
Abstract
Components of the plasminogen-plasmin system participate in a wide variety of physiologic and pathologic processes, including tumor growth, invasion and metastasis, through their effect on angiogenesis and cell migration. These components are found in most tumors and their expression not only signifies their function but also carries a prognostic value. Their expression is in turn modulated by cytokines and growth factors, many of which are up-regulated in cancer. Though both tPA and uPA are expressed in tumor cells, uPA with its receptor (uPAR) is mostly involved in cellular functions, while tPA with its receptor Annexin II on endothelial surface, regulates intravascular fibrin deposition. Among the inhibitors of fibrinolysis, PAI-1 is a major player in the pathogenesis of many vascular diseases as well as in cancer. Therapeutic interventions, either using plasminogen activators or experimental inhibitor agents against PAI-1, have shown encouraging results in experimental tumors but not been verified clinically.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The plasminogen-plasmin system is involved in not only the regulation of hemostatic balance but also a wide range of biologic processes. These include embryogenesis, development, wound healing, cell proliferation and migration. As such, the system plays an important functional role in both physiologic and pathologic conditions. When this system was first discovered, it was named the fibrinolytic system. However, with the realization that fibrin is not the only substrate for the active enzyme plasmin, the term plasminogen-plasmin system is more appropriate and will be used here. This chapter will review our current understanding of how components of this system affect tumor growth and metastasis.
2 Historical Background
The first association between the pathology of cancer and fibrin must be credited to Billroth [1], who first described the presence of tumor cells within a thrombus. Later, Iwasaki found that tumor cells within the vascular channels of recanalizing thrombi were viable [2]. The significance of fibrin within tumors was again noted by O’Meara and Jackson [3], who postulated that. if unresolved, the fibrin would induce further tumor growth and vascular proliferation. Subsequent observations by others led to experimental and clinical trials with anticoagulants [4], defibrinating agents [5], and thrombolytic agents [6]. Though the results were controversial, experimental tumor metastases were found to be enhanced by increasing the fibrin content in tumors through the use of antifibrinolytic agents, or by induction of hyperfibrinogenemia [7]. In the ensuing years, the role of the plasminogen-plasmin system gained attention not only in the pathogenesis of bleeding and thrombotic complications in the cancer patient, but also in tumor growth and metastasis [8].
3 Plasminogen-Plasmin System
The precursor of the active protease plasmin is plasminogen [9–17]. This is a single-chain glycoprotein of 92 kDa synthesized mainly in the liver. It is present in plasma and extracellular fluids at a concentration of 1–2 µM, with a biologic half-life of 2.2 days. In the native form, the amino-terminal is occupied by glutamic acid. On activation by a plasminogen activator or by plasmin, proteolytic cleavage at Lys-77 and Lys-78 results in the formation of Lys-plasminogen (Fig. 4.1). Further cleavage of the Arg-560 and Val-561 peptide bond results in the formation of a two-chain plasmin held together by two disulfide bonds.

The plasminogen-plasmin system
Its structure contains five triple-looped structures with three disulfide bonds known as kringles. These kringles are involved in binding of plasminogen to cell surfaces and to fibrin. Of interest is that the structure of a potent anti-angiogenic protein, angiostatin, is identical to the first four of the five kringles.
The lysine at the amino-terminal of plasminogen functions as the binding domain for many proteins, including fibrin, α2-antiplasmin, thrombospondin and the plasminogen receptor annexin II. It also enables plasminogen to bind to specific lysine binding sites on many cell surfaces. Furthermore, plasminogen activation is blocked by lysine binding to ε-aminocarboxylic acids (epsilon amino-caproic acid, tranexamic acid). These are clinically useful antifibrinolytic agents.
Plasminogen has no proteolytic activity prior to conversion to plasmin. On the other hand, plasmin is a serine protease with broad substrate specificity, which includes fibrin, fibrinogen and extracellular matrix (ECM) proteins such as laminin and fibronectin, either directly or indirectly through the activation of latent metalloproteinases [8, 17, 18]. Thus, it is an intermediary protease with a wide range of functions in health, such as tissue remodeling and wound healing, and involvement in pathologic processes including tumor growth and metastasis. Other substrates of importance include the pro-forms of growth factors, which can be cleaved and activated by plasmin. Plasmin can also proteolyse specific cleavage sites on plasminogen to generate angiostatin.
3.1 Plasminogen Activators
Though there are many proteases derived from bacteria, fungus, insects and other animals that can proteolyse plasminogen, only two activators are present in man.
The first one is tissue-type plasminogen activator (tPA) [19–21]. It is a 70-kDa glycoprotein which, under physiologic conditions, is synthesized mainly by endothelial cells. It is responsible for maintaining vascular patency in response to intravascular fibrin formation. However, it is also produced by neurons, keratinocytes, melanocytes and various tumor cells. Observations in transgenic mice with tPA null−/− have now shown that, in addition to maintaining vascular patency, tPA participates also in neuronal development and neurologic functions [22].
The resting plasma level of tPA is low, around 5 ng/mL, but large amounts can be released from endothelial cells under a variety of circumstances. It is then quickly bound to the circulating inhibitor PAI-1. A lesser amount is also bound to α2-macroglobulin. It is then rapidly removed from the circulation by the liver with a plasma half-life of around 5 min.
The source of the circulating tPA is believed to be the vascular endothelium. Earlier observations from our laboratory indicated that fibrinolytic activity can be released from the vascular wall by ischemia [23, 24], serotonin [25] and other vasoactive stimuli [23]. This fibrinolytic activity was later shown to be derived mostly from tPA and, to a lesser extent, from uPA. However, recently, there is evidence that tPA present in the neuronal terminals in the autonomic nervous system can also be released through the vascular wall [26]. This new finding explains our earlier observation that stimulation of a vessel wall can release fibrinolytic activity from a vessel located distally to the site of stimulation, indicating that the stimuli is transmitted via perivascular sympathetic nerves [23].
The “finger” domain present at the amino-terminal enables tPA to have a high affinity for fibrin, thus making tPA a more efficient thrombolytic agent than uPA. In addition to its high affinity for fibrin, tPA also binds to extracellular matrix (ECM) proteins, including laminin and fibronectin, and the mannose-6-phosphate/insulin-like growth factor. Recently, a cellular surface receptor for both tPA and plasminogen, termed annexin II, was found in endothelial cells, macrophages and certain tumor cells [27, 28]. The close proximity of these two ligands on this dual receptor enhances plasminogen activation. The high expression of annexin II in acute promyelocytic leukemia and other malignant conditions may explain the high bleeding risk in these disorders [29].
The second plasminogen activator in man is urokinase-type plasminogen activator (uPA). This fibrinolytic enzyme was first discovered in urine, hence its name urokinase. It originates from kidney cells [30, 31]. When first released, it is in a single-chain form, pro-urokinase, which is then rapidly converted by plasmin or kallikrein to the two-chain form connected by a disulfide bond. The single-chain glycoprotein has a molecular mass of 53 kDa. Both forms are fibrinolytic and are used as thrombolytic agents, but the single-chained uPA has a higher affinity for fibrin. The tertiary structure of uPA is composed of the amino-terminal fragment (ATF) that contains a growth factor domain, as well as a kringle domain. Both the single-chain and the two-chain uPA binds to the uPA receptor (uPAR) via the ATF, forming a uPA-uPAR complex [32]. The complex form facilitates plasminogen activation by uPA.
In addition to being a potent activator of plasminogen, uPA also directly activates procollagenase. This allows it to exert a regulatory effect on cell migration as well as tumor growth and metastasis.
uPA is secreted by many other cell types including endothelial cells and tumor cells in addition to kidney cells. The role of tumor-derived uPA in tumor growth and metastasis has been the subject of many studies and will be reviewed in a later section of this chapter.
uPAR is anchored to the cell surface by its glycosyl-phosphatidylinositol (GPI) domain at the C-terminal, and released by phosphatilyl-inositol specific phospholipase C [33]. On the cell surface, uPAR has a high affinity for uPA, and can also bind uPA which had been inactivated by PAI-1, forming a uPA-uPAR-PAI complex. This complex is rapidly internalized [34], a process facilitated by several members of the low density lipoprotein receptor (LDLR) family [35]. Following internalization, the uPA-PAI-1 portion of the complex is degraded while uPAR is recycled and emerges at a different site on the cell surface [34]. This process is believed to be directional and important in cell migration.
3.2 Inhibitors of the Plasminogen-Plasmin System
Inhibitors of plasmin are α2-antiplasmin (α2-AP), and α2-macroglobulin. α2-AP belongs to the family of serpins (serine protease inhibitors) [36]. Plasmin generated in circulating blood binds to a lysine binding site on α2-AP and is rapidly inhibited. The level of the plasmin-α2-AP complex in blood is often used as an indicator of the intensity of fibrinolytic activity. α2-AP also cross-links with the α-chain of fibrin, preventing the latter’s proteolysis by plasmin. When excess circulating plasmin in blood has saturated all the available α2-AP, a slower acting inhibitor α2-macroglobulin acts as a second line of defense. In addition, there are other serine protease inhibitors including antithrombin, α1-antitrypsin, α1-antichymotrypsin, inter-α-trypsin inhibitor and C-1 inactivator. The recently discovered thrombin activatable fibrinolytic inhibitor (TAFI) inhibits plasmin as well as both plasminogen activators, tPA and uPA.
4 Plasminogen Activator Inhibitors (PAIs)
4.1 Plasminogen Activator Inhibitor Type 1 (PAI-1)
PAI-1 is a member of the serine protease inhibitor (SERPIN) family (Table 4.1). It is perhaps the most important component of the plasminogen-plasmin system in the regulation of many physiologic processes and in the pathogenesis of many disorders including cancer [37–40]. PAI-1 is synthesized by the endothelial cells, liver, adipose tissues, vascular smooth muscle cells, and a large number of tumor cells. In addition to being a potent inhibitor of both tPA and uPA, PAI-1 inhibits plasmin directly.
PAI-1 is stored in the α-granules of platelets. Most of the PAI-1 in platelets is in a latent form, but when the active portion is released into a thrombus or into the ECM, it can exert their effects on fibrinolysis as well as ECM functions.
The plasma level of PAI-1 in resting healthy individuals is around 1 nM, an amount which is two to three times more than needed to inhibit the circulating plasminogen activators. As discussed below, multiple stimuli, including inflammatory and tumor derived cytokines, can further increase this level. The plasma level of PAI-1 is elevated in obesity, metabolic syndrome, type II diabetes, inflammatory states, and cancer [37]. PAI-1 levels are often used as a prognostic marker for thromboembolic complications in patients with cancer, type II diabetes and veno-occlusive disease in the post-bone marrow transplantation setting. PAI-1 is present in many tumor cell types and in the stromal fibroblasts of the tumor microenvironment as well as in tumor associated endothelial cells. In these settings, it may modulate tumor growth, invasion and angiogenesis.
PAI-1 is the principal inhibitor of both uPA and tPA. In addition, it has a high affinity for ECM proteins, especially vitronectin. The interactions between vitronectin, uPA, uPAR and PAI-1 modulate multiple functions of uPA. The binding of PAI-1 to vitronectin stabilizes the adhesion of cells to the ECM. Both of these processes are essential for cell migration. PAI-1 bound to the uPA-uPAR complex is internalized by endocytosis. While PAI-1 and uPA are degraded intracellularly, uPAR is secreted by the cell and recycled. This process facilitates the propelling action observed in cell migration and tissue remodelling. In addition, PAI-1 binds to glycoaminoglycans, and to low density lipoprotein receptors (LDL-R) [38]. LDL-R facilitates the internalization of the PAI-1-uPA-uPAR complex.
PAI-1 added to cell cultures inhibits apoptosis of both normal vascular smooth muscle cells and tumor cells, and may thus contribute to tumor proliferation and to angiogenesis [39, 40].
4.2 Regulation of PAI-1
As the PAI-1 gene is expressed in almost every cell type in the body, transcription of this gene is regulated by numerous signals generated by cellular responses to various stimuli [41, 42] (Table 4.1). These responses include inflammatory cytokines such as TNFα and IL-1, growth factors, including TGFβ1, EGF, and FGF, hormones such as insulin and glucocorticoids, angiotensin II and angiotensin IV [43] and hypoxia-inducing factor and reactive oxygen species. Through these pathways, PAI-1 is up-regulated in obesity and metabolic syndrome, type II diabetes, hypertension and many types of cancer. These observations led to the concept that PAI-1 not only plays a major role in thrombogenesis by inhibiting fibrinolysis, but is also involved in the pathogenesis of many other disorders by its modulation of cellular interactions.
Several anti-PAI-1 agents are being developed for possible therapeutic use in cancer and other disorders affected by PAI-1 [3, 37].
4.3 Plasminogen Activator Inhibitor Type 2 (PAI-2)
PAI-2 is synthesized by the placenta, monocytes [44, 45], eosinophils and keratinocytes as well as by ovarian tumors and myeloid leukemic cells. PAI-2, as in the case of PAI-1, is an inhibitor of both uPA and tPA [46]. Its expression by the placenta is believed to contribute to the increased prothrombotic risk in late pregnancy. Also, like PAI-1, it binds the uPA-uPAR complex on the cell surface and is then internalized. However, unlike PAI-1, most PAI-2 exists within the cell in the cytosol and only a small fraction is secreted. The plasma level is barely detectable. While the secreted portion takes part with PAI-1 in the inhibition of the plasminogen activators, the main functions of PAI-2 within the cell are not clear. One known intracellular function is the protection of cells against TNFα-mediated apoptosis [47]. It is notable that the PAI-2 gene is located in chromosome 8, less than 300 mbp from the apoptosis-inhibiting BCL-2 gene. In follicular lymphoma with the t(14;18) translocation, BCL-2 is over-expressed, resulting in inhibited apoptosis of the lymphoma cells [48] . However, it is not known whether PAI-2 has a similar effect on lymphoma.
There are a number of observations of a tumorogenic effect of PAI-2. Studies with oligonucleotide microarrays indicate there is at least a 12-fold increase in PAI-2 genes in ovarian serous papillary carcinoma compared to normal ovarian tissues [49]. The level of PAI-2 has been correlated with a poor prognosis for ovarian, and colorectal carcinomas, while low expression in epithelial carcinoma such as head and neck squamous cell carcinoma signifies invasion [50]. In contrast, a high PAI-2 level indicates a favorable prognosis in breast carcinoma. Recently, high expression of PAI-2 was found in the gastric mucosa of patients with Helocobacter pylori infection, suggesting another possible link between the proliferative and apoptotic inhibitory actions of PAI-2 and the ultimate gastric cancer formation [51].
4.4 Thrombin-Activatable Fibrinolytic Inhibitor (TAFI)
Thrombin-activatable fibrinolytic inhibitor, also known as procarboxypeptidase U, is a 55-kDa carboxypeptidase synthesized in the liver that has been shown to play an important role in fibrinolysis. It is initially produced as a proenzyme and is converted to an active, zinc ion dependent carboxypeptidase B-like enzyme (TAFIa) through cleavage at the Arg92-Ala93 bond. This process can be mediated by trypsin or plasmin. However, these activators are much less efficient than thrombin. Thrombin conversion of TAFI to TAFIa is accelerated 1250-fold in the presence of thrombomodulin [52], and the activation is calcium-ion dependent. TAFIa production occurs in two peaks, with the first peak occurring shortly after initiation of clot formation and the second stimulated by thrombin generation. The half-life of the enzyme is approximately 10 min and therefore the first increase in concentration is transient. The second peak is prompted by plasmin formation and unlike the first, has little effect on fibrinolysis [53].
TAFIa is able to attenuate fibrinolysis by preventing plasmin formation through removal of carboxyterminal arginine and lysine residues from fibrin and fibrin cleavage products. This blocks the binding of tPA and plasminogen, thereby interfering with the positive feedback of plasmin formation [54]. Removal of the arginine and lysine residues from fibrin also removes the inhibitory effect fibrin has on antiplasmin. Fibrinolysis is then prevented by the greater resulting influence of antiplasmin on free plasmin [55]. Plasmin may also be directly inhibited by TAFI, to impair fibrinolysis further. The increased amounts of TAFIa generated by activation of the intrinsic system continue to propagate the downregulation of fibrinolysis. There have been no reported inhibitors of TAFIa to date, and the activity appears to decay spontaneously.
Due to its effect on fibrinolysis, levels of TAFIa may correspond to an increased risk of thrombosis or bleeding. Increased levels have been shown to be a weak risk factor in incident (twofold increased risk) [56] and recurrent (twofold increased risk) [57] venous thromboembolism. Concurrent elevations in factor VIII may enhance this thrombogenic risk. On the other hand, low TAFI activity is seen in patients with acute promyelocytic leukemia, and this finding may in part explain the hyperfibrinolysis and propensity for hemorrhage seen in this disease [58]. TAFI levels have also been investigated in a number of other disease states where perturbations of clotting may play a central role. Examples include demonstration of increased TAFI levels in ischemic stroke, in patients with angina, and in men requiring coronary artery bypass grafting [59–61].
It is unclear what role, if any, TAFI has in malignant disease states. A mouse model failed to show any affect of TAFI deficiency on growth or metastasis of different tumor cell types [62]. Malignancy may increase TAFI expression through a cytokine-mediated process. Theoretically, increased levels of TAFI may promote growth and spread of tumor cells through intra-tumoral fibrin deposition, and may accentuate the several prothrombotic features of various malignant states. Clinical data in humans supporting these possibilities is lacking.
5 Role of the Plasminogen – Plasmin System in Tumor Growth and Metastasis
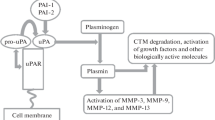
The relationship between the plasminogen-plasmin system and tumor biology is complex [8, 42, 63, 64] (Fig. 4.2). It involves several important steps as shown in Fig. 4.2. The major ones are cell proliferation, apoptosis, cell migration and invasion and angiogenesis. In all these steps, one or more of the components of the plasminogen-plasmin system participate in the process.

Plasminogen-plasmin system in tumor growth and metastasis
5.1 Tumor Cell Proliferation and Apoptosis
uPA bound to uPAR is mitogenic. On the surface of tumor cells, initiation of intracellular signaling follows the assembly of the amino-terminal fragment of uPA (containing an epidermal growth factor-like domain), and uPAR, along with an ECM protein (such as vitronectin or fibronectin) and the epidermal growth factor receptor, leads to growth stimulation [65, 66]. The same events occur with stromal cells, vascular smooth cells and endothelial cells in the tumor microenvironment. These characteristics of uPA form the basis for a high uPA expression in tumors signifying a poor prognosis. On the other hand, we had shown previously that, in vitro and in animals, high expression of PAI-1, by inhibiting uPA, impairs tumor growth, angiogenesis and metastasis [67]. When PAI-1 was transfected into an aggressive human prostate cancer cell, PC-3, tumors with transfected cells that over-expressed PAI-1 had a slower growth rate in vitro. Tumors in athymic mice given PC-3 clones with high PAI-1 expression were smaller, less metastatic and contained less vasculature. This anti-tumorogenic effect is attributed to the inhibitory action of uPA on tumor invasion, and on uPA activation of plasmin. However, in a different setting, PAI-1 has an opposite effect by being inhibitory to apoptosis. When the stable form of PAI-1 is added to tumor cell cultures, or endothelial cell culture, both spontaneous and induced apoptosis in tumor cells are inhibited [39]. In this respect, PAI-1 enhances tumor growth. PAI-2 added to tumor cell cultures also has an anti-apoptotic action. As tumor growth involves both cell proliferation and apoptosis, PAI-1 may favor tumor growth by inhibiting apoptosis. In certain tumors, a high PAI-1 content signifies a poor prognosis. These observations of stimulated and inhibited tumor growth were confirmed by others [38]. Which of these opposing effects of PAI-1 are acting on the tumor must be dependent on a number of factors, many of which are still not fully understood. PAI-1 injected into animals or added to tumor cell cultures likely inhibits uPA on the tumor cell surface, where the assembly of PAI-1 and the uPA-uPAR complex are internalized, leading to the degradation of uPA. Also, it is likely that the effect may be dose-dependent. One determinant may also be the tumor type. For example, in carcinoma of the breast, the known tumorogenic effect of estradiol may in part be mediated through the down-regulation of tPA, uPA, uPAR and PAI-1 [68].
The activation of pro-forms of growth factors is another way in which plasmin, tPA or uPA can participate in tumorogenesis. One example is that plasmin can activate latent β-FGF. Plasmin has also been shown to activate the propeptide of VEGF-C and VEGF-D, both angiogenic and lymphangiogenic factors [69]. Of interest, tPA can directly activate the latent form of PDGF-CC by proteolytic removal of the CUB domains [70].
5.2 Tumor Cell Migration and Invasion
The regulation of cell migration by uPA, uPAR and PAI-1 in cell migration is important in wound healing as well as in cancer cell invasion, and metastasis. uPA bound to uPAR is present at focal adhesion sites on the cell surface [71, 72]. Locally generated plasmin from the activation of plasminogen at these sites in turn activates latent metalloproteinases and latent growth factors [5]. It is believed that the resulting proteolysis of the extracellular matrix (ECM) frees the cell from its adhesion site allowing cell migration. Since uPA bound to uPAR is a much more potent activator of plasminogen, the location of uPAR determines the direction of the cell migration. There is also another major cellular process, involving vitronectin in the ECM, uPA-uPAR complex, PAI-1 and the recycling of uPAR [34]. Both monomeric and dimeric forms of uPAR are present on the cell surface [33, 73]. Vitronectin in the ECM binds to uPAR, preferentially the dimeric form [74], and affects the distribution of uPAR on the cell surface. The complex of uPA, uPAR and PAI-l binds to vitronectin on the cell surface, resulting in changes in the cell shape and cell adhesiveness to the ECM. With the endocytosis of the complex assembly, de-adhesion occurs. Following the recycling of uPAR, readhesion takes place [33]. These steps regulate cell migration and propulsion.
5.3 Tumor Angiogenesis
The plasminogen-plasmin system is involved in tumor angiogenesis in two aspects [75–79]. First, along with tumor derived cytokines and VEGF, uPA and PAI-1 modulate endothelial cell proliferation. Second, plasmin and plasminogen activators proteolyse plasminogen and, acting with several cofactors, release one or more of the kringle structures. These kringles possess inhibitor effects on tumor angiogenesis, best exemplified by angiostatin. Plasmin, derived from uPA activation of plasminogen, can be reduced by plasmin reductase (phosoglycerate kinase) in the presence of co-factors, including glutathione and cysteine. The disulfide bonds between kringles are further proteolysed to form kringle peptides with potent inhibitory activity against the proliferation of microvascular endothelial cells. Despite encouraging results of tumor reduction in animals, these results were not reproduced in early human clinical trials [79].
6 Role of the Plasminogen-Plasmin System in Thrombotic Complications in Cancer
Though procoagulants and their activation in cancer play a major role in thrombogenesis, changes in fibrinolytic components are also important contributory factors. Evidence for this is provided by in vitro studies as well as observations of inhibited fibrinolysis in vivo. The inhibited fibrinolysis is primarily due to increased in PAI-1 activity in the plasma of cancer patients. Both uPA and tPA are regulated by PAI-1. While uPA acts on cell proliferation and migration and thus plays an important role in cancer progress, tPA, on the other hand, is the body’s defense against extension of intravascular fibrin thrombi, and thus a high PAI-1 level is thrombogenic.
7 The Plasminogen-Plasmin System in Acute Leukemia
Both thrombotic and bleeding complications are linked to perturbation of the plasminogen-plasmin system in acute leukemia [80, 81]. In acute promyelocytic leukemia (FAB: M-3), annexin II, a dual receptor for tPA and for plasminogen [27, 28], is highly expressed in the leukemic cells [29, 82]. Its level is also increased in a small number of patients with AML (FAB: M4-5) or with ALL. Annexin II is a cell membrane surface protein found in endothelial cells, macrophages and several malignant cell lines. It is a protein with a molecular weight of 40 kDa. In addition to binding tPA, it is a co-receptor for plasminogen, with tPA binding at the amino-terminal of the core 1 domain, while plasminogen binding occurs at the lysine binding site in the core 4 domain. The close proximity of the two ligands on the cell surface facilitates their interaction in plasmin generation, with in vitro enhancement of 60-fold. Annexin II on endothelial cell surface has the highest expression on the microvascular endothelial cells in the brain [83]. In acute promyelocytic leukemia (APL), this location of a higher Annexin II expression is of clinical significance. APL cells in the bone marrow and peripheral blood as well as the APL cell line NB4, express both uPA and tPA [84]. After treatment with trans-retinoic acid, the uPA expression is further dysregulated in NB4 cells for 24 h before returning to normal levels as these cells undergo differentiation [85]. Thus, an increased expression of uPA by APL cells, in conjunction with the expression of tPA as well as the presence of annexin II contributes to the excessive fibrinolysis [29]. In addition, TAFI level is low in APL and thus there is less inhibitory control over fibrinolysis [59]. Larger amounts of plasmin were also found to be generated in vitro by the brain endothelial cells [84]. This may provide an explanation for the relatively higher incidence of intracranial hemorrhage in APL. In a recent analysis of the early deaths seen in patients with APL, hemorrhagic complications were found to be the major cause of early deaths, accounting for over 50% of patients with or without ATRA therapy [86–90]. Intracranial bleeding accounts for most of the fatal hemorrhages, with other sites including diffuse pulmonary alveolar and gastrointestinal hemorrhage. In addition to plasmin induced fibrinolysis, elastase and chymotrypsin released by leukemic blasts may also contribute to the impaired hemostasis by proteolysis of von Willebrand factor [91].
8 Clinical Observations of the Plasminogen-Plasmin System in Cancer
As discussed, the plasminogen-plasmin system has potential biological effects extending beyond thrombosis and hemorrhage. Tumor growth and metastasis may be promoted by uPA-mediated enhancement of cell proliferation, adhesion, migration, and degradation of the extracellular matrix. Alterations of this system in cancer could lead to a variety of altered clinical outcomes, which has been demonstrated in a number of cancer types.
The majority of the clinical literature evaluating upregulation of the plasminogen-plasmin system has focused on prognosis through the enhancement of tumor growth and progression. As mentioned in detail previously, this may come about through inhibition of apoptosis and through increased release of TGFβ, FGF2, ILGF-1, and hepatocyte growth factor. Degradation of the extracellular matrix and promotion of cellular adhesion may assist in the development of metastatic disease. These features of the plasminogen-plasmin system have been correlated with its over-expression, leading to adverse outcomes in a number of malignancies (Table 4.2). High uPA and/or PAI-1 levels have been shown to be adverse prognostic markers in breast, colorectal, esophageal, gastric, ovarian, prostate, renal, and endometrial cancers. These findings may prove to be a vital addition to previously known prognostic markers and potentially assist in individualizing cancer treatments.
The prognostic significance of the plasminogen-plasmin system has best been demonstrated in breast cancer. Levels of uPA and PAI-1 were more predictive of both disease-free and overall survival than ER status and tumor size in a pooled analysis of over 8,000 patients with breast cancer [92]. Increased uPA and PAI-1 were associated with a worse prognosis. Application of this finding to clinical practice was demonstrated in a prospective study of 761 patients [93]. Based on the hypothesis that poor outcome is associated with high PAI-1 and uPA, patients with lymph node negative disease, but with high PAI-1 and uPA, were given adjuvant cyclophosphamide-methotrexate-5-fluorouracil (CMF) chemotherapy. Another large trial of over 3,000 women showed increased levels of PAI-1 and uPA correlated with greater benefit from adjuvant chemotherapy vs those with lower levels [94]. Similar findings are present with regard to adjuvant hormonal treatment, with uPA and PAI-1 negative breast tumors responding better to intervention with tamoxifen than those with high expression, independent of ER/PR status [95].
Application of the prognostic information given by the plasminogen-plasmin system to clinical care has also been extended to other malignant diseases. For example, it has already been shown that higher TAFI levels in lung cancer directly correlate with a more favorable response to chemotherapy [96]. Such observations allow better individualization of cancer care, with administration of more aggressive treatment to those patients who are likely to benefit most. In addition, those who are unlikely to benefit could be spared the toxicities associated with many therapeutic interventions. Additional data is needed in this area to justify its wider application.
Available evidence indicates a pathophysiologic role of the plasminogen-plasmin system in the prothrombotic nature of malignant disease, with high tumor expression of PAI-1 and resultant inhibition of fibrinolysis potentially exacerbating the hypercoagulability associated with malignancy. To date, clinical studies demonstrating this correlation have largely been lacking. Whether components of this system can be used successfully in the treatment or prevention of thrombosis in cancer remains to be established. It is certainly plausible that alterations in the expression of the various components of the uPA system may predict risk for thrombosis in the same way as with disease outcome. This type of information may prove useful in targeting those at highest risk, with greater surveillance and possibly prophylactic treatment for those patients whose thrombotic potential is greatest.
9 Conclusion
Though fibrin was found in cancer tissues as early as the late nineteenth century, the active investigation of the role of the plasminogen-plasmin system in cancer has accelerated only in the past two decades. This has greatly increased our understanding of how the components of this system, especially uPA, uPAR and PAI-1, affect tumor growth, invasion and angiogenesis. Undoubtedly these findings have contributed to the elucidation of the pathogenesis of many forms of malignant disorders. However, little progress has been made in translating the findings from in vitro studies and animal experiments into innovative therapeutic approaches. In experimental tumors in animals, perturbation of uPA and of PAI-1 has been found to impair tumor growth and metastasis, while only a few anecdotal results have been reported in humans. As the pathogenesis of cancer is complex, one would expect that the influence of uPA and PAI-1 is only one part of this process. New agents are being designed to interdict these effects, especially those of PAI-1. Whether they will be effective remains to be determined by clinical trials. Questions to be addressed in future clinical trials will concern the effect of anti-PAI-1 or anti-uPA agents by themselves, or whether effectiveness will require a combination with cyto-reductive measures including chemotherapy or radiation, and in addition, the combination with anti-angiogenic agents or with hormonal therapy wherever applicable.
References
Billroth T. Lectures on surgical pathology and therapeutics. In: A handbook for students and practitioners. London: The New Sydenham Society; 1877–1878;1878.
Iwasaki T. Histological and experimental observations on the destruction of tumor cells in the blood vessels. J Pathol Bacteriol 1912;20:85–104.
O'Meara RA, Jackson RD. Cytological observations on carcinoma. Irish J Med Sci 1958;171(391):327–8.
Zacharski LR, Henderson WG, Rickles FR, et al. Effect of warfarin on survival in small cell carcinoma of the lung. Veterans Administration Study No. 75. Jama 1981;245(8):831–5.
DeWys WD, Kwaan HC, Bathina S. Effect of defibrination on tumor growth and response to chemotherapy. Cancer Res 1976;36(10):3584–7.
Meehan KR, Zacharski LR, Maurer LH, et al. Studies of possible mechanisms for the effect of urokinase therapy in small cell carcinoma of the lung. Blood Coagul Fibrinolysis 1995;6(2):105–12.
Agostino D, Agostino N. Trauma, fibrinogen levels and metastasis formation in experimental oncology. Sangre 1982;27(3):301–7.
Kwaan HC. The plasminogen-plasmin system in malignancy. Cancer Metastasis Rev 1992;11(3–4):291–311.
Petersen TE, Martzen MR, Ichinose A, Davie EW. Characterization of the gene for human plasminogen, a key proenzyme in the fibrinolytic system. J Biol Chem 1990;265(11):6104–11.
Summaria L, Spitz F, Arzadon L, Boreisha IG, Robbins KC. Isolation and characterization of the affinity chromatography forms of human Glu- and Lys-plasminogens and plasmins. J Biol Chem 1976;251(12):3693–9.
Robbins KC, Bernabe P, Arzadon L, Summaria L. NH2-terminal sequences of mammalian plasminogens and plasmin S-carboxymethyl heavy (A) and light (B) chain derivatives. A re-evaluation of the mechanism of activation of plasminogen. J Biol Chem 1973;248(20):7242–6.
Summaria L, Arzadon L, Bernabe P, Robbins KC. Isolation, characterization, and comparison of the S-carboxymethyl heavy (A) and light (B) chain derivatives of cat, dog, rabbit, and bovine plasmins. J Biol Chem 1973;248(18):6522–7.
Robbins KC, Summaria L. Isoelectric focusing of human plasminogen, plasmin, and derived heavy (A) and light (B) chains. Ann N Y Acad Sci 1973;209:397–404.
Robbins KC, Bernabe P, Arzadon L, Summaria L. The primary structure of human plasminogen. II. The histidine loop of human plasmin: light (B) chain active center histidine sequence. J Biol Chem 1973;248(5):1631–3.
Robbins KC, Bernabe P, Arzadon L, Summaria L. The primary structure of human plasminogen. I. The NH 2 -terminal sequences of human plasminogen and the S-carboxymethyl heavy (A) and light (B) chain derivatives of plasmin. J Biol Chem 1972;247(21):6757–62.
Myohanen H, Vaheri A. Regulation and interactions in the activation of cell-associated plasminogen. Cell Mol Life Sci 2004;61(22):2840–58.
Kwaan HC. The biologic role of components of the plasminogen-plasmin system. Progress Cardiovasc Dis 1992;34(5):309–16.
Vassalli JD, Sappino AP, Belin D. The plasminogen activator/plasmin system. J Clin Invest 1991;88(4):1067–72.
Bachmann F, Kruithof IE. Tissue plasminogen activator: chemical and physiological aspects. Semin Thromb Hemost 1984;10(1):6–17.
Blasi F, Riccio A, Sebastio G. Human plasminogen activators. Genes and proteins structure. Horiz Biochem Biophys 1986;8:377–414.
Collen D, Lijnen HR. Tissue-type plasminogen activator: a historical perspective and personal account. J Thromb Haemost 2004;2(4):541–6.
Carmeliet P, Schoonjans L, Kieckens L, et al. Physiological consequences of loss of plasminogen activator gene function in mice. Nature 1994;368(6470):419–24.
Kwaan HC, Lo R, McFadzean AJ. On the production of plasma fibrinolytic activity within veins. Clin Sci (Lond) 1957;16(2):241–53.
Kwaan HC, McFadzean AJ. On plasma fibrinolytic activity induced by ischaemia. Clin Sci (Lond) 1956;15(2):245–57.
Kwaan HC, Lo R, McFadzean AJ. The production of plasma fibrinolytic activity in vivo by serotonin (5-hydroxytryptamine) creatinine sulphate. Clin Sci (Lond) 1957;16(2):255–9.
O'Rourke J, Jiang X, Hao Z, Cone RE, Hand AR. Distribution of sympathetic tissue plasminogen activator (tPA) to a distant microvasculature. J Neurosci Res 2005;79(6):727–33.
Hajjar KA, Menell JS. Annexin II: a novel mediator of cell surface plasmin generation. Ann N Y Acad Sci 1997;811:337–49.
Hajjar KA, Krishnan S. Annexin II: a mediator of the plasmin/plasminogen activator system. Trends Cardiovasc Med 1999;9(5):128–38.
Menell JS, Cesarman GM, Jacovina AT, McLaughlin MA, Lev EA, Hajjar KA. Annexin II and bleeding in acute promyelocytic leukemia. N Engl J Med 1999;340(13):994–1004.
Bernik MB, Kwaan HC. Origin of fibrinolytic activity in cultures of the human kidney. J Lab Clin Med 1967;70(4):650–61.
Bernik MB, Kwaan HC. Plasminogen activator activity in cultures from human tissues. An immunological and histochemical study. J Clin Invest 1969;48(9):1740–53.
Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev 2002;3(12):932–43.
Caiolfa VR, Zamai M, Malengo G, et al. Monomer dimer dynamics and distribution of GPI-anchored uPAR are determined by cell surface protein assemblies. J Cell biol 2007;179(5):1067–82.
Nykjaer A, Conese M, Christensen EI, et al. Recycling of the urokinase receptor upon internalization of the uPA:serpin complexes. EMBO J 1997;16(10):2610–20.
Gliemann J. Receptors of the low density lipoprotein (LDL) receptor family in man. Multiple functions of the large family members via interaction with complex ligands. Biol Chem 1998;379(8–9):951–64.
Aoki N, Harpel PC. Inhibitors of the fibrinolytic enzyme system. Semin Thromb Hemost 1984;10(1):24–41.
Vaughan DE, De Taeye BM, Eren M. PAI-1 antagonists: predictable indications and unconventional applications. Curr Drug Targets 2007;8(9):962–70.
Stefansson S, McMahon GA, Petitclerc E, Lawrence DA. Plasminogen activator inhibitor-1 in tumor growth, angiogenesis and vascular remodeling. Curr Pharm Des 2003;9(19):1545–64.
Kwaan HC, Wang J, Svoboda K, Declerck PJ. Plasminogen activator inhibitor 1 may promote tumour growth through inhibition of apoptosis. Br J Cancer 2000;82(10):1702–8.
Chen Y, Kelm RJ Jr, Budd RC, Sobel BE, Schneider DJ. Inhibition of apoptosis and caspase-3 in vascular smooth muscle cells by plasminogen activator inhibitor type-1. J Cell Biochem 2004;92(1):178–88.
Nagamine Y, Medcalf RL, Munoz-Canoves P. Transcriptional and posttranscriptional regulation of the plasminogen activator system. Thromb Haemost 2005;93(4):661–75.
Durand MK, Bodker JS, Christensen A, et al. Plasminogen activator inhibitor-I and tumour growth, invasion, and metastasis. Thromb Haemost 2004;91(3):438–49.
Vaughan DE, Lazos SA, Tong K. Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J Clin Invest 1995;95(3):995–1001.
Medcalf RL, Stasinopoulos SJ. The undecided serpin. The ins and outs of plasminogen activator inhibitor type 2. FEBS J 2005;272(19):4858–67.
Swartz JM, Bystrom J, Dyer KD, Nitto T, Wynn TA, Rosenberg HF. Plasminogen activator inhibitor-2 (PAI-2) in eosinophilic leukocytes. J Leukocyte Biol 2004;76(4):812–9.
Kruithof EK, Baker MS, Bunn CL. Biological and clinical aspects of plasminogen activator inhibitor type 2. Blood 1995;86(11):4007–24.
Dickinson JL, Norris BJ, Jensen PH, Antalis TM. The C-D interhelical domain of the serpin plasminogen activator inhibitor-type 2 is required for protection from TNF-alpha induced apoptosis. Cell Death Differ 1998;5(2):163–71.
Weiss LM, Warnke RA, Sklar J, Cleary ML. Molecular analysis of the t(14;18) chromosomal translocation in malignant lymphomas. N Engl J Med 1987;317(19):1185–9.
Santin AD, Zhan F, Bellone S, et al. Gene expression profiles in primary ovarian serous papillary tumors and normal ovarian epithelium: identification of candidate molecular markers for ovarian cancer diagnosis and therapy. Int J Cancer 2004;112(1):14–25.
Hasina R, Hulett K, Bicciato S, Di Bello C, Petruzzelli GJ, Lingen MW. Plasminogen activator inhibitor-2: a molecular biomarker for head and neck cancer progression. Cancer Res 2003;63(3):555–9.
Varro A, Noble PJ, Pritchard DM, et al. Helicobacter pylori induces plasminogen activator inhibitor 2 in gastric epithelial cells through nuclear factor-kappaB and RhoA: implications for invasion and apoptosis. Cancer Res 2004;64(5):1695–702.
Bajzar L, Morser J, Nesheim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem 1996;271(28):16603–8.
Leurs J, Wissing BM, Nerme V, Schatteman K, Bjorquist P, Hendriks D. Different mechanisms contribute to the biphasic pattern of carboxypeptidase U (TAFIa) generation during in vitro clot lysis in human plasma. Thromb Haemost 2003;89(2):264–71.
Wang W, Boffa MB, Bajzar L, Walker JB, Nesheim ME. A study of the mechanism of inhibition of fibrinolysis by activated thrombin-activable fibrinolysis inhibitor. J Biol Chem 1998;273(42):27176–81.
Schneider M, Nesheim M. A study of the protection of plasmin from antiplasmin inhibition within an intact fibrin clot during the course of clot lysis. J Biol Chem 2004;279(14):13333–9.
van Tilburg NH, Rosendaal FR, Bertina RM. Thrombin activatable fibrinolysis inhibitor and the risk for deep vein thrombosis. Blood 2000;95(9):2855–9.
Eichinger S, Schonauer V, Weltermann A, et al. Thrombin-activatable fibrinolysis inhibitor and the risk for recurrent venous thromboembolism. Blood 2004;103(10):3773–6.
Meijers JC, Oudijk EJ, Mosnier LO, et al. Reduced activity of TAFI (thrombin-activatable fibrinolysis inhibitor) in acute promyelocytic leukaemia. Br J Haematol 2000;108(3):518–23.
Morange PE, Juhan-Vague I, Scarabin PY, et al. Association between TAFI antigen and Ala147Thr polymorphism of the TAFI gene and the angina pectoris incidence. The PRIME Study (Prospective Epidemiological Study of MI). Thromb Haemost 2003;89(3):554–60.
Montaner J, Ribo M, Monasterio J, Molina CA, Alvarez-Sabin J. Thrombin-activable fibrinolysis inhibitor levels in the acute phase of ischemic stroke. Stroke 2003;34(4):1038–40.
Santamaria A, Oliver A, Borrell M, et al. Risk of ischemic stroke associated with functional thrombin-activatable fibrinolysis inhibitor plasma levels. Stroke 2003;34(10):2387–91.
Reijerkerk A, Meijers JC, Havik SR, Bouma BN, Voest EE, Gebbink MF. Tumor growth and metastasis are not affected in thrombin-activatable fibrinolysis inhibitor-deficient mice. J Thromb Haemost 2004;2(5):769–79.
Dano K, Andreasen PA, Grondahl-Hansen J, Kristensen P, Nielsen LS, Skriver L. Plasminogen activators, tissue degradation, and cancer. Adv Cancer Res 1985;44:139–266.
Mignatti P, Rifkin DB. Biology and biochemistry of proteinases in tumor invasion. Physiol Rev 1993;73(1):161–95.
Kirchheimer JC, Wojta J, Hienert G, et al. Effect of urokinase on the proliferation of primary cultures of human prostatic cells. Thromb Res 1987;48(3):291–8.
Kirchheimer JC, Wojta J, Christ G, Binder BR. Proliferation of a human epidermal tumor cell line stimulated by urokinase. Faseb J 1987;1(2):125–8.
Soff GA, Sanderowitz J, Gately S, et al. Expression of plasminogen activator inhibitor type 1 by human prostate carcinoma cells inhibits primary tumor growth, tumor-associated angiogenesis, and metastasis to lung and liver in an athymic mouse model. J Clin Invest 1995;96(6):2593–600.
Levenson AS, Kwaan HC, Svoboda KM, Weiss IM, Sakurai S, Jordan VC. Oestradiol regulation of the components of the plasminogen-plasmin system in MDA-MB-231 human breast cancer cells stably expressing the oestrogen receptor. Br J Cancer 1998;78(1):88–95.
McColl BK, Baldwin ME, Roufail S, et al. Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J Exp Med 2003;198(6):863–8.
Fredriksson L, Li H, Fieber C, Li X, Eriksson U. Tissue plasminogen activator is a potent activator of PDGF-CC. EMBO J 2004;23(19):3793–802.
Burridge K, Fath K, Kelly T, Nuckolls G, Turner C. Focal adhesions: transmembrane junctions between the extracellular matrix and the cytoskeleton. Ann Rev Cell Biol 1988;4:487–525.
Takahashi K, Ikeo K, Gojobori T, Tanifuji M. Local function of urokinase receptor at the adhesion contact sites of a metastatic tumor cell. Thromb Res 1990;10:55–61.
Madsen CD, Ferraris GM, Andolfo A, Cunningham O, Sidenius N. uPAR-induced cell adhesion and migration: vitronectin provides the key. J Cell Biol 2007;177(5):927–39.
Cunningham O, Andolfo A, Santovito ML, Iuzzolino L, Blasi F, Sidenius N. Dimerization controls the lipid raft partitioning of uPAR/CD87 and regulates its biological functions. EMBO J 2003;22(22):5994–6003.
O'Reilly MS, Holmgren L, Shing Y, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994;79(2):315–28.
Gately S, Twardowski P, Stack MS, et al. The mechanism of cancer-mediated conversion of plasminogen to the angiogenesis inhibitor angiostatin. Proc Natl Acad Sci USA 1997;94(20):10868–72.
Geiger JH, Cnudde SE. What the structure of angiostatin may tell us about its mechanism of action. J Thromb Haemost 2004;2(1):23–34.
Stathakis P, Fitzgerald M, Matthias LJ, Chesterman CN, Hogg PJ. Generation of angiostatin by reduction and proteolysis of plasmin. Catalysis by a plasmin reductase secreted by cultured cells. J Biol Chem 1997;272(33):20641–5.
Soff GA, Wang H, Cundiff DL, et al. In vivo generation of angiostatin isoforms by administration of a plasminogen activator and a free sulfhydryl donor: a phase I study of an angiostatic cocktail of tissue plasminogen activator and mesna. Clin Cancer Res 2005;11(17):6218–25.
Kwaan HC. Double hazard of thrombophilia and bleeding in leukemia. Hematology Am Soc Hematol Educ Program 2007;2007:151–7.
Tallman MS, Kwaan HC. Intravascular clotting activation and bleeding in patients with hematologic malignancies. Rev Clin Exp Hematol 2004;8(1):E1.
Kwaan HC, Wang J, Boggio LN. Abnormalities in hemostasis in acute promyelocytic leukemia. Hematol Oncol 2002;20(1):33–41.
Kwaan HC, Wang J, Weiss I. Expression of receptors for plasminogen activators on endothelial cell surface depends on their origin. J Thromb Haemost 2004;2(2):306–12.
Tapiovaara H, Alitalo R, Stephens R, Myohanen H, Ruutu T, Vaheri A. Abundant urokinase activity on the surface of mononuclear cells from blood and bone marrow of acute leukemia patients. Blood 1993;82(3):914–9.
Tapiovaara H, Matikainen S, Hurme M, Vaheri A. Induction of differentiation of promyelocytic NB4 cells by retinoic acid is associated with rapid increase in urokinase activity subsequently downregulated by production of inhibitors. Blood 1994;83(7):1883–91.
Fenaux P, Le Deley MC, Castaigne S, et al. Effect of all transretinoic acid in newly diagnosed acute promyelocytic leukemia. Results of a multicenter randomized trial. European APL 91 Group. Blood 1993;82(11):3241–9.
Tallman MS, Abutalib SA, Altman JK. The double hazard of thrombophilia and bleeding in acute promyelocytic leukemia. Semin Thromb Hemost 2007;33(4):330–8.
Tallman MS, Andersen JW, Schiffer CA, et al. All-trans retinoic acid in acute promyelocytic leukemia: long-term outcome and prognostic factor analysis from the North American Intergroup protocol. Blood 2002;100(13):4298–302.
Fenaux P, Chastang C, Chevret S, et al. A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood 1999;94(4):1192–200.
Sanz MA, Martin G, Gonzalez M, et al. Risk-adapted treatment of acute promyelocytic leukemia with all-trans-retinoic acid and anthracycline monochemotherapy: a multicenter study by the PETHEMA group. Blood 2004;103(4):1237–43.
Federici AB, Falanga A, Lattuada A, Di Rocco N, Barbui T, Mannucci PM. Proteolysis of von Willebrand factor is decreased in acute promyelocytic leukaemia by treatment with all-trans-retinoic acid. Br J Haematol 1996;92(3):733–9.
Look MP, van Putten WL, Duffy MJ, et al. Pooled analysis of prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in 8377 breast cancer patients. J Natl Cancer Inst 2002;94(2):116–28.
Harbeck N, Kates RE, Schmitt M. Clinical relevance of invasion factors urokinase-type plasminogen activator and plasminogen activator inhibitor type 1 for individualized therapy decisions in primary breast cancer is greatest when used in combination. J Clin Oncol 2002;20(4):1000–7.
Harbeck N, Kates RE, Look MP, et al. Enhanced benefit from adjuvant chemotherapy in breast cancer patients classified high-risk according to urokinase-type plasminogen activator (uPA) and plasminogen activator inhibitor type 1 (n = 3424). Cancer Res 2002;62(16):4617–22.
Foekens JA, Look MP, Peters HA, van Putten WL, Portengen H, Klijn JG. Urokinase-type plasminogen activator and its inhibitor PAI-1: predictors of poor response to tamoxifen therapy in recurrent breast cancer. J Natl Cancer Inst 1995;87(10):751–6.
Hataji O, Taguchi O, Gabazza EC, et al. Increased circulating levels of thrombin-activatable fibrinolysis inhibitor in lung cancer patients. Am J Hematol 2004;76(3):214–9.
Janicke F, Prechtl A, Thomssen C, Harbeck N, Meisner C, Untch M, Sweep CG, Selbmann HK, Graeff H, Schmitt M. Randomized adjuvant chemotherapy trial in high-risk, lymph node-negative breast cancer patients identified by urokinase-type plasminogen activator and plasminogen activator inhibitor type 1. J Natl Cancer Inst 2001;93:913–20.
Foekens JA, Buessecker F, Peters HA, Krainick U, van Putten WL, Look MP, Klijn JG, Kramer MD. Plasminogen activator inhibitor-2: Prognostic relevance in 1012 patients with primary breast cancer. Cancer Res 1995;55:1423–7.
Sternlicht MD, Dunning AM, Moore DH, Pharoah PD, Ginzinger DG, Chin K, Gray JW, Waldman FM, Ponder BA, Werb Z. Prognostic value of pai1 in invasive breast cancer: Evidence that tumor-specific factors are more important than genetic variation in regulating pai1 expression. Cancer Epidemiol Biomarkers Prev 2006;15:2107–14.
Grondahl-Hansen J, Peters HA, van Putten WL, Look MP, Pappot H, Ronne E, Dano K, Klijn JG, Brunner N, Foekens JA. Prognostic significance of the receptor for urokinase plasminogen activator in breast cancer. Clin Cancer Res 1995;1:1079–87.
Duffy MJ, Duggan C, Mulcahy HE, McDermott EW, O’Higgins NJ. Urokinase plasminogen activator: A prognostic marker in breast cancer including patients with axillary node-negative disease. Clin Chem 1998;44:1177–83.
Demirkan B, Ozcan MA, Glu AA, Yuksel F, Undar B, Alakavuklar M. The effect of anthracycline-based (epirubicin) adjuvant chemotherapy on plasma tafi and pai-1 levels in operable breast cancer. Clin Appl Thromb Hemost 2006;12:9–14.
Wojtukiewicz MZ, Sierko E, Zacharski LR, Zimnoch L, Kudryk B, Kisiel W. Tissue factor-dependent coagulation activation and impaired fibrinolysis in situ in gastric cancer. Semin Thromb Hemost 2003;29:291–300.
Heiss MM, Babic R, Allgayer H, Gruetzner KU, Jauch KW, Loehrs U, Schildberg FW. Tumor-associated proteolysis and prognosis: New functional risk factors in gastric cancer defined by the urokinase-type plasminogen activator system. J Clin Oncol 1995;13:2084–93.
Nekarda H, Schmitt M, Ulm K, Wenninger A, Vogelsang H, Becker K, Roder JD, Fink U, Siewert JR. Prognostic impact of urokinase-type plasminogen activator and its inhibitor pai-1 in completely resected gastric cancer. Cancer Res 1994;54:2900–907.
Cho JY, Chung HC, Noh SH, Roh JK, Min JS, Kim BS. High level of urokinase-type plasminogen activator is a new prognostic marker in patients with gastric carcinoma. Cancer 1997;79:878–83.
Beyer BC, Heiss MM, Simon EH, Gruetzner KU, Babic R, Jauch KW, Schildberg FW, Allgayer H. Urokinase system expression in gastric carcinoma: Prognostic impact in an independent patient series and first evidence of predictive value in preoperative biopsy and intestinal metaplasia specimens. Cancer 2006;106:1026–35.
Luebke T, Baldus SE, Spieker D, Grass G, Bollschweiler E, Schneider PM, Thiele J, Dienes HP, Hoelscher AH, Moenig SP. Is the urokinase-type plasminogen activator system a reliable prognostic factor in gastric cancer? Int J Biol Markers 2006;21:162–9.
Skelly MM, Troy A, Duffy MJ, Mulcahy HE, Duggan C, Connell TG, O’Donoghue DP, Sheahan K. Urokinase-type plasminogen activator in colorectal cancer: Relationship with clinicopathological features and patient outcome. Clin Cancer Res 1997;3:1837–40.
Mulcahy HE, Duffy MJ, Gibbons D, McCarthy P, Parfrey NA, O’Donoghue DP, Sheahan K. Urokinase-type plasminogen activator and outcome in dukes’ b colorectal cancer. Lancet 1994;344:583–4.
Yang JL, Seetoo D, Wang Y, Ranson M, Berney CR, Ham JM, Russell PJ, Crowe PJ. Urokinase-type plasminogen activator and its receptor in colorectal cancer: Independent prognostic factors of metastasis and cancer-specific survival and potential therapeutic targets. Int J Cancer 2000;89:431–9.
Ganesh S, Sier CF, Heerding MM, Griffioen G, Lamers CB, Verspaget HW. Urokinase receptor and colorectal cancer survival. Lancet 1994;344:401–2.
Stephens RW, Nielsen HJ, Christensen IJ, Thorlacius-Ussing O, Sorensen S, Dano K, Brunner N. Plasma urokinase receptor levels in patients with colorectal cancer: Relationship to prognosis. J Natl Cancer Inst 1999;91:869–74.
Kockar C, Kockar O, Ozturk M, Dagli M, Bavbek N, Kosar A. Global fibrinolytic capacity increased exponentially in metastatic colorectal cancer. Clin Appl Thromb Hemost 2005;11:227–30.
Sciacca FL, Ciusani E, Silvani A, Corsini E, Frigerio S, Pogliani S, Parati E, Croci D, Boiardi A, Salmaggi A. Genetic and plasma markers of venous thromboembolism in patients with high grade glioma. Clin Cancer Res 2004;10:1312–7.
Landau BJ, Kwaan HC, Verrusio EN, Brem SS. Elevated levels of urokinase-type plasminogen activator and plasminogen activator inhibitor type-1 in malignant human brain tumors. Cancer Res 1994;54:1105–8.
Hsu DW, Efird JT, Hedley-Whyte ET. Prognostic role of urokinase-type plasminogen activator in human gliomas. Am J Pathol 1995;147:114–123.
Kwaan HC, Lo R, McFadzean AJ. Antifibrinolytic activity in primary carcinoma of the liver. Clin Sci 1959;18:251–61.
De Petro G, Tavian D, Copeta A, Portolani N, Giulini SM, Barlati S. Expression of urokinase-type plasminogen activator (u-pa), u-pa receptor, and tissue-type pa messenger rnas in human hepatocellular carcinoma. Cancer Res 1998;58:214–9.
Pavey SJ, Hawson GA, Marsh NA. Impact of the fibrinolytic enzyme system on prognosis and survival associated with non-small cell lung carcinoma. Blood Coagul Fibrinolysis 2001;12:51–8.
Miyake H, Hara I, Yamanaka K, Gohji K, Arakawa S, Kamidono S. Elevation of serum levels of urokinase-type plasminogen activator and its receptor is associated with disease progression and prognosis in patients with prostate cancer. Prostate 1999;39:123–9.
Hienert G, Kirchheimer JC, Pfluger H, Binder BR. Urokinase-type plasminogen activator as a marker for the formation of distant metastases in prostatic carcinomas. J Urol 1988;140:1466–9.
Shariat SF, Roehrborn CG, McConnell JD, Park S, Alam N, Wheeler TM, Slawin KM. Association of the circulating levels of the urokinase system of plasminogen activation with the presence of prostate cancer and invasion, progression, and metastasis. J Clin Oncol 2007;25:349–55.
Torzewski M, Sarbia M, Verreet P, Dutkowski P, Heep H, Willers R, Gabbert HE. Prognostic significance of urokinase-type plasminogen activator expression in squamous cell carcinomas of the esophagus. Clin Cancer Res 1997;3:2263–8.
Nekarda H, Schlegel P, Schmitt M, Stark M, Mueller JD, Fink U, Siewert JR. Strong prognostic impact of tumor-associated urokinase-type plasminogen activator in completely resected adenocarcinoma of the esophagus. Clin Cancer Res 1998;4:1755–63.
Wojtukiewicz MZ, Sierko E, Zacharski LR, Rozanska-Kudelska M, Zimnoch L. Occurrence of components of fibrinolytic pathways in situ in laryngeal cancer. Semin Thromb Hemost 2003;29:317–20.
Kuhn W, Schmalfeldt B, Reuning U, Pache L, Berger U, Ulm K, Harbeck N, Spathe K, Dettmar P, Hofler H, Janicke F, Schmitt M, Graeff H. Prognostic significance of urokinase (upa) and its inhibitor pai-1 for survival in advanced ovarian carcinoma stage figo iiic. Br J Cancer 1999;79:1746–51.
Konecny G, Untch M, Pihan A, Kimmig R, Gropp M, Stieber P, Hepp H, Slamon D, Pegram M: Association of urokinase-type plasminogen activator and its inhibitor with disease progression and prognosis in ovarian cancer. Clin Cancer Res 2001;7:1743–9.
Kwaan HC, Radosevich JA, Xu CG, Lastre C. Tissue plasminogen activator and inhibitors of fibrinolysis in malignant melanoma. Tumour Biol 1988;9:301–6.
Tecimer C, Doering DL, Goldsmith LJ, Meyer JS, Abdulhay G, Wittliff JL. Clinical relevance of urokinase-type plasminogen activator, its receptor, and its inhibitor type 1 in endometrial cancer. Gynecol Oncol 2001;80:48–55.
Kobayashi H, Fujishiro S, Terao T. Impact of urokinase-type plasminogen activator and its inhibitor type 1 on prognosis in cervical cancer of the uterus. Cancer Res 1994;54:6539–48.
Hofmann R, Lehmer A, Buresch M, Hartung R, Ulm K. Clinical relevance of urokinase plasminogen activator, its receptor, and its inhibitor in patients with renal cell carcinoma. Cancer 1996;78:487–92.
Ohba K, Miyata Y, Kanda S, Koga S, Hayashi T, Kanetake H. Expression of urokinase-type plasminogen activator, urokinase-type plasminogen activator receptor and plasminogen activator inhibitors in patients with renal cell carcinoma: Correlation with tumor associated macrophage and prognosis. J Urol 2005;174:461–5.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Kwaan, H.C., McMahon, B. (2009). The Role of Plasminogen-Plasmin System in Cancer. In: Kwaan, H., Green, D. (eds) Coagulation in Cancer. Cancer Treatment and Research, vol 148. Springer, Boston, MA. https://doi.org/10.1007/978-0-387-79962-9_4
Download citation
DOI: https://doi.org/10.1007/978-0-387-79962-9_4
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-0-387-79961-2
Online ISBN: 978-0-387-79962-9
eBook Packages: MedicineMedicine (R0)