the kidney and liver contribute significantly to the risk of postoperative bleeding.
References
Marcucci C, Chassot PG, Asmis LM, et al. Hematologic risk assessment. In: Newman MF, Fleisher LA, Fink MP, editors. Perioperative medicine: managing for outcome. Philadelphia, PA: Saunders Elsevier; 2008.
Chee YL, Crawford JC, Watson HG, et al. Guidelines on the assessment of bleeding risk prior to surgery or invasive procedures: British Committee for Standards in Hematology. Br J Hematol. 2008;140:496–504.
Girolami A, Luzzatto G, Varvarikis C, et al. Main clinical manifestations of a bleeding diathesis: an often disregarded aspect of medical and surgical history taking. Haemophilia. 2005;11:193–202.
Koscielny J, Ziemer S, Radtke H, et al. A practical concept for preoperative identification of patients with impaired primary hemostasis. Clin Appl Thromb Hemost. 2004;10:195–204.
Koscielny J, von Tempelhoff GF, Ziemer S, et al. A practical concept for preoperative management of patient with impaired primary hemostasis. Clin Appl Thromb Hemost. 2004;10:155–66.
Payne BA, Pierre RV. Pseudothrombocytopenia: a laboratory artifact with potentially serious consequences. Mayo Clin Proc. 1982;59:123–5.
Vicari A, Banfi G, Bonini PA. EDTA-dependent pseudothrombocytopenia: a 12-month epidemiological study. Scand J Clin Lab Invest. 1988;48:5370542.
Bartholomew JR, Begelman SM, Almahameed A. Heparin-induced thrombocytopenia: principles for early recognition and management. Cleve Clin J Med. 2005;72(Suppl):S31–6.
Horlocker TT, Wedel DJ, Benzon H, et al. Regional anesthesia in the anticoagulated patient: defining the Risks (Second ASRA Consensus Conference on Neuraxial Anesthesia and Anticoagulation). Reg Anesth Pain Med. 2003;28:172–97.
Ramasamy I. Inherited bleeding disorders: disorders of platelet adhesion and aggregation. Crit Rev Oncol Hematol. 2004;49(1):1–35.
Petri M. Epidemiology of the antiphospholipid antibody syndrome. J Autoimmue. 2000;15:145–51.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer New York
About this chapter
Cite this chapter
MacKenzie, C.R. (2014). Coagulation Disorders and Orthopedic Surgery. In: MacKenzie, C., Cornell, C., Memtsoudis, S. (eds) Perioperative Care of the Orthopedic Patient. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-0100-1_20
Download citation
DOI: https://doi.org/10.1007/978-1-4614-0100-1_20
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-0099-8
Online ISBN: 978-1-4614-0100-1
eBook Packages: MedicineMedicine (R0)