
Abstract
Hedgehog (HH) and protein kinase C (PKC) signaling pathways are critically involved in embryonic development, stem cells, and cancer cells. Recent evidence has shown the regulation of these phenotypes by the crosstalk between the HH and PKC pathways. Among the PKC family, PKCα and PKCδ are important isoforms involved in HH pathway regulation. In this review, we summarize the current state regarding the role of PKC signaling in mediating HH pathways.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Hedgehog (HH) signaling plays an important role in human cancers through promoting cancer cell growth and proliferation of tumor stem cells. The protein kinase C (PKC) family, which comprises at least ten isoforms, has been shown to exert multiple biological functions, including adhesion, secretion, proliferation, differentiation, and apoptosis. In this review, we will summarize recent findings demonstrating crosstalk between the HH and PKC signaling pathways during development, in stem cells and in malignant and nonmalignant cells. The role of MEK/ERK pathway in this crosstalk will also be discussed. The integration of these signaling pathways in the regulation of HH signaling provides for potentially new targets in the control of HH-dependent tumorigenesis.
Protein Kinase C
The protein kinase C (PKC) family represents a group of widely distributed serine–threonine kinases [1]. Eleven PKC isoforms have been identified and divided into three major classes: the conventional PKCs (α, βI, βII, and γ), the novel PKCs (δ, ε, θ, and η), and the atypical PKCs (ζ and ι/λ) [2]. The conventional PKC isoforms have an intact C1 diacylglycerol/phorbol ester binding domain and C2 calcium-binding domain and thus require phospholipids and calcium for activation [2]. The novel PKCs do not require calcium for their activation [2]. The atypical PKCs can be activated in the absence of diacylglycerol and calcium [2]. Upon activation, PKC isoforms often translocate to particular subcellular compartments, including the plasma membrane, Golgi complex, nuclear membrane and nucleus [3]. PKC isoforms play important roles in signal transduction of various physiological stimuli, including growth factors, hormones, and transmitters, thus PKCs are involved in many cellular processes [4].
Functions of Different PKC Isoforms in Human Cancer
Each of the PKC isoforms is unique in its contribution to cancer development and progression. The conventional PKCs are generally considered to be predominantly antiapoptotic and principally involved in promoting cell survival and proliferation. PKCα regulates multiple biological processes, including cell proliferation, apoptosis, and cell motility [5]. However, the role of PKCα in regulating tumor growth and development is complex and highly tissue dependent. PKCα can either act as a tumor promoter or a tumor suppressor [6]. Overexpression of PKCα has been demonstrated in tissue samples of prostate, endometrial, high-grade urinary bladder, hepatocellular and breast cancers, suggesting a role of PKCα as a tumor promoter. In contrast, PKCα is down-regulated in basal cell carcinoma (BCC) and colon cancers, demonstrating a possible role of PKCα as a tumor suppressor in these tumor types. PKCβI and βII function in various signal-transducing pathways for proliferation, differentiation, metabolism, and more cell-type-specific functions [7, 8].
The novel PKCs generally have a tumor suppressor function and are regarded as pro-apoptotic proteins; however, the evidence is complex. PKCδ has been implicated both as a tumor suppressor and positively or negatively regulates cell proliferation and apoptosis [9]. For example, in breast cancer, PKCδ has shown both pro-survival and pro-apoptotic effects [10]. PKCε has been shown to behave as an oncoprotein [11]. Overexpression of PKCε increased proliferation, motility, and invasion of fibroblasts or immortalized epithelial cells. In addition, transgenic animal models have clearly shown that overexpression of PKCε is tumorigenic resulting in metastatic disease. PKCθ has been proposed as a key player in T-cell activation and an attractive therapeutic target in T-cell-mediated disease processes [12].
Atypical PKCs have been implicated in the malignant behavior of transformed human cells as well. Evidence over the past few years has shown that PKCι is a human oncogene and that the oncogenic PKCι signaling is a target for novel mechanism-based cancer therapy [13]. For example, PKCι is critical for transformed growth in human non-small cell lung cancer cells. PKCζ is involved in diverse physiological functions [14]. For example, PKCζ is involved in the control of glioblastoma cell migration and invasion by regulating the cytoskeleton rearrangement, cell adhesion, and matrix metalloprotease-9 expression. These findings suggest that PKCζ is a potential therapeutic target for glioblastoma.
Crosstalk of HH and PKC
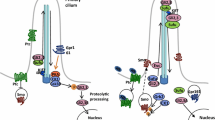
Gli proteins, including Gli1, Gli2, and Gli3, represent a family of zinc-finger transcription factors and play critical roles in the mediation and interpretation of HH signals [15]. Fused (Fu) is a serine–threonine kinase required for HH signaling and hSu(fu), a human homologue of Drosophila Su(fu), is a suppressor of Fu [16]. hSu(fu) negatively regulates Gli activity [16, 17]. hSu(fu) contains conserved PKC phosphorylation sites [16, 17], indicating that it may be subject to regulation by PKC. Information demonstrating the crosstalk between HH and PKC signaling pathways was derived from nontumor cells, such as mammalian 293T and NIH 3T3 fibroblasts. These studies mainly focused on the crosstalk of PKCα or PKCδ with HH signaling. Neill et al. [18] first demonstrated that PKCα and PKCδ-mediated Gli activity in the mammalian 293T cells. When 293T cells were cotransfected with constitutively active PKCα or PKCδ and a luciferase reporter construct containing Gli1-binding sites (GBS), the constitutively active PKCα decreased Gli1 activity by over 60%, suggesting that PKCα is a potent negative regulator of Gli1 transcriptional activity. In contrast to PKCα, constitutively active PKCδ increased the activity of Gli1, indicating a positive role of PKCδ in the regulation of Gli1 transcriptional activity.
The interaction of PKC with HH was further studied in LIGHT2 cells, a HH-responsive NIH 3T3 fibroblast cell line stably transfected with a Gli-regulated luciferase reporter containing eight tandem copies of GBS (8× GBS-luciferase) [19]. Treatment of LIGHT2 cells with phorbol 12-myristate 13-acetate (PMA), a phorbol ester, increased Gli-luciferase activity that was blocked by the PKC inhibitor GF109203X, suggesting PMA activation of Gli is mediated through PKC. Treatment with PMA increased mRNA levels of PTCH 1 and GLI1, two endogenous Gli-regulated genes, which was inhibited by GF109203X. The specificity of Gli-dependent transcription by PMA in NIH 3T3 fibroblasts was further confirmed by transfecting a wild-type 8× GBS-luciferase reporter or a mutated 8× GBS-luciferase reporter harboring a point mutation that abolishes the binding of Gli. PMA-stimulated Gli-luciferase activity was only detected in cells transfected with wild-type GBS-luciferase reporter but not the mutant reporter, indicating that PMA activity is mediated through activation of GLI transcriptional activity. Furthermore, stimulation of GLI-dependent transcription by PMA is mediated through a novel PKC. When LIGHT2 cells were treated with PMA in the presence of Gö6976 (inhibitor of classical PKCs) or rottlerin (inhibitor of novel PKCs), PMA-mediated GLI transcriptional activity was prevented by rottlerin, but not by Gö6976, suggesting this effect is mediated through a novel PKC isoform, which is likely PKCδ since NIH 3T3 fibroblasts express PKCα and PKCδ, two isoforms responsive to PMA. The involvement of PKCδ was confirmed by transfection of LIGHT2 cells with a dominant-negative mutant of PKCδ which blocked the GLI-luciferase activity. Taken together, this study demonstrates that PKCα plays a negative role, whereas PKCδ plays a positive role, in the regulation of HH signaling.
Our group further established the crosstalk of PKCα or PKCδ with HH signaling [20]. As noted above, Neill et al. [18] have shown that PKCα is a negative regulator of GLI1 transcriptional activity in 293T cells. We further confirmed the specific regulation of PKCα on GLI activity in NIH 3T3 fibroblasts. NIH 3T3 cells were cotransfected with a wild-type or a mutated GLI-luciferase reporter (point mutation that abolishes the binding of Gli) and expression plasmids, Gli1 and constitutively active PKCα. The constitutively active PKCα significantly increased the wild-type GLI-luciferase activity, but not the mutant, confirming that PKCα negatively regulates HH signaling. It has been demonstrated that PKCδ increased the activity of Gli1 in NIH 3T3 and 293T cells. Therefore, we cotransfected NIH 3T3 cells with Gli-luciferase reporter, Gli1, and either wild-type PKCδ, kinase-dead PKCδ K376R, or constitutively active PKCδΔNPS in which the N-terminal pseudosubstrate domain was deleted. Treatment with PMA increased Gli-luciferase activity only in cells cotransfected with control vector, which is consistent with the previous findings showing that the endogenous PKCδ positively regulates Gli activity [19]. In contrast, in cells transfected with wild-type PKCδ, Gli-luciferase activity was significantly decreased by PMA treatment; this effect was blocked by rottlerin. In the cells transfected with PKCδΔNPS, Gli-luciferase activity was further decreased either in the presence or absence of PMA, whereas Gli-luciferase activity was not altered in cells transfected with kinase-dead PKCδ K376R. In cells transfected with empty vector (pcDNA3) or kinase-dead PKCδ, PTCH1 mRNA expression was not altered either with or without PMA treatment. In contrast, PMA treatment decreased PTCH1 mRNA levels in cells transfected with wild-type PKCδ as well as PKCδΔNPS either in the presence or absence of PMA. Taken together, PKCδ appears to play a negative role in the regulation of Gli activity stimulated by PMA.
Crosstalk of HH and PKC in Development
The role of HH signaling in development is well known [21]. PKC isoforms have been implicated in a number of key steps during gametogenesis, fertilization, and early development [22]. However, the interaction of the two signaling pathways in the regulation of development has not been studied extensively. Lu et al. [23] tested the efficacy of the competitive inhibitors chelerythrine chloride and Gö6976 (specific inhibitors of PKC) and sphingosine (inhibits PKC and other kinases) in primary limb bud mesenchyme cultures. PKC inhibition resulted in smaller buds and truncated wings and caused complete loss of sonic hedgehog (Shh) expression in the buds, suggesting the possibility that PKC may control Shh expression. Indeed, the PKC inhibition-induced phenotype and lost Shh expression were rescued by providing ectopic Shh. These experiments demonstrated that, providing exogenous Shh to wing buds in which PKC signaling had been blocked, restored limb development and that Shh is one of the primary targets of PKC signaling.
Crosstalk of HH and PKC in Stem Cells
HH signaling has an essential role in the control of stem cell growth in embryonic tissues. Heo et al. [24] examined the effect of Shh on the self-renewal of mouse embryonic stem cells and its related mechanisms. They treated these cells with Shh and noted translocation of PKCα, δ, and ζ isoforms from the cytosol to the membrane, demonstrating the activation of these PKC isoforms by Shh stimulation. On the other hand, Shh-induced PKC activation was blocked by cyclopamine, a steroid alkaloid that blocks Shh signaling. Pretreatment with bisindolylmaleimide I (a PKC inhibitor) inhibited Shh-induced Gli1 gene expression and [3H] thymidine incorporation, demonstrating that Shh stimulated mouse ES cell proliferation through Gli1 activation as well as PKC. Consistently, in mesenchymal stem cells transfected with Shh, the expression of angiogenic and pro-survival growth factors was increased and migration and tube formation were significantly improved in a PKC-dependent manner.
Crosstalk of HH and PKC in Human Cancer
The HH signaling pathway, when mutated or dysregulated, contributes to tumorigenesis. Recent studies provide evidence demonstrating the crosstalk of HH and certain PKC isoforms in human cancer cells.
Gli1 expression is associated with the development of BCC. Gli1 is expressed in the outer root sheath (ORS) of the hair follicle which is thought to be a potential source of BCC. PKCα was expressed in the epidermis and ORS of human hair follicles and PKCδ in the inner root sheath. Neill et al. [18] found that PKCα is down-regulated in BCC, suggesting that loss of PKCα expression may be relevant to tumor formation. We screened PKCδ expression in a set of hepatocellular cancer (HCC) in which the activation status of HH signaling had previously been determined by in situ hybridization using probes against Gli1 and PTCH1 [20]. Interestingly, the expression of PKCδ was not detected in any of these specimens with activated HH signaling. These results suggest that decreased expression of PKCδ may account for activation of HH signaling in certain HCC, further demonstrating a negative role of PKCδ in the regulation of HH signaling in cancer cells. Additional evidence to support these findings was provided by in vitro studies using Hep3B cells, a human hepatoma cell line [20]. By a combination of overexpression of PKCδ and knockdown with PKCδ siRNA, we demonstrated that overexpression of wild-type or active PKCδ decreased Gli-luciferase activity, mRNA levels of PTCH, and Gli and endogenous Gli protein levels, whereas knockdown by PKCδ siRNA had opposite effects on these HH target proteins. Furthermore, PKCδ knockdown with siRNA enhanced the proliferation and significantly blocked the inhibitory effects of KAAD-cyclopamine (a potent derivative of cyclopamine). Taken together, the loss of PKCδ increased HH signaling and Gli1 protein expression and rescued the inhibitory effect of KAAD-cyclopamine on cellular proliferation, demonstrating that PKCδ negatively regulates HH signaling.
MEK/ERK Pathway in PKC-Mediated HH Signaling
The Raf/MEK/ERK signaling pathways regulate a variety of cellular activities including proliferation, differentiation, survival, and death. HH signaling exerts a positive feedback effect on these pathways; furthermore, PKC is well known as an activator of the ERK pathway. Therefore, it is very likely that the ERK pathway is involved in PKC-mediated HH signaling. Riobo et al. [19] investigated whether PKC activates Gli activity through the ERK pathway in LIGHT2 cells. They showed that PMA-induced GLI-luciferase reporter activity was blocked by the selective MEK-1 inhibitor PD98059 or the dual MEK-1/2 inhibitor U0126. These findings place MEK-1 downstream of PKC in the activation of GLI. Moreover, PKCα plays a positive role in the regulation of Gli1 activity; this effect was mediated by the MEK/ERK pathway.
Summary
We have highlighted data demonstrating evidence of crosstalk between HH and PKC. Consistently, PKCα has been shown to negatively regulate HH signaling. However, studies demonstrated that PKCδ plays either a negative or positive role in the regulation of HH signaling. We proposed that the balance between PKCα and PKCδ is important in the regulation of Gli activity. When PKCα is dominant, the negative effect of PKCδ is weak, and PMA increases Gli activity through the PKCα/MEK/ERK pathway. However, when PKCδ is dominant, PMA treatment decreases Gli activity through the activation of PKCδ. It is clear that the HH pathway plays an important role in tumor cell growth and survival. However, several issues regarding the precise role of the HH signaling pathway in human cancer remain unresolved, including the exact mechanisms of signal transduction. We anticipate that more mechanistic studies will further illuminate the conserved and divergent aspects of HH signaling. A better understanding of HH signaling and its crosstalk with other signaling pathways is of importance for developing a more precise understanding of HH-associated diseases and, furthermore, holds great promise for developing new therapies based upon this information.
References
Newton AC (2010) Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab 298:E395–E402
Steinberg SF (2008) Structural basis of protein kinase C isoform function. Physiol Rev 88:1341–1378
Shirai Y, Saito N (2002) Activation mechanisms of protein kinase C: maturation, catalytic activation, and targeting. J Biochem 132:663–668
Rosse C et al (2010) PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol 11:103–112
Nakashima S (2002) Protein kinase C alpha (PKC alpha): regulation and biological function. J Biochem 132:669–675
Konopatskaya O, Poole AW (2010) Protein kinase Calpha: disease regulator and therapeutic target. Trends Pharmacol Sci 31:8–14
Kawakami T, Kawakami Y, Kitaura J (2002) Protein kinase C beta (PKC beta): normal functions and diseases. J Biochem 132:677–682
Sledge GW Jr, Gokmen-Polar Y (2006) Protein kinase C-beta as a therapeutic target in breast cancer. Semin Oncol 33:S15–S18
Jackson DN, Foster DA (2004) The enigmatic protein kinase Cdelta: complex roles in cell proliferation and survival. FASEB J 18:627–636
Vucenik I, Ramljak D (2006) The contradictory role of PKCdelta in cellular signaling. Breast Cancer Res Treat 97:1–2
Basu A, Sivaprasad U (2007) Protein kinase Cepsilon makes the life and death decision. Cell Signal 19:1633–1642
Chaudhary D, Kasaian M (2006) PKCtheta: a potential therapeutic target for T-cell-mediated diseases. Curr Opin Investig Drugs 7:432–437
Fields AP, Frederick LA, Regala RP (2007) Targeting the oncogenic protein kinase Ciota signalling pathway for the treatment of cancer. Biochem Soc Trans 35:996–1000
Hirai T, Chida K (2003) Protein kinase Czeta (PKCzeta): activation mechanisms and cellular functions. J Biochem 133:1–7
Kasper M, Regl G, Frischauf AM, Aberger F (2006) GLI transcription factors: mediators of oncogenic Hedgehog signalling. Eur J Cancer 42:437–445
Stone DM et al (1999) Characterization of the human suppressor of fused, a negative regulator of the zinc-finger transcription factor Gli. J Cell Sci 112(Pt 23):4437–4448
Merchant M et al (2004) Suppressor of fused regulates Gli activity through a dual binding mechanism. Mol Cell Biol 24:8627–8641
Neill GW et al (2003) Loss of protein kinase Calpha expression may enhance the tumorigenic potential of Gli1 in basal cell carcinoma. Cancer Res 63:4692–4697
Riobo NA, Haines GM, Emerson CP Jr (2006) Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res 66:839–845
Cai Q, Li J, Gao T, Xie J, Evers BM (2009) Protein kinase Cdelta negatively regulates hedgehog signaling by inhibition of Gli1 activity. J Biol Chem 284:2150–2158
Nagase T, Nagase M, Machida M, Yamagishi M (2007) Hedgehog signaling: a biophysical or biomechanical modulator in embryonic development? Ann N Y Acad Sci 1101:412–438
Kalive M, Faust JJ, Koeneman BA, Capco DG (2010) Involvement of the PKC family in regulation of early development. Mol Reprod Dev 77:95–104
Lu HC, Swindell EC, Sierralta WD, Eichele G, Thaller C (2001) Evidence for a role of protein kinase C in FGF signal transduction in the developing chick limb bud. Development 128:2451–2460
Heo JS, Lee MY, Han HJ (2007) Sonic hedgehog stimulates mouse embryonic stem cell proliferation by cooperation of Ca2+/protein kinase C and epidermal growth factor receptor as well as Gli1 activation. Stem Cells 25:3069–3080
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Li, J., Evers, B.M. (2011). Hedgehog and Protein Kinase C Signaling. In: Xie, J. (eds) Hedgehog signaling activation in human cancer and its clinical implications. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-8435-7_6
Download citation
DOI: https://doi.org/10.1007/978-1-4419-8435-7_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-8434-0
Online ISBN: 978-1-4419-8435-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)