
Abstract
Neuroendocrine neoplasms occurring primary in the lung account for approximately 15 % of this family of tumors. Unfortunately, the bulk of neuroendocrine tumors are made up by small cell lung carcinoma. In recent statistics, it has been estimated that small cell carcinomas of the lung may account for approximately 30,000 new cases on a yearly basis. Paradoxically, a disease that previously was observed predominantly in older men, now appears to affect men and women almost equally. However, it is important to keep in mind that although the gamut of primary lung tumors that may show neuroendocrine differentiation is rather complex, involving tumors not only of epithelial but also of mesenchymal or neural origin, this chapter will be limited to the most conventional epithelial carcinomas that range from the low- to the high-grade types. Another important aspect in this chapter this chapter will be the discussion with another neuroendocrine tumor that, even though not considered carcinoma, may pose a significant problem in the differential diagnosis: intrapulmonary paraganglioma.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Neuroendocrine neoplasms occurring primary in the lung account for approximately 15% of this family of tumors. Unfortunately, the bulk of neuroendocrine tumors are made up by small cell lung carcinoma. In recent statistics, it has been estimated that small cell carcinomas of the lung may account for approximately 30,000 new cases on a yearly basis. Paradoxically, a disease that previously was observed predominantly in older men, now appears to affect men and women almost equally. However, it is important to keep in mind that although the gamut of primary lung tumors that may show neuroendocrine differentiation is rather complex, involving tumors not only of epithelial but also of mesenchymal or neural origin, this chapter will be limited to the most conventional epithelial carcinomas that range from the low- to the high-grade types. Another important aspect in this chapter will be the discussion with another neuroendocrine tumor that, even though not considered carcinoma, may pose a significant problem in the differential diagnosis: intrapulmonary paraganglioma.
Neuroendocrine neoplasms are ubiquitous tumors that have been the subject of much investigation for over a century. Although the low-grade tumors (carcinoid tumor) were initially described as a form of neoplasms that behave better than conventional carcinoma different studies of these tumors have focused on the concept of a family of neoplasms that may expand from the indolent and insignificant small lesion (so-called tumorlet)—more often encountered by chance—to low-, intermediate-, and high-grade malignancies. In order to provide a better understanding in terms of clinical course, behavior, and possible histogenesis, different studies including morphological, immunohistochemical, and molecular studies have attempted to shed some light on this family of neoplasms. However, a universal agreement has not been reached. Some reviews on the subject [1] are either priort to the most recent classification of lung tumors by the World Health Organization (WHO) [2] or have separated these tumors in the conventional three-way category system [3]. Although other studies have followed the WHO classification, those studies have stated the difficulty that exists in making these diagnoses on surgical biopsy specimens [4]. One of the biggest problems in achieving full agreement across all anatomic areas is the fact that the designation given to some of these tumors depends largely on the anatomic site in which they may occur. To illustrate this fact, one only has to review the WHO classification for tumors of the pleura, lung, thymus, and heart [2] in which tumors in the thymus are separated into low- and high-grade malignancies while those in the lung are separated into a four-way category system, demonstrating the inconsistency in the classification of these tumors, even in the same anatomic area—the thorax.
Siegfried Oberndorfer [5] is credited for coining the term “carcinoid tumor” in 1907. However, Bunting [6] from Johns Hopkins in 1904 had described a case under the designation of “multiple primary carcinomata” and made reference to other possible descriptions that dated back to the eighteenth century. Thus, it appears that this tumor may have been recognized well over a century ago. In 1914, Gossett and Masson [7] described similar tumors in the appendix and made an analogy to the previous description by Oberndorfer [5]. Contrary to the knowledge generated for these tumors in the gastrointestinal system, similar tumors in the respiratory tract, essentially those occurring in the bronchial wall, were being coded as bronchial adenomas [8, 9]. Gmelich et al. [10] identified the presence of Kultschitzky cells in bronchioles and established the relationship of these cells and the occurrence of these neoplasms in the lung. Interestingly, Hausman and Weimann [11] described a case with lymph node metastasis under the designation of “pulmonary tumorlet.” The authors alluded to the fact that these tumors have a low malignant potential. As per their description of the case, the tumor measured 1.5 cm, and it had spindle cell morphology with lymph node metastasis. On the other hand, Azzopardi [12] in a study of 100 cases of what he called “oat cell carcinoma,” based on 16 surgical cases and 84 cases from autopsy material, stated that oat cell carcinoma has positive structural features that identify this tumor including streams, ribbons, rosettes, and ductules. Judging by this definition and at least one of the illustrations presented in this review, it is possible that some of the cases presented in this study may not represent oat cell carcinoma as it is defined today. Thus, it is possible that some of the cases presented by Azzopardi [12] may correspond to intermediate-grade neuroendocrine carcinomas of today. Similar experience may be drawn from the 138 cases of oat cell carcinoma presented by Yukato et al. [13] where some of those tumors, although neuroendocrine in nature, may not necessarily be of the oat cell type, as it is defined today.
In order to shed light onto this difficult subject, many authors have employed different terms to explain the origin of these tumors. Terms such as histogenesis, differentiation, multidirectional differentiation, and/or divergent differentiation have been used, unfortunately not with the intended goal, as they have introduced more confusion into this field. Gould et al. [14] introduced the term “multidirectional differentiation” after observing the presence of neuroendocrine, mucosubstance-producing, and squamous cells in pulmonary carcinomas. In addition, Gould et al. [14] made observations that certain tumors may share similar patterns of differentiation. Interestingly, Gould et al. [14] also described cases, which by electron microscopy showed predominant features of squamous differentiation, thus designating those tumors as neuroendocrine carcinomas with squamous differentiation. The authors also alluded to the fact that some squamous and adenocarcinomas of the lung may show membrane-bound and dense-core granules by electron microscopy. However, Gould [15] also warned about the possibility of having cell populations with similar or identical patterns of differentiation, which may not necessarily share identical or even closely related embryogenesis. To that extent other authors had documented earlier the presence of neoplasms of the non-small cell type, which histologically may look like squamous cell carcinomas or adenocarcinomas and in which electron microscopic studies may show the presence of neurosecretory granules. Earlier denominations for those tumors have been atypical endocrine tumors. Such descriptions have raised high concerns about the true significance of the many classification systems available for neuroendocrine tumors [16]. In addition, tumor differentiation raises even higher issues such as the fact that some small cell carcinomas may lack the presence of immunohistochemical differentiation for neuroendocrine markers and/or the presence of neurosecretory granules by ultrastructural studies, at the same time showing the presence of ultrastructural features of epithelial tumors. A fact that is worth mentioning is that regardless of the histological subtype, all lung tumors are capable of showing neuroendocrine differentiation by immunohistochemistry or electron microscopy.
Recently, other authors [17] arguing that the current revised classification of tumors by the World Health Organization [2] clearly defines each one of the neuroendocrine tumors of the lung have introduced the term “divergent differentiation.” The authors state that divergent differentiation applies to a subset of non-small cell carcinomas that are not considered neuroendocrine on morphological grounds but that show neuroendocrine differentiation with immunohistochemical markers (so-called non-small cell carcinoma with neuroendocrine differentiation). In addition, Brambilla et al. [17] state that the so-called tumorlets do not differ in the cellular composition from the so-called typical carcinoid and that these tumorlets also display divergent differentiation. Although there are reports of “metastatic tumorlets”[11], those early reports are incorrect and represent the current so-called typical carcinoid. However, the authors have personally seen a few cases in which the neuroendocrine tumor in question has measured under 0.5 cm and yet metastatic disease to the lymph nodes has been observed. These cases are highly unusual and by no means represent the more likely biological behavior of the so-called tumorlet, which in the vast majority of cases shows indolent clinical behavior.
Classification Schemas
Neuroendocrine lung neoplasms have been a subject of numerous classification approaches, many of them, although logical, fail to provide a practical approach while others, although practical, fail to properly define this complex group of tumors. Herein we evaluate the most important past and present classification systems with their respective salient features and at the same time attempt to highlight their practical use or the lack of it. Table 4.1 depicts three of the most common approaches to the classification of these tumors.
In 1977, Gould [18] introduced the terms “neuroendocrinomas” and “neuroendocrine carcinomas” by drawing an analogy of these tumors with the APUD (amine precursor uptake and decarboxylation) cell system neoplasms and their aberrant secretory activities. Gould emphasized the numerous neoplasms that may belong to this APUD system, which is not limited to the respiratory tract or to a particular group of tumors, i.e., carcinoid tumor. Gould also elaborated on abandoning traditional terms such as “bronchial adenoma”—a term that does not convey the true nature of these neoplasms and that at the same time is used to encompass a diverse group of tumoral conditions. A few important issues may be highlighted from this study: first, the preferred term of bronchopulmonary neuroendocrine tumor as opposed to bronchopulmonary carcinoid; second, the concept that “oat cell” carcinomas represent the malignant counterpart of carcinoid tumor; and third, the continuous use of the term undifferentiated “oat cell” carcinoma. In 1983, Gould et al. [19] presented a new classification system for neuroendocrine pulmonary neoplasms. Novel to this classification system was the introduction of a four-way classification system instead of the conventional three-way split. Gould’s schema is as follows:
-
Bronchopulmonary carcinoid—Typical histology, locally invasive, potential for recurrence, and distant metastasis. Bronchopulmonary carcinoid is separated from neuroendocrine carcinoma (see below). The cases depicted showed penetration of the bronchial wall and mediastinal soft tissue. In addition, six cases showed direct invasion into lymph nodes at the time of initial presentation.
-
Well-differentiated neuroendocrine carcinoma—Even though it was stated that it may not represent an entirely satisfactory designation, this designation is for tumors that retain a clearly organoid pattern, moderate cellular pleomorphism, mitosis, and “true” lymph node metastasis.
-
Neuroendocrine carcinoma of intermediate-size cells—Represents a variant of small cell neuroendocrine carcinoma; the cells are twice the size of “small cell” counterparts with prominent nucleoli and abundant mitotic figures. Interestingly, the authors state that 7 of the 11 cases presented showed features of glandular or squamous differentiation.
-
Neuroendocrine carcinoma of small cell type—Typical “oat” cell carcinoma, abundant mitoses, and inconspicuous nucleoli. The authors comment that not all tumors in this category are neuroendocrine and recommend the systematic use of immunohistochemistry to separate those tumors that are neuroendocrine from those which are not.
In 1985, Warren et al. [20] presented a study of 81 cases of pulmonary neuroendocrine neoplasms assessing Gould’s classification systems and determined their usefulness for the proper classification and treatment of patients with those neoplasms.
At the same time, Paladugu [21] presented a new classification system under the designation of bronchopulmonary Kultschitzky cell carcinomas (KCC) and reverted to the three-way category of neuroendocrine neoplasms. The authors used the designation of KCC-1, KCC-II, and KCC-III for the typical carcinoid, atypical carcinoid, and small cell carcinoma, respectively. The author provided a mortality rate of 1.7% for KCC-I and 27% for KCC-II. Histologically, the tumors coded under KCC-II showed a mitotic activity of 1 mitotic figure per high-power field (hpf). The authors concluded that their nomenclature is preferred to that of Gould given the fact that they consider the K-cell as the origin of these tumors and in addition, it is simpler and less confusing.
However, it was Arrigoni et al. [22] who in 1972 laid the basis for the concept of “atypical carcinoid” by separating those tumors based on cellular atypia and mitotic activity with an average of one mitotic figure per one or two high-power fields, thus leaving a window of 5–10 mitotic figures per 10 hpf. It is of importance to note that in either Arrigoni et al. [22], Gould et al. [19], and Paladugu’s [21] classification systems, the number of mitotic figures per 10 hpf is left to some extent to interpretation, as all these authors were not dogmatic in presenting a specific number of mitotic figures to separate conventional from atypical “carcinoid tumor”. Interestingly, in 1982, Mills et al. [23] presented a study of 17 cases of atypical carcinoid tumors of the lung in which the mitotic count in those tumors varied from 2 to 28 mitotic figures (mean: 14, median: 13). Also Valli et al. [24] presented a study of 33 cases of atypical carcinoid tumors of the lung in which the mitotic activity varied from 4 to 80/1.52 mm2. In other studies [25], the reported criteria are those of increased mitotic activity, greater than one per one or two hpf. Based on those reports, one can only assume that the criteria to separate carcinoid from atypical carcinoid have been less than ideal when it comes to the issue of mitotic activity. In 1995, Capella et al. [26] revised the classification of neuroendocrine tumors of the lung, pancreas, and gut, stating the following schema:
-
Benign or low-grade malignant nonfunctioning well-differentiated tumor as the equivalent for conventional carcinoid
-
Low-grade malignant nonfunctioning well-differentiated carcinoma as equivalent for atypical carcinoid
-
High-grade malignant functioning or nonfunctioning poorly differentiated carcinoma for the large cell type and the small cell or intermediate type
The mitotic count to separate typical from atypical carcinoid was established at no more than 3 mitotic figures × 10 hpf. The authors added that if metastasis or gross invasion is present, tumors should be called low-grade neuroendocrine carcinoma.
In 1991, Travis et al. [27] presented a study of 35 cases of neuroendocrine carcinomas of the lung in which criteria were presented for large cell neuroendocrine carcinoma. In this study, previous criteria for other neuroendocrine carcinomas were followed, and the large cell carcinoma was presented as a tumor with a “neuroendocrine pattern, high mitotic activity with an average of 66 × 10 hpf, and prominent nucleoli.” The authors stated that the prognosis of large cell neuroendocrine carcinoma lies between atypical carcinoid and small cell carcinoma. However in 1998, Travis et al. [28] presented a new study of neuroendocrine neoplasms in which the authors’ goal was to provide clear definitions for the four neuroendocrine tumors, in addition to modify the criteria for the diagnosis of carcinoid and atypical carcinoid. The “new” classification schema placed large cell neuroendocrine carcinoma in the high-grade category of tumors contrary to the previous study [27]. This new approach is as follows:
-
Conventional carcinoid tumor is now restricted to no more than 2 mitoses × 10 hpf.
-
Atypical carcinoid: 2–10 mitoses × 10 hpf, or necrosis (often punctuate).
-
Large cell neuroendocrine carcinoma: tumors with “neuroendocrine morphology”, >10 mitoses × 10 hpf, cytologic features of large cell carcinoma, and positive immunohistochemical staining for neuroendocrine markers.
-
Small cell carcinoma is a tumor with the cytology of small cell tumor cells (absent nucleoli), mitotic figures of >10 × 10 hpf, and frequent necrosis.
This classification system is essentially repeated in the last version of the WHO publication for the classification of tumors of the lung, pleura, thymus, and heart [2]. However, it is important to note that this represents a classification for resected specimens. It is of interest to note that in a separate study on the reproducibility of the proposed classification of neuroendocrine lung tumors conducted by Travis et al. [29], in which 5 experienced pulmonary pathologists participated in the evaluation of 40 surgical resections of neuroendocrine tumors, a unanimous agreement was reached in only 55% of the cases. The most common disagreements were between large cell neuroendocrine carcinoma and small cell carcinoma. Lastly, in 2002, Huang et al. [30] presented the latest attempt to classify neuroendocrine tumors of the lung. The authors presented a study of 234 cases and classified them into five different categories. This system essentially follows Travis’ criteria [28] for the separation of carcinoid and atypical carcinoid, now named well- and moderately differentiated neuroendocrine carcinoma; the terms large cell neuroendocrine carcinoma and small cell carcinoma are kept with the prefix of “undifferentiated” with mitotic counts of more than 30 × 10 hpf. A new category is what the authors call “poorly differentiated neuroendocrine carcinoma,” which is conceptualized as an atypical carcinoid with an increase mitotic activity of more than 10 × 10 hpf.
Clinical Features
The clinical presentation of neuroendocrine tumors of the lung is varied. For instance, small cell carcinoma of the lung usually presents as a large hilar mass with bulky mediastinal lymphadenopathy. In many cases, patients may present with widespread metastatic disease at the time of diagnosis. Nevertheless, in a few instances, small cell carcinomas may present with a solitary peripheral nodule. Paraneoplastic syndromes including Cushing’s syndrome, inappropriate secretion of antidiuretic hormone, and carcinoid syndrome have also been reported in association with neuroendocrine carcinomas; carcinoid syndrome are associated with approximately 10% of the cases of well-differentiated histology. Other conditions that have also been associated with these tumors include Lambert-Eaton myasthenic syndrome and paraneoplastic encephalomyelitis and sensory neuropathy. In addition, depending on the location of the tumor, those in the central location may also show symptoms of pulmonary obstruction, dyspnea, cough, and/or chest pain. Patients with tumors in the periphery of the lung may be asymptomatic until the tumor reaches a larger size. Although neuroendocrine carcinomas may occur at any age, the tumors are more commonly encountered in the fifth to seventh decade of life. No gender predilection has been noted.
Macroscopic Features
Those tumors occurring in the central location may present as polypoid tumors obstructing the lumen of the airway. The tumors may measure from 1 cm to more than 10 cm in diameter. They are light brown and at cut surface appear homogenous. The presence of areas of necrosis or hemorrhage should alert for a higher grade tumor. In high-grade neuroendocrine carcinomas, it is common to encounter invasion into mediastinal structures at the time of diagnosis.
Microscopic Features
Tumorlet
The histopathological features present in the so-called tumorlet are essentially the same as those that one would find in otherwise well-differentiated neuroendocrine carcinoma (Fig. 4.1a,b). As a matter of fact, this so-called tumorlet may represent the true carcinoid tumor of the lung. However, the diagnosis of tumorlets is restricted to lesions of no more than 0.5 cm size in greatest diameter. Although this lesion is more often found incidentally in lung biopsies, nowadays with the use of more sophisticated radiological techniques, “tumorlets” are becoming more common. Even though the great majority of cases are likely to be encountered as an incidental finding in cases in which resection has taken place for other reasons, we have observed a few cases in which there has been an incidental “tumorlet carcinoid” in the lung, fulfilling all the required histopathological characteristics, but yet the tumor had metastasized to a lymph node. This occurrence should be taken into consideration when encountered with the finding of metastatic disease to the lymph node from an unknown source of disease.

(a) Carcinoid tumorlet (see arrows) with similar cellular characteristics of a well-differentiated neuroendocrine carcinoma, except that these lesions are under 0.5 cm in diameter. (b) Higher magnification showing a homogeneous cellular proliferation without mitotic activity
Well- and Moderately Differentiated Neuroendocrine Carcinomas (Carcinoid and Atypical Carcinoid)
The current designation provided by the WHO [2] defines these tumors as having neuroendocrine morphology, namely, organoid, trabecular, insular, palisading, ribbon, and rosette-like features (Figs. 4.2a–c and 4.3a–c). However, the separation of well- and moderately differentiated neuroendocrine carcinoma according to the WHO is that the former has fewer than two mitotic figures per 10 hpf while the latter has 3–10 mitotic figures per 10 hpf. Clinically, both tumors may be associated with carcinoid syndrome [31] and show a spectrum of cell differentiation that includes spindle cells, oncocytic, and melanocytic features, among others [32–37]. Although the WHO still maintains the nomenclature of carcinoid and atypical carcinoid, some authors believe that the most accurate designation for these neoplasms is that of neuroendocrine carcinoma, which conveys the true nature of these tumors, and is the one followed in this section [22, 38]. In addition, the issue of one mitosis to separate well- from moderately differentiated tumors may be an artificial designation that in practice may not hold true. Therefore, we recommend that such criteria should be used with care as in some cases the diagnosis of moderately differentiated neuroendocrine carcinoma (atypical carcinoid) may imply the use of chemotherapy. Careful communication with the oncologist is of utmost importance to convey such information. Recently, we evaluated 80 cases of low- and intermediate-grade neuroendocrine tumors using three different methods to count mitosis and encountered that the overall mean number of mitoses correlated with the recurrence-free survival [39]. Interestingly, the overall survival (OS) of patients when mitotic activity was counted in random fields was not dramatically different if the tumor showed fewer than 2 or between 2 and 10 mitotic figures per 10 high-power fields as the OS was 100 and 97%, respectively. An almost similar situation occurred when the mitotic figures were counted in mitotically active fields as the OS was that of 100 and 96%, respectively. Based on those results, it is possible that mitotic count may not be as reliable a parameter as it has been considered. On the contrary, the presence of necrosis may offer a better clue for clinical outcome.

(a) Well-differentiated neuroendocrine carcinoma showing a classical nested growth pattern, note the presence of bronchial epithelium and cartilage. (b) In other areas, the nested growth pattern is preserved with a homogeneous cellular proliferation. (c) Higher magnification showing absence of nuclear atypia and mitotic activity

(a) Moderately differentiated neuroendocrine carcinoma showing a diffuse growth pattern composed of medium-size cells. (b) In other areas, a nested growth pattern is present; however, note the presence of comedo-like necrosis. (c) High-power magnification shows the presence of mitotic activity (see arrows)
At low magnification, the tumor displays an organized growth pattern, which may show nesting, solid, pseudoglandular, and/or trabecular arrangements composed of a rather homogeneous cellular proliferation characterized by small- to medium-size cells with moderate amounts of eosinophilic cytoplasm, round to oval nuclei; nucleoli may be identified in some cells. Rosette formation may be easily identifiable. The presence of necrosis in the form of comedo-like necrosis and/or hemorrhage is an important criterium for the diagnosis of moderately differentiated neuroendocrine carcinoma.
Several histological variants have been described [40–42] for both well- and moderately differentiated neuroendocrine carcinomas including:
-
Spindle cell type: In this variant, although one may find an organoid pattern, the cell morphology is that of fusiform cells with inconspicuous nuclei and finely disperse chromatin (Figs. 4.4, 4.5, 4.6, 4.7, and 4.8). In some cases, the spindle cell proliferation may be associated with dilated blood vessels imparting a hemangiopericytic pattern. However, this variant may also display nuclear atypia and mitotic activity.
Fig. 4.4 
Well differentiated neuroendocrine carcinoma with a prominent spindle cell component; note the presence of airways and bronchial cartilage
Fig. 4.5 
(a) The cellular proliferation may show tightly packed nests of spindle cells. (b) Loosely arranged nests of spindle cells separated by fibroconnective tissue
Fig. 4.6 
(a) The tumor may spread into alveolar spaces. (b) The tumor may also show vascular permeation
Fig. 4.7 
Moderately differentiated neuroendocrine carcinoma, spindle cell type with comedo-like necrosis
Fig. 4.8 
Areas of pseudoglandular appearance may also be present
-
Oncocytic type: The growth pattern of this neoplasm is essentially similar as those of the conventional cell type. The tumors may have a diffuse growth or a glandular appearance. Cytologically, the cells are of medium size with ample eosinophilic cytoplasm, round to oval nuclei, and in some cells prominent nucleoli are seen (Figs. 4.9, 4.10, 4.11, and 4.12). In some cases, clusters of oncocytic cells are present among tumor cells—so-called oncoblasts. This variant may also display features of moderately differentiated neuroendocrine carcinoma by displaying increased mitotic activity. However, one needs to be careful in assessing the presence of mitotic activity since oncocytic tumors may display areas of nuclear atypia without mitotic activity.
Fig. 4.9 
Peripheral oncocytic neuroendocrine carcinoma showing a well-demarcated tumor
Fig. 4.10 
(a) The tumor may be formed by cords of neoplastic cells. (b) The tumors may show a prominent solid growth pattern
Fig. 4.11 
The tumor may show numerous calcifications of “psammoma-like” type (see arrows)
Fig. 4.12 
Higher magnification showing tumor cells with ample eosinophilic cytoplasm, round nuclei, and prominent nucleoli
-
Mucinous type: This is a rare occurrence in primary neuroendocrine carcinomas of the lung. The presence of mucus material may be limited to the intraluminal component in some cases of glandular arrangement, or more rare the mucinous component may be intermixed with the neoplastic cellular proliferation (Fig. 4.13a,b).
Fig. 4.13 
(a) Mucinous neuroendocrine carcinoma showing pools of mucoid material. (b) In some areas, the neoplastic cells may be embedded in pools of mucoid material
-
Melanocytic type: The growth pattern is similar as those of the conventional type; however, melanin pigment may be present in cells (Fig. 4.14a,b) or distributed along the fibroconnective tissue.
Fig. 4.14 
(a) Pigmented neuroendocrine carcinoma with oncocytic changes. Note the cluster of cells with melanin pigment (see arrow). (b) Closer view of the cluster of cells with melanin pigment
-
Clear cell type: In this variant, the neoplastic cells are characterized by the presence of clear cytoplasm (Fig. 4.15a,b). This cytoplasmic feature may be seen in either spindle cell or conventional morphology.
Fig. 4.15 
(a) Neuroendocrine carcinoma showing a classical nested pattern and composed of cells with clear cytoplasm. (b) Higher magnification showing prominent clear cell changes
-
Angiectatic type: This variant is characterized by the presence of large dilated spaces filled with blood mimicking a vascular neoplasm (Fig. 4.16). However, closer inspection of the tumor cells reveals the presence of more conventional areas of neuroendocrine morphology.
Fig. 4.16 
Neuroendocrine carcinoma with presence of ectatic spaces filled with blood imparting a pseudo-vascular appearance
-
Amyloid-like type: The morphology of these tumors is essentially the same as conventional neuroendocrine carcinoma. However, the tumor cells are admixed with areas of hyalinization imparting an amyloid-like matrix (Fig. 4.17).
Fig. 4.17 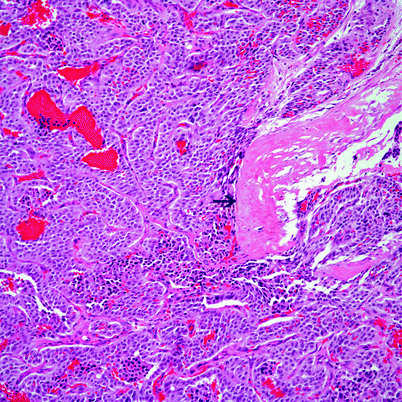
Conventional neuroendocrine carcinoma with areas of dense hyalinization (see arrow) mimicking amyloid
-
Sclerotic type: In these cases, the tumor cells may not be readily identifiable due to the extent of collagenization of the sclerosing changes. This type of neuroendocrine carcinoma may pose a challenge when dealing with small biopsies. However, in all the cases described, clusters of neuroendocrine cells have been present enough to make a diagnosis. Nevertheless, the sclerotic changes should not be considered any type of special feature in order to upgrade the tumor to a higher grade as such changes may be seen in low- as well as intermediate-grade tumors (Fig. 4.18a–c).
Fig. 4.18 
(a) Neuroendocrine carcinoma with extensive areas of hyalinization in transition with tumoral areas. (b) Extensive hyalinization with only clusters of neuroendocrine cells (arrow). (c) Higher magnification showing the neuroendocrine cellular proliferation (arrows)
-
Metaplastic bone formation: The growth pattern is of conventional neuroendocrine carcinoma in which the presence of well-formed bone (Fig. 4.19) is admixed with the neoplastic cellular proliferation.
Fig. 4.19 
Neuroendocrine carcinoma with metaplastic bone formation
















Immunohistochemical Features
The immunohistochemical features of both well- or moderately differentiated neuroendocrine carcinomas (carcinoid and atypical carcinoid) are similar in terms of sharing positive staining using the conventional neuroendocrine markers, namely, chromogranin, synaptophysin, and CD56. In addition, the use of thyroid transcription factor-1 has also been shown to be helpful in the evaluation of neuroendocrine neoplasms of the lung [43]. More recently, Tsuta et al. [44] conducted a study of neuroendocrine carcinomas of the lung in which the tumors were stained with Sox10 for sustentacular cells. The authors stated that Sox10-positive sustentacular cells were observed in well-differentiated tumors (carcinoid tumors) but not in high-grade neuroendocrine carcinomas. On the other hand, the use of proliferation markers such as KI-67 in the separation of conventional and atypical carcinoid has found that a 4% cutoff provides significant differences for the 4-year overall survival rate [45]. In this particular study, the methodology that was followed for the classification of neuroendocrine neoplasms was the one presented by Arrigoni [22] and Warren [20], and the study was performed on resected specimens. This study raises an important issue, and that is that the complete interpretation of these tumors rests on surgical resections and not on biopsy specimens, where labeling of proliferation markers may be deceiving. Since most of the initial approach to the classification of neuroendocrine tumors is performed on a biopsy specimen, it is possible that using specific mitotic markers may be of aid in difficult cases in which there is doubt about the possibility of mitotic activity. In that regard, Tsuta et al. [46], using a mitosis specific marker—the anti-phosphohistone H3 (PHH3)—to assess mitosis in neuroendocrine carcinomas of the lung, concluded that PHH3 may be a reliable aid in determining mitotic activity.
Molecular Features
Well- and moderately differentiated tumors have also been the subject of more modern techniques such as chromosomal studies in which 11q deletions appear to be shared by both tumors [47]. Losses of 10q and 13q may also be responsible for a possible aggressive behavior shown by some of these tumors.
Clinical Behavior
Due to the lack of real comparisons between previous and current schemas for the classification of these tumors, the survival rates of patients with low- and intermediate- grade neuroendocrine carcinomas are difficult to assess and address meaningfully. Wilkins et al. [48] in 1984, obviously using a different schema (most likely Arrigoni’s criteria), presented a study of 111 patients who underwent surgical resection for “bronchopulmonary carcinoid”; 11 of these patients had atypical carcinoid, and 45% of them died in a period of 33 months. Those who were still alive had been followed for 16–48 months. However, even though the authors provided a survival rate of 82% for a 10-year period, close examination of the data provided is rather limited, and no clear-cut survival rate is provided for the conventional carcinoid. A better-defined publication on “typical carcinoid” is the one presented by Schreurs [49] in a study of 93 patients and a period of 25 years. Once again, the likely criteria for classification in this study were those formulated by Arrigoni. The authors provided an important survival rate of 100% for 86 patients at 5, 10, and years (according to the authors, seven patients died of unrelated causes) for surgically treated patients. However, the selection process for this study excluded patients with distant metastasis. Thus, raising the issue that staging of the neoplasm at the time of diagnosis also plays an important role in this group of tumors and impacts heavily in the survival rate.
Small Cell Carcinoma
The latest version of the WHO [2] defines this tumor as a malignant tumor with cells with scant cytoplasm and absent or inconspicuous nucleoli; necrosis is extensive and mitotic count is high. The mitotic count provided is of 60 mitoses/2 mm2. It is obvious from this definition that the authors of the WHO are far from the real practice of surgical pathology as such criteria apply only when there is a resected specimen, which rarely happens in daily practice. Nevertheless, in a study of 100 cases of small cell carcinoma [50], the authors clearly state that in over 90% of the cases, the diagnosis of small cell carcinoma can be established on small biopsy. Needless to say, an important fact that must be emphasized is that most patients with small cell carcinoma are beyond a stage that makes them amenable for surgical resection of the tumor, as has been noted in the literature [51], thus leaving the surgical pathologist confronted with a small biopsy in order to provide a definitive diagnosis.
The topic of small cell carcinoma has not been exempted from controversy. In 1985, Vollmer et al. [52] presented a study in which the authors analyzed the issue of subclassification of small cell carcinoma into oat cell or intermediate types. However, in 1988, the Pathology Committee of the International Association for the Study of Lung Cancer recommended the use of small cell carcinoma and that such designation would include tumors previously denominated as oat cell and intermediate subtypes [53]. Even terms such as small cell neuroendocrine carcinoma [54] may fall under scrutiny, as a proportion of these tumors may not show positive reaction with antibodies against neuroendocrine markers.
Macroscopic Features
As stated before, small cell carcinomas often present in late stages of the disease. The tumors are commonly centrally located, of large size and with variable proportions of necrosis.
Microscopic Features
The tumor shows a non-cohesive cellular distribution composed of small cells with scant cytoplasm, round nucleus, and inconspicuous nucleoli. Single cell necrosis or extensive areas of necrosis are common. In addition, the presence of the so-called Azzopardi phenomenon is commonly observed in these tumors. The mitotic count in resected specimens or in open lung biopsies is more than 10 × 10 hpf. In some areas, the morphology is that of spindle cells, with oval nuclei and inconspicuous nucleoli. Mitotic activity is high (Figs. 4.20 and 4.21a–d). However, it is important to recognize that most of these features may be absent in a small transbronchial biopsy and that the established criteria for diagnosis of small cell carcinoma may not be met in such material. Small biopsies are characterized by the presence of crush artifact and little viable cells with only occasional mitotic figures.

Small cell carcinoma showing the typical cellular proliferation. Note the presence of residual normal endobronchial glands and bronchial cartilage

(a) Small cell carcinoma with extensive necrosis. (b) The so-called Azzopardi phenomenon is classic in small cell carcinomas. (c) Closer view at the neoplastic cellular proliferation showing high mitotic activity. (d) Higher magnification showing small cells with absence of nucleoli
Immunohistochemical Features
There are no comprehensive studies on the immunohistochemistry of small cell carcinoma. In a rather limited immunohistochemical study of small cell carcinoma [55], the authors found that neuroendocrine markers such as chromogranin were positive in 60% of open lung biopsies but only in 47% of transbronchial biopsies, while synaptophysin showed 5 and 19% positivity, respectively. Nevertheless, most authors concur that small cell carcinoma represents the end of the spectrum of neuroendocrine carcinomas of the lung. One additional factor that must be carefully analyzed is that in small, poorly preserved biopsies in which the intensity of neuroendocrine markers is marked, one must carefully exclude the possibility of a lower-grade neuroendocrine carcinoma as an alternative for small cell carcinoma [56].
The implications of a diagnosis of small cell carcinoma cannot be overemphasized, as the patients may undergo chemotherapy, radiation therapy, or both. In addition, large studies have estimated, that the long term survival of patients with small cell carcinoma is approximately 10% at 2 years [57]. However, even though there is little mention about staging for small cell carcinoma, the Veterans Administration Lung Group stages small cell carcinoma in a two-tier schema: (1) limited-stage disease, in which the tumor is confined to the ipsilateral hemithorax, and (2) extensive-stage disease, in which the tumor has grown beyond the ipsilateral hemithorax. However, more recently a TNM system has been presented, which appears to provide prognostic significance [58].
Large Cell Neuroendocrine Carcinoma, Large Cell Carcinoma with Neuroendocrine Morphology, and Large Cell Carcinoma with Neuroendocrine Differentiation
By definition, these tumors are poorly differentiated non-small cell carcinomas. According to the WHO [2], the main distinguishing features of large cell neuroendocrine carcinoma and small cell carcinoma are the presence of prominent nucleoli.
Macroscopic Features
Large cell neuroendocrine carcinomas (LCNEC) are large neoplasms usually in advance clinical stages at the time of diagnosis. The tumor may be in a central location, and it may present as well-demarcated, solid, grayish, with or without apparent necrosis.
Microscopic Features
Large cell neuroendocrine carcinomas will display a neuroendocrine morphology, namely, the presence of ribbons, rosettes, or nesting patterns combined with large zones of necrosis and a mitotic count higher than 11 mitotic figures/2 mm2 (average 75) of viable tumor (Figs. 4.22, 4.23, 4.24, 4.25, and 4.26). This latter definition clearly applies only to resected tumors; thus, such diagnoses can hardly be accomplished on small biopsies in which one may not get enough viable tumor to evaluate the so-called neuroendocrine pattern or evaluate the mitotic count of more than 11 mitotic figures.

Large cell neuroendocrine carcinoma adjacent to an airway. Note the nested neuroendocrine growth pattern of the tumor

(a) Areas of necrosis are commonly seen, and in some cases comedo-like necrosis is common. (b) In other areas, the tumor may show a basaloid component. (c) In more unusual tumors, areas of mucinous pools may be seen

(a) Important in the diagnosis of large cell neuroendocrine carcinoma is the identification of areas with neuroendocrine growth pattern. (b) Closer view at the neuroendocrine growth pattern showing cells larger cells with round nuclei and presence of nucleoli

Closer view at the neoplastic cells showing larger cells with presence of nucleoli and increased mitotic activity (see arrows)

Chromogranin showing positive staining in tumor cells
Mooi et al. [59] described 11 primary lung carcinomas under the rubric of non-small cell carcinoma with neuroendocrine features. These tumors had been diagnosed either as large cell carcinoma or squamous cell carcinoma, but all the tumors had similarities to bronchial carcinoid and small cell carcinoma. Six of seven cases in which electron microscopy studies were available showed dense-core granules, while all cases showed positive staining for neuron-specific enolase (NSE) (most of the antibodies that are available today may not have been available in 1988). The authors also stated that neuroendocrine features may be suspected by light microscopy. Judging by some of the illustrations provided, one can assume that some of those tumors, if not all, are similar to today’s designation of large cell neuroendocrine carcinoma. Dresler et al. [60] in 1997 analyzed 40 cases of large cell neuroendocrine carcinoma and concluded that large cell neuroendocrine carcinomas identified by histological examination have poor prognosis. The authors also suggested that lesions previously categorized as large cell carcinoma with neuroendocrine features should be regarded as “large cell carcinoma with occult neuroendocrine differentiation.” This latter suggestion specifically addresses an issue that goes to the core of these types of tumors, which is how to group tumors that have a “neuroendocrine pattern” but yet fail to show immunoreactivity for neuroendocrine markers. As the criteria are currently presented, in order to make the diagnosis of “large cell neuroendocrine carcinoma,” the tumor must have “neuroendocrine morphology” and positive staining for at least one neuroendocrine marker [2]. This issue has become even more confusing by the use of terms such as small cell-like large cell neuroendocrine carcinoma in some cytologic studies [61]. In addition, following the presence of nucleoli to separate small from non-small cell carcinoma may prove difficult just as using cell size for their differentiation. Marchevsky et al. [62] evaluated, by morphometric means, 28 surgically resected high-grade pulmonary neuroendocrine carcinomas (16 small cell carcinomas and 12 large cell neuroendocrine carcinomas). The authors concluded that there is considerable nuclear overlap between these entities, which helped the author to separate only 9 of 28 cases studied, and suggested the use of more generic terminology such as high-grade neuroendocrine carcinoma or grade III neuroendocrine carcinoma. The entire topic of “large cell neuroendocrine carcinoma” has generated a myriad of publications in order to bring light to this confusing topic. For instance, Iyoda et al. [63], in a study of 2,070 cases of large cell carcinomas, divided such tumors into four different categories: large cell neuroendocrine carcinoma, large cell carcinoma with neuroendocrine differentiation, large cell carcinoma with neuroendocrine morphology, and “classic” large cell carcinoma. In a multivariable analysis, the authors grouped the three former entities into a single entity, which the authors denominated as large cell carcinoma with neuroendocrine features separate from the “classic” large cell carcinoma, and concluded that those tumors are more aggressive. What is interesting and important in this study is the fact that tumors in which the authors did not find morphologic evidence of neuroendocrine change were grouped as large cell carcinomas with neuroendocrine morphology. Yet on multivariable analysis, the authors found that these tumors – just the same as those grouped as large cell neuroendocrine carcinoma and large cell carcinoma with neuroendocrine differentiation – behave more aggressively compared to the “classic” large cell carcinoma. Nevertheless, their point raises another issue and that is whether it is important to determine neuroendocrine “differentiation” by immunohistochemical means and whether a specific diagnosis is warranted for those tumors. Some other publications on this topic have lumped large cell neuroendocrine carcinomas and large cell carcinoma with neuroendocrine morphology under the same designation [64]. Unfortunately, in many recent publications on the subject, even as the authors claimed that they have used rigorous application of the WHO criteria [2], the emphasis has been on those tumors that fit the criteria of morphology and immunohistochemistry, leaving unanswered the question of how to handle tumors that may show the morphology but not the immunohistochemistry of a neuroendocrine carcinoma [65–75]. In other reports, it is very difficult to discern which cases were accounted for as large cell neuroendocrine carcinoma, since the terminology used appears to be ambiguous [76–79]. More recently Glisson and Moran [80] addressed the difficulties and controversies in the diagnosis and treatment of large cell neuroendocrine carcinoma and stated that because of the rarity of this tumor and vagaries in diagnosis because of the complexity of current pathologic classification, current treatment recommendations must be based on retrospective data, which are imperfect at best.
Chromosomal and loss of heterozygosity (LOH) analyses have been undertaken to separate tumors with features of large cell neuroendocrine carcinoma [81], while in others the nomenclature used is not the one assigned by the WHO classification system [82]. In some reports, these tumors have been correlated with pathologic stage, raising the issue of staging, since these tumors may present in different pathological stages [83]. In some recent publications [84, 85], an emphasis has been made to separate large cell neuroendocrine carcinoma from small cell carcinoma by using immunohistochemistry and molecular studies. If one is to apply WHO criteria or any of the other schemas presented, such distinction should not be a difficult task, since one is small cell and the other is non-small cell carcinoma with prominent nucleoli. Furthermore, the problem of separating small from non-small cell carcinoma should not require immunohistochemistry or molecular studies.
Important to note is that some reports have concentrated on non-small cell carcinomas with neuroendocrine differentiation or neuroendocrine morphology [79, 86–91]. In a study by Howe et al. [86], the authors studied 439 cases of non-small cell carcinoma, which were evaluated using immunohistochemical stains for neuroendocrine markers. The authors reported that 36% of these tumors showed at least positive staining for one neuroendocrine marker and concluded that the presence of neuroendocrine differentiation in non-small cell carcinoma is of no prognostic significance, as has also been reported by other authors [88]. More recently, Ionescu et al. [91] arrived at a similar conclusion after reviewing 609 cases of non-small cell carcinoma. Important to mention is the fact that some adenocarcinomas may show neuroendocrine differentiation, which has been suggested as an important prognostic feature [92] while others have had a more circumspect approach about this particular issue [93].
Critical review of the literature raises some important questions:
-
1.
The diagnosis of large cell neuroendocrine carcinoma in most of the series presented has been a diagnosis in retrospect after analysis of all large cell carcinomas.
-
2.
The cases included in a good number of publications, despite the “rigorous” application of the WHO criteria, suggest that some of the tumors included may not exactly belong into that designation.
-
3.
The fact that resections have been evaluated speaks volumes to the fact that these tumors have already been initially treated as non-small cell carcinomas.
-
4.
Although all authors believe that these tumors are of high grade, there is not unanimous agreement regarding the best method of treatment for patients diagnosed with large cell neuroendocrine carcinomas. In this regard, some authors have advocated that large cell neuroendocrine carcinoma is potentially treatable with surgery [71] while others advocate additional medical treatment [94].
-
5.
There is some confusion regarding the use of adjectives such as “neuroendocrine differentiation” and “neuroendocrine morphology.”
-
6.
Staging may still represent an important independent factor in the prognosis of these tumors. The entire issue of neuroendocrine morphology or differentiation can be summarized as follows:
-
Large cell neuroendocrine carcinoma: tumors with neuroendocrine pattern and positive staining for neuroendocrine markers, i.e. chromogranin, synaptophysin, or CD56.
-
Large cell carcinoma with neuroendocrine features or morphology: tumors with neuroendocrine pattern but negative staining for neuroendocrine markers.
-
Large cell carcinoma with neuroendocrine differentiation: positive staining for neuroendocrine markers but absent neuroendocrine morphology.
-
Although one would expect that the issue of large cell carcinoma may be restricted to the issue of neuroendocrine morphology or neuroendocrine differentiation, the WHO separated yet another tumor that may provide even more confusion to the current classification, the so-called basaloid carcinoma of the lung [95].
Basaloid Carcinoma
This particular neoplasm was initially described as a new morphological and phenotypical entity. However, close analysis of their histopathological description shows that 19 of the 38 cases described showed areas of squamous cell carcinoma, adenocarcinoma, and large cell carcinoma, raising the possibility that at least half of the tumors described may be grouped into one of those more specific categories. On the other hand, the WHO classification [2] recognized that the distinction between large cell neuroendocrine carcinoma and basaloid carcinoma is difficult and often requires immunohistochemical studies. Also, the WHO acknowledges that in 10% of cases of basaloid carcinoma, a neuroendocrine marker may be positive, thus raising the possibility that “basaloid carcinoma” may represent a growth pattern rather than a specific pathological entity. Other studies on basaloid carcinoma have stressed the use of immunohistochemical studies, namely, cytokeratins 1, 5, 10, and 14 (34βE12) to differentiate such tumor from large cell neuroendocrine carcinoma. However, Lyda and Weiss [96] reported an incidence of 5% positive staining for 34βE12 in neuroendocrine carcinomas. Thus, separating these tumors by immunohistochemistry may not be as straightforward as thought, if indeed they represent different neoplasms.
Microscopic Features
These tumors appear to be well defined masses destroying lung parenchyma. As the name implies, the low-power shows islands of tumor cells arranged in ribbons and forming rosettes with palisading of the nuclei. High-power magnification shows oval to spindle cells with elongated nuclei, and in some cells prominent nucleoli may be observed (Fig. 4.27a,b). The mitotic count in these tumors is high and in resected specimens is well beyond 10 × 10 hpf.

(a) Basaloid carcinoma showing classic appearance with peripheral palisading of the nuclei. (b) Higher magnification showing a subtle spindling of the neoplastic cells
Carcinomas with Mixed Histologies
It is well known that pulmonary carcinomas in a small but well-represented number of cases may show combined histologies (Fig. 4.28a,b), namely, of the small cell/non-small cell categories [97–101]. It appears that the general consensus with these types of tumors is that the behavior is more aggressive than in pure small cell carcinomas, leading to a shorter survival rate. However, an important issue that needs further investigation is the possible association of small cell carcinoma and large cell neuroendocrine carcinoma or large cell carcinoma with neuroendocrine differentiation. Whether that distinction has a practical value in the treatment of these patients may be just an academic exercise since both of those tumors belong to the high-grade category and may be treated with chemotherapeutic regimens for high-grade neuroendocrine carcinomas.

(a) Combined small cell carcinoma (see arrow) and non-small cell carcinoma. (b) Combined large cell neuroendocrine carcinoma and conventional adenocarcinoma
Molecular Biology
Genetic studies have been performed showing a gain of 3q in about 66% of small cell carcinoma while deletions of 10q, 16q, and 17p were less frequent in large cell neuroendocrine carcinoma than in small cell carcinoma [102]. Other studies have shown that gene expression profiling has failed to distinguish small cell from large cell neuroendocrine carcinomas and has led some authors to suggest that both entities should be grouped under the designation of high-grade neuroendocrine tumors [103]. Other studies have placed more emphasis on the issue of the spectrum of differentiation of neuroendocrine tumors and have found through studies of expression of gene products that there is no evidence linking carcinoids and small cell carcinoma [104]. Similar findings have been encountered by other authors [105], leading to the suggestion that small cell carcinomas are derived from epithelial cells and that bronchial carcinoids are related to neural crest-derived tumors. It also appears that human p19ARF protein encoded by the beta transcript of the p161NK4a gene is more commonly lost in high-grade neuroendocrine carcinomas than in conventional non-small cell carcinomas [106].
Analysis of p53, K-ras-2, and C-raf-1 by some authors has led to the suggestion that typical and atypical carcinoids are genetically distinct from high-grade neuroendocrine carcinomas [107]. Regarding atypical carcinoids, it has been documented that loss of heterozygosity at 3p14.2–p21.3 is significantly more extensive in atypical carcinoids, while typical carcinoids are strongly positive for the cytoplasmic Fhit protein, similar to normal lung epithelia [108]. Interestingly, studies on the expression of Bcl-2 have concluded that the expression of Bcl-2 is involved in the pathogenesis of small cell carcinoma and carcinoid tumors of the lung [109]. The expression of retinoblastoma (RB) protein in neuroendocrine tumors has disclosed its presence in carcinoids and atypical carcinoids, while it is absent in small cell and large cell neuroendocrine carcinoma [110, 111]. It has also been suggested that hASH-1 (human homologue of Mas1) is involved in the neuroendocrine differentiation of small and non-small cell carcinomas [112].
Conceptual Practical Approach to the Diagnosis of Neuroendocrine Carcinomas
Based on current concepts and approaches, a practical approach to the diagnosis of neuroendocrine carcinomas may be conceptualized as follows:
Surgical Resections
-
Carcinoid tumorlet (neuroendocrine lesions under 0.5 cm in size)
-
Well differentiated neuroendocrine carcinoma (typical carcinoid tumor): 0–3 mitotic figures ´ 10hpf, only focal punctuate necrosis
-
Moderately differentiated neuroendocrine carcinoma (atypical carcinoid tumor): 4–10 mitotic figures ´ 10 hpf, and necrosis
-
High-grade neuroendocrine carcinoma:
-
Small cell carcinoma: with or without positive neuroendocrine markers, tumors with > 10 mitotic figures ´ 10 hpf, and necrosis
-
Large cell neuroendocrine carcinoma: large cell carcinoma with positive neuroendocrine morphology and positive neuroendocrine markers and large cell carcinoma with positive morphology and negative staining for neuroendocrine markers, large tumor cells with nucleoli, >10 mitotic figures ´ 10 hpf, and necrosis
-
Biopsy Specimens
-
Neuroendocrine carcinoma—if the tumor is in the spectrum of well- or moderately differentiated neuroendocrine carcinoma (carcinoid or atypical carcinoid) and the tumor radiologically is > 0.5 cm in greatest diameter (specify the possibilities)
-
Small cell carcinoma
-
Squamous cell carcinoma
-
Adenocarcinoma
-
Large cell carcinoma
Pulmonary Paraganglioma
Pulmonary paragangliomas will be discussed in this chapter as part of the spectrum of neuroendocrine tumors; however, their clinical behavior is distinct to that of the conventional neuroendocrine carcinoma. Also, important to note is the fact that intrapulmonary paragangliomas are rare tumors in this location and every effort has to be made in order to determine the possibility of a metastatic lesion to the lung. In general, these tumors are exceedingly rare, and only a few well-described cases are reported in the literature. The tumors appear to occur in adult individuals, who clinically may present with hypertension, increase serum norepinephrine, or Cushing’s syndrome [113–117].
Macroscopic Features
These tumors vary in size from under 1 cm to more than 3 cm in greatest dimension. The tumors are endobronchial and may obstruct the bronchial lumen producing symptoms of cough, wheezing, and dyspnea. Macroscopically, they cannot be separated from low- or intermediate-grade neuroendocrine carcinomas.
Microscopic Features
Low-power magnification shows that the tumor may appear as a polypoid mass filling the bronchial lumen. The classic low-power appearance of paragangliomas is the so-called Zellballen pattern. The cells are arranged in a nesting pattern in which the nests are separated by thin fibroconnective tissue and ectatic blood vessels. The tumor may also show oncocytic changes with a homogeneous cellular proliferation (Figs. 4.29, 4.30, 4.31, and 4.32). At high-power magnification, it is common to identify the presence of cells with macronuclei or bizarre nuclei but no associated mitotic activity [113–117]. Also common is the presence of large dilated vessels or the presence of extensive areas of hyalinization. In some cases, ganglion cells, represented by larger cells with eosinophilic cytoplasm, and prominent nucleoli accompany the cellular proliferation. In other cases, the tumor may show spindle cell morphology. In a few cases, lymph node involvement has been reported.

Primary pulmonary paraganglioma showing the conventional nested growth pattern. Note the presence of bronchial cartilage

Pulmonary paraganglioma showing a nested growth pattern composed of a homogenous cellular proliferation

(a) Cords of neuroendocrine cells separated by areas of fibrocollagen. (b) Dilated vascular structures are commonly seen. Note the presence of the neoplastic cells around the vessel

Higher magnification of a pulmonary paraganglioma showing large nuclei but absence of mitotic activity
Immunohistochemical Features
The same as neuroendocrine carcinomas, paragangliomas react positively with neuroendocrine markers such as chromogranin, synaptophysin, and/or CD56. However, keratin is negative in paragangliomas, while positive in neuroendocrine carcinomas. S-100 protein is positive in the sustentacular cells of paragangliomas.
Clinical Behavior
Surgical treatment appears to be the treatment of choice and in the majority of cases is the only treatment accompanied by close follow-up. However, due to the rarity of these tumors in an intrapulmonary location, it is difficult to meaningfully determine the clinical behavior of these tumors especially when these tumors can involve lymph nodes.
References
Muller NL, MIller RR. Neuroendocrine carcinomas of the lung. Semin Roentgenol. 1990;XXV(1):96–104.
Beasley MB, Thunnissen FB, Hasleton PS, et al. Tumours of the lung, pleura, thymus and heart. Lyon: IARC Press; 2004.
Cerilli LA, Ritter J, Mills SE, Wick MR. Neuroendocrine neoplasms of the lung. Am J Clin Pathol. 2001;116 Suppl 1:S65–96.
Flieder DB. Neuroendocrine tumors of the lung: recent developments in histopathology. Curr Opin Pulm Med. 2002;8:275–80.
Oberndorfer S. Karzinoide Tumoren des Dunndarms. Frankfurter Zeischrift fur Pathologie. 1907;1:425–32.
Bunting CH. Multiple primary carcinomata of the ileum. Johns Hopkins Hosp Bull. 1904;165(December):389–94.
Gosset A, Masson P. Tumeurs endocrines de l’appendice. La Presse Medicale. 1914;25:237–40.
Kinney FJ, Kovarik JL. Bone formation in bronchial adenoma. Am J Clin Pathol. 1965;44(1):52–6.
Heimburger IL, Kilman JW, Battersby JS. Peripheral bronchial adenomas. J Thorac Cardiovasc Surg. 1966;52(4):542–9.
Gmelich JT, Bensch KG, Liebow AA. Cells of Kultschitzky type in bronchioles and their relation to the origin of peripheral carcinoid tumor. Lab Invest. 1967;17(1):88–98.
Hausman DH, Weimann R. Pulmonary tumorlet with hilar lymph node metastasis. Cancer. 1967;20(9):1515–9.
Azzopardi JG. Oat cell carcinoma of the bronchus. J Pathol Bacteriol. 1959;78(2):513–9.
Yutaka K, Ferguson TB, Bennett DE, Burford TH. Oat cell carcinoma of the lung. Cancer. 1969;23(3):517–24.
Gould VE, Memoli VA, Dardi LE. Multidirectional differentiation in human epithelial cancers. J Submicrosc Cytol. 1981;13(1):97–115.
Gould VE. Histogenesis and differentiation: a re-evaluation of these concepts as criteria for the classification of tumors. Hum Pathol. 1986;17(3):212–5.
Sheppard MN. Neuroendocrine differentiation in lung tumours. Thorax. 1991;46:843–50.
Brambilla E, Lantuejoul S, Sturm N. Divergent differentiation in neuroendocrine tumors. Semin Diagn Pathol. 2000;17(2):138–48.
Gould VE. Neuroendocrinomas and neuroendocrine carcinomas: APUD cell system neoplasms and their aberrant secretory activities. Pathol Annu. 1977;12:33–62.
Gould VE, Linnoila RI, Memoli VA, Warren WH. Neuroendocrine cells and neuroendocrine neoplasms of the lung. Pathol Annu 1983;18:287–330.
Warren WH, Gould VE, Faber LP, Kittle CF, Memoli VA. Neuroendocrine neoplasms of the bronchopulmonary tract. J Thorac Cardiovasc Surg. 1985;89(6):819–25.
Paladugu RR, Benfield JR, Pak HY, Ross RK, Teplitz RL. Bronchopulmonary Kultschitzky cell carcinomas: a new classification scheme for typical and atypical carcinoids. Cancer. 1985;55(6):1303–11.
Arrigoni MG, Woolner LB, Bernatz PE. Atypical carcinoid tumors of the lung. J Thorac Cardiovasc Surg. 1972;64(3):4113–421.
Mills SE, Cooper PH, Walker AN, Kron IL. Atypical carcinoid tumor of the lung. Am J Surg Pathol. 1982;6(7):643–54.
Valli M, Fabris GA, Dewar A, Hornall D, Sheppard MN. Atypical carcinoid tumour of the lung: a study of 33 cases with prognostic features. Histopathology. 1994;24:363–9.
Grote TH, Macon WR, Davis B, Greco FA, Johnson DH. Atypical carcinoid of the lung: a distinct clinicopathologic entity. Chest. 1988;93(2):370–5.
Capella C, Heitz PU, Hofler H, Solcia E, Kloppel G. Revised classification of neuroendocrine tumours of the lung, pancreas and gut. Virchows Arch. 1994;425:547–60.
Travis WD, Linnoila RI, Tsokos MG, et al. Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma An ultrastructural, immunohistochemical, and flow cytometric study of 35 cases. Am J Surg Pathol. 1991;15(6):529–53.
Travis WD, Rush W, Flieder DB, et al. Survival analysis of 200 pulmonary neuroendocrine tumors with clarification of criteria for atypical carcinoid and its separation from typical carcinoid. Am J Surg Pathol. 1998;22(8):934–44.
Travis WD, Gal AA, Colby TV, Klimstra DS, Falk R, Koss MN. Reproducibility of neuroendocrine lung tumor classification. Hum Pathol. 1998;29:272–9.
Huang Q, Muzitansky A, Mark EJ. Pulmonary neuroendocrine carcinomas: a review of 234 cases and statistical analysis of 50 cases treated at one institution using a simple clinicopathologic classification. Arch Pathol Lab Med. 2002;126:545–53.
Ricci C, Patrassi N, Massa R, Mineo C, Benedetti-Valentini F. Carcinoid syndrome in bronchial adenoma. Am J Surg. 1973;126(11):671–7.
Sklar JL, Churg A, Bensch KG. Oncocytic carcinoid tumor of the lung. Am J Surg Pathol. 1980;4(3):287–92.
Cebelin MS. Melanocytic bronchial carcinoid tumor. Cancer. 1980;46(8):1843–8.
Wise WS, Bonder D, Aikawa M, Hsieh CL. Carcinoid tumor of lung with varied histology. Am J Surg Pathol. 1982;6(3):261–7.
Grazer R, Jacobs SB, Lucas P. Melanin-containing peripheral carcinoid of the lung. Am J Surg Pathol. 1982;6(1):73–8.
Gal AA, Koss MN, Hochholzer L, DeRose PB, Cohen C. Pigmented pulmonary carcinoid tumor. Arch Pathol Lab Med. 1993;117:832–6.
Tsutsumi Y, Yazaki K. Atypical carcinoid tumor of the lung, associated with giant-cell transformation in bone metastasis. Acta Pathol Jpn. 1990;40(8):609–15.
Memoli VA. Well-differentiated neuroendocrine carcinoma: a designation comes of age. Chest. 1991;10:892.
Tsuta K, Raso MG, Kalhor N, Liu DD, Wistuba I, Moran CA. Histologic features of low-grade and intermediate grade neuroendocrine carcinoma (typical and atypical carcinoid tumors) of the lung. Lung Cancer. 2011;71:34–41.
Tsuta K, Kalhor N, Raso MG, Wistuba I, Moran CA. Oncocytic neuroendocrine tumors of the lung: histologic spectrum and immunohistochemical analysis of 15 cases. Hum Pathol. 2011;42:578–85.
Tsuta K, Kalhor N, Wistuba I, Moran CA. Clinicopathological and immunohistochemical analysis of spindle cell carcinoid tumour of the lung. Histopathology. 2011;59:526–36.
Kalhor N, Suster S, Moran CA. Primary sclerosing neuroendocrine carcinomas of the lung: a clinicopathologic and immunohistochemical study of 10 cases. Am J Clin Pathol. 2010;133:618–22.
Sturm N, Rossi G, Lantuejoul S, et al. Expression of thyroid transcription factor-1 in the spectrum of neuroendocrine cell lung proliferations with special interest in carcinoids. Hum Pathol. 2002;33:175–82.
Tsuta K, Raso MG, Kalhor N, Liu DC, Wistuba II, Moran CA. Sox10-positive sustentacular cells in neuroendocrine carcinoma of the lung. Histopathology. 2011;58:276–85.
Costes V, Marty-Ane C, Picot MC, et al. Typical and atypical bronchopulmonary carcinoid tumors: a clinicopathologic and Ki-67-labeling study. Hum Pathol. 1995;26(7):740–5.
Tsuta K, Liu DC, Kalhor N, Wistuba II, Moran CA. Using the mitosis-specific marker anti-phosphohistone H3 to assess mitosis in pulmonary neuroendocrine carcinomas. Am J Clin Pathol. 2011;136:252–9.
Walch AK, Zitzelsberger HF, Bauchinger M, et al. Typical and atypical carcinoid tumors of the lung are characterized by 11q deletions as detected by comparative genomic hybridization. Am J Pathol. 1998;153:1089–98.
Wilkins EW, Grillo HC, Moncure AC, Scannell JG. Changing times in surgical management of bronchopulmonary carcinoid tumor. Ann Thorac Surg. 1984;38(4):339–44.
Schreurs JM, Westermann JJ, Bosch JMM, Vanderschueren RGJRA, Riviere AB, Knaepen PJ. A twenty-five-year follow-up of ninety-three resected typical carcinoid tumors of the lung. J Thorac Cardiovasc Surg. 1992;104(5):1470–5.
Nicholson S, Beasley MB, Brambilla E, et al. Small cell carcinomas (SCLC): a clinicopathologic study of 100 cases with surgical specimens. Am J Surg Pathol. 2002;26(9):1184–97.
Zakowski MF. Pathology of small cell carcinoma of the lung. Semin Oncol. 2003;30(1):3–8.
Vollmer RT, Birch R, Ogden L, Crissman JD. Subclassification of small cell cancer of the lung: the southern cancer study group experience. Hum Pathol. 1985;16(3):247–52.
Hirsch FR, Matthews MJ, Aisner S, et al. Histopathologic classification of small cell lung cancer: changing concepts and terminology. Cancer. 1988;62(5):973–7.
Warren WH, Memoli VA, Jordan AG, Gould VE. Reevaluation of pulmonary neoplasms resected as small cell carcinomas. Cancer. 1990;65:1003–10.
Guinee DG, Fishback NF, Koss MN, Abbondanzo SL, Travis WD. The spectrum of immunohistochemical staining of small-cell lung carcinoma in specimens from transbronchial and open lung biopsies. Cancer. 1994;102(4):406–14.
Warren WH, Gould VE. Differential diagnosis of small cell neuroendocrine carcinoma of the lung. Chest Surg Clin N Am. 1997;7(1):49–63.
Spiegelman D, Maurer LH, Ware JH, et al. Prognostic factors in small cell carcinoma of the lung: an analysis of 1,521 patients. J Clin Oncol. 1989;7(3):344–54.
Kalemkerian GP, Akerley W, Bogner P, et al. Small cell lung cancer: clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2011;9(10):1086–110.
Mooi WJ, Dewar A, Springall D, Polak JM, Addis BJ. Non-small cell lung carcinomas with neuroendocrine features. A light microscopic, immunohistochemical and ultrastructural study of 11 cases. Histopathology. 1988;13:329–37.
Dresler CM, Ritter JH, Patterson GA, Ross E, Bailey MS, Wick MR. Clinical-pathological analysis of 40 patients with large cell neuroendocrine carcinoma of the lung. Ann Thorac Surg. 1997;63:180–5.
Yang YJ, Steele CT, Ou XL, Snyder KP, Kochman LJ. Diagnosis of high-grade pulmonary neuroendocrine carcinoma by fine-needle aspiration biopsy: non-small-cell or small-cell type? Diagn Cytopathol. 2001;25:292–300.
Marchevsky AM, Gal AA, Shah S, Koss MN. Morphometry confirms the presence of considerable nuclear size overlap between “small cells” and “large cells” in high-grade pulmonary neuroendocrine neoplasms. Am J Clin Pathol. 2001;116:466–72.
Iyoda A, Hiroshima K, Toyozaki T, Haga Y, Fujisawa T, Ohwada H. Clinical characterization of pulmonary large cell neuroendocrine carcinoma and large cell carcinoma with neuroendocrine morphology. Cancer. 2001;91:1992–2000.
Zacharias J, Nicholson AG, Ladas GP, Goldstraw P. Large cell neuroendocrine carcinoma and large cell carcinomas with neuroendocrine morphology of the lung: prognosis after complete resection and systematic nodal dissection. Ann Thorac Surg. 2003;75:348–52.
Takei H, Asamura H, Maeshima A, et al. Large cell neuroendocrine carcinoma of the lung: a clinicopathologic study of eighty-seven cases. J Thorac Cardiovasc Surg. 2002;124(2):285–92.
Mazieres J, Daste G, Molinier L, et al. Large cell neuroendocrine carcinoma of the lung: pathological study and clinical outcome of 18 resected cases. Lung Cancer. 2002;37:287–92.
Jung K-J, Lee KS, Han J, et al. Large cell neuroendocrine carcinoma of the lung: clinical, CT, and pathological findings in 11 patients. J Thorac Imaging. 2001;16:156–62.
Filosso PL, Ruffini E, Oliaro A, et al. Large cell neuroendocrine carcinoma of the lung: a clinicopathologic study of eighteen cases and the efficacy of adjuvant treatment with octreotide. J Thorac Cardiovasc Surg. 2005;129(4):819–24.
Hage R, Seldenrijk K, Bruin PD, Swieten HV, Bosch JVD. Pulmonary large cell neuroendocrine carcinoma (LCNEC). Eur J Cardiothorac Surg. 2003;23:457–60.
Yamanishi M, Takeuchi S, Kurashina R, et al. High survival rate of 6 cases of pulmonary large cell neuroendocrine carcinoma formerly classified as small cell carcinoma. J Nippon Med Sch. 2001;68:335–9.
Doddoli C, Barlesi F, Chetaille B, et al. Large cell neuroendocrine carcinoma of the lung: an aggressive disease potentially treatable with surgery. Ann Thorac Surg. 2004;77:1168–72.
Paci M, Cavazza A, Annessi V, et al. Large cell neuroendocrine carcinoma of the lung: a 10-year clinicopathologic retrospective study. Ann Thorac Surg. 2004;77:1163–7.
Jiang S-X, Kameya T, Shoji M, Dobashi Y, Shinada J, Yoshimura H. Neuroendocrine carcinoma of the lung: a histologic and immunohistochemical study of 22 cases. Am J Surg Pathol. 1998;22(5):526–37.
Skuladottir H, Hirsch FR, Hansen HH, Olsen JH. Pulmonary neuroendocrine tumors: incidence and prognosis of histological subtypes. A population-based study in Denmark. Lung Cancer. 2002;37:127–35.
Cooper WA, Thourani VH, Gal AA, Lee RB, Mansour KA, Miller JI. The surgical spectrum of pulmonary neuroendocrine neoplasms. Chest. 2001;119:14–8.
Veronesi G, Morandi U, Alloisio M, et al. Large cell neuroendocrine carcinoma of the lung: a retrospective analysis of 144 surgical cases. Lung Cancer. 2006;53:111–5.
Masuya D, Liu D, Ishikawa S, Yamamoto Y, Huang C-l, Yokomise H. Large cell carcinoma with neuroendocrine morphology of the lung. Jpn J Thorac Cardiovasc Surg. 2006;54:31–4.
Battafarano RJ, Fernandez FG, Ritter J, et al. Large cell neuroendocrine carcinoma: an aggressive form of non-small cell lung cancer. J Thorac Cardiovasc Surg. 2005;130:166–72.
Iyoda A, Hiroshima K, Baba M, Saitoh Y, Ohwada H, Fujisawa T. Pulmonary large cell carcinoma with neuroendocrine features are high-grade neuroendocrine tumors. Ann Thorac Surg. 2002;73:1049–54.
Glisson BS, Moran CA. Large cell neuroendocrine carcinoma: controversies in diagnosis and treatment. J Natl Compr Canc Netw. 2011;9(10):1122–9.
Peng W-X, Sano T, Oyama T, Kawashima O, Nakajima T. Large cell neuroendocrine carcinoma of the lung: a comparison with large cell carcinoma with neuroendocrine morphology and small cell carcinoma. Lung Cancer. 2005;47:225–33.
Garcia-Yuste M, Matilla JM, Alvarez-Gago T, et al. Prognostic factors in neuroendocrine lung tumors: a Spanish multicenter study. Ann Thorac Surg. 2000;70:258–63.
Iyoda A, Hiroshima K, Moriya Y, et al. Prognostic impact of large cell neuroendocrine histology in patients with pathologic stage Ia pulmonary non-small cell carcinoma. J Thorac Cardiovasc Surg. 2006;132:312–5.
Hiroshima K, Iyoda A, Shida T, et al. Distinction of pulmonary large cell neuroendocrine carcinoma from small cell lung carcinoma: a morphological, immunohistochemical, and molecular analysis. Mod Pathol. 2006;19:1358–68.
Nitadori JI, Ishii G, Tsuta K, et al. Immunohistochemical differential diagnosis between large cell neuroendocrine carcinoma and small cell carcinoma by tissue microarray analysis with a large antibody panel. Am J Clin Pathol. 2006;125:682–92.
Howe MC, Chapman A, Kerr K, Dougal M, Anderson H, Hasleton PS. Neuroendocrine differentiation in non-small cell lung cancer and its relation to prognosis and therapy. Histopathology. 2004;46:195–201.
Carey FA, Save VE. Neuroendocrine differentiation in lung cancer. J Pathol. 1997;182:9–10.
Wertzel H, Grahmann PR, Bansbach S, Lange W, Hasse J, Bohm N. Results after surgery in undifferentiated large cell carcinoma of the lung: the role of neuroendocrine expression. Eur J Cardiothorac Surg. 1997;12:698–702.
Jensen SM, Gazdar AF, Cuttitta F, Russell EK, Linnoila I. A comparison of synaptophysin, chromogranin, and L-Dopa decarboxylase as markers for neuroendocrine differentiation in lung cancer cell lines. Cancer Res. 1990;50:6068–74.
Burnett HE, Zakhour HD, Walker C. Neuroendocrine and epithelial markers in diagnostic bronchial lung cancer biopsy specimens. Eur J Cancer. 1992;28A(4/5):853–5.
Ionescu D, Treaba D, Gilks CB, et al. Nonsmall cell lung carcinoma with neuroendocrine differentiation - an entity of no clinical or prognostic significance. Am J Surg Pathol. 2007;31(1):26–32.
Hiroshima K, Iyoda A, Shibuya K, et al. Prognostic significance of neuroendocrine differentiation in adenocarcinoma of the lung. Ann Thorac Surg. 2002;73:1732–5.
Souhami RL. Neuroendocrine phenotype, chemosensitivity and prognosis in adenocarcinoma of the lung. Ann Oncol. 1991;2:323–4.
Iyoda A, Hiroshima K, Toyozaki T, et al. Adjuvant chemotherapy for large cell carcinoma with neuroendocrine features. Cancer. 2001;92:1108–12.
Brambilla E, Moro D, Veale D, et al. Basal cell (Basaloid) carcinoma of the lung: a new morphologic and phenotypic entity with separate prognostic significance. Hum Pathol. 1992;23(9):993–1003.
Lyda MH, Weiss LM. Immunoreactivity for epithelial and neuroendocrine antibodies are useful in the differential diagnosis of lung carcinomas. Hum Pathol. 2000;331:980–7.
McDowell EM, Trump BF. Pulmonary small cell carcinoma showing tripartite differentiation in individual cells. Hum Pathol. 1981;12(3):286–94.
Tsubota YT, Kawaguchi T, Hoso T, Nishino E, Travis WD. A combined small cell and spindle cell carcinoma of the lung: report of a unique case with immunohistochemical and ultrastructural studies. Am J Surg Pathol. 1992;16(11):1008–115.
Adelstein DJ, Tomashefski JF, Snow NJ, Horrigan TP, Hines JD. Mixed small cell and non-small cell lung cancer. Chest. 1986;89(5):699–704.
Radice PA, Matthews MJ, Ihde DC, et al. The clinical behavior of “mixed” small cell/large cell bronchogenic carcinoma compared to “pure” small cell subtypes. Cancer. 1982;50(12):2894–902.
Ruffini E, Rena O, Oliaro A, et al. Lung tumors with mixed histologic pattern. Clinico-pathologic characteristics and prognostic significance. Eur J Cardiothorac Surg. 2002;22:701–7.
Ullmann R, Petzmann S, Sharma A, Cagle PT, Popper HH. Chromosomal aberrations in a series of large-cell neuroendocrine carcinomas: unexpected divergence from small-cell carcinoma of the lung. Hum Pathol. 2001;32:1059–63.
Jones MH, Virtanen C, Honjoh D, et al. Two prognostically significant subtypes of high-grade lung neuroendocrine tumours independent of small cell and large cell neuroendocrine carcinomas identified by gene expression profiles. Lancet. 2004;363:775–81.
Sampietro G, Tomasic G, Collini P, et al. Gene product immunophenotyping of neuroendocrine lung tumors. Appl Immunohistochem Mol Morphol. 2000;8(1):49–56.
Anbazhagan R, Tihan T, Bornman DM, et al. Classification of small cell lung cancer and pulmonary carcinoid by gene expression profiles. Cancer Res. 1999;59(10):5119–22.
Gazzeri S, Valle VD, Chaussade L, Brambilla C, Larsen CJ, Brambilla E. The human p19ARF protein encoded by the beta transcript of the p16INK4a gene is frequently lost in small cell lung cancer. Cancer Res. 1998;58(17):3926–31.
Przygodzki RM, Finkelstein SD, Langer JC, et al. Analysis of p53, K-ras-2, and C-raf-1 in pulmonary neuroendocrine tumors: correlation with histological tumors. Am J Pathol. 1996;148(5):1531–41.
Kovatich A, Friedland DM, Druck T, et al. Molecular alterations to human chromosome 3p loci in neuroendocrine lung tumors. Cancer. 1998;83:1109–17.
Wang DG, Johnston CF, Sloan JM, Buchanan KD. Expression of Bcl-2 in lung neuroendocrine tumours: comparison with p53. J Pathol. 1998;184:247–51.
Cagle PT, Naggar AK, Xu HJ, Xu SX, Benedict WF. Differential retinoblastoma protein expression in neuroendocrine tumors of the lung. Potential diagnostic implications. Am J Pathol. 1997;150:393–400.
Rusch VW, Klimstra DS, Venkatraman ES. Molecular markers help characterized neuroendocrine lung tumors. Ann Thorac Surg. 1996;62:798–810.
Ito T, Udaka N, Okudela K, Yazawa T, Kitamura H. Mechanisms of neuroendocrine differentiation in pulmonary neuroendocrine cells and small cell carcinoma. Endocr Pathol. 2003;14(2):133–9.
Shibara J, Goto A, Niki T, et al. Primary pulmonary paraganglioma: report of a functioning case with immunohistochemical and ultrastructural study. Am J Surg Pathol. 2004;28(6):825–9.
Kee AR, Forrest CH, Brennan BA, Papadimitriou JM, Glancy RJ. Gangliocytic paraganglioma of the bronchus: a case report with follow-up and ultrastructural assessment. Am J Surg Pathol. 2003;27:1380–5.
Palau MA, Merino MJ, Quezado M. Corticotropin-producing pulmonary gangliocytic paraganglioma associated with Cushing’s syndrome. Hum Pathol. 2006;37:623–6.
Aubertine CL, Flieder DB. Primary paraganglioma of the lung. Ann Diagn Pathol. 2004;8:237–41.
Dahir KM, Gonzales A, Revelo MP, et al. Ectopic adrenocorticotropin hormone hypertension due to a primary pulmonary paraganglioma. Endocr Pract. 2004;10:424–8.
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Weissferdt, A., Moran, C.A. (2013). Neuroendocrine Carcinomas. In: Diagnostic Pathology of Pleuropulmonary Neoplasia. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-0787-5_4
Download citation
DOI: https://doi.org/10.1007/978-1-4419-0787-5_4
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-0786-8
Online ISBN: 978-1-4419-0787-5
eBook Packages: MedicineMedicine (R0)