
Abstract
Bicuspid aortic valve (BAV) disease is a common congenital malformation associated with significant morbidity and mortality, mostly related to valvular dysfunction. BAV has been associated with a dilated proximal aorta and a risk of dissection and rupture. Uncertainty remains about whether this aortopathy is caused by molecular dysregulation during embryonic development, or whether aortic root dilation is a function of abnormal hemodynamics. This has therapeutic ramifications especially since the optimal timing of aortic root replacement in BAV is not known and the etiology of aortic root dilation will be instrumental in devising a definitive answer to this problem. In this chapter, we will discuss the genetics and molecular biology of BAV, especially as they pertain to aortic root dilation. We will also present the two competing theories of BAV aortopathy. Attention is then directed to current guidelines for surveillance and treatment of BAV aortopathy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Background
Estimates of the prevalence of bicuspid aortic valves (BAV) range from 0.5 to 1.3 % [1–3], making it the most common congenital heart disease. Morbidity and mortality attributable to BAV outweigh all other congenital heart diseases combined [4]. Forty-three percent of people who either die of or undergo surgery for aortic valve disease have congenital BAV [5]. As early as the late nineteenth century, William Osler realized that distinct developmental abnormalities may underlie BAV development and may be related to development of the aortic root [6]. Maude Abbott first described the association between BAV and coarctation of the aorta, thus establishing a link between BAV and aortic pathology [7]. In 1972, McKusick described the association between BAV and the process of cystic medial necrosis that is known to contribute to aneurysmal dilation of the aortic root [8]. These early studies led to our understanding of BAV as a disease entity with extra-valvular manifestations. Table 3.1 lists other congenital malformations which are associated with BAV [9–17]. In this chapter, we describe the genetics and molecular pathophysiology of BAV disease. We will also review the management and treatment of the aortopathy of BAV.
Embryologic Development
Initially a hollow, linear structure, the primordial heart tube loops and bends at around 4–5 weeks of gestation such that the common atrium is situated posteriorly (dorsal) and rightward compared to the common ventricle. Focal swelling of the extracellular matrix occurs, leading to formation of the atrioventricular (AV) canal and the outflow tract (OFT). The extracellular matrix and endocardial cells in the AV canal and OFT form the endocardial cushion in a process that involves complex signal transduction [18]. The endocardial cushions thicken and fuse to form primordial valves. Some of the mesenchymal cells in the OFT are derived from neural crest cells [19]. These neural crest cells are instrumental in the septation of the OFT into the right ventricular and left ventricular OFT and in forming their connections to the great vessels [19, 20]. Neural crest cell migration and endocardial cushion development thus play a fundamental role in valvular morphogenesis and can contribute to congenital valve defects including the development of a BAV [21].
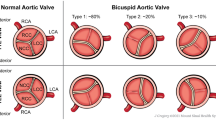
The endocardial cushion in the OFT forms three swellings, or cusps, per valve. After septation, these primordial valves remodel to form three thin leaflets [22, 23]. In the aortic valve, the leaflets are named the left, right and non-coronary cusps, based on their orientation to the coronary arteries. BAV is caused by fusion of two leaflets, resulting in asymmetric leaflet sizes often with a raphe in the larger leaflet signifying this fusion [24]. BAV can be divided into subtypes based on leaflet orientation with fusion of the right and left coronary cusps (RL) being the dominant morphology, found in around 70 % of patients in one retrospective study [25]. Distinctive morphologies have import for the development of valvular and aortic pathology; significant aortic stenosis and aortic regurgitation predominate in patients with right and non-coronary cusp fusion (RN), while 90 % of patients with coexisting aortic coarctation have a RL fusion pattern [25]. Therefore, both valvular hemodynamic profiles and separate aortic lesions can influence the potential for aortic root dilation depending on the BAV phenotype.
Defining the Scope of the Problem: BAV Aortopathy
While most clinical manifestations of BAV revolve around the abnormal aortic valve, approximately 50 % of patients have nonvalvular findings associated with BAV [14]. Dilation of the thoracic aorta is the most common nonvalvular manifestation of BAV disease (Fig. 3.1). In two prospective cohorts of BAV, the prevalence of aortic sinus dilation was 28 and 15 % at the start of each study [26, 27]. The aortopathy of BAV seems to exist even without significant valvular dysfunction. Echocardiographic studies of children with well-functioning BAV revealed dilated aortas with a faster rate of aortic root dilation when compared to trileaflet controls [28–30]. Studies in adults with well-functioning BAV have also revealed larger aortic root and ascending aortic dimensions when compared to age and sex matched trileaflet cohorts [31–34].

Transthoracic echocardiogram of a BAV with severe aortic root dilation and aortic dissection. (a) Parasternal long axis view with severe aortic root dilation (arrows ). (b) Apical five chamber view demonstrating the dilated aortic root with aortic dissection (arrow). (c) Short axis view demonstrating the BAV (Reprinted with permission from Braverman et al. [24])
Estimates of the rate of aortic root dilation with BAV range from 0.2 to 1.9 mm per year [35–38]. The rate of dilation increases with increasing aortic size in BAV so that larger aortic roots tend to dilate faster than smaller aortas. This is not unique to BAV aortopathy [30, 36, 39, 40]. However, for any given aortic root size, patients with BAV tend to have faster aortic root dilation than patients with trileaflet aortic valves [38, 41]. Moreover, patients with BAV who present with aortic dilation are significantly younger with an average age of 49 years than patients with trileaflet aortic valves and dilated aortic roots who have an average age of 61–64 years at presentation [38].
Aortic Dissection
The most feared complication of rapid aortic dilation is aortic dissection or rupture with a high mortality rate. Studies of thoracic aortic aneurysms have revealed that aortic diameter is directly proportional to the risk of aortic dissection and rupture, especially with aortic diameters of 6 cm and larger [42–44]. One large database study of 1,600 thoracic aneurysms reported a cumulative event rate of dissection, rupture and aorta-related deaths of 14.1 % for aneurysms >6 cm, as compared to an event rate of 6.5 % for aneurysms between 5.5 and 6 cm [44]. This is reflected in another study in which the median ascending aortic diameter at the time of rupture was 6 cm [42].
Catastrophic aortic syndromes associated with BAV arise at a relatively young age [38, 45–47]. In a study of aortic dissections in people under the age of 40, 24 % were associated with BAV [47]. Similar to patients with idiopathic thoracic aortic aneurysms, patients with BAV who have aortic dissections have a mean aortic diameter of 6 cm [48]. The risk of aortic dissection in patients with BAV, as assessed by autopsy and surgical pathology studies, was estimated to be around 4 % and was nine times that of patients with trileaflet aortic valves [49, 50]. In contrast, two recent cohort studies reported a much lower risk with the incidence of aortic dissection being 1 per 1,000 patient-years in one study [26] and 3.1 per 10,000 patient-years in the other [51]. In 416 patients with BAV in Olmstead County, MN with a mean follow-up of 16 years, the calculated age-adjusted relative risk of aortic dissection was 8.4 (95 % CI 2.1–33.5) when compared to the general population [51]. Thus, while the absolute rate of aortic dissection in patients with BAV appears low, the risk is significantly increased when compared to the general population.
Genetics
Heritability of BAV has been suggested by its increased prevalence amongst first degree relatives of probands with BAV [52–55]. While some studies have suggested an autosomal dominant inheritance pattern with incomplete penetrance for BAV, [54, 56] mutations in diverse genes with different inheritance patterns are likely responsible for BAV [57]. Chromosomal linkage analysis in families with BAV has implicated loci on chromosomes 18q, 15q and 13q, but specific genes in these loci have not been identified [58]. Animal models have been used to probe the contribution of specific genes to the development of BAV, along with other congenital cardiovascular conditions [59]. The first mouse model of BAV was developed by deletion of the endothelial nitric oxide synthase (eNOS) gene. All eNOS null mice had BAV due to RN fusion morphology, thus suggesting specific genetic alterations for various BAV phenotypes [60]. Further confirmation of the role of eNOS came from a study showing reduced eNOS expression in the aortic wall of patients with BAV as compared to trileaflet controls [61]. There was also a significant inverse correlation with eNOS expression and aortic diameter in patients with BAV, but not in the trileaflet control group. Thus eNOS signaling may contribute to aortic aneurysm formation in BAV patients, but specific gene mutations in this signal transduction pathway have not been identified.
Other genetic studies involving two families with an autosomal dominant form of BAV have revealed genetic mutations in the NOTCH1 gene on chromosome 19. These mutations may also be associated with accelerated aortic valve calcification [62]. NOTCH1 mutations have also been identified in BAV patients with thoracic aortic aneurysms [63]. One study has reported a missense mutation in the transforming growth factor beta receptor (TGFβ[beta]R2) in a patient with BAV and aortic aneurysm [64]. Enhanced TGF-β[beta] expression has been described in Marfan syndrome and may have therapeutic implications as discussed below [65–67].
Certain gene mutations have syndromic associations with aortic valve malformations including BAV. These include mutations in the HOXA1 gene seen in the Bosley-Salih-Alorainy syndrome and Athabascan Brainstem Dysgenesis syndrome [68]. Another example of this is a point mutation in the KCNJ2 gene which codes for the inward-rectifying potassium channel in patients with Anderson syndrome, who also have a higher prevalence of BAV [69]. Finally, genetic linkage analysis of family members with thoracic aortic aneurysm, three of whom had BAV, has revealed mutations in the ACTA2 gene which codes for vascular smooth muscle α[alpha]-actin. Whether this mutation plays a role in the morphogenesis of BAV is unknown [70].
BAV Aortopathy: Genes or Hemodynamics?
While the aortopathy of BAV has been well characterized, its etiology remains controversial. Two main theories have been proposed [71]. The genetic theory postulates that a common developmental defect involving the aortic valve extends into the ascending aorta, resulting in aortic wall fragility and aortic dilation. The hemodynamic theory emphasizes that a BAV causes abnormal ascending aortic flow mechanics and shear stress inducing post-stenotic aortic dilation. Both these theories have therapeutic ramifications since the timing of aortic root replacement in patients with BAV is controversial. If ascending aortic dilation was solely due to hemodynamic factors, then replacement of the pathologic BAV should be sufficient to stem further aortic root dilation. However, if congenital aortic fragility contributes to aortic dilation, then isolated valve replacement would not suffice to prevent catastrophic aortic syndromes like rupture and dissection.
Molecular Biology of BAV Aortopathy
The genetic theory of BAV aortopathy postulates that a developmental abnormality influences changes in the molecular ultrastructure of the aorta with a predilection for aortic root dilation. A clinical correlate of this postulate was observed by Hahn and colleagues in their retrospective analysis of a mixed population of 83 patients with normally functioning as well as stenotic and regurgitant aortic valves [33]. Echocardiographic measures revealed dilation of the aortic root and ascending aorta in patients with BAV across the spectrum of valve function (from normally functioning BAV to stenotic and regurgitant valves) as compared to trileaflet controls. Similar findings were also reported by Keane and colleagues [34]. These investigators found that BAV patients had dilated aortic roots and ascending aortas compared to trileaflet controls with similar degrees of valvular dysfunction. Their findings also established the link between aneurysmal dilation of the aorta and BAV, although worsening aortic regurgitation (but not aortic stenosis) was associated with greater degrees of aortic dilation.
These clinical studies support the plausibility of an intrinsic aortic wall defect which leads to aortic dilation in patients with BAV, regardless of the degree of valvular disease. Ascending aortic aneurysm formation is histopathologically defined by the process of Erdheim’s cystic medial necrosis (CMN), which is characterized by the triad of noninflammatory vascular smooth muscle cell (VSMC) loss, fragmentation of elastic fibers and accumulation of basophilic ground substance in the cell depleted vessel wall media (Fig. 3.2) [72, 73]. This seems to be a final common mechanism for ascending aortic aneurysm formation in both acquired disorders like aortitis, atherosclerosis and enhanced shear stress as well as in inherited syndromes of aortic wall weakness like Marfan syndrome and Ehlers Danlos syndrome [74, 75]. The process of CMN appears to be a function of programmed cell death (apoptosis) of VSMC rather than necrosis and is found in the aorta of patients with BAV as well as those with Marfan syndrome [8, 76, 77]. Furthermore, this process of CMN appears to be active in the nondilated aortas of patients with BAV, [76] particularly in the aortic convexity [78]. In contrast, the VSMC population appears to be preserved in patients with idiopathic ascending aortic aneurysms [79].

Elastic tissue stain of aortic media from a normal aorta of a trileaflet aortic valve patient demonstrating normal elastic fiber orientation (labeled normal aorta); and elastic stain from a patient with BAV and aortic root aneurysm demonstrating cystic medial necrosis (labeled aortic aneurysm. Courtesy of Robert Thompson, MD) (Reprinted with permission from Braverman et al. [24])
VSMC appear to play a role in the remodeling and maintenance of the extracellular matrix (ECM) of the aortic media. They are responsible for secreting proteins which comprise the ECM, including collagen, elastin, laminin, proteoglycan, fibrillin, fibronectin and tenascin [77, 80]. In addition to the VSMC loss that occurs in the aorta of patients with BAV, there appears to be a defect in ECM protein transport which adversely affects the structural integrity of the aortic media and probably also promotes VSMC apoptosis [77].
Another important mechanism by which VSMC maintain ECM homeostasis is by secretion of proteinases called matrix metalloproteinases (MMP) which maintain the integrity of the ECM and regulate its turnover [81, 82]. The action of MMP is regulated by the tissue inhibitors of metalloproteinases (TIMP) which control the activity of MMP in specific tissues [83, 84]. Additionally, MMPs are also regulated by metallothionein, a metal-binding protein that serves as an anti-oxidant [85]. Downregulation of metallotheinein may expose aortic VSMCs to oxidative stress and may contribute to the breakdown of the aortic ECM [85]. Many studies have reported increased activity [86, 87] and expression [88–90] of specific MMPs in the ascending aorta of patients with BAV and aortic aneurysms as compared to patients with idiopathic thoracic aortic aneurysms. MMP-9 (collagenase B) is secreted by macrophages and is active in the ascending aortas of patients with abdominal aortic aneurysms as well as idiopathic thoracic aortic aneurysms, thus suggesting that these aneurysms are formed by an inflammatory process [91, 92]. In contrast, increased MMP-2 activity is seen in thoracic aortic aneurysms in patients with BAV, [88] thus underscoring the non-inflammatory nature of ECM degradation in BAV aortopathy. While earlier studies suggested increased MMP expression as responsible for increased ECM degradation, it seems now that the total proteolytic activity in the ascending aorta is more a function of the balance between specific MMP and TIMP. Ikonomidis and colleagues analyzed MMP expression and activity and TIMP activity in the ascending aortas of patients with BAV and thoracic aortic aneurysms [93]. While MMP expression remained largely unchanged, total MMP activity was increased with a parallel decrease in TIMP activity, thus reflecting an enhanced proteolytic environment in the ECM of BAV patients with dilated ascending aortas. A calculated MMP/TIMP score appraised the proteolytic environment in different BAV phenotypes. Patients with fusion of the right-left coronary cusps (RL) had the highest score and thus were thought to reflect the most aggressive proteolytic environment. Thus, dysregulation of the balance between MMP and TIMP appears to play an important role in ECM degradation contributing to aortic aneurysm formation in patients with BAV. More recent evidence suggests that patients with BAV and dilated aortas show signs of generalized endothelial dysfunction and arterial stiffness, in addition to increased MMP-2 plasma levels, thus linking these biochemical abnormalities to diffuse changes in endothelial functioning and arterial stiffness [94].
Micro RNAs (miRs) have recently also been implicated in the breakdown of the ECM and aortic aneurysm formation. MiRs are short, noncoding RNAs that regulate gene expression by degrading mRNA and blocking protein translation. MiR-29 has recently been shown to downregulate expression of ECM proteins and may sensitize the thoracic aorta to dilation in patients with trileaflet aortic valves as well as BAV [95]. In addition, increased miR-29b expression in transgenic Marfan mice resulted in increased apoptosis of ECM component proteins and augmented MMP-2, thus contributing to thoracic aortic dilation [96]. The therapeutic utility of inhibiting miR-29 still needs to be conclusively established.
Hemodynamic Theory of BAV Aortopathy
The three dimensional aortic wall has complex vascular mechanics [97, 98]. The histological structure of the aorta varies according to its size and function. The proximal aorta serves as a reservoir, absorbing the stress of each systolic impulse and thus has a greater proportion of elastic fibers. More distally, the aorta serves as a conductance vessels with a higher proportion of collagen [97]. The tensile stress generated by ventricular contraction and blood flow through the aortic valve is exerted perpendicularly and evenly around the circumference of the aorta. Tensile stress is explained by LaPlace’s Law and is directly proportional to the diameter of the aorta. Thus, aortas with higher tensile stress will be expected to be more dilated than aortas with lower tensile stress. In contrast, aortic wall shear stress (WSS) is a product of blood viscosity and velocity and is exerted in parallel to the vessel wall. WSS exerts friction on the aortic wall and leads to activation of cellular signaling cascades which in turn lead to activation of MMP and growth factors that cause degradation of the extracellular matrix and VSMC apoptosis [99]. Aortic dilation with focal wall weakness increases the likelihood of dissection and rupture.
The above explanation supports a mechanistic theory of increased aortic wall stress and aortic dilation in patients with turbulent flow in the ascending aorta due to a stenotic or regurgitant aortic valve. Observations that patients with well-functioning BAV also have dilated aortas were felt to detract from this hemodynamic explanation of BAV aortopathy [28, 100]. However, emerging data now suggests that even patients with well-functioning BAV have abnormal flow patterns, asymmetric aortic WSS and abnormal proximal aortic stiffness [101–106]. Robicsek et al. used cryopreserved aortic roots from patients with BAV to show that even apparently well-functioning BAV are inherently stenotic and have abnormal load bearing characteristics throughout the cardiac cycle [101]. This may be due to excessive folding and creasing of the BAV leaflets and leads to abnormal flow patterns in the proximal aorta. This turbulence likely leads to abnormal aortic WSS and may predispose the aortic root of even well-functioning BAV to abnormal shear forces.
Considerable heterogeneity exists in the structure of congenital BAV. Cardiac MRI studies reveal that 86 % of patients with BAV have a raphe [107]. In this population, 84 % of patients with a raphe had fusion of the right and left cusps (RL) and 15 % had fusion of the right and noncoronary cusps (RN). Time resolved 3D MRI studies (also known as 4D MRI) have further studied eccentric blood flow in the ascending aorta in BAV patients, correlating BAV leaflet phenotypes and aortic WSS [102, 103]. Hope and colleagues [103] used 4D MRI to describe nested helical blood flow in the ascending aortas of 15/20 patients with BAV and in none of 25 patients with trileaflet aortic valves. Patients with RL cusp fusion morphology had right-anterior flow jets in the ascending aorta. In contrast, patients with RN cusp fusion pattern had left-handed helical flow with left-posterior flow jets in the ascending aorta. Figure 3.3 is an example of MR angiography and 4D MRI showing ascending aortic dilation and right-sided helical flow in the ascending aorta in a patient with BAV. These findings may help explain why BAV patients with RL fusion patterns have more dilated aortic roots in some studies [108] while BAV patients with RN fusion patterns have increased aortic arch dimensions [109]. However, other studies have not revealed a difference in aortic sizes amongst BAV patients with different leaflet fusion patterns [107]. Nevertheless, these studies do create a biophysical platform for even well-functioning BAV to be associated with turbulent flow and eccentric WSS in the ascending aorta, thus lending credence to the theory that abnormal hemodynamics may play some role in the distinct aortopathy of BAV.

Images in a patient with a BAV and a focal ascending aortic aneurysm. (a) MR angiographic and (b) T1-weighted spin-echo MR images show focal aneurysm of proximal ascending aorta (up to 4.8 cm). Four-dimensional flow MR imaging data in an oblique-sagittal orientation with 3D streamline analysis (color-coded for velocity, see key) from (c) right and (d) left sides of thoracic aorta shows dramatic systolic right-handed helical flow in the aortic root (Reproduced with permission from Hope et al. [103])
The hemodynamic and genetic theories of BAV aortopathy can be further scrutinized by considering the fate of the aortic root in patients with BAV after isolated aortic valve replacement (AVR). Unfortunately the data in this particular area is scarce and conflicting [41, 110–112]. Yasuda and colleagues evaluated 13 patients with BAV and 15 trileaflet controls before and after isolated aortic valve replacement (AVR) [41]. Serial measurements of aortic dimensions revealed progressive dilation of the proximal ascending aorta in BAV patients after isolated AVR when compared to trileaflet controls. This rate of dilation was similar to that seen in 18 BAV patients who did not undergo AVR. However, no aortic complications were seen in the BAV group after isolated AVR. These findings were similar to the findings of Gorland and colleagues who followed a larger cohort of 252 patients for a median time period of nearly 9 years with very few aortic complications [112]. By contrast, Russo and colleagues found an unfavorable post-operative course in a cohort of 50 patients with BAV after isolated AVR, with eight late aortic events and seven sudden cardiac deaths.[110] More recently, Girdauskas and colleagues analyzed their institutional database of 153 patients with stenotic BAV and dilated aortic roots (between 4 and 5 cm in diameter) who underwent isolated AVR [113]. The 15 year actuarial survival rate was 78 % with a 94 % rate of freedom from aortic interventions at 15 years and a 93 % rate of freedom from adverse aortic events. The freedom from adverse events was significantly lower in a smaller subset of patients with aortic regurgitation. These authors concluded that the rate of adverse aortic events in BAV patients with aortic stenosis and mild to moderate aortic root dilation was very low after isolated AVR [113]. Heterogeneity of this data makes it difficult to draw definitive conclusions about the fate of the proximal aorta in BAV patients after isolated AVR, but suggest an overall low aortic event rate. This may also support the hemodynamic, rather than the genetic theory of aortic dilation in BAV [71].
Therefore, the BAV is inherently abnormal, even in patients without clinically or echocardiographically evident aortic stenosis or regurgitation. This valvular morphology in turn creates highly eccentric and turbulent jets through the ascending aorta with resultant eccentric aortic WSS. These eccentric forces may lead to the characteristic patterns of ascending aortic dilation seen in various aortic leaflet morphologies in BAV patients. However, it does seem that 10–15 % of patients with BAV may have isolated dilation of the aortic root at the sinuses of Valsalva without dilation of the proximal aorta. This is mostly seen in young, male patients and is associated with varying degrees of aortic insufficiency [100]. This subgroup of patients with pure aortic regurgitation also appears to have differential transcription of type 1 collagen when compared to stenotic BAV [114]. The aortopathy associated with this particular phenotype of BAV may be a function of purely genetic rather than hemodynamic factors.
Imaging the Aortic Root
The 2006 ACC/AHA Guidelines on the management of patients with valvular heart disease recommend that patients with known BAV undergo an initial transthoracic echocardiogram to assess the size of the aortic root and the ascending aorta [115]. Aortic root size in a population varies with age, gender and body size [116]. A normal diameter for the ascending aorta is typically defined as 20–37 mm [76]. Using regression formulae and nomograms for body size, the upper limit of the aortic root has been defined as 2.1 cm/m2 at the level of the sinuses of Valsalva, when indexed to the body surface area (BSA) [117]. An increase in indexed aortic diameter beyond the upper limit of normal represents aortic root dilation. An aortic aneurysm is defined as an increase in aortic root size to 50 % more than the upper limit of normal [118].
Transthoracic echocardiography (TTE) depicts aortic valve function, left ventricular function, pulmonary pressures, and provides measurements of the thoracic aorta. Generally, a good correlation exists between aortic root measurements by TTE and by ECG-gated multidetector CT or cardiac MRI [119, 120]. Echo imaging of the ascending aorta can be difficult to obtain and is affected by body habitus [121]. Thus, the ACC/AHA guidelines recommend using cardiac magnetic resonance imaging (MRI) or cardiac computed tomography (CT) in cases where TTE does not provide adequate measurements of the aortic root or ascending aorta [115].
Multidetector cardiac CT (MDCT) with three dimensional reconstruction can be useful for analyzing the anatomy of the aortic root. ECG-gating can be used to limit radiation exposure with MDCT or standard spiral CT can be used for aortic root diameter surveillance [21, 122, 123]. MDCT reconstructions may also be used to assess aortic wall stress which is increased in patients with BAV for a given aortic root size when compared to patients with trileaflet aortic valves [124].
Sizing of the ascending aorta in patients with BAV by cardiac MRI utilizes ECG-gated black blood pulse sequences [125]. Steady state free precession (SSFP) imaging sequences can be very effective in quantifying the degree of aortic valve dysfunction [125, 126]. Besides aortic root diameter, CMR techniques can also be applied to the assessment of elastic properties of the aortic root in BAV [127]. These novel metrics may become useful in analyzing the biophysical properties of the aortic root in BAV, regardless of the degree of aortic dilation. More recently, time-resolved, three dimensional MRI (4D MRI) has been utilized to evaluate asymmetric blood flow in the ascending aorta which, by contributing to differential shear stress on different points of the aortic wall, may lead to distinctive aortic root dilation [128]. While this imaging modality offers much promise in the visualization of the complex hemodynamics in the ascending aorta in BAV, its current clinical use is limited and still experimental.
Despite these advances in newer imaging modalities, transthoracic (and on occasion, transesophageal) echocardiography remains the most commonly utilized imaging modality in BAV. TTE can readily appraise the aortic valve, root and ascending aorta. Cardiac CT and cardiac MRI image the ascending aorta if this is not well seen on echo and can also be used to supplement echo findings and provide a better assessment of aortic root size. Patients with ascending aorta or aortic root diameter >4 cm should undergo surveillance imaging with echo, CT or MRI on a yearly basis [115].
Medical Therapy
Limited data exist for medical therapy in BAV patients with aortic dilation. Consensus guidelines suggest using beta blockers in patients with BAV and aortic diameter >4 cm who are not candidates for surgical therapy and who do not have severe aortic regurgitation [115]. Data for the use of beta blockers to slow the rate of aortic root dilation comes from studies involving patients with Marfan syndrome. Beta blockers are thought to slow the rate of aortic root dilation by reducing the rate of pressure increase in the aorta (dP/dT) and by reductions in heart rate which reduces the number of systolic impulses encountered by the aorta per minute [129]. While some studies showed that beta blockers reduced the rate of aortic root dilation and the need for aortic root replacement in children with Marfan syndrome [130, 131], other studies, including a meta-analysis showed no benefit of beta blocker use on aortic root size [132, 133]. Studies examining the use of beta blockers in patients with BAV are lacking.
Angiotensin II receptor and angiotensin II concentrations are increased in the ascending aortas of children with Marfan syndrome and cell culture studies show that ace inhibitors can reduce VSMC apoptosis [134]. Furthermore, the use of ace inhibitors and angiotensin receptor blockers have been shown to reduce the rate of aortic dilation in small cohorts of children with Marfan syndrome [135, 136]. Losartan inhibits aortic dilation by reducing TGF-β[beta] (and concomitant MMP) expression and, along with doxycycline, may also inhibit downstream signaling by inhibiting the phosphorylation of the Erk 1/2 proteins involved in the TGF-β[beta] signaling cascade [137]. The applicability of these results to patients with BAV and dilated aortic roots is unknown.
Statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) may inhibit MMP expression and have been shown to slow the rate of progression of abdominal aortic aneurysms [138]. A recent retrospective cohort study of patients with BAV who were undergoing pre-operative coronary angiography before AVR showed that the aortic root size was significantly smaller in patients with BAV and severe aortic stenosis who were on statin therapy [138]. However, no prospective studies have assessed the effect of statins on the progression of aortic root dilation in BAV.
Surgical Replacement of the Aortic Root
The risk of thoracic aortic dissection or rupture increases with progressive dilation of the aorta and patients with an ascending aortic aneurysm of 6 cm or larger have a 34 % lifetime risk of rupture with a 4 % yearly risk of dissection or rupture [139]. For patients with idiopathic ascending aortic aneurysms, aortic root replacement is recommended for an aortic root or ascending aorta diameter of >5.5 cm [44]. The aortic root in patients with BAV tends to dilate more rapidly and thus places these patients at risk for aortic dissection and rupture at a younger age [140]. Optimal threshold for aortic root replacement in BAV patients is controversial. Expert consensus opinion supports aortic root replacement in patients with BAV with an ascending aorta >5.0 cm [115]. This is similar to the recommendations for aortic root replacement in patients with Marfan syndrome [141]. Aortic root replacement is recommended in BAV patients with an aortic diameter of >4.5 cm if the patient is undergoing concomitant aortic valve replacement. Aortic root replacement should also be considered if the root size is increasing >0.5 cm/year [115]. Variability exists in the risk for aortic dissection and rupture, however, and aortic size criteria offer only an incomplete characterization of this risk. Borger and colleagues reviewed their database of 201 patients with isolated AVR for BAV who were followed for a median of 10 years [111]. The 15 year survival free of ascending aorta related complications was 43 % for patients with ascending aorta diameters of 4.5–4.9 cm as compared to 86 % for patients with aortic diameters of less than 4.0 cm. This led the authors to conclude that aortic root replacement can be considered for patients with BAV and with ascending aorta diameter greater than 4.5 cm [111]. Similarly, the International Registry of Aortic Dissection (IRAD) database revealed an average aortic diameter of 5.3 cm at the time of ascending aortic dissection in the 591 patients examined and 50 % of the patients, including 6 of 11 with BAV, had aortic dissection with ascending aortic diameter of <5.5 cm [142]. While aortic root size cutoffs can help guide management in patients with BAV and a dilated ascending aorta, the full magnitude of the risk of aortic dissection is not captured by strict size criteria alone.
Clinical comorbidities may contribute to the risk of thoracic aortic disruption. Patients with BAV and coarctation of the aorta may represent a subgroup of BAV patients with more diffuse aortopathy, [143, 144] and thus may benefit from earlier intervention on the aortic root. A family history of aortic dissection or rupture can suggest a heritable defect in the aortic root and may prompt consideration or earlier aortic root replacement [21]. Smoking is another risk factor that may expedite aortic root dilation. Smoking promotes fragmentation of elastin in the ECM [21]. In addition, recent data from a transgenic mouse model has revealed that nicotine activates the AMP-activated kinase α[alpha]2 subunit leading to upregulation of MMP-2 and formation of abdominal aortic aneurysms [145]. This mechanism has not been validated in thoracic aortic aneurysms, but remains a potential pathway for smoking mediated aortic root dilation. As such, patients with a smoking history and especially those with COPD have dilated aortas and a more rapid rate of aortic root dilation than nonsmokers [146–148]. It has been suggested that BAV patients with the risk factors mentioned above should be considered for aortic root replacement when their aortic diameter is >4.5 cm [21]. Since aortic root sizes vary with body size and smaller patients (especially women) may have smaller aortic roots at baseline, some experts have recommended using a body surface area (BSA) indexed aortic root size of 2.5 cm/m2 as a threshold for aortic root replacement [149].
Conclusion
Patients with BAV may also have dilated aortic roots and are at risk for aortic dissection and rupture. The mechanism of aortic root dilation has not been completely clarified, but likely involves a complex interplay between genetic and molecular biologic factors affecting gene expression in the aortic root and hemodynamic stressors borne out of abnormal valvular structure and function. The aortic root should be imaged with transthoracic or transesophageal echocardiography in all patients with BAV. If the aortic root is not well seen with these modalities, cardiac CT and cardiac MRI can provide better estimates of aortic root size. Patients with an aortic root diameter >4 cm should undergo yearly surveillance of the aortic root with echocardiography, CT or MRI. While aortic size is an incomplete predictor of the risk of aortic dissection, consensus guidelines recommend replacement of the aortic root at a diameter of >5 cm in patients with BAV. Patients who belong to higher risk groups may benefit from aortic root replacement for aortic diameters >4.5 cm and patients with BAV undergoing aortic valve replacement should also have their aortic root or ascending aorta replaced for aortic diameter >4.5 cm. Smoking cessation must be aggressively advocated in patients with BAV and aortic dilation. Medical therapy has not been well studied in the care of patients with BAV, but the use of beta blockers, ace inhibitors and statins may be useful in slowing the rate of aortic root and ascending aortic dilation.
References
Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39(12):1890–900.
Nistri S, Basso C, Marzari C, Mormino P, Thiene G. Frequency of bicuspid aortic valve in young male conscripts by echocardiography. Am J Cardiol. 2005;96(5):718–21.
Tutar E, Ekici F, Atalay S, Nacar N. The prevalence of bicuspid aortic valve in newborns by echocardiographic screening. Am Heart J. 2005;150(3):513–5.
Ward C. Clinical significance of the bicuspid aortic valve. Heart. 2000;83(1):81–5.
Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol. 1970;26(1):72–83.
Osler W. The bicuspid condition of the aortic valves. Transactions of the Association of American Physicians. Philadelphia: Wm. J. Dornan; 1886. p. 185–92.
Abbott ME. Coarctation of the aorta of the adult type. Am Heart J. 1928;3:578–618.
McKusick VA. Association of congenital bicuspid aortic valve and erdheim’s cystic medial necrosis. Lancet. 1972;1(7758):1026–7.
Roos-Hesselink JW, Schölzel BE, Heijdra RJ, Spitaels SE, Meijboom FJ, et al. Aortic valve and aortic arch pathology after coarctation repair. Heart. 2003;89(9):1074–7.
Hinton Jr RB, Martin LJ, Tabangin ME, Mazwi ML, Cripe LH, Benson DW. Hypoplastic left heart syndrome is heritable. J Am Coll Cardiol. 2007;50(16):1590–5.
Bolling SF, Iannettoni MD, Dick 2nd M, Rosenthal A, Bove EL. Shone’s anomaly: operative results and late outcome. Ann Thorac Surg. 1990;49(6):887–93.
Sybert VP. Cardiovascular malformations and complications in Turner syndrome. Pediatrics. 1998;101(1):E11.
De Rubens Figueroa J, Rodríguez LM, Hach JL, Del Castillo RV, Martínez HO. Cardiovascular spectrum in Williams-Beuren syndrome: the Mexican experience in 40 patients. Tex Heart Inst J. 2008;35(3):279–85.
Siu SC, Silversides CK. Bicuspid aortic valve disease. J Am Coll Cardiol. 2010;55(25):2789–800.
Higgins CB, Wexler L. Reversal of dominance of the coronary arterial system in isolated aortic stenosis and bicuspid aortic valve. Circulation. 1975;52(2):292–6.
Rashid A, Saucedo JF, Hennebry TA. Association of single coronary artery and congenital bicuspid aortic valve with review of literature. J Interv Cardiol. 2005;18(5):389–91.
Hutchins GM, Nazarian IH, Bulkley BH. Association of left dominant coronary arterial system with congenital bicuspid aortic valve. Am J Cardiol. 1978;42(1):57–9.
Chakraborty S, Combs MD, Yutzey KE. Transcriptional regulation of heart valve progenitor cells. Pediatr Cardiol. 2010;31(3):414–21.
Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220(4601):1059–61.
Rothenberg F, Fisher SA, Watanabe M. Sculpting the cardiac outflow tract. Birth Defects Res C Embryo Today. 2003;69(1):38–45.
Tadros T, Klein MD, Shapira OM. Ascending aortic dilation associated with bicuspid aortic valve: pathophysiology, molecular biology and clinical implications. Circulation. 2009;119(6):880–90.
Hurle JM, Ojeda JL. Cell death during the development of the truncus and conus of the chick embryo heart. J Anat. 1979;129(pt2):427–39.
Hurle JM, Colvée E, Blanco AM. Development of mouse semilunar valves. Anat Embryol (Berl). 1980;160(1):83–91.
Braverman AC, Güven H, Beardsless MA, Makan M, Kates AM, Moon MR. The bicuspid aortic valve. Curr Probl Cardiol. 2005;30(9):470–522.
Fernandes SM, Sanders SP, Khairy P, Jenkins KJ, Gauvreau K, et al. Morphology of bicuspid aortic valves in children and adolescents. J Am Coll Cardiol. 2004;44(8):1648–51.
Tzemos N, Therrien J, Yip J, Thanassoulis G, Tremblay S, et al. Outcomes in adults with bicuspid aortic valves. JAMA. 2008;300(11):1317–25.
Michelena HI, Desjardins VA, Avierinos JF, Russo A, Nkomo VT, et al. Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community. Circulation. 2008;117(21):2776–84.
Beroukhim RS, Kruzick TL, Taylor AL, Gao D, Yetman AT. Progression of aortic dilation in children with a functionally normal bicuspid aortic valve. Am J Cardiol. 2006;98(6):828–30.
Gurvitz M, Chang RK, Drant S, Allada V. Frequency of aortic dilation in children with a bicuspid aortic valve. Am J Cardiol. 2004;94(10):1337–40.
Holmes KW, Lehmann CU, Dalal D, Nasir K, Dietz HC, et al. Progressive dilation of the ascending aorta in children with isolated bicuspid aortic valve. Am J Cardiol. 2007;99(7):978–83.
Pachulski RT, Weinberg AL, Chan KL. Aortic aneurysm in patients with functionally normal or minimally stenotic bicuspid aortic valve. Am J Cardiol. 1991;67(8):781–2.
Nistri S, Sorbo MD, Marin M, Palisi M, Scognamigilio R, Thiene G. Aortic root dilation in young men with normally functioning bicuspid aortic valves. Heart. 1999;82(1):19–22.
Hahn RT, Roman MJ, Mogtader AH, Devereux RB. Association of aortic dilation with regurgitant, stenotic and functionally normally bicuspid aortic valves. J Am Coll Cardiol. 1992;19(2):283–8.
Keane MG, Wiegers SE, Plappert T, Pochettino A, Bavaria JE, Sutton MG. Bicuspid aortic valves are associated with aortic dilation out of proportion to coexistant valvular lesions. Circulation. 2000;102(19 Suppl):III35–9.
Ferencik M, Pape LA. Changes in size of ascending aorta and aortic valve function with time in patients with congenitally bicuspid aortic valves. Am J Cardiol. 2003;92(1):43–6.
Dore A, Brochu MC, Baril JF, Guertin MC, Mercier LA. Progressive dilation of the diameter of the aortic root in adults with a bicuspid aortic valve. Cardiol Young. 2003;13(6):526–31.
Novaro GM, Griffin BP. Congenital bicuspid aortic valve and rate of ascending aortic dilatation. Am J Cardiol. 2004;93(4):525–6.
Davies RR, Kaple RK, Mandapati D, Gallo A, Botta Jr DM, et al. Natural history of ascending aortic aneurysms in the setting of an unreplaced bicuspid aortic valve. Ann Thorac Surg. 2007;83(4):1338–44.
Shimada I, Rooney SJ, Pagano D, Farneti PA, Davies P, et al. Prediction of thoracic aortic aneurysm expansion: validation of formulae describing growth. Ann Thorac Surg. 1999;67(6):1968–70.
Coady MA, Rizzo JA, Hammond GL, Kopf GS, Elefteriades JA. Surgical intervention criteria for thoracic aortic aneurysms: a study of growth rates and complications. Ann Thorac Surg. 1999;67(6):1922–6.
Yasuda H, Nakatani S, Stugaard M, Tsujita-Kuroda Y, Bando K, et al. Failure to prevent progressive dilation of ascending aorta by aortic valve replacement in patients with bicuspid aortic valve: comparison with tricuspid aortic valve. Circulation. 2003;108 Suppl 1:II291–4.
Coady MA, Rizzo JA, Hammond GL, Mandapati D, Darr U, et al. What is the appropriate size criterion for resection of thoracic aortic aneurysms? J Thorac Cardiovasc Surg. 1997;113(3):476–91.
Davies RR, Goldstein LJ, Coady MA, Tittle SL, Rizzo JA, et al. Yearly rupture or dissection rates for thoracic aortic aneurysms: simple prediction based on size. Ann Thorac Surg. 2002;73(1):17–27.
Elefteriades JA. Natural history of thoracic aortic aneurysms: indications for surgery, and surgical versus nonsurgical risks. Ann Thorac Surg. 2002;74(5):S 1877–80.
La Canna G, Ficarra E, Tsagalau E, Nardi M, Morandini A, et al. Progression rate of ascending aortic dilation in patients with normally functioning bicuspid and tricuspid aortic valves. Am J Cardiol. 2006;98(2):249–53.
Edwards ED, Leaf DS, Edwards JE. Dissecting aortic aneurysm associated with congenital bicuspid aortic valve. Circulation. 1978;57(5):1022–5.
Gore I. Dissecting aneurysms of the aorta in persons under forty years of age. AMA Arch Pathol. 1953;55(1):1–13.
Svensson LG, Kim KH, Lytle BW, Cosgrove DM. Relationship of aortic cross-sectional area to height ratio and risk of aortic dissection in patients with bicuspid aortic valves. J Thorac Cardiovasc Surg. 2003;126(3):892–3.
Larson EW, Edwards WD. Risk factors for aortic dissection: a necropsy study of 161 cases. Am J Cardiol. 1984;53(6):849–55.
Roberts CS, Roberts WC. Dissection of the aorta associated with congenital malformation of the aortic valve. J Am Coll Cardiol. 1991;17(3):712–6.
Michelena HI, Khanna AD, Mahoney D, Margaryan E, Topilsky Y, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA. 2011;306(10):1104–12.
Gale AN, McKusick VA, Hutchins GM, Gott VL. Familial congenital bicuspid aortic valve: secondary calcific aortic stenosis and aortic aneurysm. Chest. 1977;72(5):668–70.
Emanuel R, Withers R, O’Brien K, Ross P, Feizi O. Congenitally bicuspid aortic valves. Clinicogenetic study of 41 families. Br Heart J. 1978;40(12):1402–7.
Clementi M, Notari L, Borghi A, Tenconi R. Familial congenital bicuspid aortic valve: a disorder of uncertain inheritance. Am J Med Genet. 1996;62(4):336–8.
Huntington K, Hunter AG, Chan KL. A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J Am Coll Cardiol. 1997;30(7):1809–12.
McDonald K, Maurer BJ. Familial aortic valve disease: evidence for a genetic influence? Eur Heart J. 1989;10(7):676–7.
Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol. 2004;44(1):138–43.
Martin LJ, Ramachandran V, Cripe LH, Hinton RB, Adelfinger G, et al. Evidence in favor of linkage to human chromosomal regions 18q, 5q and 3q for bicuspid aortic valve and associated cardiovascular malformations. Hum Genet. 2007;121(2):275–84.
Garg V. Molecular genetics of aortic valve disease. Curr Opin Cardiol. 2006;21(3):180–4.
Fernández B, Durán AC, Fernández-Gallego T, Fernández MC, Such M, et al. Bicuspid aortic valves with different spatial orientations of the leaflets are distinct etiological entities. J Am Coll Cardiol. 2009;54(24):2312–8.
Aicher D, Urbich C, Zeiher A, Dimmeler S, Schäfers HJ. Endothelial nitric oxide synthase in bicuspid aortic valve disease. Ann Thorac Surg. 2007;83(4):1290–4.
Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270–4.
McKellar SH, Tester DJ, Yagubyan M, Majumdar R, Ackerman MJ, Sundt 3rd TM. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J Thorac Cardiovasc Surg. 2007;134(2):290–6.
Girdauskas E, Schulz S, Borger MA, Mierzwa M, Kuntze T. Transforming growth factor-beta receptor type II mutation in a patient with bicuspid aortic valve disease and intraoperative aortic dissection. Ann Thorac Surg. 2011;91(5):e70–1.
Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–11.
Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, et al. TGF-beta-dependant pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–92.
Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–61.
Tischfield MA, Bosley TM, Salih MA, Alorainy IA, Sener EC, et al. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat Genet. 2005;37(10):1035–7.
Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George Jr AL, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. 2002;71(3):663–8.
Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39(12):1488–93.
Girdauskas E, Borger MA, Secknus MA, Girdauskas G, Kuntze T. Is aortopathy in bicuspid aortic valve disease a congenital defect or a result of abnormal hemodynamics? A critical reappraisal of a one-sided argument. Eur J Cardiothorac Surg. 2011;39(6):809–14.
Erdheim J. Medionecrosis aortae idiopathica. Virchows Arch. 1929;273:454–79.
Erdheim J. Medionecrosis aortae idiopathica cystica. Virchows Arch. 1930;276:187–229.
Carlson RG, Lillehei CW, Edwards JE. Cystic medial necrosis of the ascending aorta in relation to age and hypertension. Am J Cardiol. 1970;25(4):411–5.
Francke U, Berg MA, Tynan K, Brenn T, Liu W, et al. A Gly1127Ser mutation in an EGF-like domain of the fibrillin 1 gene is a risk factor for ascending aortic aneurysm and dissection. Am J Hum Genet. 1995;56(6):1287–96.
Bonderman D, Gharehbaghi-Schnell E, Wollenek G, Maurer G, Baumgartner H, Lang IM. Mechanisms underlying aortic dilation in congenital aortic valve malformation. Circulation. 1999;99(16):2138–43.
Nataatmadja M, West M, West J, Summers K, Walker P, et al. Abnormal extracellular matrix protein transport associated with increased apoptosis of vascular smooth muscle cells in marfan syndrome and bicuspid aortic valve thoracic aortic aneurysm. Circulation. 2003;108 Suppl 1:II329–34.
Della Corte A, Quarto C, Bancone C, Castaldo C, Di Meglio F, et al. Spatiotemporal patterns of smooth muscle cell changes in ascending aortic dilatation with bicuspid and tricuspid aortic valve stenosis: focus on cell-matrix signaling. J Thorac Cardiovasc Surg. 2008;135(1):8–18.
Tang PC, Coady MA, Lovoulos C, Dardik A, Aslan M, et al. Hyperplastic cellular remodeling of the media in ascending thoracic aortic aneurysms. Circulation. 2005;112(8):1098–105.
Schmid FX, Bielenberg K, Schneider A, Haussler A, Keyser A, Birnbaum D. Ascending aortic aneurysm associated with bicuspid and tricuspid aortic valve: involvement and clinical relevance of smooth muscle cell apoptosis and expression of cell death-initiating proteins. Eur J Cardiothorac Surg. 2003;23(4):537–43.
Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92(8):827–39.
Hiller O, Lichte A, Oberpichler A, Kocourek A, Tschesche H. Matrix metalloproteinases collagenase-2, macrophage elastase, collagenase-3, and membrane type 1-matrix metalloproteinase impair clotting by degradation of fibrinogen and factor XII. J Biol Chem. 2000;275(42):33008–13.
Stolow MA, Bauzon DD, Li J, Sedgwick T, Liang VC, et al. Identification and characterization of a novel collagenase in Xenopus laevis: possible roles during frog development. Mol Biol Cell. 1996;7(10):1471–83.
Patterson ML, Atkins SJ, Knäuper V, Murphy G. Specific collagenolysis by gelatinase A, MMP-2, is determined by the hemopexin domain and not the fibronectin-like domain. FASEB Lett. 2001;503(2–3):158–62.
Phillippi JA, Klyachko EA, Kenny 4th JP, Eskay MA, Gorman RC, Gleason TG. Basal and oxidative stress-induced expression of metallothionein is decreased in ascending aortic aneurysms of bicuspid aortic valve patients. Circulation. 2009;119(18):2498–506.
Fedak PW, de Sa MP, Verma S, Nili N, Kazemian P, et al. Vascular matrix remodeling in patients with bicuspid aortic valve malformations: implications for aortic dilatation. J Thorac Cardiovasc Surg. 2003;126(3):797–806.
Boyum J, Fellinger EK, Schmoker JD, Trombley L, McPartland K, et al. Matrix metalloproteinase activity in thoracic aortic aneurysms associated with bicuspid and tricuspid aortic valves. J Thorac Cardiovasc Surg. 2004;127(3):686–91.
Ikonomidis JS, Jones JA, Barbour JR, Stroud RE, Clark LL, et al. Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with bicuspid or tricuspid aortic valves. J Thorac Cardiovasc Surg. 2007;133(4):1028–36.
Koullias GJ, Korkolis DP, Ravichandran P, Psyrri A, Hatzaras I, Elefteriades J. Tissue microarray detection of matrix metalloproteinases, in diseased tricuspid and bicuspid aortic valves with or without pathology of the ascending aorta. Eur J Cardiothorac Surg. 2004;26(6):1098–103.
LeMaire SA, Wang X, Wilks JA, Carter SA, Wen S, et al. Matrix metalloproteinases in ascending aortic aneurysms: bicuspid versus trileaflet aortic valves. J Surg Res. 2005;123(1):40–8.
Newman KM, Ogata Y, Malon A, Irizarry E, Gandhi RH, et al. Identification of matrix metalloproteinases 3 (stromelysin-1) and 9 (gelatinase B) in abdominal aortic aneurysm. Arterioscler Thromb. 1994;14(8):1315–20.
Sakalihasan N, Delvenne P, Nusgens BV, Limet R, Lapière CM. Activated forms of MMP2 and MMP9 in abdominal aortic aneurysms. J Vasc Surg. 1996;24(1):127–33.
Ikonomidis JS, Ruddy JM, Benton Jr SM, Arroyo J, Brinsa TA, et al. Aortic dilatation with bicuspid aortic valves: cusp fusion correlates to matrix metalloproteinases and inhibitors. Ann Thorac Surg. 2012;93(2):457–63.
Tzemos N, Lyseggen E, Silversides C, Jamorski M, Tong JH, et al. Endothelial function, carotid-femoral stiffness, and plasma matrix metalloproteinase-2 in men with bicuspid aortic valve and dilated aorta. J Am Coll Cardiol. 2010;55(7):660–8.
Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, et al. MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res. 2011;109(10):1115–9.
Merk DR, Chin JT, Dake BA, Maegdefessel L, Miller MO, et al. MiR-29b participates in early aneurysm development in Marfan syndrome. Circ Res. 2012;110(2):312–24.
Lee RT, Kamm RD. Vascular mechanics for the cardiologist. J Am Coll Cardiol. 1994;23(6):1289–95.
O’Rourke MF, Staessen JA, Vlachopoulos C, Duprez D, Plante GE. Clinical applications of arterial stiffness; definitions and reference values. Am J Hypertens. 2002;15(5):426–44.
Lehoux S, Tedgui A. Cellular mechanics and gene expression in blood vessels. J Biomech. 2003;36(5):631–43.
Della Corte A, Bancone C, Quarto C, Dialetto G, Covino FE, et al. Predictors of ascending aortic dilatation with bicuspid aortic valve: a wide spectrum of disease expression. Eur J Cardiothorac Surg. 2007;31(3):397–404.
Robicsek F, Thubrikar MJ, Cook JW, Fowler B. The congenitally bicuspid aortic valve: how does it function? Why does it fail? Ann Thorac Surg. 2004;77(1):177–85.
Hope MD, Meadows AK, Hope TA, Ordovas KG, Reddy GP, et al. Images in cardiovascular medicine. Evaluation of bicuspid aortic valve and aortic coarctation with 4D flow magnetic resonance imaging. Circulation. 2008;117(21):2818–9.
Hope MD, Hope TA, Meadows AK, Ordovas KG, Urbania TH, et al. Bicuspid aortic valve: four-dimensional MR evaluation of ascending aortic systolic flow patterns. Radiology. 2010;255(1):53–61.
Conti CA, Della Corte A, Votta E, Del Viscovo L, Bancone C, et al. Biomechanical implications of the congenital bicuspid aortic valve: a finite element study of aortic root function from in vivo data. J Thorac Cardiovasc Surg. 2010;140(4):890–6.
den Reijer PM, Sallee 3rd D, van der Velden P, Zaaijer ER, Parks WJ, et al. Hemodynamic predictors of aortic dilatation in bicuspid aortic valve by velocity-encoded cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2010;12:4.
Nistri S, Grande-Allen J, Noale M, Basso C, Siviero P, et al. Aortic elasticity and size in bicuspid aortic valve syndrome. Eur Heart J. 2008;29(4):472–9.
Buchner S, Hülsmann M, Poschenrieder F, Hamer OW, Fellner C, et al. Variable phenotypes of bicuspid aortic valve disease: classification by cardiovascular magnetic resonance. Heart. 2010;96(15):1233–40.
Schaefer BM, Lewin MB, Stout KK, Gill E, Prueitt A, et al. The bicuspid aortic valve: an integrated phenotypic classification of leaflet morphology and aortic root shape. Heart. 2008;94(12):1634–8.
Fazel SS, Mallidi HR, Lee RS, Sheehan MP, Liang D, et al. The aortopathy of bicuspid aortic valve disease has distinctive patterns and usually involves the transverse aortic arch. J Thorac Cardiovasc Surg. 2008;135(4):901–7.
Russo CF, Mazzetti S, Garatti A, Ribera E, Milazzo A, et al. Aortic complications after bicuspid aortic valve replacement: long-term results. Ann Thorac Surg. 2002;74(5):S1773–6.
Borger MA, Preston M, Ivanov J, Fedak PW, Davierwala P, et al. Should the ascending aorta be replaced more frequently in patients with bicuspid aortic valve disease? J Thorac Cardiovasc Surg. 2004;128(5):677–83.
Goland S, Czer LS, De Robertis MA, Mirocha J, Kass RM, et al. Risk factors associated with reoperation and mortality in 252 patients after aortic valve replacement for congenitally bicuspid aortic valve disease. Ann Thorac Surg. 2007;83(3):931–7.
Girdauskas E, Disha K, Raisin HH, Secknus MA, Borger MA, Kuntze T. Risk of late aortic events after an isolated aortic valve replacement for bicuspid aortic valve stenosis with concomitant ascending aortic dilation. Eur J Cardiothorac Surg. 2012;42(5):832–7.
Cotrufo M, Della Corte A, De Santo LS, Quarto C, De Feo M, et al. Different patterns of extracellular matrix protein expression in the convexity and the concavity of the dilated aorta with bicuspid aortic valve: preliminary results. J Thorac Cardiovasc Surg. 2005;130(2):504–11.
Bonow RO, Carabello BA, Kanu C, de Leon Jr AC, Faxon DP, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 1998 guidelines for the Management of Patients With Valvular Heart Disease): developed in collaboration with the Society of Cardiovascular Anesthesiologists: endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. Circulation. 2006;114(5):e84–231.
Vasan RS, Larson MG, Levy D. Determinants of echocardiographic aortic root size. The Framingham Heart Study. Circulation. 1995;91(3):734–40.
Roman MJ, Devereux RB, Kramer-Fox R, O’Loughlin J. Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am J Cardiol. 1989;64(8):507–12.
Johnston KW, Rutherford RB, Tilson MD, Shah DM, Hollier L, Stanley JC. Suggested standards for reporting on arterial aneurysms. Subcommittee on Reporting Standards for Arterial Aneurysms, Ad Hoc Committee on Reporting Standards, Society for Vascular Surgery and North American Chapter, International Society for Cardiovascular Surgery. J Vasc Surg. 1991;13(3):452–8.
Tamborini G, Galli CA, Maltagliati A, Andreini D, Pontone G, et al. Comparison of feasibility and accuracy of transthoracic echocardiography versus computed tomography in patients with known ascending aortic aneurysm. Am J Cardiol. 2006;98(7):966–9.
Meijboom LJ, Groenink M, van der Wall EE, Romkes H, Stoker J, Mulder BJ. Aortic root asymmetry in marfan patients; evaluation by magnetic resonance imaging and comparison with standard echocardiography. Int J Card Imaging. 2000;16(3):161–8.
Hartnell GG. Imaging of aortic aneurysms and dissection: CT and MRI. J Thorac Imaging. 2001;16(1):35–46.
Morgan-Hughes GJ, Ca R, Owens PE, Marshall AJ. Dilatation of the aorta in pure, severe, bicuspid aortic valve stenosis. Am Heart J. 2004;147(4):736–40.
Ocak I, Lacomis JM, Deible CR, Pealer K, Parag Y, Knollmann F. The aortic root: comparison of measurements from ECG-gated CT angiography with transthoracic echocardiography. J Thorac Imaging. 2009;24(3):223–6.
Nathan DP, Xu C, Plappert T, Desjardins B, Gorman 3rd JH, et al. Increased ascending aortic wall stress in patients with bicuspid aortic valves. Ann Thorac Surg. 2011;92(4):1384–9.
Cooney JR, Ho VB. Answer to last month's radiology case and image: bicuspid aortic valve. Mil Med. 2006;171(8):iv–v.
Myerson SG. Heart valve disease: investigation by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2012;14:7.
Donato Aquaro G, Ait-Ali L, Basso ML, Lombardi M, Pingitore A, Festa P. Elastic properties of aortic wall in patients with bicuspid aortic valve by magnetic resonance imaging. Am J Cardiol. 2011;108(1):81–7.
Hope MD, Hope TA, Crook SES, Ordovas KG, Urbania TH, et al. 4D flow CMR in assessment of valve-related ascending aortic disease. JACC Cardiovasc Imaging. 2011;4(7):781–7.
Prokop EP, Palmer RF, Wheat Jr MW. Hydrodynamic forces in dissecting aneurysms. In-vitro studies in a Tygon model and in dog aortas. Circ Res. 1970;27(1):121–7.
Shores J, Berger KR, Murphy E, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. N Engl J Med. 1994;330(19):1335–41.
Ladouceur M, Fermanian C, Lupoglazoff JM, Edouard T, Dulac Y, et al. Effect of beta-blockade on ascending aortic dilatation in children with the Marfan syndrome. Am J Cardiol. 2007;99(3):406–9.
Selamet Tierney ES, Feingold B, Printz BF, Park SC, Graham D, et al. Beta-blocker therapy does not alter the rate of aortic root dilation in pediatric patients with Marfan syndrome. J Pediatr. 2007;150(1):77–82.
Gersony DR, McClaughlin MA, Jin Z, Gersony WM. The effect of beta-blocker therapy on clinical outcome in patients with Marfan’s syndrome: a meta-analysis. Int J Cardiol. 2007;114(3):303–8.
Nagashima H, Sakomura Y, Aoka Y, Uto K, Kameyama K, et al. Angiotensin II type 2 receptor mediates vascular smooth muscle cell apoptosis in cystic medial degeneration associated with Marfan's syndrome. Circulation. 2001;104(12 Suppl 1):I282–7.
Yetman AT, Bornemeier RA, McCrindle BW. Usefulness of enalapril versus propranolol or atenolol for prevention of aortic dilation in patients with the Marfan syndrome. Am J Cardiol. 2005;95(9):1125–7.
Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz 3rd HC. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358(26):2787–95.
Xiong W, Meisinger T, Knispel R, Worth JM, Baxter BT. MMP-2 regulates Erk 1/2 phosphorylation and aortic dilation in Marfan syndrome. Circ Res. 2012;110:e92–101.
Nagashima H, Aoka Y, Sakomura Y, Sakuta A, Aomi S, et al. A 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, cerivastatin, suppresses production of matrix metalloproteinase-9 in human abdominal aortic aneurysm wall. J Vasc Surg. 2002;36(1):158–63.
Elefteriades JA. Indications for aortic replacement. J Thorac Cardiovasc Surg. 2010;140(6 Suppl):S5–9.
Braverman AC. Aortic involvement in patients with a bicuspid aortic valve. Heart. 2011;97(6):506–13.
Milewicz DM, Dietz HC, Miller DC. Treatment of aortic disease in patients with Marfan syndrome. Circulation. 2005;111(11):e150–7.
Pape LA, Tsai TT, Isselbacher EM, Oh JK, O’gara PT, et al. Aortic diameter >or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation. 2007;116(10):1120–7.
Oliver JM, Gallegro P, Gonzalez A, Aroca A, Bret M, Mesa JM. Risk factors for aortic complications in adults with coarctation of the aorta. J Am Coll Cardiol. 2004;44(8):1641–7.
Rosenthal E. Coarctation of the aorta from fetus to adult: curable condition or life long disease process? Heart. 2005;91(11):1495–502.
Wang S, Zhang C, Zhang M, Liang B, Zhu H, et al. Activation of AMP-activated protein kinase α[alpha]2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med. 2012;18:902–10.
Agmon Y, Khandheria BK, Meissner I, Schwartz GL, Sicks JD, et al. Is aortic dilatation an atherosclerosis-related process? Clinical, laboratory, and transesophageal echocardiographic correlates of thoracic aortic dimensions in the population with implications for thoracic aortic aneurysm formation. J Am Coll Cardiol. 2003;42(6):1076–83.
Cambria RA, Gloviczki P, Stanson AW, Cherry Jr KJ, Bower TC, et al. Outcome and expansion rate of 57 thoracoabdominal aortic aneurysms managed nonoperatively. Am J Surg. 1995;170(2):213–7.
Muluk SC, Gertler JP, Brewster DC, Cambria RP, LaMuraglia GM, et al. Presentation and patterns of aortic aneurysms in young patients. J Vasc Surg. 1994;20(6):880–6.
McDonald ML, Smedira NG, Blackstone EH, Grimm RA, Lytle BW, Cosgrove DM. Reduced survival in women after valve surgery for aortic regurgitation: effect of aortic enlargement and late aortic rupture. J Thorac Cardiovasc Surg. 2000;119(6):1205–12.
Trimarchi S, Nienaber CA, Rampoldi V, Myrmel T, Suzuki T, Mehta RH, Bossone E, Cooper JV, Smith DE, Menicanti L, Frigiola A, Oh JK, Deeb MG, Isselbacher EM, Eagle KA, International Registry of Acute Aortic Dissection Investigators. Contemporary results of surgery in acute type A aortic dissection: the International Registry of Acute Aortic Dissection experience. J Thorac Cardiovasc Surg. 2005;129(1):112–22.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag London
About this chapter
Cite this chapter
Siddiqi, O.K., Klein, M.D. (2014). The Aortopathy of Bicuspid Aortic Valves. In: Bonser, R., Pagano, D., Haverich, A., Mascaro, J. (eds) Controversies in Aortic Dissection and Aneurysmal Disease. Springer, London. https://doi.org/10.1007/978-1-4471-5622-2_3
Download citation
DOI: https://doi.org/10.1007/978-1-4471-5622-2_3
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-5621-5
Online ISBN: 978-1-4471-5622-2
eBook Packages: MedicineMedicine (R0)