
Abstract
A wide variety of core–shell electrocatalysts have been investigated in recent years, showing benefits for the oxygen reduction reaction (ORR) in acid electrolytes. Particularly high values of activity per gram of Pt are often measured for core–shell systems in rotating disc electrode (RDE) measurements; however, fewer systems have been tested for performance and durability in membrane electrode assemblies (MEAs) under realistic proton exchange membrane fuel cell (PEMFC) conditions. This chapter discusses the various approaches, both electrochemical and chemical, used to prepare core–shell materials at both small and gram scales and highlights some of the methods used to assess the uniformity of the Pt shell and activity and durability. Available data from MEA testing is reviewed along with some of the implications on overall cost of the use of precious metals within the core. So far, a limited number of core–shell materials have been tested in MEAs, and these data tend to show a lower activity compared to testing at microgram scale under more idealized conditions, due to the combination of catalyst scale-up issues and differences in testing protocols, test conditions (temperature, pH), and catalyst instability. Thus, an increasing focus on the validation of the core–shell approach under realistic MEA test conditions is necessary, to demonstrate their true benefits as cost-effective cathode catalysts for PEMFCs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Oxygen Reduction Reaction
- Proton Exchange Membrane Fuel Cell
- Electron Energy Loss Spectroscopy
- Rotate Disc Electrode
- Oxygen Reduction Reaction Activity
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
19.1 Introduction
At present, the commercial large-scale use of PEM fuel cell technology, particularly in automotive applications, is limited by the relatively high cost and insufficient durability of the currently available electrocatalyst materials used for the oxygen reduction reaction (ORR) at the cathode [1–3]. The core–shell electrocatalyst approach deliberately deposits the high-cost, high-activity precious metal component (usually Pt) as a thin layer on the surface of a lower-cost substrate/nanoparticle core, as only atoms at the nanoparticle surface actively participate in the electrochemical reactions. This approach can provide significant benefits in precious metal thrifting, depending on the choice of core type, compared with Pt nanoparticle catalysts, as precious metal within the center of the nanoparticle (core) is replaced with lower-cost elements. To date a wide range of different types of core have been investigated including Pd, Pd alloys, Ir, Ir alloys, Au, Ag, and various first-row transition metals and their oxides [4–7]. In addition to cost reduction achieved by reducing/removing precious metal content within the particle core, the geometric and electronic effects induced by the underlying substrate core can enhance the activity of the Pt overlayer for the oxygen reduction reaction, leading to greater activity per mass of Pt than that expected based on thrifting alone.
Pt-based mass activities from 0.5 to over 2 A/mgPt at 900 mV [8–10], significantly exceeding the US Department of Energy Targets of 0.44 A/mgPt for automotive fuel cell applications, have been reported for a wide variety of core–shell electrocatalyst systems in measurements of oxygen reduction activity using the rotating disc electrode (RDE) setup, at room temperature in liquid electrolytes. These high activities also significantly exceed the typical mass activity of commercial carbon-supported Pt nanoparticles of around 0.15 A/mgPt [1]. However, these very high activities reported for core–shell materials are yet to be translated to such high performances in membrane electrode assemblies (MEAs), with the highest Pt-based mass activity reported in MEAs using US DoE-recommended protocols remaining a much more modest 0.3–0.4 A/mgPt [11, 12]. The discrepancies between the oxygen reduction activity observed in MEAs compared with that under more ideal conditions may be caused by a range of factors, such as differences in catalyst layer preparation and utilization, test protocol, temperature, and electrolyte type [13]. However, these discrepancies often appear larger in the case of core–shell catalyst materials compared to pure Pt and Pt alloy nanoparticles on carbon [14]. Electrochemical cycles and oxidizing conditions are expected to segregate core atoms toward the surface of the nanoparticles, disrupting the uniformity and stability of the Pt overlayer [15], and such processes may be more aggressive in the MEA environment. In addition, the highest activity values reported for core–shell material are, for example, prepared by electrochemical Pt deposition in situ on the RDE tip, while the preparation of sufficient material for MEA testing often requires adjustments to the methods used and tends to give lower activities both in MEA and RDE test environments, presumably as a consequence of poorer shell uniformity in the scaled-up samples [9, 16]. Therefore, multiple factors can influence the performance and durability of core–shell catalyst materials within the MEA environment, and these will be discussed in the subsequent sections of this chapter.
This chapter will focus mainly on core–shell materials prepared by the addition of a Pt monolayer (PtML) to a preformed core, by various approaches (chemical and electrochemical), and consider the uniformity, stability, and activity of materials of this structure. Chapter 18 in this book describes alternative methods used to produce a core–shell or surface-segregated structures, such as annealing treatments and dealloying/electrochemical cycling of base-metal-rich Pt alloy particles to produce a Pt-rich surface, so these approaches will not be discussed in detail here. As many reported core–shell electrocatalysts use alloys of alternative precious metals to Pt within the core, such as Pd, Au, and Ir, the implications of core type on catalyst cost reduction will also be considered.
19.2 Background
The preparation of core–shell catalysts for the ORR was pioneered by Adzic et al. Initially, this group investigated the preparation and behavior of Pt monolayers deposited on a range of different single crystal surfaces, prepared by galvanic displacement of a copper monolayer that had been deposited by Cu under potential deposition (Cu UPD). The effects of different underlying metal substrates on the O2 dissociation and OH formation energies on the Pt overlayer [17] and correlation with the oxygen reduction activity of these different surfaces were observed. This balance between ease of O2 dissociation and ease of OHads formation (required to react further with H+ and generate water) produces a typical volcano plot, illustrated in Fig. 19.1. The slight contraction in the Pt overlayer on a Pd substrate results in enhanced ORR activity for PtML/Pd(111) over Pt only, while PtML/Au(111) binds Oads too strongly and PtML/Ir(111) binds Oads too weakly for these examples to give enhanced activity.

Models of pseudomorphic monolayers of Pt on three different substrates inducing compressive strain (Ir(111) and Pd(111)) and expansive strain (Au(111)) and activation energies for O2 dissociation and OH formation on different PtML/X surfaces as a function of oxygen binding energy. Figure redrawn by the author with permission based on a diagram provided by R. Adzic, based on diagrams in [17]
Both geometric and electronic effects from the underlying substrate create shifts in the d-band center of the core–shell electrocatalyst compared to Pt only, which influence the activity for the ORR. The Pt monolayer deposition via Cu UPD displacement is described in the next section in comparison with other core–shell preparation methods.
This approach was then applied to produce Pt monolayer catalysts on Pd nanoparticles [18] and a wide range of different nanoparticle cores supported on carbon, as well as other single crystal surfaces. Various Pd alloy cores have been investigated, aiming to fine-tune the strength of the Pt–O bond in PtML/Pd y X(1 − y) (X = Co, Fe, Cu, Cr) and further enhance the activity for the ORR by progressing toward the peak of the activity volcano [8, 14]. The effects of mixing other elements such as Ir and Re into the Pt overlayer and the use of full and partial interlayers of Pd, Ir, and Au between the main part of the core and Pt overlayer have also been explored [6, 19, 20] and shown to influence both the activity and stability of the core–shell electrocatalysts.
Various electroless preparation methods have also been investigated, and the different synthetic approaches will be reviewed in the next section. Synthesis of shaped core particles such as tetrahedra, cubes, and nanowires has also been attempted to increase the fraction of the particle surface that is present as particularly active crystal facets, such as (111).
19.3 Overview of Core–Shell Preparation Methods, Characterization, and Factors Affecting Shell Uniformity
19.3.1 Pt Shell Deposition Methods: Electrochemical
The deposition of a Pt monolayer via the galvanic displacement of an under potentially deposited (UPD) Cu monolayer has been widely used to deposit Pt shells onto various substrate cores. Figure 19.2 illustrates a schematic of this process for the deposition of Cu onto a Pd@Co core followed by displacement of the Cu layer with Pt [21].

Schematic illustrating the process of Cu under potential deposition followed by galvanic displacement by Pt for a PdCo nanoparticle core [21]
The precursor core material, Pd or Pd alloy nanoparticles supported on carbon, for example, is dispersed in solvent + Nafion ionomer solution and coated onto a glassy carbon RDE and dried. The electrode is then immersed in a 50 mM H2SO4 + 50 mM CuSO4 solution and held at or cycled to potentials positive of the bulk Cu deposition potential of 0.36 V vs. RHE to achieve Cu monolayer deposition. Initial descriptions of this procedure describe removal and washing of the electrode before transferring to a 1.0 mM K2PtCl4 + 50 mM H2SO4 for the Pt galvanic displacement step [21] where Cu0 is spontaneously oxidized by Pt2+ to leave Pt on the nanoparticle surface and the removal of Cu. However, more recent reports use a slow injection of K2PtCl4 at lower concentrations and repeated cycles between 0.37 and 0.67 V in the presence of both the Cu and Pt salts. This is thought to lead to an increased uniformity of the Pt overlayer, as the repeated Cu UPD and stripping cycles slow the rate of Pt deposition and allow time for the surface diffusion of Pt adatoms so smooth layers may be formed [22]. A final sweep to 1 V is used to completely remove Cu. A combination of high-angle annular dark-field (HAADF) imaging and Pd electron energy loss spectroscopy (EELS) used to map the nanoparticles and presence of Pd within them suggested a 0.5 nm difference in radius consistent with the formation of a two-layer-thick Pt shell [22] (Fig. 19.3).

LHS diagram illustrates the cyclic voltammetry changes during repeated cycles of Cu-mediated Pt deposition onto Pd cores, while central diagram shows HAADF for a single particle and the RHS Pd EELS for the same particle, indicating difference in radius between entire particle and Pd-rich center [22]
In addition to the preparation of microgram samples on the RDE tip, this approach has been modified to scale up to gram-size quantities using an electrochemical cell where core nanoparticles are dispersed as powder in the electrolyte and a RuO2-coated Ti cylinder acts as the working electrode (WE) [9].
The uniformity of the Pt overlayer is of great importance in determining the activity and stability of the final catalyst material. While original studies were performed using uniform single crystal surfaces, small nanoparticles contain a range of particle sizes and coordination sites/surface facets. As described above, high-resolution TEM (HAADF) and EELS may be combined to map the structure of individual particles in some core–shell systems, but such analysis is limited by the small fraction of the total number of particles studied (and the EELS signal associated with core atoms). X-ray absorption spectroscopy (XAS) and X-ray diffraction (XRD) measurements provide information on the average coordination of different elements within the structure, giving a more global view of the electrocatalyst structure.
Recently XAS investigations were used to explore the Cu UPD process as a function of applied potential on carbon-supported Au nanoparticles [23]. Analysis of both the Cu and Au extended X-ray absorption fine structure (EXAFS) indicated only partial monolayer coverage at potentials where a complete Cu monolayer is predicted for single crystals. The structure of deposited Cu was found to be more consistent with Cu cluster formation at defects in the Au surface than the presence of a smooth monolayer due to lower than expected numbers of Au–Cu neighbors and Cu–Cu neighbors fitted in EXAFS analysis. The absence of a uniform monolayer of Cu on the nanoparticles implies that subsequent displacement of Cu by Pt is unlikely to result in the formation of a uniform Pt shell [23].
The presence of Pt nanoparticles/clusters compared to the desired uniform shell has also been inferred from changes to synchrotron XRD patterns under He and H2 for PtML/Pd core–shell samples via Cu UPD at gram scales [24]. The similar lattice constants of Pt (0.389 Å) and Pd (0.391 Å) result in XRD peak at similar positions; however, Pd nanoparticles absorb H2 resulting in an increase in lattice constant causing the XRD peak to shift to lower 2θ values. While a thin Pt monolayer on the Pd nanoparticle surface would be expected to also expand in the presence of H2, isolated Pt clusters on the Pd surface/detached Pt nanoparticles will be uninfluenced by the change to an H2 atmosphere. This technique has been used as a diagnostic to observe the benefits of modifications to a preparation procedure [24, 25]. The effect of reducing potentials and H absorption into the Pd core lattice have also been explored via EXAFS on PtML/Pd/C core–shell samples of varying Pt shell thickness prepared by a chemical deposition procedure [26]; these results are discussed in Sect. 19.3.2.
The Cu UPD approach has recently been applied to produce Pt–Pd core–shell nanowire catalysts [27]. Ultrathin Pd nanowires with diameters of ~2 nm were prepared and deposited onto XC-72R carbon, followed by surfactant removal by either acetic acid wash or a novel UV–ozone process, then selective CO adsorption to remove undesirable organic residue. Pt was then deposited via a Cu UPD galvanic displacement step using a sample of the treated Pd nanowires deposited on an RDE tip. At 900 mV, high mass activities for the ORR reaction of 1.83 A/mgPt and 1.47 A/mgPt were achieved for the ozone-treated and acid-treated samples, respectively. Pt monolayers have also been prepared on Pd tetrahedra using the Cu UPD galvanic displacement approach [28].
In addition to analysis by HAADF and elemental line scans, changes to the X-ray absorption near edge structure (XANES) may be investigated as a function of potential to investigate the structure of core–shell materials. Figure 19.4 shows results of these types of analysis for PtML/IrNi/C core–shell particles, where smaller changes in the XANES spectra vs. reference spectra at 0.41 V [plotted as (Δμ − Δμ 0.41)/Δμ 0.41 y axis, Fig. 19.4c] are observed for the core–shell compared to a Pt/C reference catalyst material. These data may be interpreted as a smaller change in Pt surface oxidation for the core–shell system, indicative of lower OH coverage and O binding energy, correlating with a higher activity for ORR vs. Pt only (Fig. 19.4d) [29].

(a) and (b) Dark-field TEM line scan and elemental mapping of PtML/IrNi core–shell particle, (c) variation in mass activity in RDE testing for RDE and g-scale Cu UPD scale up, and (d) Δμ XANES of PtML/IrNi compared with Pt-only nanoparticles [29]
Pt deposition via galvanic displacement of a Cu UPD layer has been achieved for a variety of nanoparticle cores/substrates such as Nb2O5 nanoparticles, Au nanoparticles, Ir and Ir alloy [29], and Pd and Pd alloys [11, 30]. The activity of some of these systems is summarized in Table 19.1 in Sect. 19.4 of this chapter. Direct deposition of Pt onto lower-cost transition metal cores such as Co and Ni is challenging, due to the highly oxidizable nature of these cores and the presence of a more noble metal as an interlayer, or preparation of a surface-segregated alloy core, have been explored. Voltage cycling of the core in the presence of Br− anions has been used to remove low coordination sites in Pd3Co cores and increase Pt shell uniformity and activity; however, Co is still leached from the core under operational conditions [31]. The use of Pd9Au alloy cores has been shown to significantly enhance the durability in MEA potential cycling tests (see Sect. 19.4) compared to PtML/Pd/C where substantial Pd dissolution was observed [11]. The same group has also reported similar benefits for a hydrogen adsorption/desorption core–shell preparation method (although full details are yet to be published) [12].
In all cases where direct comparisons are reported, the scale-up of Cu UPD-based preparations from RDE tip to gram scale results in an associated drop in measured activity [29, 30] (as illustrated in Fig. 19.4d) for the PtML/IrNi/C system and summarized in the table at the end of Sect. 19.4. Successful scale-up of electrochemical Pt overlayer deposition to the tens to hundreds of grams required for commercial MEA production remains a challenge with various groups investigating different electrochemical cell designs, additives, and procedures to improve shell uniformity in large batches of material. Japan-based company N.E. Chemcat recently licensed BNL core–shell catalyst scale-up technology including apparatus used in gram-scale manufacture [32].
19.3.2 Pt Shell Deposition Methods: Chemical
Core–shell catalyst materials may also be prepared by non-electrochemical routes. Core–shell nanoparticles may be produced in solution using colloidal methods, by sequential deposition of the core and shell components [33], or Pt layers may be deposited chemically or via displacement reactions onto preprepared core nanoparticles, but in contrast to approaches described in Sect. 19.3.1, no applied potential is required; typically core particles or colloidal core–shell particles are deposited onto carbon supports.
In colloidal preparation routes using various metal salts, either aqueous or organic solvents, a surfactant and a reducing agent can be used to produce nanoparticles and core–shell materials of varying types. A recent review by Peng covers many of these approaches and discusses how the choice of metal salt, solvent, surfactant, reducing agent, and reaction conditions may be used to control the size, shape, and metal shell growth mode [34] of various types of nanoparticles. Where surfactants are used to control electrocatalyst shape, size, and morphology, the steps required to later remove these organic species from the catalyst surface should also be considered, as such species may act to poison the surface sites.
An example of such a core–shell preparation is the synthesis of PtML/PdPt core–shell nanoparticles using an organic colloidal method. The PtPd cores were prepared by premixing palladium chloride (precursor metal salt) with sodium citrate (surfactant) and ethylene glycol (both solvent and reducing agent), followed by addition of hexachloroplatinate, pH adjustment, and heating at 160 °C in an autoclave for 8 h; then further Pt salt was added via a similar procedure to produce the Pt shell [33]. PtML/Pd3Fe/C [35] and PtML/Au/C core–shells [36, 37] have been prepared using similar types of approaches. The nanoparticles may be added to a carbon support after synthesis, or in some case, the carbon support is present during the autoclave/reflux step. The concentration and choice of surfactant used influence both the size and shape of the nanoparticles formed [34].
A chemical preparation route was also used to prepare Pt on Ag nanoparticles, where silver trifluoroacetate was mixed with oleylamine in isoamyl ether to synthesize Ag nanoparticles which were isolated, washed, then further reacted with Pt(acac)2 [38]. UV–vis spectroscopy was used to monitor the growth of Pt onto the Ag nanoparticle, with a gradual decrease in the intensity of the Ag plasmon band indicating increasing coverage by Pt nanoparticles. HAADF-STEM and elemental mapping were used to confirm the presence of an Ag core almost completely covered with multiple 3 nm Pt nanoparticle rather than a continuous Pt overlayer (Fig. 19.5). Two types of treatment were investigated to remove the surface-capping agents on the Pt–Ag nanoparticles: (1) reaction with acetic acid where the Pt–Ag nanoparticle structure remained intact and (2) heat treatment in air to 300 °C followed by exposure to a reducing atmosphere where XRD evidence suggests mixing to form an alloy had occurred; the effect of electrochemical leaching of Ag to generate hollow Pt spheres was also explored—see Sect. 19.3.3 [38].

Representative HAADF-STEM images and their corresponding elemental maps of Pt-on-Ag nanoparticles at (a–d) low and (e–h) high magnifications [38]
Pt–Pd bimetallic nanodendrites produced by a chemical route exhibit a somewhat similar morphology to the Pt–Ag samples described above; however, in the case of the Pt–Pd nanodendrites, Pt branches appear to grow epitaxially on faceted Pd nanocrystal seeds. Reduction of Na2PdCl4 with l-ascorbic acid in aqueous solution was used to generate these truncated octahedral Pd nanocrystals, followed by addition of K2PtCl4, nucleation of Pt on the Pd facets, and further growth of Pt on these nuclei to form the nanodendrites [39]. These systems showed ORR mass activity of 0.241 A/mgPt at 900 mV in RDE tests.
A series of PtML/Pd/C core–shell samples with varying nominal Pt shell thickness have also been prepared via a proprietary chemical method and explored using XAS and electrochemical techniques [26]. Analysis of EXAFS at the Pd K and Pt L3 edges for catalyst pellet samples revealed the expected increase in Pt–Pt and decrease in Pd–Pt and Pt–Pd neighbors with increasing nominal Pt coverage from 0.5 to 2 monolayers of Pt (calculated based on catalyst surface area of the Pd/C cores). Further EXAFS measurements under electrochemical control in liquid electrolytes revealed an increase in average Pd–Pd bond distance to 2.780 Å at 0.0 V for the 0.5 of a monolayer nominal coverage compared to 2.740 Å at 0.525 V (vs. RHE) associated with absorption of H atoms into the Pd lattice with low potentials. This effect was particularly pronounced for the 0.5 monolayer coverage sample but still present in a catalyst with nominal coverage of two monolayers of Pt, although to a lesser degree, implying higher Pt coverage creates a barrier or constraint to H adsorption into the Pd core [26].
Preparation of core–shell nanoparticles of PtML/Pd core on carbon has also been attempted via an electroless Cu deposition intermediate step, where preformed carbon-supported Pd nanoparticles are suspended in a solution containing CuSO4 and ethylenediaminetetraacetic acid (EDTA) to deposit Cu, then Pt salt added to galvanically displace the Cu. XPS and HAADF-STEM of the intermediate Cu/Pd nanoparticles confirmed the successful deposition of Cu via electroless deposition. The Pt and Pd distributions in the final sample tracked one another in TEM elemental analysis, and ~2.5 times Pt mass activity improvement was observed in RDE tests [40].
The behavior of PtML/Pd3Co nanoparticles on carbon prepared by (1) electrochemical (Cu UPD) and (2) chemically deposited (proprietary) methods has been compared using the same precursor core batch [41, 42]. Figure 19.6 shows STEM of a single particle and Pd EELS and HAADF signals from line scans in different directions for a scaled catalyst prepared by a chemical route, providing structural evidence for the formation of a Pt shell. Variations in the apparent thickness of the Pt shell may relate to thinning at the Pd3Co–carbon interface and thickening of the Pt shell on the opposing side of the particle. In addition, certain faces or defects on the Pd3Co nanoparticle core may be preferred sites for Pt deposition. Similar variations in Pt shell thickness from 0.5 to 1.2 nm are also reported for gram-scale PtML/Pd2Co/C samples prepared by Cu UPD (Pd:Co 2:1 atomic ratio cores). A comparison of activity for ORR in the RDE activity comparison of samples prepared on the RDE tip, gram-scale Cu UPD preparations, and gram-scale chemical route preparations shows ~1 A/mgPt for microgram-scale RDE tip preparations, falling to 0.7 A/mgPt for both scaled preparation routes, again highlighting the difficulties of effective core–shell catalyst scale-up using either electrochemical or chemical routes.

STEM micrograph of 10 nm PtML/Pd3Co/C particle and Pd EELS and HAADF signals from line scans, providing structural evidence for the formation of a Pt shell [41]
Preparation of Pt shells on Co cores has also been explored via a redox-transmetalation process, where Pt2+ in Pt(hfac)2 is reduced to Pt, while surface Co atoms on the Co core are oxidized via migration of the hfac ligand; repeated cycles of the reaction are required to generate the Pt shell [43] (hfac = hexafluoroacetylacetonate).
19.3.3 Other Approaches: Surface Segregation and Dealloying
Core–shell structures may also be achieved via annealing of alloy particles under particular atmospheres or dealloying of the more oxidizable elements from alloy nanoparticles. Electrochemical dealloying approaches are generally used to generate a Pt-rich shell and base-metal-rich core and are reviewed in detail in Chap. 18.
Annealing has been used to segregate particular elements to the surfaces of nanoparticle alloys, where relative atomic size, energies of formation, and the choice of gas environment influence which of the component elements segregates to the surface. Annealing of alloy catalyst powders in the gas phase in certain gas environments may be used to increase the Pt surface content for PtCo and other Pt-containing alloys. Annealing of PtCo alloy particles in CO gas results in segregation of Pt to the surface as a consequence of the increased absorption enthalpy of CO on Pt compared to Co [44]. These approaches have also been used to produce surface-segregated alloy core particles for subsequent Pt deposition via the electrochemical and chemical described in Sects. 19.3.1 and 19.3.2. Annealing in H2/Ar atmospheres is used to produce Ir-rich shells on IrFe [45] and IrNi [46] cores used to prepare PtML/IrX core–shell systems, with greater uniformity of Pt deposition observed on the Ir-rich surface. Electrochemical annealing in CO-saturated electrolyte has been shown to segregate Pt to the particle surface and enhance activity for the ORR reaction [44].
Electrochemical dealloying of Pt–Ag [38] and other Pt-metal core–shell systems has been used to produce hollow Pt spheres. This is a manifestation of the nanoscale Kirkendall effect whereby less stable core metal atoms are lost from the center of the particle via vacancy-mediated diffusion. Although this effect essentially dooms all core–shell structures to the gradual loss of the activity-enhancing elements from the core, hollow Pt sphere catalyst particles may be generated that retain a lattice contraction and enhanced activity over standard Pt electrocatalysts, as illustrated in Fig. 19.7 [47].

(LHS) (a and b) High-resolution STEM images of Pt hollow spheres and (c and d) line scans parallel and perpendicular to the lattice planes (RHS) comparison of the ESA of solid and hollow Pt spheres based on hydrogen desorption charges and ORR-specific and Pt mass activities measured at 0.9 V in RDE measurements [47]
Porous/hollow Pd–Cu nanoparticles have also been prepared on the RDE tip by selectively dealloying Cu from PdCu6/C, and subsequent Pt electrochemical deposition via Cu UPD used to create highly active surfaces for the ORR. Very high oxygen mass activities of ~2.8 A/mgPt were reported, and HAADF-TEM imaging confirms much lower Cu content in porous particle compared with the uncycled precursor [10].
Physical processes may also be used to deposit Pt onto various types of supports. An example of this type of approach is the preparation of Pt-metal monolayers supported on low-cost transition metal carbides, prepared by magnetron sputtering of Pt onto thin films of W and Mo carbides. While Pt monolayers were achieved on the thin-film electrode geometry used in this study, uniform deposition of Pt onto high surface area particulate materials or mesoporous structures by these methods remains challenging [48].
19.4 Performance and Stability of Core–Shell Catalysts
19.4.1 Performance of Core–Shell Materials in Membrane Electrode Assemblies and Comparison with Rotating Disc Electrode
It has been repeatedly observed by various researchers that the very high oxygen reduction activities reported for core–shell catalyst materials measured with the RDE setup are not reproduced within the MEA operating environment. While very high Pt-based mass activities in the range from 0.5 to 1.5 A/mgPt at 900 mV may be observed under rotating disc operating conditions depending on the core–shell type, the highest Pt-based mass activity reported in MEAs using US DoE-recommended protocols remains a much more modest 0.3–0.4 A/mgPt. A range of factors can contribute to this so-called RDE-MEA gap, and these will now be discussed.
Table 19.1 summarizes some of the available published data for oxygen mass activity values at 900 mV iR-free under RDE and MEA test conditions and where possible also shows the effect of catalyst scale-up vs. preparation on the RDE tip on the mass activity. The reported activities for carbon-supported conventional Pt and PtCo alloy catalysts are shown for comparison purposes. Many core–shell systems also exhibit high specific activities (activity corrected per active catalyst surface area typically reported as μA/cm2 Pt) when compared to state of the art Pt/C. Increasing catalyst particle size has been shown to enhance specific activity for carbon-supported Pt and Pt alloys, as larger particles have a lower proportion of edge and corner sites, fewer defects, and more faceted surfaces, leading to greater intrinsic activity (the particle size effect). However, as the mass activity [A/mgPt or A/mgPGM (PGM = platinum group metal)] is of greater importance in determining the usefulness of the core–shell approach in MEAs as a cost reduction measure, specific activity improvements are not discussed further here. Specific activity values for many of the core–shell systems can be found in the references cited in Table 19.1 and elsewhere in this chapter.
As alluded to in Sect. 19.3, the scale-up of electrochemically prepared core–shell catalyst materials from preparations on the RDE tip to gram-scale batches results in a decrease in the mass activity, from over 1 A/mgPt to ~0.5–0.7 A/mgPt for PtML/PdxCo and PtML/IrNi systems where comparative data is available. While no detailed information on the reasons for this discrepancy is provided, variations in the uniformity and Pt shell thickness between RDE and gram-scale preparations are likely to be responsible. In addition, activity values are subject to reasonably large error bars due to the small amounts of Pt present. While gram-scale preparations allow an accurate determination of the Pt assay of the final product via ICP-MS, the exact Pt content of samples prepared on the RDE tip may be less accurately known if calculated from cyclic voltammetry Cu stripping charges [22]. In some cases, activity is quoted per g of PGM as well as per g of Pt, and these values should scale by the same factor for samples prepared by the different methods, if the same precursor cores are used and the same Pt thickness (Pt to core atom ratio) is achieved. However, Pt/IrNi/C cores prepared on the RDE tip show activity of 1.4 A/mgPt and 0.78 A/mgPGM, while versions from the gram-scale preparations show 0.77 A/mgPt and 0.22 A/mgPGM, indicating some variations in overall composition between the RDE and gram-scale-prepared samples [16].
PtML/Pd3Co/C systems show similar RDE activity values for gram-scale samples prepared by either electrochemical or chemical routes, but samples prepared by both of these methods are lower in activity (0.7 A/mgPt at 900 mV) compared with the sample prepared on the RDE tip [41]. On transfer to the MEA, PtML/Pd3Co/C catalysts (prepared either electrochemically or chemically) show substantially lower activity than in the RDE environment (falling from 0.7 A/mgPt to ~0.15 A/mgPt) [42], and PtML/Pd/C (electrochemically prepared) samples showed a more modest halving of activity from 0.6 A/mgPt [9] to 0.3 A/mgPt [11]. During tests in liquid electrolytes, PtML/Pd3Co/C catalysts, both electrochemically and chemically prepared versions, showed at least 25 % Pd loss on exposure to 1 M H2SO4 at 80 °C (analogous to the MEA environment) via ICP-MS measurements on the electrolyte. However, no Pd loss was observed in 0.1 M HClO4 at RT (equivalent to RDE conditions) [14].
PtML/Pd/C also clearly shows loss of Pd in the MEA environment, via the formation of a Pd band within the membrane following voltage cycles (Sect. 19.4) [11]. The increased temperature and more aggressive environment within the MEA are therefore thought to contribute to the lower activities observed (compared to the RDE environment) as a consequence of the propensity of the less noble elements to dissolve from the core.
The variation in RDE and MEA activity is also partially due to the different testing protocols used. ORR activity measurements in the MEA often use the protocols defined by the US DOE, involving a polarization curve using pure oxygen, 150 kPaabs, 100 % RH at 80 °C, running the measurement from low to high current with 15 min holds at each measurement point [2]. Cell voltage is then corrected for ohmic resistance and current values corrected for the cathode Pt load, such that mass activity at 900 mV may be determined. In contrast, values quoted from RDE measurements are typically measured in O2-saturated 0.1 M HClO4 at RT, based on data measured during a sweep from low to high voltage at ~10 mV/s. Data are then corrected using the Koutechy–Levich equation and corrected for Pt mass and in some cases ohmic resistance and capacitative effects, to yield a mass activity at 900 mV [49, 50]. Aside from the obvious differences in temperature, pressure, and electrolyte, the difference in the direction of the potential sweep and time at each potential appear to alter the observed electrocatalyst activity, presumable due to variations in the degree of Oads/spectator species present on the catalyst surface.
Figure 19.8 illustrates the effect of hold time at each current density point in MEA tests using different nanostructured thin film (NSTF) MEAs, showing increased MEA mass activity at 900 mV using 5 s holds (matching RDE values more closely) compared with lower MEA mass activity using 1,050 s holds at each point in the polarization curve [13]; similar variations in activity between the MEA and RDE techniques may also be expected for core–shell catalyst materials.

Plot of RDE mass activity at 900 mV and 60 °C for nanostructured thin film (NSTF) catalysts (3 M) vs. fuel cell mass activity at 900 mV and 80 °C using (1) 5 s holds (black-filled squares) and (2) 1,050 s holds (unfilled squares) in MEA polarization curves [13]
Typically MEA performances for core–shell cathode catalysts report mass activity at 900 mV on oxygen, rather than the full polarization data, and minimal information is available on the performance of such systems using air at the cathode. Low absolute voltages are generally achieved at high current densities using pure oxygen [51], and even lower values might therefore be expected when using air, due to mass transport effects. Figure 19.9 illustrates the H2/air performance of a PtML/Pd/C core–shell system (scaled up using a spray pyrolysis approach by Cabot) in 50 cm2 MEAs [52] albeit at 30 psig (~200 kPa back pressure) rather than the lower operating pressure of 50 kPag recommended by the US DOE. Oxygen mass activity of 0.55 A/mgPt at 900 mV is reported in this testing.

Fuel cell testing of PtML/Pd/C catalyst from scale-up synthesis by Cabot, 50 cm2 MEAs, 80 °C, 100 % RH, 30 psig back pressure, and 10 min/point [52]
To the author’s knowledge, these data, along with the high activity achieved using PtML/AuNi5/C (same high-pressure test conditions) and MEA mass activities for PtML/Pd/C and PtML/Pd9Au/C, represent the best performances achieved so far in MEA testing of core–shell samples. These systems all show 2–3.5 times activity enhancement over Pt/C comparative samples in MEA tests.
Thus, a wide variety of core–shell systems have been reported as showing high ORR activity when prepared and tested in the RDE setup, including those already discussed and others such as PtML/AuNi0.5Fe/C [53], PtML/Pd3Fe/C [8], PtML/Au/C [54], PtML/Pd/Au/C [11], and PtML/Pd/IrCo/C [55] shown in Table 19.1 (PtML/Pd/core/C denotes the use of Pd interlayer). (Table 19.1 also shows activity values for Pt3Co/C in the RDE [56] and Pt3Co/C in MEAs [57] and data for Pt/C for reference purposes.) However, of these systems, only PtML/Pd/C, PtML/Pd9Au/C, and PtML/AuNi5/C have been successfully scaled up and translated to MEAs, and all show a reduction in activity compared to samples prepared at small scale. So for many published core–shell systems (Table 19.1), successful catalyst scale-up and the translation of high performances to the MEA environment are yet to be achieved. Further optimization is therefore required to translate the high oxygen mass activities observed in the RDE setup for other types of core–shell systems to the high mass activity and performance required under realistic conditions in MEAs.
19.4.2 Stability of Core–Shell Materials: MEA and RDE Environments
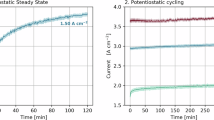
Although the stability of many core–shell electrocatalysts have been investigated via the RDE using multiple rapid voltage cycles at room temperature in 0.1 M HClO4, only a few systems have been assessed in MEAs at the cathode, by using voltage cycling to simulate extended operation in an automotive environment. Data reported by Sasaki et al. show the best durability reported in MEA voltage cycling to date using PtML/Pd/C and PtML/Pd9Au/C core–shell materials [11, 12]. Figure 19.10 shows results from these tests, illustrating the changes in mass activity at 900 mV, O2, 50 kPaabs, 100 % RH using the DoE protocol, at different stages of a cycling test, using cycles from 0.7 to 0.9 V square wave (DOE protocol). Figure 19.10a illustrates results from 2010 [12, 58], while Fig. 19.10b shows more recent data extending testing to an increased number of cycles (100,000) and improvements to activity and stability for PtML(H)Pd/C, an improvement to the core–shell catalyst preparation procedure yet to be disclosed [12].

All these core–shell electrocatalysts show an improved Pt-based mass activity over Pt only which is maintained throughout the cycling test. However, the mass activities/g total PGM are not disclosed; therefore, the overall cost reduction that might be expected from the use of these core–shell materials in MEAs is not clear.
Postmortem analysis of MEA samples using PtML/Pd/C after 100,000 potential cycles showed a Pd band precipitated within the membrane, a clear indication of Pd dissolution from the cores of the nanoparticles during operation [11]. This phenomenon had not been previously reported for core–shell electrocatalysts but is well documented for cycled MEAs containing Pt/C cathodes, resulting from Pt dissolution during voltage cycles and re-precipitation within the membrane on encountering hydrogen diffusing from the anode. In the case of PtML/Pd/C, the loss of Pd from the core is described as a means of protecting the Pt shell via sacrificial dissolution, and this observation is consistent with the higher diffusivity of less stable species from the core, a nanoscale Kirkendall effect [47]. Segregation of Pd and other less noble metals to the surface of a core–shell material in the presence of Oads is also consistent with findings of the modeling studies by Balbuena et al. [15].
The use of a PtML/Pd9Au core–shell system resolved these stability issues, with no Pd band observed within the membrane and hence minimal dissolution of Pd. Alloying of Pd with Au is thought to stabilize Pd by causing a positive shift of the Pd oxidation potential, with less Pd–OH formation observed in cyclic voltammetry and EXAFS spectra of the Pd9Au/C cores compared to Pd/C [11]. Excellent stability of the Pt-based mass activity was achieved with a mass activity in MEAs of 0.2 A/mgPt remaining after 200,000 voltage cycles in MEAs; however, mass activity per g PGM is not quoted, so the possible effect of the additional PGM used on the overall catalyst cost is not clear.
So far limited durability data exists on core–shell catalysts within MEAs, and often cycling tests in the RDE setup are used to investigate catalyst stability. Recent RDE testing on PtML/IrFe and PtML/IrNi shows minimal change in ORR activity following 30,000 and 50,000 cycles, respectively, 0.6–1.0 V at 50 mV/s, RT, 0.1 M HClO4 electrolyte [29, 45]. HAADF-STEM line scans of PtML/IrNi after 50,000 cycles also show that Ni is retained within the core of the particles after cycling, providing good evidence of the stability of the core under the RDE test conditions, attributed to the use of precursor cores with a Ni-rich center and Ir-rich outer region [29] (Fig. 19.11).

HAADF-STEM and Pt, Ir, and Ni line scans for PtML/IrNi core–shell catalyst before testing (a and c) and after 50,000 cycles (b and d) 0.6–1.0 V at 50 mV/s, RT, 0.1 M HClO4 electrolyte [29]
RDE testing of PtML/Pd3Co catalysts has shown varying degrees of stability depending on the preparation route used. Adzic et al. observed improved stability after applying a bromide treatment to remove low coordination sites on the Pd3Co core before Pt deposition. No change in ORR and surface area was observed after 25,000 cycles between 0.6 and 1.0 V using Br− treated cores compared to a substantial shift in half wave potential after only 30 mV after 5,000 cycles on untreated examples. Other testing using treated PtML/Pd3Co/C (no Br− treatment) catalyst [41, 42]-coated gas diffusion electrodes in liquid electrolytes was performed under two different test conditions, (1) 0.1 M HClO4, RT, and (2) 80 °C 1 M H2SO4, using 0.4–0.95 V square wave, 10 s per potential in both cases [14, 59]. Under the RDE-type test conditions, only minimal dissolution of Pd into the electrolyte (~10 % Pd loss) was observed, and HAADF-TEM measurements showed that the core–shell structure remained intact (bright outer regions were observed around the core due to heavier Pt atoms). However, under the test conditions in (1) 80 °C, 1 M H2SO4, substantial Pd dissolution (around 75 % removed after 1,000 cycles) occurred and TEM showed the formation of hollow spheres and some alloy, showing greater disruption to the structure in test (2) thought to be closer to MEA test conditions [59]. Significant loss of Co occurred under both test conditions, and similar ECA change and metal loss with cycles were observed using PtML/Pd3Co catalysts prepared by either the Cu UPD route or via a chemical method [14].
The substantial Co loss and disruption of the core–shell structure in more aggressive environment are consistent with expectations that less noble elements are prone to leaching when used in the core. Furthermore, the significant differences in Pd dissolution found between the RDE- and MEA-type test conditions imply that some caution may be required when extrapolating the stability of core–shell systems from the RDE to MEA. Therefore, more MEA activity and stability testing of the various types of core–shell systems is required to probe their true stability under realistic test conditions.
19.5 Use of Core–Shell Approach to Reduce Catalyst’s Precious Metal Content and Cost
Many of the core–shell systems discussed so far in this chapter use precious metals such as Pd, Au, and Ir within the core or as interlayers within the structure. Clearly, where alternative metals (Au, Ag, or PGMs such as Pd or Ir) are used to replace some of the Pt within electrocatalysts used for the ORR, the impact of this on the overall catalyst cost should also be considered. The activity targets for PEMFC cathodes of 0.44 A/mgPt at 900 mV are based around the requirement for a 0.2 gPt/kW linked to a target cost per kW for large-scale automotive MEA production. Various studies have shown that the world platinum resources are sufficient to support the large-scale use of fuel cell electric vehicles, provided the targets relating to the Pt content in fuel cell stacks are achieved and PGM producers have sufficient visibility of the increase in demand [60]. Studies by TIAX concluded sufficient Pt availability for long-term demands of automotive fuel cells, assuming a gradual increase in the number of vehicles over several decades, a 95 % efficient recycling of PGM, and an achievement of a 0.2 gPt/kW by 2025 in their calculations [61]. However, as a large number of reported core–shell systems achieve reductions in Pt content by including alternative precious metals within the core, the impact of this on the overall catalyst, MEA, and automotive fuel cell stack should also be taken into consideration.
Where Pt is replaced by alternative precious metals, activity values have often been quoted as A/mgPGM as well as A/mgPt; however, this type of analysis does not reflect any cost differences between the different precious metals. The factors affecting metal prices in the market are complex, with supply and demand varying due to the interplay of the various technological and industrial uses (particularly vehicle emission control catalysts in the case of Pt and Pd), purchasing for investment purposes, use in jewelry, etc., against the availability and abundance of the different metals from primary refining of ore and also from recycling. The precious metals Pd, Ir, Ru, and Rh are produced as by-products of Pt mining. Figure 19.12 shows the relative costs of Pt, Pd, Ir, Ru [62], and Au [63] precious metals over the last 10 years. A plot of the Rh price is not included in Fig. 19.12, as plotting Rh price on the same axes limits the visibility of subtle changes in the costs of the other precious metals, due to the significantly higher cost of Rh (ranging from two to five times that of Pt depending on the time scale viewed). Also significant use of Rh in ORR core–shell catalysts has not been reported to date, so discussion of the implications of Rh use is less relevant here.

Average monthly price of Pt, Pd, Ir, Ru, and Au precious metals since Jan 2002. Data from http://www.platinum.matthey.com/pgm-prices/ and http://www.kitco.com. One Troy oz = 31.1035 g of metal [62, 63]
Figure 19.12 shows some significant trends. Firstly, an overall general increase in price is observed for all the metals with time, in line with rising industrial demand and increasing GDP (gross domestic product), broken by a sharp drop in July 2008 linked to the global recession. The global downturn had a significant effect on the prices of Pt and Pd due to the drop in industrial metals’ usage, particularly a drop in automotive manufacturing, reducing demand for Pt and Pd used in catalysts for vehicle exhaust cleanup and hence impacting the price. Over the time period shown, changes in Pd price tend to track those of Pt but with Pd prices typically around 70 % lower. Au prices remained robust during the recession, as a result of purchasing for investment purposes [64].
The factors affecting the costs of Ru and Ir are more subtle. Ru is used in the electronics industry in chip resistors, flat screen displays, and the hard disc manufacturing process. In 2006–2007, an increase in Ru demand from the electronics industry due to changes in the technology in hard disc drives resulted in a price spike, but only for a limited period. Once purchased or leased, a large proportion of Ru metal used in hard disc manufacturing is subsequently recycled, and minimal amounts are actually present in the consumer products, so increased demand was only temporary and prices have therefore decreased again in subsequent years. Ir is used in catalysts used for acetic acid manufacture and electrochemical use in the chlor-alkali process and to make crucibles for producing high-quality sapphire single crystals used in the manufacture of LED televisions. Increased demand for Ir as a consequence of consumer demand for LED televisions and refitting of the Chinese chlor-alkali industry have both influenced the substantial increase in Ir price over the last 2 years [64].
While Pd prices have tended to retain a relative value of ~30 % with respect to the price of Pt over the time period shown in Fig. 19.9, the relative prices of other precious metals with respect to Pt are more variable. Table 19.2 shows the costs of various precious metals with respect to Pt over the last 1-, 5-, and 10-year periods calculated from the data in Fig. 19.12 and prices for Rh over the same time period.
In addition to the A/mgPt and A/mgPGM typically quoted, the relative precious metal cost values can be used to determine an activity per relative PGM cost for different core–shell materials. While Pd cost remains around 0.3 or 0.4 times that of Pt, use of Pd within the core of core–shell catalysts appears viable, depending on the performance achieved and the metal amount used. Pd is produced as a by-product of Pt mining in South Africa and Ni mining in Russia [64]. Newer mined deposits in South Africa in the UG2 reef in the Eastern Bushveld contain a higher proportion of Pd:Pt compared to previously exploited Merensky Reef ores which are higher in Pt content [65]. However, in the past (1999–2001) Pd prices have maintained parity or exceeded those of Pt, as a consequence of Pd supply issues and changes in the technology used in car exhaust catalysts to use more Pd. Also in recent years, Pd prices have been influenced by Russian state stocks being supplied into the markets, and these stocks are becoming depleted. So it is likely that Pd demand will exceed supply in coming years leading to increasing Pd prices. So although the MEA mass activities of ~0.3 A/mgPt achieved using PtML/Pd and PtML/Pd9Au[11] are promising, once Pd and Au content/cost are included and considered, these become more similar in activity/PGM cost to conventional PEMFC catalysts.
In contrast to Pd and Pt, there is a lower natural abundance of Ir in metal ores [66], so using a substantial amount of Ir within the core of a core–shell catalyst would be likely to increase Ir cost and make such a catalyst inappropriate as a long-term use in automotive PEMFC systems. The effect of Ir content on likely cost of both alloy and core–shell catalysts should therefore be scrutinized as well as the activity per mgPt.
Although the complex interplay of various factors in the precious metal market does not allow simple predictions of future costs based on abundance alone, the price increase in Ir in recent years due to increased use in LED manufacturing for flat screen televisions illustrates the likely outcome of increased Ir demand on the metal price; the Ru price spike in 2006–2007 associated with increased use in hard disc manufacture is another example. Increasing demand for Pd would also lead to increasing price, affecting the long-term viability of using Pd as a core for core–shell catalyst materials. Overall, minimizing the amount of total PGM used within core–shell materials and achieving a true reduction in overall precious metal content appear to be the best way forward in the long term. However, alternative nonprecious metal lower-cost core constituents are typically of lower stability in the electrochemical environment.
19.6 Outlook
A wide variety of core–shell systems have been explored for use at the PEMFC cathode, and many systems show great promise in the form of high activity when measured in the RDE environment (see Table 19.1). Often these high activity values relate to samples made at small scales or in situ on the RDE tip, and only a few groups have generated data using scaled materials tested in the MEA environment. Notable efforts by Sasaki and colleagues to scale the Cu UPD galvanic displacement method for core–shell preparation and perform long-term MEA testing have shown promising results using PtML/Pd/C and PtML/Pd9Au/C. Otherwise there is so far a conspicuous lack of other reliable MEA test data on these systems. Differences in activity are observed between small-scale core–shell catalyst preparations and the larger-scale batches necessary for MEA testing (Table 19.1) highlighting the difficulties associated with catalyst scale-up. The long-term effect on overall catalyst cost of using significant amounts of alternative precious metals such as Pd, Ir, or Au within the cores of core–shell catalysts must also be considered. Also, MEA activity and durability testing may reveal different performance and aging of catalyst materials compared to tests under more ideal, milder conditions. Thus, further development of scalable materials and evaluation in MEAs are needed to demonstrate the full promise of core–shell systems for use in the PEMFC environment.
References
Gasteiger HA, Kocha SS, Sompalli B, Wagner FT (2005) Activity benchmarks and requirements for Pt, Pt-alloy and non-Pt oxygen reduction catalysts for PEMFCs. Appl Catal B 46:9–35
Borup R et al (2007) Scientific aspects of polymer electrolyte membrane fuel cell durability and degradation. Chem Rev 107:3904–3951
Adzic RR, Zhang J, Sasaki K, Vukmirovic MB, Shao M, Wang JX, Nilekar AU, Mavrikakis M, Valerio JA, Uribe F (2007) Platinum monolayer fuel cell electrocatalysts. Top Catal 46:249–262
Adzic RR, Lima FHB (2009) Platinum monolayer oxygen reduction electrocatalysts, Chapter 1. In: Vielstich W, Yokokawa H, Gasteiger HA (eds) Handbook of fuel cells, fundamentals, technology and applications, Advances in electrocatalysis, materials diagnostics and durability, vol 5, part 1. Wiley, West Sussex
Cai Y, Adzic RR (2011) Platinum monolayer electrocatalysts for the oxygen reduction reaction; improvements induced by surface and subsurface modification of cores. Adv Phys Chem 11:16. doi:10.1155/2011/530397, Article ID 530397
Sasaki K, Zhang L, Adzic RR (2008) Niobium oxide-supported platinum ultra-low amount electrocatalysts for oxygen reduction. Phys Chem Chem Phys 10:159–167
Shao M, Sasaki K, Liu P, Adzic RR (2007) Pd3Fe and Pt monolayer-modified Pd3Fe electrocatalysts for oxygen reduction. Z Phys Chem 221:1175–1190
Sasaki K, Wang JX, Naohara H, Marinkovic N, More K, Inada H, Adzic RR (2010) Recent advances in platinum monolayer electrocatalysts for the oxygen reduction reaction: scale-up synthesis, structure and activity of Pt shells on Pd cores. Electrochim Acta 55:2645–2652
Shao M, Shoemaker K, Peles A, Kaneko K, Protsailo L (2010) Pt monolayer on porous Pd-Cu alloys as oxygen reduction electrocatalysts. J Am Chem Soc 132:9253–9255
Sasaki K, Naohara H, Cai Y, Choi YM, Liu P, Vukmirovic MB, Wang JX, Adzic RR (2010) Core-protected platinum monolayer shell high stability electrocatalysts for fuel-cell cathodes. Angew Chem Int Ed 49:1–7
Sasaki K, Naohara H, Choi YM, Cai Y, Chen WF, Marinkovic N, Liu P, Adzic RR (2011) Enhanced stability of platinum monolayer on palladium-gold nanoparticle electrocatalysts in long-term fuel cell tests. In: Abs 1056 220th meeting of the Electrochemical Society
Debe M (2009) Advanced cathode catalysts and supports for PEM fuel cells, DoE Hydrogen Program Review Project FC_17
Murthi V (2010) Highly dispersed alloy catalysts for durability, DoE Hydrogen Program Reviews FC-002
Ramirez-Caballero GE, Ma Y, Callejas-Tovar R, Balbeuna P (2010) Surface segregation and stability of core-shell alloy catalysts for oxygen reduction in acid medium. Phys Chem Chem Phys 12:2209–2218
Kuttiyeil K, Sasaki K, Adzic RR (2010) Bimetallic Ni-Ir core platinum monolayer shell electrocatalysts for the O2 reduction reaction. In: Abs 744, 218th meeting of the Electrochemical Society
Zhang J, Vukmirovic MB, Xu Y, Mavrikakis M, Adzic RR (2005) Controlling the catalytic activity of platinum monolayer electrocatalysts for oxygen reduction with different substrates. Angew Chem Int Ed 44:2132–2135
Zhang J, Mo Y, Vukmirovic MB, Klie R, Sasaki K, Adzic RR (2004) Platinum monolayer electrocatalysts for O2 reduction: Pt monolayer on Pd(111) and on carbon-supported Pd nanoparticles. J Phys Chem B 108:10955–10964
Zhang J, Shao MH, Sasaki K, Vukmirovic MB, Uribe F, Adzic RR (2006) Platinum and mixed platinum metal monolayer fuel cell electrocatalysts: design, activity and long-term performance stability. ECS Trans 3(1):31–36
Xing Y, Cai Y, Vukmirovic MB, Zhou W-P, Karan H, Wang JX, Adzic RR (2010) enhancing oxygen reduction reaction activity via Pd-Au alloy sublayer mediation of the Pt monolayer electrocatalysts. J Phys Chem Lett 1:3238–3242
Shao M, Sasaki K, Marinkovic NS, Zhang L, Adzic RR (2007) Synthesis and characterisation of platinum monolayer oxygen-reduction electrocatalysts with Co-Pd core-shell nanoparticle supports. Electrochem Commun 9:2848–2853
Wang JX, Inada H, Wu L, Zhu Z, Choi Y, Liu P, Zhou W-P, Adzic RR (2009) Oxygen reduction on well-defined core-shell nanocatalysts: particle size, facet and Pt shell thickness effects. J Am Chem Soc 131:17298–17302
Price SWT, Speed JD, Kannan P, Russell AE (2011) Exploring the first steps in core-shell electrocatalyst preparation: in situ characterisation of the underpotential deposition of Cu on supported Au nanoparticles. J Am Chem Soc 133:19448–19458
Shao M, Humbert M, Smith B (2011) Synthesis and characterisation of core-shell electrocatalysts for fuel cells. In: Abs 941, 220th meeting of the Electrochemical Society
Humbert M, Smith B, Wang Q, Ehrlich S, Shao M (2012) Synthesis and characterization of palladium–platinum core–shell electrocatalysts for oxygen reduction. Electrocatalysis 3:298–303
Russell AE, Tessier BC, Wise AM, Rose A, Price SWT, Richardson PW, Ball SC, Theobald B, Thompsett D, Crabb EM (2011) In situ XAS studies of core-shell PEM fuel cell catalysts: the opportunities and challenges. ECS Trans 41(1):55–67
Koenigsmann C, Santulli AC, Gong K, Vukmirovic MB, Zhou W-P, Sutter E, Wong SS, Adzic RR (2011) Enhanced electrocatalytic performance of processed, ultrathin supported Pd-Pt core-shell nanowire catalysts for the oxygen reduction reaction. J Am Chem Soc 133:9783–9795
Gong K, Sasaki K, Vukmirovic MB, Adzic RR (2011) Tetrahedral palladium nanocrystals – a new support of platinum monolayer electrocatalysts having improving activity and stability in oxygen reduction reaction. In: Abs 1941, 219th meeting of the Electrochemical Society
Kuttiyeil K, Sasaki K, Choi Y, Su D, Liu P, Adzic RR (2012) Bimetallic IrNi core platinum monolayer shell electrocatalysts for the oxygen reduction reaction. Energy Environ Sci 5:5297–5304
Zhou W-P, Sasaki K, Su D, Zhu Y, Wang JX, Adzic RR (2010) Gram-scale-synthesised Pd2Co-supported Pt monolayer electrocatalysts for oxygen reduction reaction. J Phys Chem C 114:8950–8957
Cai Y, Ma C, Zhu Y, Wang JX, Adzic RR (2011) Low coordination sites in oxygen-reduction electrocatalysis: their roles and removal. Langmuir 27(13):8540–8547
http://www.greencarcongress.com/2012/01/chemcat-20120103.html
Wu Y-N, Liao S-J, Liang Z-X, Yang L-J, Wang RF (2009) High performance core-shell PdPt@Pt/C catalysts via decorating PdPt alloy cores with Pt. J Power Sources 194(2):805–810
Peng Z, Yang H (2009) Designer platinum nanoparticles: control of shape, composition in alloy, nanostructure and electrocatalytic property. Nano Today 4:143–164
Wang H, Ji S, Wang W, Linkov V, Pasupathi S, Wang R (2012) Pt decorated PdFe/C: extremely high electrocatalytic activity for methanol oxidation. Int J Electrochem Sci 7:3390–3398
Peng Z, Yang H (2009) PtAu bimetallic heteronanostructures made by post-synthesis modification of Pt-on-Au nanoparticles. Nano Res 2:406–415
Kristian N, Wang X (2008) Ptshell-Aucore/C electrocatalyst with a controlled shell thickness and improved Pt utilisation for fuel cell reactions. Electrochem Commun 10:12–15
Peng Z, Wu J, Yang H (2010) Synthesis and oxygen reduction electrocatalytic property of platinum hollow and platinum-on silver nanoparticles. Chem Mater 22:1098–1106
Lim B, Jiang M, Camargo PHC, Cho EC, Tao J, Lu X, Zhu Y, Xia Y (2009) Pt-Pd bimetallic nanodendrites with a high activity for oxygen reduction. Science 5:1302–1305
Choi I, Ahn SH, Kim JJ, Kwon OJ (2011) Preparation of Ptshell-Pd core nanoparticle with electroless deposition of copper for polymer electrolyte membrane fuel cell. Appl Catal B Environ 102:608–613
Ball S, Burton SL, Fisher J, O’Malley R, Tessier B, Theobald BRC, Thompsett D, Zhou WP, Su D, Zhu Y, Adzic RR (2009) Structure and activity of novel Pt-core-shell catalysts for the oxygen reduction reaction. ECS Trans 25(1):1023–1036
Ball S, Burton SL, Christian E, Davies A, Fisher J, O’Malley R, Passot S, Tessier B, Theobald BRC, Thompsett D (2009) Activity and stability of Pt monolayer core-shell catalysts. ECS Trans 25(1):1011–1022
Park J, Kim MG, Jun Y, Lee JS, Lee W, Cheon J (2004) Characterisation of superparamagnetic “core-shell” nanoparticles and monitoring of their anisotropic phase transition to ferromagnetic “solid-solution” nanoalloys. J Am Chem Soc 126(29):9072–9078
Mayrhofer KJJ, Hanzlik M, Arenz M (2009) The influence of electrochemical annealing in CO saturated solution on the catalytic activity of Pt nanoparticles. Electrochim Acta 54(22):5018–5022
Sasaki K, Kuttiyiel KA, Su D, Adzic RR (2011) Platinum monolayer IrFe core-shell nanoparticle electrocatalysts for the oxygen reduction reaction. Electrocatalysis 2:134–140
Sasaki K, Kuttiyiel KA, Barrio L, Su D, Frenkel AI, Marinkovic N, Mahajan D, Adzic RR (2011) Carbon-supported IrNi core-shell nanoparticles: synthesis, characterisation and catalytic activity. J Phys Chem C 115:9894–9902
Wang JX, Ma C, Choi Y-M, Su D, Zhu Z, Liu P, Si R, Vukmirovic MB, Zhang Y, Adzic RR (2011) Kirkendall effect and lattice contraction in nanocatalysts: a new strategy to enhance sustainable activity. J Am Chem Soc 133:13551–13557
Esposito D, Hunt ST, Kimmel YC, Chen JG (2011) A new class of electrocatalyst for hydrogen production from water electrolysis: metal monolayers supported on low-cost transition metal carbides. J Am Chem Soc 134:3025–3033
Paulus UA, Schmidt TJ, Gasteiger HA, Behm RJ (2001) Oxygen reduction on a high-surface area Pt:Vulcan carbon catalyst: a thin-film rotating ring-disk electrode study. J Electroanal Chem 495:134–145
van der Vliet D, Strmcnik DS, Wang C, Stamenkovic V, Markovic NM, Koper MTM (2010) On the importance of correcting for the uncompensated ohmic resistance in model experiments of the oxygen reduction reaction. J Electroanal Chem 647:29–34
Zelenay P (2008) Advanced cathode catalysts, DoE Hydrogen Program Review Project FC_003
Zelenay P (2009) Advanced cathode catalysts, DoE Hydrogen Program Review Annual Project Progress Report V.E.5
Gong K, Su D, Adzic RR (2010) Platinum-monolayer shell on AuNi0.5Fe nanoparticle core electrocatalyst with high activity and stability for the oxygen reduction. J Am Chem Soc 132(41):14364–14366
Shao M, Peles A, Shoemaker K, Gumalla M, Njoki PN, Luo J, Zhong C-J (2010) Enhanced oxygen reduction activity of platinum monolayer on gold nanoparticles. J Phys Chem Lett 2:67–72
Gong K, Chen W-F, Sasaki K, Su D, Vukmirovic MB, Zhou W, Izzo E, Perez-Acosta C, Hirunsit P, Balbuena P, Adzic RR (2009) Platinum-monolayer electrocatalysts: palladium interlayer on IrCo alloy core improves activity in oxygen reduction reaction. J Electroanal Chem 649(1–2):232–237
Chen S, Sheng W, Yabuuchi N, Ferereira PJ, Allard LF, Shao-Horn Y (2009) Origin of oxygen reduction reaction activity on “PtCo” nanoparticles: atomically resolved chemical compositions and structures. J Phys Chem C 133:1109–1125
Matsutani K, Hayakawa K, Tada T (2010) Effect of particle size of platinum and platinum-cobalt catalysts on stability against load cycling. Platinum Met Rev 4:223–232
Naohara H, Sasaki K, Adzic RR (2010) Fuel cell performance and long term stability of platinum monolayer electrocatalysts. In: 216th ECS meeting, Abs 980
Ozkaya D, Ball S, Davies A, Fisher J, O’Malley R, Theobald BRC (2010) Unpublished work
Jaffray C, Hards G (2003) Precious metal supply requirements. In: Vielstich W, Gasteiger HA, Lamm A (eds) Hand book of fuel cells – fundamentals, technology and applications, Fuel cell technology and applications. Wiley, New York
Tiax (2003) Platinum availability and economics for PEMFC commercialisation, Report to US Department of Energy, http://www1.eere.energy.gov/hydrogenandfuelcells/pdfs/tiax_platinum.pdf
Pt, Pd, Ru, Ir and Rh prices taken from http://www.platinum.matthey.com/pgm-prices/
Au prices taken from http://www.kitco.com
PGM market reviews, from 1999 to present, http://www.platinum.matthey.com/publications/pgm-market-reviews/
Cawthorn RG (2010) The platinum group element deposits of the Bushveld Complex in South Africa. Platinum Met Rev 54(4):205–215
African Rainbow Minerals (2006) Annual report (showing values of PGM split in the two rivers platinum mine). http://www.arm.co.za/im/files/annual/2006/ar_2006.pdf, p 23
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag London
About this chapter
Cite this chapter
Ball, S. (2013). Core–Shell Catalysts in PEMFC Cathode Environments. In: Shao, M. (eds) Electrocatalysis in Fuel Cells. Lecture Notes in Energy, vol 9. Springer, London. https://doi.org/10.1007/978-1-4471-4911-8_19
Download citation
DOI: https://doi.org/10.1007/978-1-4471-4911-8_19
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-4910-1
Online ISBN: 978-1-4471-4911-8
eBook Packages: EnergyEnergy (R0)