
Abstract
Juvenile localized scleroderma (JLS), known as morphea, comprises a group of conditions which involve essentially the skin and subcutaneous tissues. They have various features and range from very small plaques to extensive fibrotic lesions which may cause significant functional changes and cosmetic deformities.
Although JLS is relatively uncommon, it is far more common than systemic sclerosis in childhood, by a ratio of at least 10:1. The disease onset is usually during late infancy, although cases with onset at early infancy and even at birth have been described.
An accurate clinical examination associated with instrumental assessment with thermography, ultrasonography, and skin scoring is crucial to address the appropriate treatment.
During the last few years, methotrexate (MTX) in combination with corticosteroids has shown to be safe and effective, particularly in patients with significant risk for disability.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Computerized skin scoring
- disability
- localized scleroderma
- methotrexate
- morphea
- phototherapy
- Thermography
- ultrasonography
Juvenile localized scleroderma (JLS), also known as morphea, comprises a group of conditions in which the process of fibrosis involves essentially the skin and subcutaneous tissues. They may range from very small plaques to extensive indurate lesions which cause significant functional and cosmetic deformity.
The most widely used classification divides JLS into five general types: plaque morphea, generalized morphea, bullous morphea, linear scleroderma, and deep morphea [1]. Some conditions, such as atrophoderma of Pasini and Pierini, eosinophilic fasciitis, or lichen sclerosus et atrophicus, are sometimes classified among the subtypes of JLS, but this aspect is still controversial. This classification does not include the mixed forms of JLS where different types of lesions occur in the same individual. This subtype is more common than previously recognized, accounting for 15% of the whole group [2].
A proposal for a new classification includes five subtypes: circumscribed morphea (CM), linear scleroderma, generalized morphea (GM), pansclerotic morphea, and the new mixed subtype where a combination of two or more of the previous subtypes is present (Table 9.1) [3].
Epidemiology
Although JLS is relatively uncommon, it is far more common than systemic sclerosis in childhood, by a ratio of at least 10:1 [4]. There is a mild female predilection being the F:M ratio 2.4:1 [2]. The mean age at disease onset is 7.3 years and a few cases with onset at birth, so called congenital localized scleroderma, have been also described [5].
Clinical Manifestations
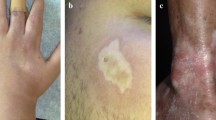
Circumscribed morphea (CM) is characterized by oval or round circumscribed areas of induration surrounded by a violaceous halo (Fig. 9.1). It is confined to the dermis with only occasional involvement of the superficial panniculus.

Circumscribed morphea of the right shoulder, characterized by an area of induration with waxy consistence and ivory color, surrounded by an inflammatory edge
When there are four or more individual plaques that are larger than 3 cm and they become confluent involving at least two out of seven anatomic sites (head-neck, right upper extremity, left upper extremity, right lower extremity, left lower extremity, anterior trunk, posterior trunk), it is called generalized morphea (GM) (Fig. 9.2). Unilateral GM has been proposed as an uncommon variant, usually beginning in childhood [1].

Generalized morphea involving, symmetrically, the trunk in a 8-year-old boy. Note the bluish halo of the active lesions, named lilac ring
Linear scleroderma, the most common subtype in children and adolescents, is characterized by one or more linear streaks that can extend through the dermis, subcutaneous tissue, and muscle to the underlying bone, causing significant deformities (Fig. 9.3). Not only the upper or lower extremities can be affected but also the face or scalp, as in the en coup de sabre variety (ECDS). The Parry-Romberg syndrome (PRS), characterized by hemifacial atrophy of the skin and tissue below the forehead, with mild or absent involvement of the superficial skin, is considered the severe end of the spectrum of ECDS and for this reason is included in subtype of linear scleroderma [6]. Evidence for this close relationship is the presence of associated disorders, including seizures, CNS abnormalities, and dental and ocular abnormalities, reported with similar prevalence in both conditions [6–9].

Linear scleroderma at the right side of the trunk
Pansclerotic morphea, an extremely rare but severe subtype, is characterized by generalized full-thickness involvement of the skin of the trunk, extremities, face, and scalp with sparing of the fingertips and toes (Fig. 9.4). It is more common in children than adults. Recent reports raised the attention on the possible evolution of chronic ulcers, frequently complicating pansclerotic morphea, to squamous cell carcinoma, a threatening complication already reported in LS [10–12].

Pansclerotic morphea involving abdomen and right lower limb in a 5-year-old girl. Note that the margins of the lesion are hardly visible
Conversely to what has been reported for many years, JLS is not exclusively confined to the skin but can present many extracutaneous features. A recent multinational study reported that almost one-fourth of the patients present extracutaneous manifestations [13]. The overall distribution of these manifestations includes arthritis 19%, neurological findings 4%, associated autoimmune conditions 3%, vascular changes (i.e., Raynaud’s phenomenon, deep vein thrombosis) 2%, and ocular or gastrointestinal abnormalities 2%.
Articular involvement is the most frequent finding, especially in linear scleroderma. Children who develop arthritis often have a positive rheumatoid factor (RF), and sometimes an elevated erythrocyte sedimentation rate (ESR) and circulating autoantibodies [13]. The most frequent neurological conditions are seizures and headaches, although behavioral changes and learning disabilities have been also described [13, 14]. Abnormalities on magnetic resonance imaging (MRI), such as calcifications, white matter changes, vascular malformations, and vasculitis, also have been reported [15, 16]. Although most of the imaging abnormalities have little clinical relevance, biopsy findings have shown sclerosis, fibrosis, gliosis, as well as vasculitis [16–17].
Gastroesophageal reflux (GER) is the only gastrointestinal complication reported so far in JLS [2, 18, 19].
Autoantibodies
Antinuclear antibodies (ANA) are present in more than 40% of patients with JLS [2]. This frequency is lower than in adult with LS [20] but is higher than in normal population. In children, there is no correlation between the presence of ANA and a particular subtype or disease course [2].
Of interest, anti-topoisomerase I antibodies (anti-Scl 70), a marker of SSc in adults, were found to be positive in 2–3% of children with JLS but not in adults with LS [2, 20]. Conversely, anti-centromere antibodies (ACA) were found in 12% of adults with LS but only in 1.7% of children [2, 21]. Whether these antibodies are markers that reflect the immunological component of the disease process or can have a prognostic significance is unclear. It should be noted that none of SCL-70 or ACA positive patients in a series of 750 JLS patients presented signs or symptoms of internal organ involvement after a mean follow-up of 3.4 years [2].
Rheumatoid factor (RF) has been detected, at low titer, in 16% of the patients with JLS, and significantly correlated with the presence of arthritis [2].
One of the major autoantigens for ANA in JLS is nuclear histone. Anti-histone antibodies (AHA) have been detected in 47% of patients with JLS with a different prevalence in the various subtypes, higher in GM, and lower in circumscribed morphea [20].
Diagnosis and Disease Assessment
The management of JLS is challenging, and the detection of disease activity and progression remains a fundamental problem. Clinical examination is subjective, and so classical skin scoring methods, utilized in the assessment of systemic sclerosis, cannot be applied. Among the new tools which have been proposed for the assessment of the skin lesions, infrared thermography (IRT), computerized skin score (CSS), ultrasound (US), and magnetic resonance imaging (MRI) are those most frequently used.
Infrared thermography (IRT) is able to detect areas of increased temperature caused by the inflammatory process, revealing, in this way, active lesions [22]. This technique has shown to have a very high reproducibility but yields false-positive results in the assessment of old lesions characterized by marked atrophy of the skin and subcutaneous tissues. In these cases, an accurate clinical examination can help differentiate these lesions from the active ones.
The computerized skin score (CSS) consists in the demarcation of hyperemic and indurate borders of the lesions on an adhesive transparent film with different colors [23]. The film, transferred over a cardboard, is scanned and recorded in a computer. Calculation of the affected area is performed by computer software.
Ultrasonography (USG) is another technique that has been proposed for monitoring JLS. USG can detect several abnormalities such as increased blood flow, increased echogenicity due to fibrosis, and loss of subcutaneous fat. The first two parameters appear to be signs of active lesions, which disappear in the remission phase. Loss of subcutaneous tissue was found in both active and stable patients [24]. The two main limits of USG are represented by its operator-dependent value and the lack of validation as outcome measure in prospective studies.
MRI is also an important tool in the clinical management of JLS. MRI is clearly most useful when CNS or eye involvement is suspected but is also able to demonstrate the true depth of soft tissue lesions and the degree to which different tissues are involved in other sites [25].
In comparison to USG, MRI has two main disadvantages: the need for sedation in younger patients and the presence of possible artifacts.
Treatment
Over the years, many treatments have tried for localized scleroderma [26]. Decisions for management must be based upon the particular subtype of disease and the realization that these disorders may spontaneously enter remission after 3–5 years (Fig. 9.5).

Proposed flowchart for the management of localized scleroderma
Circumscribed morphea generally is of cosmetic concern only, and therefore, treatments with potentially significant toxicity are not justified. In general, these lesions will spontaneously remit with residual pigmentation as the only abnormality. Therefore, treatment should be directed mainly at topical therapies such as moisturizing agents, topical glucocorticoids, or calcipotriene [27].
Phototherapy with ultraviolet (UV) represents another possible therapeutic choice for localized scleroderma [28–33]. The use of ultraviolet (UV) light therapy, with or without chemical agents such as psoralen, has been reported to be beneficial for localized or superficial lesions in a number of studies [28–32]. UVA1 phototherapy upregulates specific matrix mRNA metalloproteinases, depletes skin-infiltrating T cells, and inhibits the production of pro-inflammatory cytokines, namely IL-1, IL-6, and IL-8 [33].
Since the rate of relapse after UV phototherapy discontinuation is not known, the need for prolonged maintenance therapy, leading to a high cumulative dosage of irradiation, and the increased risk for potential long-term effects such as skin aging and carcinogenesis [34, 35] are clear limitations for its use especially in children.
Use of vitamin D or its analogs (topically and systemically) has been reported in several case series with encouraging results [27, 36]. However, in the only controlled trial, results indicated it was no more effective than placebo [37].
When there is a significant risk for disability, such as in progressive linear scleroderma crossing joint lines and generalized or pansclerotic morphea, systemic treatment, particularly with methotrexate, should be considered (Table 9.2) [38–43].
A weekly regimen of methotrexate of 10–15 mg/m2 as a single oral or subcutaneous dose per week for at least 1 year is recommended. During the first 2–3 months of therapy, a course of glucocorticoids may be used as adjunctive bridge therapy. Recently, a randomized trial comparing a 12-month course of oral methotrexate (15 mg/m2) for 12 months with a 3-month course of oral prednisone (1 mg/kg/day, maximum dose 50 mg) showed that methotrexate was effective and well tolerated in more than two-thirds of the patients with morphea [43]. New lesions appeared in only 6.5% of methotrexate-treated patients compared with 16.7% of the prednisone group. In addition, the skin score rate, which evaluates lesions’ extension changes and the mean target lesion temperature as evaluated by infrared thermography, significantly decreased in the methotrexate group.
Patients who do not respond to this treatment approach may be treated with mycophenolate mofetil at a dose of 500–1,000 mg/m2 [44].
Surgical reconstruction may be required if the disease has not been adequately controlled. Surgery should only be performed after the active phase of the disease has abated and when the child’s growth is complete [45]. Facial recontouring is a surgical treatment option that may improve quality of life in adolescents with facial asymmetry due to en coup de sabre morphea [46].
Prognosis
Information on the long-term outcome of children with JLS is very few and based on small series of patients. However, it is common experience that adults with childhood-onset localized scleroderma suffer from long-term disease sequelae that significantly impact quality of life, including permanent functional and cosmetic impairment. In addition, some continue to have episodes of active disease throughout life.
References
Peterson LS, Nelson AM, Su WPD, et al. Subspecialty clinics: rheumatology and dermatology. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70:1068–76.
Zulian F, Athreya BH, Laxer RM, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology (Oxford). 2006;45:614–20.
Laxer RM, Zulian F. Localized scleroderma. Curr Opin Rheumatol. 2006;18:606–13.
Herrick AL, Ennis H, Bhushan M, et al. Incidence of childhood linear scleroderma and systemic sclerosis in the UK and Ireland. Arthritis Care Res (Hoboken). 2010;62(2):213–8.
Zulian F, Vallongo C, de Oliveira SKF, et al. Congenital localized scleroderma. J Pediatr. 2006;149:248–51.
Jablonska S, Blaszczyk M. Long-lasting follow-up favours a close relationship between progressive facial hemiatrophy and scleroderma en coup de sabre. J Eur Acad Dermatol Venereol. 2005;19:403–4.
Menni S, Marzano AV, Passoni E, et al. Neurologic abnormalities in two patients with facial hemiathrophy and sclerosis coexisting with morphea. Pediatr Dermatol. 1997;14:113–6.
Blaszczyk M, Jablonska S. Linear scleroderma En coup de sabre: relationship with progressive facial hemiatrophy. Adv Exp Med Biol. 1999;455:101–4.
Sommer A, Gambichler T, Bacharach-Buhles M, et al. Clinical and serological characteristics of progressive facial hemiatrophy: a case series of 12 patients. J Am Acad Dermatol. 2006;54:227–33.
Wollina U, Buslau M, Weyers W, et al. Squamous cell carcinoma in pansclerotic morphea of childhood. Pediatr Dermatol. 2002;19:151–4.
Parodi PG, Roberti G, Draganic Stinco D, et al. Squamous cell carcinoma arising in a patient with long-standing pansclerotic morphea. Br J Dermatol. 2001;144:417–9.
Maragh SH, Davis MD, Bruce AJ, et al. Disabling pansclerotic morphea: clinical presentation in two adults. J Am Acad Dermatol. 2005;53:115–9.
Zulian F, Vallongo C, Woo P, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum. 2005;52:2873–81.
Blaszczyk M, Krolicki L, Krasu M, et al. Progressive facial hemiatrophy: central nervous system involvement and relationship with scleroderma en coup de sabre. J Rheumatol. 2003;30:1997–2004.
DeFelipe J, Segura T, Arellano JI, et al. Neuropathological findings in a patient with epilepsy and the Parry-Romberg syndrome. Epilepsia. 2001;42:1198–203.
Flores-Alvarado DE, Esquivel-Valerio JA, Garza-Elizondo M, et al. Linear scleroderma en coup de sabre and brain calcification: is there a pathogenic relationship? J Rheumatol. 2003;30:193–5.
Holland KE, Steffes B, Nocton JJ, et al. Linear scleroderma en coup de sabre with associated neurologic abnormalities. Pediatrics. 2006;117:132–6.
Weber P, Ganser G, Frosch M, et al. Twenty-four hour intraesophageal pH monitoring in children and adolescents with scleroderma and mixed connective tissue disease. J Rheumatol. 2000;27:2692–5.
Guariso G, Conte S, Galeazzi F, et al. Esophageal involvement in juvenile localized scleroderma: a pilot study. Clin Exp Rheumatol. 2007;25:786–9.
Takehara K, Sato S. Localized scleroderma is an autoimmune disease. Rheumatology (Oxford). 2005;44:274–9.
Ruffatti A, Peserico A, Glorioso S, et al. Anticentromere antibody in localized scleroderma. J Am Acad Dermatol. 1986;15:637–42.
Martini G, Murray KJ, Howell KJ, et al. Juvenile-onset localized scleroderma activity detection by infrared thermography. Rheumatology (Oxford). 2002;41:1178–82.
Zulian F, Meneghesso D, Grisan E, et al. A new computerized method for the assessment of skin lesions in localized scleroderma. Rheumatology (Oxford). 2007;46:856–60.
Li SC, Liebling MS, Haines KA, et al. Ultrasonography is a sensitive tool for monitoring localized scleroderma. Rheumatology (Oxford). 2007;46:1316–9.
Liu P, Uziel Y, Chuang S, et al. Localized scleroderma: imaging features. Pediatr Radiol. 1994;24:207–9.
Zulian F. New developments in localized scleroderma. Curr Opin Rheumatol. 2008;20:601–7.
Cunningham BB, Landells ID, Langman C, et al. Topical calcipotriene for morphea/linear scleroderma. J Am Acad Dermatol. 1998;39:211–5.
Kerscher M, volkenandt M, Gruss C, et al. Low dose UVA phototherapy for treatment of localized scleroderma. J Am Acad Dermatol. 1998;38:21–3.
Kreuter A, Gambichler T, Avermaete A, et al. Combined treatment with calcipotriol ointment and low-dose ultraviolet A1 phototherapy in childhood morphea. Pediatr Dermatol. 2001;18:241–5.
Camacho NR, Sánchez JE, Martin RF, et al. Medium-dose UVA1 phototherapy in localized scleroderma and its effect in CD34-positive dendritic cells. J Am Acad Dermatol. 2001;45:697.
De Rie MA, Bos JD. Photochemotherapy for systemic and localized scleroderma. J Am Acad Dermatol. 2000;43:725–6.
Kreuter A, Hyun J, Stucker M, et al. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma. J Am Acad Dermatol. 2006;54:440–7.
Kreuter A, Hyun J, Skrygan M, et al. Ultraviolet A1-induced downregulation of human beta-defensins and interleukin-6 and interleukin-8 correlates with clinical improvement in localized scleroderma. Br J Dermatol. 2006;155:600.
Staberg B, Wulf HC, Klemp P, et al. The carcinogenic effect of UVA irradiation. J Invest Dermatol. 1983;81:517–9.
Setlow RB, Grist E, Thompson K, et al. Wave-lengths effective in induction of malignant melanoma. Proc Nat Acad Sci USA. 1992;90:6666–70.
Caca-Biljanovska NG, Vlckova-Laskoska MT, Dervendi DV, et al. Treatment of generalized morphea with oral 1,25-dihydroxyvitamin D3. Adv Exp Med Biol. 1999;455:299.
Hulshof MM, Bouwes BJN, Bergman W, et al. Double-blind, placebo-controlled study of oral calcitriol for the treatment of localized and systemic scleroderma. J Am Acad Dermatol. 2000;43:1017.
Seyger MM, de Boo Theo, van den Hoogen FHJ, et al. Low-dose methotrexate in the treatment of widespread morphea. J Am Acad Dermatol. 1998;39:220–5.
Uziel Y, Feldman BM, Krafchik BR, et al. Methotrexate and corticosteroid therapy for pediatric localized scleroderma. J Pediatr. 2000;136:91–5.
Kreuter A, Gambichler T, Breuckmann F, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005;141:847–52.
Fitch PG, Retting P, Burnham JM, et al. Treatment of pediatric localized scleroderma with methotrexate. J Rheumatol. 2006;33:609–14.
Weibel L, Sampaio MC, Visentin MT, et al. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphea) in children. Br J Dermatol. 2006;155:1013–20.
Zulian F, Martini G, Vallongo C, et al. Methotrexate in juvenile localized scleroderma: a randomised, double-blind, placebo-controlled trial. Arthritis Rheum. 2011;63(7):1998–2006.
Martini G, Ramanan AV, Falcini F, et al. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil. Rheumatology (Oxford). 2009;48:1410–3.
Lapiere JC, Aasi S, Cook B, Montalvo A. Successful correction of depressed scars of the forehead secondary to trauma and morphea en coup de sabre by en bloc autologous dermal fat graft. Dermatol Surg. 2000;26:793–7.
Palmero ML, Uziel Y, Laxer RM, et al. En coup de sabre scleroderma and parry-romberg syndrome in adolescents: surgical options and patient-related outcomes. J Rheumatol. 2010;37:2174–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Zulian, F. (2012). Juvenile Localized Scleroderma. In: Varga, J., Denton, C., Wigley, F. (eds) Scleroderma. Springer, Boston, MA. https://doi.org/10.1007/978-1-4419-5774-0_9
Download citation
DOI: https://doi.org/10.1007/978-1-4419-5774-0_9
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4419-5773-3
Online ISBN: 978-1-4419-5774-0
eBook Packages: MedicineMedicine (R0)