
Peritoneal dialysis (PD) has now been utilized for more than 25 years as a first-line therapy for the treatment of end-stage renal failure. It is now accepted that patient survival on PD is similar to hemodialysis when comparable analyses are made [1, 2]. As we have gathered new knowledge over the past decade, however, about the potential complications associated with long-term therapy, such as structural and functional alterations to the peritoneal membrane and end-stage sclerosing syndromes such as encapsulating peritoneal sclerosis (EPS), there remain legitimate concerns as to whether this mode of therapy can provide adequate treatment for end-stage renal disease in the longer term [3]. Despite advances in treatment guidelines there still remains in PD a considerable dropout rate in the early years of therapy, due mainly to infective episodes and membrane dysfunction [4].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Vascular Endothelial Growth Factor
- Peritoneal Dialysis Patient
- Dialysis Solution
- Peritoneal Membrane
- Encapsulate Peritoneal Sclerosis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Peritoneal dialysis (PD) has now been utilized for more than 25 years as a first-line therapy for the treatment of end-stage renal failure. It is now accepted that patient survival on PD is similar to hemodialysis when comparable analyses are made [1, 2]. As we have gathered new knowledge over the past decade, however, about the potential complications associated with long-term therapy, such as structural and functional alterations to the peritoneal membrane and end-stage sclerosing syndromes such as encapsulating peritoneal sclerosis (EPS), there remain legitimate concerns as to whether this mode of therapy can provide adequate treatment for end-stage renal disease in the longer term [3]. Despite advances in treatment guidelines there still remains in PD a considerable dropout rate in the early years of therapy, due mainly to infective episodes and membrane dysfunction [4].
There has been a considerable increase during the past few years in our understanding of the physiology of peritoneal transport, and the basic pathophysiology of the peritoneal membrane during PD [5–8] and those factors that impact on it during the therapy (Fig. 27.1 ). This understanding has gone hand-in-hand with (and to some extent driven by) the development and utilization of a new generation of “biocompatible” dialysis solutions whose design has been based on rational improvements in their physiological nature [9–13]. The sum of this research and developmental endeavour has been a much greater understanding about the pathophysiology of the dialysis process and both its local (in the peritoneum) and systemic consequences as well as a clearer view of how inflammatory processes that are driven by infective events or by exposure to dialysis solutions impact on the peritoneum during its life as a dialyzing organ (Fig. 27.1). Central in these findings have been significant advances based on improved basic science knowledge (and its application from other organ systems) and careful clinical observation of the precise histopathological changes in the structure of this membrane that occur as a result of uremia and how these are modulated by periods of PD therapy [14–17]. During this same period, information on what was initially referred to as peritoneal host defense, but is now understood in the context of the wider peritoneal inflammatory response, has developed into a significant understanding of how the interplay between the resident cells of the peritoneal membrane (the mesothelium, peritoneal fibroblasts, and resident leukocyte populations) and infiltrating leukocytes (of all phenotypes; polymorphonuclear neutrophilic leukocytes [PMN], monocyte/macrophages, and T cells) contribute to a complex series of tightly controlled processes, mediated by different classes of secreted cytokines, inflammatory mediators, surface and soluble receptors, and distinct intracellular signalling cascades (Fig. 27.2 ). These serve and are designed to orchestrate responses to infectious stimuli and effect acute and resolving peritoneal inflammation [18–22]. We have also developed our understanding of what occurs when these processes become dysregulated resulting in the genesis of a more “chronic inflammatory scenario” and to define the relationship between this dysregulated inflammation, tissue damage, and membrane dysfunction. Equally, it has become clear that we are dealing with a process complicated by the interaction of several factors that result in the “net inflammatory state” in PD patients. These include uremia, the cellular and secreted components of inflammation (cytokines and growth factors), the dialysis solution components, and the cellular targets of their action (Fig. 27.2). In the context of the peritoneal membrane this is, in effect, all the cells in the peritoneal membrane and functional structures, compact zone and the vascular bed (and adipocytes) [23] that are impacted upon.

Factors responsible for driving alterations to peritoneal structure and function

Cellular interactions and inflammatory response in the peritoneal membrane exposed to peritoneal dialysis. The major mediators released by each cellular component of the peritoneal membrane are indicated: GFs, growth factors; HYA, hyaluronan; IGs, immunoglobulins; ILs, interleukins; MCPs, monocyte chemoattractant proteins; NO, nitric oxide; O2 –, superoxide anion; PGs, prostaglandins. The release of these mediators is influenced by exposure to the dialysate and the uremic milieu
Another key development in recent years has been the identification of the potential importance of the plasticity of cellular phenotype (epithelial to mesenchymal transition; EMT) as a key process whereby resident cells, under the influence of inflammation and/or dialysis solution exposure, contribute to detrimental alterations in peritoneal membrane structure and function [24–28].
The aim of this chapter is to provide an updated overview of current understanding of peritoneal membrane structure and function, and how this is altered by PD therapy with its associated inflammatory changes, chemical insults, and intermittent infective episodes. This will range from a review of the pathophysiological changes induced by PD therapy to an exploration of the (intrinsic and extrinsic) mechanisms that drive peritoneal membrane structural and functional changes. The review will be based on the available published evidence and will utilize data from clinical observational studies, longitudinal characterization of changes in PD patients, as well as recent in vitro findings and data from animal models of inflammation and dialysis solution exposure. We will attempt to describe the molecular mechanisms that are involved in driving membrane structural and functional changes.
Peritoneal Structural Changes over Time
Central in the development of theories about how the process of PD induces changes to the peritoneum have been those studies that have sought to characterize the histopathological changes that occur in the peritoneal membrane both from standpoint of changes driven by uremia per se but most importantly those that are a feature of the process of the PD therapy. Observations on structural alterations in the peritoneal membrane in PD patients date back to the early observations made by Dobbie and DiPaolo [29–37], which were the forerunners of more contemporary studies by Honda and eventually the peritoneal biopsy registry [15–17, 38, 39]. These have provided the basis for our increased understanding of what is actually occurring in the peritoneum during PD and has begun to indicate those factors that might be responsible (or at least associated with) the observed structural alterations (Fig. 27.3 ).

Structural changes in the peritoneal membrane exposed to long-term peritoneal dialysis. Panel A shows the structure at the beginning of therapy, whereas panel B shows the structural alterations – loss of mesothelial integrity, submesothelial fibrosis, vasculopathy, and vascular proliferation – after 5 years on peritoneal dialysis. Brown staining indicates immunoreactivity for Factor VIII indicating the presence of blood vessels. Bar: 100 microns
Peritoneal Biopsy Data
Early observations on peritoneal membrane alterations were made in samples taken at catheter replacement or removal or at autopsy. The sum of these observations suggested alterations in the mesothelium (denudation, loss of microvilli), interstitium (various degrees of “fibrosis”), and alterations in the vascular bed [40–44]. These early studies showed clear evidence, albeit without strong linkage to particular clinical events other that PD exposure, of interstitial and vascular alterations in the peritoneal membrane. These were significantly extended by the landmark studies of Honda et al., who, although studying limited amounts of biopsy material, made significant observations on the nature of changes in the vasculature of the peritoneal membrane induced by PD therapy [15, 16]. In a small group of patients with clinically defined ultrafiltration (UF) failure, these authors described extensive interstitial fibrosis, loss of mesothelium, and vascular changes. The changes to the vasculature included severe fibrosis and hyalinization of the media of venules with extensive deposition of type IV collagen and laminin in the vascular wall and degeneration of smooth muscle cells in the media [15]. In more studies, these authors demonstrated that changes in the peritoneal vasculature were associated with accumulation of the advanced glycosylation end-products (AGEs) as assessed by staining with an anti-CML antibody [16]. Despite the relatively small size of the study, AGE accumulation correlated with both interstitial fibrosis and vascular degeneration.
The Peritoneal Biopsy Registry
In 1999, the Peritoneal Biopsy Registry was established in an attempt to systematically examine and characterize changes in membrane structure in a large cohort of PD patients. A large series of parietal peritoneal samples collected at the time of transplantation (and thereby not selected for reasons of treatment failure or other complications of PD) was examined by an independent pathologist and compared to control biopsies obtained from uremic predialysis patients (those being preemptively transplanted) or hemodialysis patients (prior to transplantation). In addition, a cohort of “true” normal samples was collected from the living related donor population. This extensive study allowed for the first time a thorough evaluation of those changes induced by PD (time on therapy, infection history, and dialysis solution exposure) compared to relevant control samples.
The key observations of these studies suggest that time on PD using conventional dialysis solutions results in progressive (if highly variable) thickening of the submesothelial cell compact zone and progressive vascular alterations (characterized as a hyalinizing vasculopathy with eventual vascular obliteration) [17, 38, 39]. Evidence of significant increases in vessel numbers, as suggested by previous studies [45] and by several animal studies, was only observed in patients with UF failure, although it is now becoming clear that this may be related to anatomical location (parietal versus visceral peritoneum) [46] and its inherent susceptibility to angiogenesis. Importantly, uremia per se appears to play a significant contributory role in these alterations as biopsies from predialysis and hemodialysis patients already show significant interstitial and vascular alterations compared to the normal controls. This effect of uremia corroborates effects on peritoneal membrane structure and function seen in experimental animals rendered uremic following nephrectomy [14, 47]. In addition, when examining the origin of the biopsies, those patients with clinical complications requiring catheter removal or suffering from UF failure had the greatest degree of peritoneal pathology [17].
Although such cross-sectional studies cannot provide direct mechanistic evidence of those factors directly responsible for the observed pathological changes, strong correlations were observed between duration of dialysis, total glucose exposure and previous infection history, and the observed alterations in both fibrosis and vascular degeneration (vasculopathy) (Fig. 27.4 ) [17, 38, 39].

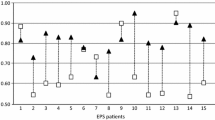
Relationship between previous infection history and the degree of submesothelial cell compact zone thickening in peritoneal biopsies from PD patients. Data presented with the permission of the Peritoneal Biopsy Registry. P<0.01
However, there remain significant gaps in our knowledge. While the data suggest that progressive structural changes do occur, the severity of which are linked to the length of time the membrane is exposed to PD, the correlations are based on small numbers of patients and do not provide mechanistic insight. The implication of those factors as being directly responsible for, or a result of the structural changes, although logical and attractive and, to some extent, supported by the emerging literature in animal models, must nevertheless be interpreted with caution. Likewise, the emerging understanding of encapsulating peritoneal sclerosis (EPS), which is beyond the scope of this chapter, and its relationship to the described observations made from biopsy studies is equally unclear. As yet, no unifying hypothesis for the relationship between “simple” fibrosis and vasculopathy and EPS exists and, in fact, current thinking suggests that they may indeed be diseases of separate peritoneal compartments (visceral versus parietal peritoneum). Increasing our understanding of peritoneal pathology must be an area of priority and intense investigation over the coming years.
Peritoneal Functional Changes over Time
Solute Transport
Although many PD patients may have stable transport rates for 5 years of more [48, 49], cross-sectional studies and longitudinal cohort studies have now clearly established that, on average, there is a slowly increasing small solute transport rate over time [8, 50–55]. Detailed analyses of the 574 PD patients available from the Stoke cohort revealed that solute transport increased significantly in the 6 months following the initiation of PD, and continued throughout the course of treatment [53]. It has been suggested that the increase in the average small solute transport could be due to a minority of patients experiencing a rise, whereas the majority have stable function. This interpretation is probably false, since patients with high small solute transport will leave CAPD more rapidly because high transport status is associated with a higher technique failure and higher mortality [8, 56, 57]. The cause is not clear, but an association with decreased ultrafiltration is possible since an increased transport will lead to a more rapid absorption of glucose with abolition of the osmotic gradient and reduced UF. In turn, a reduced UF will lead to fluid overload and thus a role of cardiovascular problems is increasingly suggested [56, 58, 59]. As discussed below, the morphological cause for a progressive increase in small solute transport may be an enlarged vascular surface area, reflecting the increased number of capillaries demonstrated in biopsies from long-term PD patients [17].
The factors believed to cause these structural changes with time on PD include repeated episodes of peritonitis and long-term exposure to bioincompatible dialysates [17, 49, 60]. Hypertonic glucose-based solutions are particularly suspected, due to their hyperosmolality, high glucose content, and associated levels of glucose degradation products (GDPs). Circumstantial evidence supports the role of dialysate glucose exposure in membrane damage, both in morphologic studies of the membrane [38, 39] and longitudinal studies showing that increased use of hypertonic glucose precedes changes in membrane function [49]. The residual renal function must also be considered, since its loss will require increased use of hypertonic glucose to sustain UF and will change the clearance of cytokines, thereby increasing systemic inflammation.
Ultrafiltration
One of the commonest changes with time on PD is a gradual decline in UF when using glucose dialysate, which represents a major cause for technical failure and increased mortality/morbidity [8, 53]. It has been suggested that, by 3 years, some 10% of patients have UF failure [61], and by 6 years this has increased to 30% [62]. A study from Japan reported that up to 50% of those individuals who had survived at least 6 years on PD would have technique failure due to fluid overload from UF loss [63]. Besides catheter-related problems, mechanisms operating in the membrane play an important role in UF failure. As discussed above, PD patients develop over time an increased transport rate for small solutes, which induces a faster absorption of glucose and an early dissipation of the osmotic gradient [8, 53]. This mechanism, responsible for an increased mass transfer area coefficient (MTAC) for small solutes, is considered as the most frequent cause of UF failure [64, 65]. However, the longitudinal studies by Davies et al. revealed that the changes in UF capacity are not exactly reflecting those in small solute transport [53]. Indeed, the fall in UF capacity observed in long-term patients (>4 years of PD) is disproportionately large for the rise in solute transport. These differences suggest that causes other than increased solute transport participate in the genesis of UF failure. A reduced sodium sieving during a hypertonic dwell, suggesting impaired free water transport, has been documented in long-term PD patients with UF failure [66, 67]. Subsequent cross-sectional studies suggested that a reduced osmotic conductance to glucose, causing a reduction of free-water transport, was involved in UF failure [62, 65, 68]. Although the mechanism of this reduced conductance remains unknown, it could be due to a decreased reflection coefficient (sigma), itself reflecting an alteration of the function of the aquaporin-1 (AQP1) water channels in the endothelium lining peritoneal capillaries [68, 69]. It must be pointed, however, that the expression of AQP1 is apparently unchanged in patients with long-term PD and UF failure [70, 71]. Furthermore, a recent study of 50 stable PD patients with UF failure showed no differences in the sigma coefficient between stable PD patients with or without UF failure and according to the duration of PD [64]. Thus, the potential role of functional impairment of AQP1 in UF failure remains to be substantiated. Other causes of UF dysfunction include increased effective fluid reabsorption by lymphatics, causing a decrease in intraperitoneal volume [62, 64] and modifications of the hydraulic conductance of the interstitium [72].
It must be emphasized that changes in intrinsic peritoneal transportwith time are rarely measured. In one case-matched study comparing patients treated for 5 years with PD compared to those starting treatment there appeared to be a significant reduction in peritoneal permeability with time [52], despite increased effective peritoneal surface area. This would be in keeping with an increase in peritoneal fibrosis, but longitudinal cohort studies have not confirmed this as yet. Clerbaux et al. showed no modifications of the sodium sieving in subgroups of 35 and 18 PD patients followed for 1 year and 2 years, respectively [55]. The recent description and validation of simple methods to determine the free water transport and the osmotic conductance to glucose [73, 74] is an important step in obtaining longitudinal informations on these parameters.
Macromolecular Clearance
Very few studies have assessed the changes in intrinsic permeability of the peritoneum to macromolecules such as albumin, IgG, and alpha 2-macroglobulin during PD. The issue is of importance, since longitudinal studies have shown a significant decrease in serum albumin concentration with time on PD [50, 55], which could play a role in the co-morbidity and the outcome of PD patients [75]. The macromolecule clearance can be assessed by the restriction coefficient of the membrane [76] that is inversely correlated with the permeability through the large pores. The restriction coefficients for macromolecules apparently do not change during the first 2 years of PD [77]. However, they may increase after a longer period of treatment, indicating that permeability to macromolecules could decrease with time on PD [77]. A reduced macromolecular clearance could be caused by a reduction in the large pore radius or by alterations in the interstitial tissue, for instance, due to the increased thickening of the submesothelial layer with collagen deposition [17].
The large pore fluid flux (JvL) can be assessed by the Peritoneal Dialysis Capacity (PDC) program [78]. A high JvL at the start of PD is related to comorbidity [79] but also to the development of hypoalbuminemia and survival on PD [80]. Indeed, patients with a high JvL showed an important fall in plasma albumin immediately after initiation of PD, suggesting peritoneal albumin loss. The correlation of JvL with older age, smoking, and atherosclerosis suggests that increased large pore permeability reflects generalized capillary disease [80]. Longitudinal studies showed no significant change in this parameter for up to 4 years on PD [79, 80].
Long-Term Effects of Automated Peritoneal Dialysis (APD)
Many APD individuals have been switched to this therapy from CAPD because of problems with achieving solute adequacy targets and/or difficulties with fluid removal. These patients may have variable residual function, and there is little information about the longitudinal changes in membrane function under this treatment modality. The European Automated Peritoneal Dialysis Outcome Study (EAPOS) examined longitudinal changes in the solute transport and UF capacity in a prospective cohort of 177 functionally anuric patients. The cohort experienced an increase in small solute transport and a reduction in UF capacity at 1 year and 2 years. These changes were more severe in patients using hypertonic glucose or in those not using icodextrin at baseline. Importantly, these differences could not be explained by informative censoring nor by age, comorbidity, or peritonitis rate [6]. This study yielded important mechanistic information, since residual renal function was eliminated as a confounding factor. It supports the link between exposure to higher glucose concentrations and accelerated changes in solute transport, as well as the potential benefit of using icodextrin in that respect [81–83].
Mechanisms Driving Structural and Functional Damage to the Membrane
The Link Between Structural Changes and Ultrafiltration Failure in PD
The endothelium lining peritoneal capillaries is considered as the major functional barrier to water and small solutes transport during PD. The amount of perfused capillaries within the peritoneum determines the so-called “effective peritoneal surface area” (EPSA) that is the functional area of exchange between blood and dialysate [84]. The transport of water and solutes across the capillary endothelium is best described by the three-pore model, which includes transcellular, ultrasmall pores (radius: 3–5 Å) exclusively permeable to water, small pores (radius: 40–50 Å) permeable to water and small solutes, and large pores (radius >150 Å) permeable to macromolecules [85, 86]. Studies in transgenic mice have demonstrated that the water channel AQP1 is the molecular counterpart of the ultrasmall pores, which mediate up to 50% of UF during a hypertonic dwell [87, 88]. Colloid osmosis (e.g., with icodextrin) occurs at the level of interendothelial small pores that allow the diffusion of water and small solutes [89]. Numerous studies have shown that long-term PD is associated with modifications of the peritoneal membrane, including submesothelial fibrosis, vascular proliferation, vascular diabetiform changes, and alterations of the mesothelium (Fig. 27.3) [16, 29, 45, 90]. In the Perotoneal Biopsy Registry, Williams et al. reported a positive correlation between the extent of submesothelial fibrosis and vascularization. This suggests an interaction between the two structural changes [17, 45]. However, fibrosis and angiogenesis may represent two independent responses to peritoneal injury. For instance, in a rat model of dialysate exposure, Margetts et al. were able to dissociate the fibrotic and angiogenic responses by using adenovirus-mediated gene therapy targeting either fibrosis (decorin) or angiogenesis (angiostatin) [91]. Importantly, these data showed that reduction of angiogenesis improved UF, whereas decreasing fibrosis had little impact on transport [91]. Submesothelial vascularization, vasodilatation, and increased reactivity for nitrotyrosine secondary to peroxynitrite release have also been observed in rat and mouse models of acute peritonitis [70, 92, 93]. Thus, vascular proliferation and, possibly, vasodilatation or recruitment of preexisting vessels, might represent the structural basis for increased EPSA encountered in acute peritonitis and long-term PD [70, 90, 93].
Over the past few years, several experimental models have given new insights into the pathophysiology of UF failure in PD. These models include rats and mice with acute peritonitis [70, 92–94], chronic exposure to diabetes [95, 96] or uremia [14, 28, 97], and chronic dialysate exposure [91, 98]. A common feature of these models is the local release of growth factors such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and transforming growth factor β (TGF-β) in the peritoneal membrane, leading to the development of areas of neovascularization and/or submesothelial fibrosis. These structural modifications, which have been documented at the morphological and molecular level, are associated with functional changes including higher permeability for glucose and small solutes, decreased sodium sieving, and reduced UF. Thus, an inverse relationship between the vascular density in the peritoneum and the amount of UF has been demonstrated in rats [91], and such a correlation also exists in the human peritoneal membrane [17]. It must be pointed out that, to date, there is no molecular evidence for a functional or structural modification of the water channel AQP1 in the peritoneum exposed to PD.
Epithelial to Mesenchymal Transition (EMT)
There is now a body of emerging evidence that suggests that changes to the peritoneal membrane are at least in part driven by chronic alterations to the mesothelium. Although not yet causatively implicated, it is suggested that dialysis solution-driven chronic activation results in the activation of the process of epithelial to mesenchymal transition (EMT) whereby epithelioid cells alter their phenotype and become myofibroblast in form and function [24–26].
This hypothesis is based on studies of mesothelial cells isolated from PD where with time on dialysis there appeared to be morphological alterations to a more fibroblastoid phenotype accompanied with loss of epithelioid markers such as cytokeratin expression. This switch in phenotype from epithelioid to fibroblastic confers on these cells a migratory phenotype, allowing them to move into the submesothelial stroma (compact zone), where they may contribute directly to the fibrotic process [24]. Although definitive proof that such a process occurs in PD patients is still lacking, it remains an attractive hypothesis that might partly explain the morphological alterations seen in the mesothelium in the biopsy registry although this cannot explain the observation of complete denudation of mesothelium. This attractive hypothesis does gain support from recent studies from animal models that implicates both EMT and AGE-RAGE-driven EMT in driving peritoneal fibrosis [27, 28, 99, 100].
Dialysis Solutions and Membrane Structural and Functional Changes
Over the past 30 years, the focus of PD research has changed from the technical issues related to the establishment of clinical peritoneal dialysis to complex problems of peritoneal membrane pathobiology. Up to the mid-1980s, dialysis fluids and methods of access had to be developed along with the clinical procedures. Investigations were directed primarily at treatment efficacy and infection control. The first research articles on PD fluid biocompatibility were published, most using peripheral blood leukocytes as model system to assess the impact of dialysis solution components on cell viability and function [101, 102]. After this, peritoneal cell cultures were developed, and used extensively as preclinical testing for the development of new peritoneal dialysis solutions. Description of this extensive data is beyond the scope of this chapter as reviewed in [20, 103–105]. Instead, in the context of what factors are responsible for structural changes in the dialyzed peritoneum, we will focus on more recent data generated by an increasing number of research groups using animal models of PD in an effort to more closely mimic the specific in vivo environment of the dialyzed peritoneum [106]. These studies have cast significant light on the mechanisms that contribute to peritoneal membrane damage and those factors responsible for the initiation of peritoneal fibrosis.
As described above, longitudinal studies of PD patients have shown that small solute transport increases with time on PD [8, 53]. These data suggest that long-term exposure to conventional, glucose-based dialysis fluids plays a central role in the pathogenesis of the modifications of the peritoneal membrane. This hypothesis is supported by the observation that an early and high cumulative glucose exposure is associated with higher small solute transport across the peritoneum of PD patients [49] and by the results of the EAPOS study on anuric patients [6].
Animal Models and the Impact of Dialysis Solutions
Obtaining peritoneal biopsies in patients is not simple procedure, and these cannot be obtained electively. As described earlier, data from peritoneal biopsies although vital to our understanding of peritoneal pathology [17, 38, 39] only provide a cross-sectional snapshot of membrane changes (and they are limited to small areas of parietal or visceral peritoneum). Animal models, however, while failing to totally mimic human PD [107], do provide the opportunity to study longitudinal changes in peritoneal membrane pathology and thereby delve into the fundamental pathophysiological processes that drive membrane damage. In the field of PD, animal models have for a long time been underdeveloped and it is only in the past decade that significant progress has been made [108–113].
A number of research groups now have sophisticated animal models to more closely simulate the specific environment of the dialyzed peritoneum [89]. Descriptions of these different animals, with their advantages, limitations, and technical aspects have been reviewed on several occasions [110, 113]. Methodological differences such as study duration, species and strain of experimental animals, methods of peritoneal exposure, assessment of solute transport and ultrafiltration, and histological assessment differ substantially among the various research groups and to some extent make interpretation difficult. There have nevertheless been some substantial advances in understanding peritoneal membrane changes.
Most experimental models of PD that have been described make use of rats, and the majority of models are short-time (in general no longer than 4–6 weeks) [114]. These more chronic rat models were introduced to investigate biocompatibility of dialysis solutions in nonuremic animals in which PD was performed twice daily for 4 weeks [115]. Most models are peritoneal exposure models in which there is no fluid exchange. Peritoneal dialysis fluid is instilled at volumes ranging from 10 mL [116, 117] to 25 mL [118]. The longest long-term peritoneal exposure models in Wistar rats allow peritoneal infusion for up to 20 weeks [119, 120].
Some groups have tried to establish uremic models in rats [121] and rabbits [122], but difficulties still exist because nephrectomy results in technical problems and leads to a high dropout rate [123]. Nevertheless, this in vivo research has provided important breakthroughs in understanding peritoneal pathophysiology [107]. These breakthroughs are 1) the observation and integration of the long-term structural and functional alterations of the peritoneal membrane, 2) a better understanding of the pathophysiology of peritoneal solute transport and ultrafiltration, and 3) a detailed description of the impact of standard dialysis solutions and inflammation on morphological and functional alterations of the peritoneal membrane that may in the future contribute to the development of improved dialysis solutions.
The traditional PD solutions are deemed to be bioincompatible because of their low pH, high glucose and lactate concentrations, hyperosmolality, and the presence of glucose degradation products formed during heat sterilization [106]. Glucose in high concentrations is toxic for the mesothelium both in in vitro and in animal studies [124]. Degradation of glucose results in formation of glucose degradation products and finally in formation of AGEs that have been described to accumulate in peritoneal tissues of patients [90, 125]. Glucose is also likely to be involved in the development of peritoneal neoangiogenesis. This is supported by the “diabetiform” alterations of the microvessels that are present in patients [15, 16, 45] and that can be induced to some extent in animal models [120]. Glucose degradation products are toxic to peritoneal cells and induce the formation of AGEs at a faster rate than glucose itself [126, 127]. The relative importance of glucose degradation products to glucose has not been clarified. It has been suggested that lactate may contribute to the toxicity of glucose [128].
The Impact of Angiogenesis and Fibrosis
Long-term exposure to conventional glucose/lactate-based dialysis solutions can induce peritoneal alterations [17, 31, 34, 38, 39, 45]. The morphologic changes consist of an increased thickness of the submesothelial compact collagenous zone of the parietal peritoneum, sometimes accompanied by loss of surface mesothelium (Fig. 27.3). Interstitial fibrosis can also be found in omental tissue. Extensive vascular abnormalities have been described. These include not only subendothelial hyalinosis of arterioles, but also of the venules and small veins [15–17]. Also, an increased number of vessels have been found [45], especially in patients with UF failure [17]. The thickness of the submesothelial compact zone was related to the duration of PD, the absence of mesothelium and the prevalence of vasculopathy. A correlation has also been described between the number of peritoneal vessels and fibrotic alterations [17].
As described earlier, angiogenesis appears to be a key feature of dialysis solution–driven changes in peritoneal morphology. Although this concept is primarily based on animal studies, there is clear evidence of increased vessel formation in PD patients (particularly those with fibrosis and membrane dysfunction) [17, 45, 90].
Long-term PD is associated with alterations in peritoneal permeability and loss of UF. In view of the role of increased peritoneal surface area, modifications of activity and/or expression of nitric oxide synthase (NOS) isozymes might play a role in these modifications, via enhanced local production of nitric oxide (NO). This hypothesis was supported by the studies of Combet et al. [90], who determined NOS activities in peritoneal biopsies from control subjects, uremic patients immediately before the onset of PD, and uremic patients on short- or long-term PD. Peritoneal NOS activity is increased fivefold in long-term PD patients compared with control subjects. In uremic patients, NOS activity is positively correlated with the duration of PD. Increased NOS activity is mediated solely by Ca2+-dependent NOS and, as shown by immunoblotting, an upregulation of endothelial NOS. The biologic relevance of increased NOS in long-term PD was demonstrated by enhanced nitrotyrosine immunoreactivity and a significant increase in vascular density and endothelial area in the peritoneum. Immunoblotting and immunostaining studies demonstrated an upregulation of VEGF, mostly along the endothelium lining peritoneal blood vessels in long-term PD patients. In the latter, VEGF co-localized with deposits of the AGE product pentosidine. These data provided a morphologic (angiogenesis and increased endothelial area) and molecular (enhanced NOS activity and endothelial NOS upregulation) basis for explaining the permeability changes observed in long-term PD. They also supported the implication of local AGE product deposits and liberation of VEGF in that process.
The use of animal models of PD has allowed to elucidate some of the molecular mechanisms involved in these degenerative processes. The important roles of aquaporins [13, 129, 130], VEGF [95], nitric oxide [13, 131], AGE formation and their receptor (RAGE) upregulation [28, 90, 100], and TGF-β [99] in the fibrotic alterations of the membrane as observed in patients on long-term PD have been investigated. More recently, genetically modified mice have emerged as an important tool to investigate the molecular basis of peritoneal changes during dialysis and during acute peritonitis [92, 93, 113, 132] (Fig. 27.5 ). The effects of RAGE in modulating fibrosis were not so clearcut in studies in RAGE-deficient mice, although this might be related to the nature of the genetic defect in these animals and its significant impact on inflammatory responses [98].

Use of genetically modified mice to elucidate the impact of eNOS on vascular density and peritoneal transport. Wild-type mice (eNOS WT-p) or littermates lacking eNOS (eNOS KO-p) were submitted to a catheter-induced peritonitis model for 5 days. The lack of eNOS is reflected by a significant reduction in the vascular proliferation (panel A, staining with the endothelial marker CD31; panel C, morphometry analyses) and a marked reduction in the transport of small solutes (panel B, dialysate-over-plasma (D/P) ratio for urea during a 2-h dwell, n = 6 mice in each group; panel C, area under curve, AUC) and in the loss of protein in the dialysate (panel C). *P<0.05; **P<0.01.Data compiled from [93]
The Effect of GDP, RCOs, AGEs, and RAGE
Heat sterilization of conventional, glucose-based dialysis solutions generates the formation of GDP and reactive carbonyl species (RCOs) such as glyoxal, methylglyoxal (MGO), 3-deoxyglucosone, and 3,4-dideoxyglucosone-3-ene (3,4-DGE) [133, 134]. Furthermore, high levels of RCOs are also present in uremic plasma [135]. Multiple studies have demonstrated that both GDPs and RCOs accelerate the formation of AGEs in the peritoneal membrane, where they localize both in the mesothelium and endothelium [90, 127, 136, 137]. Both RCOs and AGEs modify proteins and/or interact with receptors, which may in turn initiate a range of cellular responses including stimulation of monocytes, secretion of inflammatory cytokines, proliferation of vascular smooth muscle cells, stimulation of growth factors, secretion of matrix proteins and alterations of mesothelial cell function [135, 138, 139]. The link between RCOs/AGEs and VEGF is also supported by the demonstration of increased VEGF expression in the peritoneum of rats chronically injected with MGO [136], as well as by the co-localization of pentosidine and VEGF in peritoneal capillaries from long-term PD patients [13, 90, 140] (Fig. 27.6 ). Recently, Kakuta et al. used a uremic rat model on PD to demonstrate that the combination of uremia and dialysis solution exposure generated vascular proliferation with ensuing transport abnormalities, AGE genesis, and upregulation of the angiogenic VEGF and bFGF. These modifications were significantly improved by giving pyridoxamine, an inhibitor of AGE, to the uremic rats, pointing to the pathophysiological importance of carbonyl stress in the alterations of the peritoneal membrane [97].

Integrative approach to understanding the influence of factors that drive. Structural and functional changes in the peritoneal membrance (PM). See text for details
Studies on human peritoneal mesothelial cells (HPMC) in culture also yielded important insights into the pathophysiology of GDP in PD. Witowski et al. used HPMC to demonstrate the cytotoxic effect of GDP, which decreased cell growth and viability [141], while more recently Morgan et al., have shown that specific GDP species (notably 3,4-DGE) modulate mesothelial cell repair processes [138]. Lai et al. showed that incubation of HPMC with GDPs or dialysate fluids resulted in increased AGE synthesis and VEGF expression, as well as the expression of RAGE and other AGE receptors, in a dose-dependent manner [142]. Boulanger et al. showed that heat-sterilized dialysates (containing high glucose and GDP concentrations) reduced HPMC proliferation by inducing mesothelial cell apoptosis and oncosis [143]. These changes in cell function were significantly attenuated by blocking AGE-RAGE interaction using recombinant soluble-RAGE. Using a co-culture system, these investigators further demonstrated that mesothelial RAGE activation by AGEs enhanced VEGF release, which potentialized capillary tube formation by endothelial cells, a process that was attenuated by using blocking antibodies directed against RAGE or VEGF. The pathogenic role of AGE-RAGE interaction and its potential to drive EMT has been substantiated in a rat infusion model (see earlier) [28, 100].
The growth factor VEGF is a potent regulator of angiogenesis and vascular permeability [144]. The VEGF gene codes for several VEGF isoforms, which arrange into disulfide-linked homodimers. The binding of VEGF dimers to tyrosine-kinase receptors (VEGFR-1 and VEGFR-2) located in endothelial cells initiates a signal transduction cascade responsible for endothelial proliferation and migration, activation of plasminogen and collagenase, and vasodilation, resulting in physiological angiogenesis [144]. In addition, VEGF binds to the extracellular matrix, thereby inducing the release of bFGF, another potent angiogenic factor [145]. Stimuli for VEGF expression include hypoxia, hypoglycemia, cytokines such as interleukin-6 (IL-6), growth factors, and hormones. VEGF is expressed in the human peritoneal membrane, where it binds to the endothelium lining peritoneal capillaries [90]. Thus, by analogy with other angiogenic diseases, upregulation of VEGF may trigger vascular proliferation in the peritoneal membrane in long-term PD (Fig. 27.6). In this respect it is of interest that plasma and dialysate concentrations of VEGF and IL-6 have been shown to be associated with high peritoneal solute transport rate [146].
Nitric Oxide and Oxidative Stress
Nitric oxide is an attractive candidate to regulate EPSA and UF during PD, given its crucial role in the regulation of vascular tone and permeability [147], and its interactions with angiogenic growth factors [148]. The paradigm has been provided by the loss of UF in a rat model of acute peritonitis, characterized by a major upregulation of the endothelial and inducible NOS isoforms and a parallel increase in the permeability for glucose and small solutes [70]. Addition of the NOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME) to the dialysate was able to restore most of the UF capacity in this rat model [94] and a similar effect was observed in a LPS-induced peritonitis mouse model [92]. In addition to its vascular effects, NO might also affect the peritoneum by generating peroxynitrite (a powerful, cytotoxic oxidant) and by modifying critical residues, as suggested by an increased reactivity for nitrotyrosine [70] or nitrosocysteine [94].
Multiple interactions between NO, endothelial NOS, and VEGF occur within endothelial cells (Fig. 27.6 ). Both NO and the endothelial NOS are required for VEGF-driven angiogenesis and vascular permeability [149]. On the other hand, VEGF is known to activate endothelial cells and upregulate the production of NO [150]. In turn, NO modulates and even suppresses the hypoxic induction of VEGF, which creates a negative feedback between NO and VEGF induction [148]. Interestingly, such crosstalk exists in the peritoneum, since upregulation of eNOS in a rat model of acute peritonitis is associated with down-regulation of VEGF [70].
The link between oxidative stress and peritoneal damage was substantiated by the studies of Lee et al. [151] who demonstrated that high glucose increased reactive oxygen species (ROS) in HPMC through the activation of a signaling cascade involving protein kinase C (PKC). In turn, the ROS generated were able to upregulate the expression of fibronectin by HPMC. A subsequent study by the same group provided in vivo evidence for the damaging consequences of ROS generated by conventional dialysates. Rats exposed to such dialysates for 12 weeks showed increased small solute transport, reduced UF, increased membrane thickness, and increased expression of eNOS, TGF-β1, VEGF, collagen I, and angiotensin II [152]. All of these changes were prevented by the administration of N-acetylcysteine or losartan, suggesting that these agents may play a protective role in long-term PD [152].
Contribution of Uremia and Glycemic Control
By analogy with the increased permeability of serosal membranes such as the pleura or the pericardium, it has been suggested that uremia per se might increase the permeability of the peritoneum [153]. That hypothesis has been supported by the association of several molecular mechanisms – upregulation of NOS, high levels of circulating RCOs and AGEs, increased growth factors – with higher peritoneal permeability in a chronic uremic rat model [14]. Subsequent studies confirmed that uremia alone induces AGE formation, upregulation of growth factors including VEGF and TGF-β1, myofibroblast transdifferentiation and development of interstitial fibrosis, as well as vascular proliferation and increased transport for small solutes [28, 97]. The attenuation of these changes by neutralizing anti-RAGE antibodies [28] or the RAGE-inhibitor pyridoxamine [97] suggests a role for the AGE-RAGE interaction in these processes (Fig. 27.6).
Diabetes may represent another factor that affects the peritoneal membrane. The CANUSA prospective study showed a greater proportion of diabetics among high transporter PD patients [57], and it has been suggested that diabetic patients have higher permeability for creatinine and lower UF than nondiabetic patients [48, 55, 154]. Studies performed in a streptozotocin-induced diabetic rat model [95, 96] showed that chronic hyperglycemia alone is sufficient to induce functional (increased transport for small solutes) and structural (areas of vascular proliferation) changes in the peritoneum. It is interesting to note that these modifications were associated with the selective regulation of NOS isoforms and AGEs deposits [96]. All the alterations were prevented by chronic insulin treatment, demonstrating that adequate control of glycemia in this diabetic rat model is sufficient to preserve the integrity of the peritoneum. Taken together, these data suggest an independent contribution of uremia and hyperglycemia in peritoneal changes during PD.
Peritoneal Inflammation and Membrane Dysfunction
As mentioned previously, inflammation appears to play a central role in modulating the function and possibly structure of the peritoneal membrane. Over the past decade, information gleaned from measurements of intraperitoneal inflammatory mediator levels (in PD effluent isolated from patients during stable PD and during peritonitis), as well as a large body of in vitro cell culture data has identified an outline of the process by which peritoneal inflammation is initiated, amplified, and how it resolves [155]. These data suggest that, following bacterial contamination of the peritoneal cavity, a coordinated train of events is set in motion to eradicate the invading organisms and resume normal tissue homeostasis. Also clear is that both the resident and infiltrating cells (both tissue cells and resident macrophages and infiltrating leukocytes) participate in these processes [103, 156, 157].
Inflammation in the dialyzed peritoneum involves an initiation phase resulting from the activation of resident phagocytes, and probably the mesothelium, by invading micro-organisms or their secreted products [19, 20]. Next, there is an amplification phase in which mesothelial cell activation by peritoneal macrophage-derived pro-inflammatory cytokines (such as IL-1β and TNFα) appears to play a key role. This process results in the generation of chemotactic signals, via the creation of a gradient of chemotactic cytokines (specific for individual leukocyte subpopulations) leading to the recruitment of these inflammatory cells to the site of activation [21, 22, 158–161]. This infiltration process is facilitated by the upregulation of leukocyte specific adhesion molecules of the immunoglobulin superfamily (ICAM-1 and VCAM-1/2) on the mesothelial surface [156, 162–164]. There is also the involvement of other pathways, including CD40-CD154 ligation, which also appears to regulate chemokine secretion and leukocyte trafficking [165–169]. The process of leukocyte infiltration is tightly controlled, such that initially polymorphonuclear leukocytes predominate (6–24 h) and are subsequently replaced by mononuclear cells (mononuclear phagocytes and T and B lymphocytes) [22]. This switch in leukocyte phenotype during peritonitis (Fig. 27.7 ), is known to be controlled by a sophisticated and differential regulation of chemokine secretion and polymorphonuclear leukocytes apoptosis regulated by IL-6/sIL-6R signaling through its signal transducer gp130 [21, 22, 160, 161] as reviewed by Jones et al. [170, 171].

Changes in recruited leukocyte phenotype and IL-6 family cytokines during the whole time course of bacterial peritonitis. Data (left panel) from flow cytometry using markers specific for PMN (CXCR1), monocytes (CD14), and T lymphocytes (CD3). Data (right panel) of IL-6, soluble IL-6 receptor (sIL-6R), and soluble gp130 (sgp130) measured by ELISA in 12 episodes of Gram-positive peritonitis
The Link Between Inflammation and Membrane Dysfunction
Given the number of factors that potentially impact on the peritoneal membrane (Figs. 27.2 and 27.6), it is perhaps difficult to separate out cause and effect and particularly difficult to attribute the degree of importance of single processes. That being said, there is little doubt that the process of inflammation in all its guises likely plays a significant role in peritoneal physiology and pathophysiology. In the former case, as was discussed earlier, alterations in cytokine or growth factor production (under genetic control) influence membrane function [131].
Each year, the understanding of inflammation and inflammatory process moves on in massive leaps and bounds in all aspects of disease and in all organ systems, and the same is the case for the peritoneal cavity. In the 1980s and early 1990s, we coined the phrase “the peritoneal cytokine network” and were primarily concerned with peritoneal host defense [20, 103] and the impact upon it of “bioincompatible” dialysis solutions [18]. We are now in a totally different era. The current paradigm suggests that inflammation comes in many guises and has both beneficial and detrimental effects. It is good if it promotes host defense and resolves inflammation, protecting the host from “insult” and restoring tissue homeostasis. It is bad if it loses control of itself, drives a more “chronic” phenotype, and contributes to tissue and end organ damage.
In the context of the peritoneum, our current understanding of the situation is equally if not more complex as there are both intrinsic and extrinsic factors impacting on the inflammatory process (Fig. 27.1). While it is clear that acute inflammation associated with infective episodes is a normal response to insult and, in the case of the peritoneum designed to promote bacterial removal, it is becoming equally clear that each insult is not the same, and this may compromise the ability of acute inflammation to resolve and in fact the nature of each individual response. This might result in the cumulative severity of acute inflammation changing with each subsequent insult. Although this is speculation, our basic understanding of inflammation makes it a likely scenario. While we now understand the nature of some of the factors that control acute inflammation (eg,. chemokine and IL-6 trans-signaling) and have defined these in the context of single episodes of peritoneal inflammation in animal models and man, we are some way from being able to demonstrate their dysregulation, although preliminary animal data (in peritonitis and arthritis) suggests that this is the case [21, 22, 160, 161, 170, 171]. It is equally important that we do not limit our description to acute inflammation (or innate immunity) as these regulatory mechanisms also responsible for the recruitment and retention of both macrophage and T-cell populations in the peritoneal cavity.
To date, the definition of chronic inflammation is loosely defined as the retention of activated leukocytes (monocyte/macrophages and lymphocytes) within an organ system. Does a similar scenario appertain to the dialyzed peritoneal cavity? There is indeed the continuous retention (and indeed recruitment) of mononuclear cells and lymphocytes, and evidence of altered phenotype (Roberts and Topley, unpublished observations). We are only just beginning, however, to understand what this means in terms of peritoneal membrane longevity, and significant further study is required to define the activation status of these cells and how dialysis impacts upon them and whether they contribute to membrane dysfunction [172, 173]. Given the role of IL-6 as a marker of poor outcome in many diseases and its central role in regulating cellular inflammation, it is not a huge conceptual leap to suggest that there may indeed be some inter-relationship between inflammation and disease outcome, which is of relevance to the peritoneal cavity. This is especially relevant when one considers that trials of new dialysis solutions have shown variable alterations in local IL-6 levels following conversion to the use of more “biocompatible” dialysis [174, 175].
While we are only beginning to understand the full nature of local inflammation in the peritoneum and its long-term impact on membrane survival, we are also trying to get to grips with the impact of systemic inflammation in PD patients and the potential inter-relationships between them. Although factors such as IL-6 and C-reactive protein (CRP) predict poor outcome [176–178], the mechanism that drives these poor prognoses are likely extraperitoneal. It is nevertheless important to understand whether local effects (within the peritoneum) can impact positively on systemic inflammatory parameters and patient survival. In this respect, recent data that suggest alterations in plasma CRP in patients converted to new dialysis solutions are intriguing but require corroboration in much larger cohorts [179].
The scope of this review does not allow a detailed description of the immunology of the peritoneal cavity, although this is a rapidly expanding area of research. There are many questions to address that, by and large, will require human studies, as the rodent peritoneum does not mimic all of the subtleties and sophistications of the human immune system. The dialyzed peritoneal cavity provides us with unique access for this type of endeavour, and one hopes that over the coming decade we will be able to understand how inflammation in its various guises contributes to membrane structural and functional changes in PD patients so that we can modulate therapeutic interventions to limit its detrimental but accentuate its positive attributes.
Reversibility of Structural Changes in the Peritoneum
If the nature of the structural changes that occur in the peritoneal membrane exposed to long-term PD is now becoming better defined, the intriguing question of their reversibility upon peritoneal rest (i.e., terminating PD) or indeed specific therapy remains an important question. Since dialysis solutions can never be physiological (in order to perform there dialytic function), it may be that membrane protective strategies are an essential feature of the therapy [106, 180]. The issue is important, not only for technique survival but also in terms of potential benefits resulting from the different therapeutic approaches. Early human studies showed that peritoneal rest may result in a recovery of ultrafiltration in PD patients with hyperpermeability [181, 182]. In a limited study, Zhe et al. showed an improved UF capacity due to decreased solute transport rate in PD patients transferred to daytime PD with overnight membrane rest [183]. Kim et al. used a rat model exposed for 3 weeks to glucose dialysate to show that a 4-week-period of rest resulted in a significant reversibility of transport abnormalities, with improved UF and reduction of peritoneal thickness [184]. Similar conclusions were reached by Zareie et al. [46], who showed that a long peritoneal rest (12 weeks) in rats exposed to PD fluids resulted in a significant reduction of angiogenesis and fibrosis.
Individual Variation in Membrane Transport : Genetic versus Clinical Factors
The transport properties of the peritoneal membrane (i.e., the three populations of pores) are usually assessed by the peritoneal equilibration test (PET) or alternative methods such the standard peritoneal permeability analysis (SPA) or the Peritoneal Dialysis Capacity (PDC) tests [185, 186]. Assessing the individual variability in peritoneal transport has a major clinical importance [185]. The CANUSA study first documented the association between higher transport for small solutes and lower combined patient and technical survival [57], an association that has been confirmed in various populations [56]. Indeed, a high D/P ratio for creatinine is paralleled by a low D/D0 ratio for glucose with, as a consequence, reduction in the osmotic gradient, loss of UF, and fluid retention. A high transport status may also be associated with increased peritoneal albumin losses and, thus, potential denutrition. Furthermore, individual variability in peritoneal transport status also influences PD prescription: high transporters benefit from short dwells and icodextrin, while low transporters are prescribed longer dwells.
Twardowski et al. first showed that approximately two-thirds of patients have average transport rate, the remaining one third being almost equally distributed between high and low transporters [186]. This considerable patient variability renders it difficult to extrapolate mean values in cohort studies to the individual.
Subsequent series confirmed the existence of a significant interpatient variability in the baseline solute transport characteristics of the peritoneum, and showed that a high co-morbidity score, including hypoalbuminemia, older age, diabetes, and ACE inhibitor prescription were likely influencing this individual variation [53, 57, 187–190]. In particular, systemic inflammation, associated with co-morbid states and hypoalbuminemia as surrogate marker, may result in structural changes such as neoangiogenesis, leading to modifications in transport. Of note, one should remain cautious in interpreting albumin levels in relation to solute transport: rather than being a marker of inflammation, lower systemic albumin concentration may reflect the fluid overload encountered in high transporters. The latter may also lose albumin in the dialysate. At any rate, the complexity of the relationships between UF and small solute transport [190], as well as the fact that independent clinical variables account for only ˜20% of the individual variability of solute transport [189], strongly suggest that factors that are not routinely measured do play a role in the baseline functional characteristics of the peritoneum.
The small solute transport rate depends mainly on the amount of perfused capillaries within the peritoneum, the blood flow, and the physical area of membrane contact with the dialysate. Accordingly, an increased EPSA is associated with higher transport of small solutes and, eventually, UF failure. As discussed above, growth factors such as VEGF and TGF-β1, cytokines such as IL-6, together with the release of NO by endothelial cells, play a central role in angiogenesis and fibrosis changes in the peritoneal membrane exposed to PD. Furthermore, polymorphisms within the regulatory region of the genes coding for VEGF, IL-6, and eNOS modify the amount of gene expression in vitro, and they have been associated with diseases – such as diabetic retinopathy and/or nephropathy – that are potentially relevant for the peritoneal membrane. Based on the finding that most of the variability in peritoneal transport remains unexplained by clinical factors, recent studies have investigated whether genetic variants (polymorphisms) could influence transport properties at baseline [191].
Wong et al. were the first to observe a positive association between a variable number of tandem repeats in the intron 4 of ENOS (ENOS4a/b) and transport properties in incident PD patients [192]. Of note, the ENOS genotype remained an independent predictor for peritoneal transport after adjustment for clinical parameters (gender, age, diabetes) by multivariate analysis. Szeto et al. investigated whether two promoter polymorphisms of VEGF may influence peritoneal transport in a series of 135 incident PD patients [193]. There was no association between VEGF polymorphisms and transport at baseline. However, in a subanalysis restricted to 83 patients for which a 12-month follow-up was available, there was a positive association between the two variants and the longitudinal changes in D/P creatinine and the VEGF mRNA level in a subset of dialysate effluent samples. Recently, Gillerot et al. investigated the respective contributions of common polymorphisms in ENOS, VEGF, and IL6 to the small solute transport rate in a multicentric series of 152 incident PD patients [189]. Their studies identified a common polymorphism of IL6 as an independent predictor of peritoneal transport, together with co-morbidity and serum albumin level. The effect was reflected by significant changes in IL-6 at the mRNA and protein levels, pointing to the role of local and systemic inflammation in regulating small solute transport [189] (Fig. 27.8 ).

Potential influence of genetic factors on alterations in peritoneal transport during inflammation. A biologically active variant in the promoter of the IL6 gene regulates the systemic and local expression of the pro-inflammatory cytokine interleukin-6 (IL-6). The upregulation of IL-6 contributes, via local and systemic effects, to the increase in the transport of small solute and the loss of albumin in the dialysate, and to the general state of malnutrition-inflammation. Modified from [189]
Taken together, these studies support the hypothesis that inherited genetic variants might regulate specific mediators and, in association with clinical factors, affect the transport properties of the peritoneal membrane. Conflicting results in association studies are common and the methodological limitations have been clearly delineated [194], leading to specific guidelines [195]. Confirming the strength of the positive associations and deciphering the genetic influence on peritoneal transport will require well-designed, adequately powered studies, in different populations and different settings, as well as a detailed assessment of the biological role of the polymorphisms. In addition to the candidate gene approach, future studies will probably benefit from large genome-wide association studies currently performed in diabetic nephropathy or retinopathy.
Future Therapeutic Strategies and Perspectives for Membrane Preservation in PD
The introduction of glucose-free PD solutions including icodextrin, glycerol, and amino-acids solutions as well as their potential clinical benefits have the clear goal of reducing the deleterious effect of long-term exposure to glucose and its associated deleterious local and systemic effects [89]. The parallel development of two-chamber or dual-chamber bags has allowed a dramatic reduction in dialysate GPD concentrations by reducing GDP formation during heat sterilization [196]. The two-chamber system separates concentrated glucose from other components, allowing sterilization of glucose at a very low pH. Mixing of the two compartments results in a solution characterized by low levels of GDPs and also allows the production of a solution with a more physiologic pH [197–202]. When tested in vitro, such biocompatible dialysates have been shown to reduce AGE formation [203], decrease acute vasoactive effects on the peritoneal circulation [12, 204, 205], and improve ex vivo peritoneal macrophage function [202]. Two short-term randomized trials using such solutions have shown no significant modifications of peritoneal transport parameters but an increase in dialysate CA125 (taken as a marker of mesothelial cell mass) and a decrease in dialysate hyaluronan (taken as a marker of peritoneal inflammation) [201, 206].
As discussed earlier, the development of animal models has provided a rationale for other therapeutic strategies against structural and functional alterations of the peritoneum. Inhibition of the AGE formation [97] with compounds such as aminoguanidine, OPB-9195, or pyridoxamine are being currently evaluated, as well as the possibility of detoxifying RCOs by the glyoxalase pathway [97, 207]. Inhibition of the L-arginine:NO pathway, for instance, with L-arginine analogues has been shown to dramatically improve UF in rat model of acute peritonitis [94]. The potential usefulness of NOS inhibitors in PD patients will have to take into account the current lack of specificity of most inhibitors, as well as the potential problems inherent with inhibition of a ubiquitous mediator such as NO [208]. Inhibition of the oxidative stress by N-acetylcysteine or angiotensin II receptor blocker (losartan) was shown to be effective in preserving membrane structure and function in a rat model [152]. The antisclerosis and anti-inflammatory potential of atorvastatin, lisinopril, and valsartan has been suggested in rat models [209–212], with further demonstration of the effect of ACE-inhibitors and angiotensin II receptor blockers in cultured HPMC [213, 214]. The anticalcic agent diltiazem has been shown to decrease collagen synthesis and IL-1β-induced production of TGF-β1 in HPMC [215].
Modulation of angiogenesis with agents that inhibit endothelial cell growth, adhesion, or migration, or interfere with growth factors such as VEGF and bFGF and their receptors have been proposed. Margetts et al. first demonstrated the validity of the antiangiogenic approach by showing improvement of structural and functional parameters in a chronic infusion rat model of PD treated with adenovirus-mediated gene transfer of angiostatin [91]. However, it should be kept in mind that i) different molecular pathways are involved in various types of angiogenesis; ii) angiogenic growth factors may participate in other physiological processes; and iii) there is little information on safety, long-term side-effects, and impact of antiangiogenic therapy on processes such as healing [216]. Elegant studies showed the potential of the antiangiogenic compounds TNP-470 [217] or endostatin [218] to reduce peritoneal sclerosis – together with angiogenesis – in the chlorhexidine gluconate mouse EPS model. The pharmacologic induction of AQP1 may also provide a target for manipulating water permeability and treating some cases of UF failure [96]. Finally, one may predict that a better knowledge of the genetic determinants potentially involved in the individual variability in peritoneal transport at the onset of PD may pave the way for individualized therapeutic approaches.
Conclusions and Perspectives
This chapter has attempted to describe what is currently known about peritoneal membrane structure and functional change during peritoneal dialysis and to provide insight into the mechanisms that drive these alterations. This has borrowed heavily on both clinical data as well as results from developing work an extensive array of experimental models. Our knowledge of the peritoneal membrane and its alterations during (and preceding) dialysis have increased tremendously over the past 5 years. This has undoubtedly increased our understanding of potential membrane preservation strategies but also of the natural history of changes in the membrane. Hopefully, this, combined with the increasing number of treatment options available, should make it possible to enhance treatment quality, reduce the incidence of infection and membrane failure, and improve patient and technique survival on this treatment modality.
References
Burkart J, Piraino B, Kaldas H, et al. Why is the evidence favoring hemodialysis over peritoneal dialysis misleading? Semin Dial 2007; 20 (3): 200–202.
Vonesh EF, Snyder JJ, Foley RN, Collins AJ. Mortality studies comparing peritoneal dialysis and hemodialysis: what do they tell us? Kidney Int 2006; 70 (suppl. 103): S3–S11.
Giannattasio M, Buemi M, Caputo F, Viglino G, Verrina E. Can peritoneal dialysis be used as a long term therapy for end stage renal disease? Int Urol Nephrol 2003; 35 (4): 569–577.
Maiorca R, Cancarini G. Thirty years of progress in peritoneal dialysis. J Nephrol 1999; 12 (suppl. 2): S92–S99.
Engel B, Davies SJ. Achieving euvolemia in peritoneal dialysis. Perit Dial Int 2007; 27 (5): 514–517.
Davies SJ, Brown EA, Frandsen NE, et al. Longitudinal membrane function in functionally anuric patients treated with APD: data from EAPOS on the effects of glucose and icodextrin prescription. Kidney Int 2005; 67 (4): 1609–1615.
Davies SJ. Mitigating peritoneal membrane characteristics in modern peritoneal dialysis therapy. Kidney Int 2006; 70 (suppl. 103): S76–S83.
Davies SJ, Phillips L, Griffiths AM, Russell LH, Naish PF, Russell GI. What really happens to people on long-term peritoneal dialysis? Kidney Int 1998; 54 (6): 2207–2217.
ter Wee PM, van Ittersum FJ. The new peritoneal dialysis solutions: friends only, or foes in part? Nat Clin Pract Nephrol 2007; 3 (11): 604–612.
McIntyre CW. Update on peritoneal dialysis solutions. Kidney Int 2007; 71 (6): 486–490.
Diaz-Buxo JA. Clinical use of and experience with neutral-pH solutions. Adv Perit Dial 2006; 22: 167–170.
Mortier S, Faict D, Gericke M, Lameire N, De Vriese A. Effects of new peritoneal dialysis solutions on leukocyte recruitment in the rat peritoneal membrane. Nephron 2005; 101 (4): e139–e145.
Devuyst O, Topley N, Williams JD. Morphological and functional changes in the dialysed peritoneal cavity: impact of more biocompatible solutions. Nephrol Dial Transplant 2002; 17 (suppl. 3): 12–15.
Combet S, Ferrier ML, Van Landschoot M, et al. Chronic uremia induces permeability changes, increased nitric oxide synthase expression, and structural modifications in the peritoneum. J Am Soc Nephrol 2001; 12 (10): 2146–2157.
Honda K, Nitta K, Horita H, Yumura W, Nihei H. Morphological changes in the peritoneal vasculature of patients on CAPD with ultrafiltration failure. Nephron 1996; 72: 171–176.
Honda K, Nitta K, Horita S, et al. Accumulation of advanced glycation end products in the peritoneal vasculature of continuous ambulatory peritoneal dialysis patients with low ultra-filtration. Nephrol Dial Transplant 1999; 14 (6): 1541–1549.
Williams JD, Craig KJ, Topley N, et al. Morphologic changes in the peritoneal membrane of patients with renal disease. J Am Soc Nephrol 2002; 13 (2): 470–479.
Topley N. Biocompatibility of peritoneal dialysis solutions and host defense. Adv Ren Replace Ther 1996; 3 (4): 309–311.
Topley N. The host's initial response to peritoneal infection: the pivotal role of the mesothelial cell. Perit Dial Int 1995; 15 (2): 116–117.
Topley N. The cytokine network controlling peritoneal inflammation. Perit Dial Int 1995; 15 (7) :S35–S39; discussion S39–S40.
McLoughlin RM, Witowski J, Robson RL, et al. Interplay between IFN-gamma and IL-6 signaling governs neutrophil trafficking and apoptosis during acute inflammation. J Clin Invest 2003; 112 (4): 598–607.
Hurst SM, Wilkinson TS, McLoughlin RM, et al. IL-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001; 14 (6): 705–714.
Leung JC, Chan LY, Tang SC, Chu KM, Lai KN. Leptin induces TGF-beta synthesis through functional leptin receptor expressed by human peritoneal mesothelial cell. Kidney Int 2006; 69 (11): 2078–2086.
Yanez-Mo M, Lara-Pezzi E, Selgas R, et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N Engl J Med 2003; 348 (5): 403–413.
Selgas R, Bajo A, Jimenez-Heffernan JA, et al. Epithelial-to-mesenchymal transition of the mesothelial cell – its role in the response of the peritoneum to dialysis. Nephrol Dial Transplant 2006; 21 (suppl. 2): ii2–ii7.
Aroeira LS, Aguilera A, Sanchez-Tomero JA, et al. Epithelial to mesenchymal transition and peritoneal membrane failure in peritoneal dialysis patients: pathologic significance and potential therapeutic interventions. J Am Soc Nephrol 2007; 18 (7): 2004–2013.
Margetts PJ, Bonniaud P, Liu L, et al. Transient overexpression of TGF-{beta}1 induces epithelial mesenchymal transition in the rodent peritoneum. J Am Soc Nephrol 2005; 16 (2): 425–436.
De Vriese AS, Tilton RG, Mortier S, Lameire NH. Myofibroblast transdifferentiation of mesothelial cells is mediated by RAGE and contributes to peritoneal fibrosis in uraemia. Nephrol Dial Transplant 2006; 21 (9): 2549–2555.
Dobbie JW, Lloyd JK, Gall CA. Categorization of ultrastructural changes in peritoneal mesothelium, stroma and blood vessels in uremia and CAPD patients. Adv Perit Dial 1990; 6: 3–12.
Dobbie JW, Anderson JD, Hind C. Long-term effects of peritoneal dialysis on peritoneal morphology. Perit Dial Int 1994; 14 (suppl. 3): S16–S20.
Dobbie JW. Ultrastructure and pathology of the peritoneum in peritoneal dialysis. In: Gokal R, Nolph KD, eds. The Textbook of Peritoneal Dialysis. Dordrecht: Kluwer Academic Publishers; 1994: 17–44.
Dobbie JW. The role of peritoneal biopsy in clinical and experimental peritoneal dialysis. Perit Dial Int 1993; 13 (suppl. 2): S23–S26.
Dobbie JW. The biopsy registry as a quality control mechanism in the development of continuous ambulatory peritoneal dialysis. Perit Dial Int 1993; 13 (suppl. 2): S583–S584.
Dobbie JW. Peritoneal ultrastructure and changes with continuous ambulatory peritoneal dialysis. Perit Dial Int 1993; 13 (suppl. 2): S585–S587.
Di Paolo N, Sacchi G, De Mia M, et al. Morphology of the peritoneal membrane during continuous ambulatory peritoneal dialysis. Nephron 1986; 44: 204–211.
Di Paolo N, Sacchi G, Buoncristiani V. The morphology of the human peritoneum in CAPD patients. In: Maher J, ed. Frontiers in Peritoneal Dialysis. New York: Field Rich; 1985: 11–19.
Di Paolo N, Sacchi G. Peritoneal vascular changes in continuous ambulatory peritoneal dialysis (CAPD): an in vivo model for the study of diabetic microangiopathy. Perit Dial Int 1989; 9 (41): 41–45.
Williams JD, Craig KJ, von Ruhland C, Topley N, Williams GT. The natural course of peritoneal membrane biology during peritoneal dialysis. Kidney Int 2003; 64 (suppl. 88): S43–S49.
Williams JD, Craig KJ, Topley N, Williams GT. Peritoneal dialysis: Changes to the structure of the peritoneal membrane and potential for biocompatible solutions. Kidney Int 2003; 63 (suppl. 84): 158–161.
Rubin J, Herrara GA, Collins D. An autopsy study of the peritoneal cavity from patients on continuous ambulatory peritoneal dialysis. Am J Kidney Dis 1991; 17: 97–102.
Verger C, Brunschvicg O, Charpentier YL, Lavergne A, Vantelon J. Structural and ultrastructural peritoneal membrane changes and permeability alterations during continuous ambulatory peritoneal dialysis. Proc Eur Dial Transplant Assoc 1981; 18: 199–205.
Verger C, Luger A, Moore HL, Nolph KD. Acute changes in peritoneal morphology and transport properties with infectious peritonitis and mechanical injury. Kidney Int 1983; 23: 823–831.
Gotloib L, Shostack A. Ultrastructural morphology of the peritoneum: new findings and speculations on transfer of solutes and water during peritoneal dialysis. Perit Dial Bull 1987; 7: 119–129.
Gotloib L, Shostak A. The functional anatomy of the peritoneum as a dialysing membrane. In: Twardowski ZJ, Nolph KD, Khanna R, eds. Peritoneal Dialysis. New York: Churchill Livingstone; 1990: 1–27.
Mateijsen MA, van der Wal AC, Hendriks PM, et al. Vascular and interstitial changes in the peritoneum of CAPD patients with peritoneal sclerosis. Perit Dial Int 1999; 19 (6): 517–525.
Zareie M, Keuning ED, ter Wee PM, Beelen RH, van den Born J. Peritoneal dialysis fluid-induced changes of the peritoneal membrane are reversible after peritoneal rest in rats. Nephrol Dial Transplant 2005; 20 (1): 189–193.
Zareie M, De Vriese AS, Hekking LH, et al. Immunopathological changes in a uraemic rat model for peritoneal dialysis. Nephrol Dial Transplant 2005; 20 (7): 1350–1361.
Selgas R, Bajo MA, Castro MJ, et al. Risk factors responsible for ultrafiltration failure in early stages of peritoneal dialysis. Perit Dial Int 2000; 20 (6): 631–636.
Davies SJ, Phillips L, Naish PF, Russell GI. Peritoneal glucose exposure and changes in membrane solute transport with time on peritoneal dialysis. J Am Soc Nephrol 2001; 12 (5): 1046–1051.
Struijk DG, Krediet RT, Koomen GC, Boeschoten EW, Hoek FJ, Arisz L. A prospective study of peritoneal transport in CAPD patients. Kidney Int 1994; 45 (6): 1739–1744.
Selgas R, Fernandez-Reyes MJ, Bosque E, et al. Functional longevity of the human peritoneum: how long is continuous peritoneal dialysis possible? Results of a prospective medium long-term study. Am J Kidney Dis 1994; 23 (1): 64–73.
Struijk DG, Krediet RT, Koomen GC, et al. Functional characteristics of the peritoneal membrane in long-term continuous ambulatory peritoneal dialysis. Nephron 1991; 59 (2): 213–220.
Davies SJ. Longitudinal relationship between solute transport and ultrafiltration capacity in peritoneal dialysis patients. Kidney Int 2004; 66 (6): 2437–2445.
Fussholler A, zur Nieden S, Grabensee B, Plum J. Peritoneal fluid and solute transport: influence of treatment time, peritoneal dialysis modality, and peritonitis incidence. J Am Soc Nephrol 2002; 13 (4): 1055–1060.
Clerbaux G, Francart J, Wallemacq P, Robert A, Goffin E. Evaluation of peritoneal transport properties at onset of peritoneal dialysis and longitudinal follow-up. Nephrol Dial Transplant 2006; 21 (4): 1032–1039.
Brimble KS, Walker M, Margetts PJ, Kundhal KK, Rabbat CG. Meta-analysis: peritoneal membrane transport, mortality, and technique failure in peritoneal dialysis. J Am Soc Nephrol 2006; 17 (9): 2591–2598.
Churchill DN, Thorpe KE, Nolph KD, Keshaviah PR, Oreopoulos DG, Page D. Increased peritoneal membrane transport is associated with decreased patient and technique survival for continuous peritoneal dialysis patients. The Canada-USA (CANUSA) Peritoneal Dialysis Study Group. J Am Soc Nephrol 1998; 9 (7): 1285–1292.
Konings CJ, Kooman JP, Schonck M, et al. Assessment of fluid status in peritoneal dialysis patients. Perit Dial Int 2002; 22 (6): 683–692.
Ates K, Nergizoglu G, Keven K, et al. Effect of fluid and sodium removal on mortality in peritoneal dialysis patients. Kidney Int 2001; 60 (2): 767–776.
Davies SJ, Bryan J, Phillips L, Russell GI. Longitudinal changes in peritoneal kinetics: the effects of peritoneal dialysis and peritonitis. Nephrol Dial Transplant 1996; 11: 498–506.
Davies SJ, Brown B, Bryan J, Russell GI. Clinical evaluation of the peritoneal equilibration test: a population- based study. Nephrol Dial Transplant 1993; 8 (1): 64–70.
Heimbürger O, Waniewski J, Werynski A, Tranaeus A, Lindholm B. Peritoneal transport in CAPD patients with permanent loss of ultrafiltration capacity. Kidney Int 1990; 38: 495–506.
Kawaguchi Y, Hasegawa T, Nakayama M, Kubo H, Shigematu T. Issues affecting the longevity of the continuous peritoneal dialysis therapy. Kidney Int 1997; 62: S105–S107.
Parikova A, Smit W, Struijk DG, Krediet RT. Analysis of fluid transport pathways and their determinants in peritoneal dialysis patients with ultrafiltration failure. Kidney Int 2006; 70 (11): 1988–1994.
Smit W, Struijk DG, Ho-Dac-Pannekeet MM, Krediet RT. Quantification of free water transport in peritoneal dialysis. Kidney Int 2004; 66 (2): 849–854.
Monquil MC, Imholz AL, Struijk DG, Krediet RT. Does impaired transcellular water transport contribute to net ultrafiltration failure during CAPD? Perit Dial Int 1995; 15 (1): 42–48.
Ho-dac-Panekeet MM. Assesment of Peritoneal Permeability and Mesothelial Cell Mass in Peritoneal Dialysis Patients : Effects of Non-Glucose Solutions [Ph.D.]. Amsterdam: University of Amsterdam; 1998.
Smit W, Schouten N, van den Berg N, Langedijk MJ, Struijk DG, Krediet RT. Analysis of the prevalence and causes of ultrafiltration failure during long-term peritoneal dialysis: a cross-sectional study. Perit Dial Int 2004; 24 (6): 562–570.
Smit W, Parikova A, Struijk DG, Krediet RT. The difference in causes of early and late ultrafiltration failure in peritoneal dialysis. Perit Dial Int 2005; 25(suppl. 3): S41–S45.
Combet S, Van Landschoot M, Moulin P, et al. Regulation of aquaporin-1 and nitric oxide synthase isoforms in a rat model of acute peritonitis. J Am Soc Nephrol 1999; 10 (10): 2185–2196.
Devuyst O, Nielsen S, Cosyns JP, et al. Aquaporin-1 and endothelial nitric oxide synthase expression in capillary endothelia of human peritoneum. Am J Physiol 1998; 275 (1 Pt 2): H234–H242.
Flessner MF. The transport barrier in intraperitoneal therapy. Am J Physiol 2005; 288 (3): F433–F442.
Coester AM, Struijk DG, Smit W, de Waart DR, Krediet RT. The cellular contribution to effluent potassium and its relation with free water transport during peritoneal dialysis. Nephrol Dial Transplant 2007; 22 (12): 3593–3600.
La Milia V, Di Filippo S, Crepaldi M, et al. Mini-peritoneal equilibration test: a simple and fast method to assess free water and small solute transport across the peritoneal membrane. Kidney Int 2005; 68 (2): 840–846.
Davies SJ, Phillips L, Naish PF, Russell GI. Quantifying comorbidity in peritoneal dialysis patients and its relationship to other predictors of survival. Nephrol Dial Transplant 2002; 17 (6): 1085–1092.
Krediet RT, Struijk DG, Koomen GC, et al. Peritoneal transport of macromolecules in patients on CAPD. Contrib Nephrol 1991; 89: 161–174.
Ho-dac-Pannekeet MM, Koopmans JG, Struijk DG, Krediet RT. Restriction coefficients of low molecular weight solutes and macromolecules during peritoneal dialysis. Adv Perit Dial 1997; 13: 72–76.
Haraldsson B. Assessing the peritoneal dialysis capacities of individual patients. Kidney Int 1995; 47 (4): 1187–1198.
Johansson AC, Haraldsson B. Physiological properties of the peritoneum in an adult peritoneal dialysis population over a three-year period. Perit Dial Int 2006; 26 (4): 482–489.
Heaf JG, Sarac S, Afzal S. A high peritoneal large pore fluid flux causes hypoalbuminaemia and is a risk factor for death in peritoneal dialysis patients. Nephrol Dial Transplant 2005; 20 (10): 2194–2201.
Brown EA, Davies SJ, Rutherford P, et al. Survival of functionally anuric patients on automated peritoneal dialysis: the European APD Outcome Study. J Am Soc Nephrol 2003; 14 (11): 2948–2957.
Davies SJ, Brown EA. EAPOS: what have we learned? Perit Dial Int 2007; 27 (2): 131–135.
Davies SJ, Brown EA, Reigel W, et al. What is the link between poor ultrafiltration and increased mortality in anuric patients on automated peritoneal dialysis? Analysis of data from EAPOS. Perit Dial Int 2006; 26 (4): 458–465.
Krediet RT. The peritoneal membrane in chronic peritoneal dialysis. Kidney Int 1999; 55 (1): 341–356.
Rippe B, Simonsen O, Stelin G. Clinical implications of a three-pore model of peritoneal transport. Adv Perit Dial 1991; 7: 3–9.
Rippe B, Stelin G, Haraldsson B. Computer simulations of peritoneal fluid transport in CAPD. Kidney Int 1991; 40 (2): 315–325.
Devuyst O, van Ypersele de Strihou C. Nitric oxide, advanced glycation end products, and uremia. Kidney Int 2000; 58 (4): 1814–1815.
Ni J, Verbavatz JM, Rippe A, et al. Aquaporin-1 plays an essential role in water permeability and ultrafiltration during peritoneal dialysis. Kidney Int 2006; 69 (9): 1518–1525.
Krediet RT, van Westrhenen R, Zweers MM, Struijk DG. Clinical advantages of new peritoneal dialysis solutions. Nephrol Dial Transplant 2002; 17 (suppl. 3): 16–18.
Combet S, Miyata T, Moulin P, Pouthier D, Goffin E, Devuyst O. Vascular proliferation and enhanced expression of endothelial nitric oxide synthase in human peritoneum exposed to long-term peritoneal dialysis. J Am Soc Nephrol 2000; 11 (4): 717–728.
Margetts PJ, Gyorffy S, Kolb M, et al. Antiangiogenic and antifibrotic gene therapy in a chronic infusion model of peritoneal dialysis in rats. J Am Soc Nephrol 2002; 13 (3): 721–728.
Ni J, Cnops Y, McLoughlin RM, Topley N, Devuyst O. Inhibition of nitric oxide synthase reverses permeability changes in a mouse model of acute peritonitis. Perit Dial Int 2005; 25 (suppl. 3): S11–S14.
Ni J, Moulin P, Gianello P, Feron O, Balligand JL, Devuyst O. Mice that lack endothelial nitric oxide synthase are protected against functional and structural modifications induced by acute peritonitis. J Am Soc Nephrol 2003; 14 (12): 3205–3216.
Ferrier ML, Combet S, van Landschoot M, et al. Inhibition of nitric oxide synthase reverses changes in peritoneal permeability in a rat model of acute peritonitis. Kidney Int 2001; 60 (6): 2343–2350.
De Vriese AS, Tilton RG, Stephan CC, Lameire NH. Vascular endothelial growth factor is essential for hyperglycemia- induced structural and functional alterations of the peritoneal membrane. J Am Soc Nephrol 2001; 12 (8): 1734–1741.
Stoenoiu MS, Ni J, Verkaeren C, et al. Corticosteroids induce expression of aquaporin-1 and increase transcellular water transport in rat peritoneum. J Am Soc Nephrol 2003; 14 (3): 555–565.
Kakuta T, Tanaka R, Satoh Y, et al. Pyridoxamine improves functional, structural, and biochemical alterations of peritoneal membranes in uremic peritoneal dialysis rats. Kidney Int 2005; 68 (3): 1326–1336.
Schwenger V, Morath C, Salava A, et al. Damage to the peritoneal membrane by glucose degradation products is mediated by the receptor for advanced glycation end-products. J Am Soc Nephrol 2006; 17 (1): 199–207.
Margetts PJ, Kolb M, Galt T, Hoff CM, Shockley TR, Gauldie J. Gene transfer of transforming growth factor-beta1 to the rat peritoneum: effects on membrane function. J Am Soc Nephrol 2001; 12 (10): 2029–2039.
De Vriese AS, Flyvbjerg A, Mortier S, Tilton RG, Lameire NH. Inhibition of the interaction of AGE-RAGE prevents hyperglycemia-induced fibrosis of the peritoneal membrane. J Am Soc Nephrol 2003; 14 (8): 2109–2118.
Duwe AK, Vas SI, Weatherhead JW. Effects of the composition of peritoneal dialysis fluid on chemiluminescence, phagocytosis and bactericidal activity in vitro. Infect Immun 1981; 33 (130): 130–135.
Topley N, Alobaidi HM, Davies M, Coles GA, Williams JD, Lloyd D. The effect of dialysate on peritoneal phagocyte oxidative metabolism. Kidney Int 1988; 34 (3): 404–411.
Topley N. The host's initial response to peritoneal infection: the pivotal role of the mesothelial cell. Perit Dial Int 1995; 15 (2): 116–117.
Topley N, Jörres A, Mackenzie R, Coles GA, Williams JD. Interactions of macrophages and mesothelial cells in peritoneal host defence. Nieren-und Hochdruckkrankheiten 1994; 23: S88–S91.
Topley N. Membrane longevity in peritoneal dialysis: impact of infection and bio-incompatible solutions. Adv Ren Replace Ther 1998; 5 (3): 179–184.
Jorres A, Witowski J. Lessons from basic research for PD treatment. Perit Dial Int 2005; 25 (suppl. 3): S35–S38.
Topley N. Animal models in peritoneal dialysis: more questions than answers? Perit Dial Int 2005; 25 (1): 33–34.
Lameire N, Topley N. Animal models of peritoneal dialysis. Nephrol Dial Transplant 2001; 16 (3): 647.
Mortier S, Lameire NH, De Vriese AS. Animal models in peritoneal dialysis research: a need for consensus. Perit Dial Int 2005; 25 (1): 16–24.
Lameire N, Van Biesen W, Mortier S, De Vriese A. What did we learn from animal models in peritoneal dialysis? Contrib Nephrol 2006; 150: 70–76.
ter Wee PM, Beelen RH, van den Born J. The application of animal models to study the biocompatibility of bicarbonate-buffered peritoneal dialysis solutions. Kidney Int 2003; 64 (suppl. 88): S75–S83.
Fabbrini P, Zareie M, ter Wee PM, Keuning ED, Beelen RH, van den Born J. Peritoneal exposure model in the rat as a tool to unravel bio(in)compatibility of PDF. Nephrol Dial Transplant 2006; 21 (suppl. 2): ii8–ii11.
Nishino T, Ni J, Devuyst O. Transgenic mouse models. Perit Dial Int 2007; 27 (6): 625–633.
Hekking LH, Aalders MC, Van Gelderop E, et al. Effect of peritoneal dialysis fluid measured in vivo in a rat-model of continuous peritoneal dialysis. Adv Perit Dial 1998; 14: 14–18.
Wieczorowska-Tobis K, Korybalska K, Polubinska A, et al. Long-term effects of glycylglycine peritoneal dialysis solution with neutral pH on peritoneum in rats. Adv Perit Dial 1997; 13: 42–46.
Hekking LH, Huijsmans A, Van Gelderop E, et al. Effect of PD fluid instillation on the peritonitis-induced influx and bacterial clearing capacity of peritoneal cells. Nephrol Dial Transplant 2001; 16 (3): 679–682.
Hekking LH, Zareie M, Driesprong BA, et al. Better preservation of peritoneal morphologic features and defense in rats after long-term exposure to a bicarbonate/lactate-buffered solution. J Am Soc Nephrol 2001; 12 (12): 2775–2786.
Kim YL, Cho S, Kim JC, et al. Effect in a rat model of heparinized peritoneal dialysis catheters on bacterial colonization and the healing of the exit site. Perit Dial Int 2001; 21 (suppl. 3): S357–S358.
Mortier S, Faict D, Lameire NH, De Vriese AS. Benefits of switching from a conventional to a low-GDP bicarbonate/lactate-buffered dialysis solution in a rat model. Kidney Int 2005; 67 (4): 1559–1565.
Zweers MM, Splint LJ, Krediet RT, Struijk DG. Ultrastructure of basement membranes of peritoneal capillaries in a chronic peritoneal infusion model in the rat. Nephrol Dial Transplant 2001; 16 (3): 651–654.
Lameire N, Van Biesen W, Van Landschoot M, et al. Experimental models in peritoneal dialysis: a European experience. Kidney Int 1998; 54 (6): 2194–2206.
Gotloib L, Crassweller P, Rodella H, et al. Experimental model for studies of continuous peritoneal dialysis in uremic rabbits. Nephron 1982; 31 (3): 254–259.
Van Biesen W, Vanholder R, Lameire N. Animal models in peritoneal dialysis: a story of kangaroos and ostriches. Perit Dial Int 2006; 26 (5): 571–573.
Breborowicz A, Rodela H, Oreopoulos DG. Toxicity of osmotic solutes on human mesothelial cells in vitro. Kidney Int 1992; 41: 1280–1285.
Nakayama M, Kawaguchi Y, Yamada K, et al. Immunohistochemical detection of advanced glycosylation end-products in the peritoneum and its possible pathophysiological role in CAPD. Kidney Int 1997; 51 (1): 182–186.
Thornalley PJ. Measurement of protein glycation, glycated peptides, and glycation free adducts. Perit Dial Int 2005; 25 (6): 522–533.
Thornalley PJ. Glycation free adduct accumulation in renal disease: the new AGE. Pediatr Nephrol 2005; 20 (11): 1515–1522.
Wong TY, Phillips AO, Witowski J, Topley N. Glucose-mediated induction of TGF-beta1 and MCP-1 in mesothelial cells in vitro is osmolality and polyol pathway dependent. Kidney Int 2003; 63 (4): 1404–1416.
Devuyst O, Ni J, Verbavatz JM. Aquaporin-1 in the peritoneal membrane: implications for peritoneal dialysis and endothelial cell function. Biol Cell 2005; 97 (9): 667–673.
Carlsson O, Nielsen S, Zakaria el R, Rippe B. In vivo inhibition of transcellular water channels (aquaporin-1) during acute peritoneal dialysis in rats. Am J Physiol 1996; 271 (6 Pt 2): H2254–H2262.
Devuyst O, Topley N. Peritoneal membrane transport: driving under the influence. Perit Dial Int 2006; 26 (1): 35–37.
Ni J, Cnops Y, Debaix H, Boisde I, Verbavatz JM, Devuyst O. Functional and molecular characterization of a peritoneal dialysis model in the C57BL/6 J mouse. Kidney Int 2005; 67 (5): 2021–2031.
Miyata T, van Ypersele de Strihou C, Kurokawa K, Baynes JW. Alterations in nonenzymatic biochemistry in uremia: origin and significance of “carbonyl stress” in long-term uremic complications. Kidney Int 1999; 55 (2): 389–399.
Erixon M, Linden T, Kjellstrand P, et al. PD fluids contain high concentrations of cytotoxic GDPs directly after sterilization. Perit Dial Int 2004; 24 (4): 392–398.
Miyata T, Horie K, Ueda Y, et al. Advanced glycation and lipidoxidation of the peritoneal membrane: respective roles of serum and peritoneal fluid reactive carbonyl compounds. Kidney Int 2000; 58 (1): 425–435.
Inagi R, Miyata T, Yamamoto T, et al. Glucose degradation product methylglyoxal enhances the production of vascular endothelial growth factor in peritoneal cells: role in the functional and morphological alterations of peritoneal membranes in peritoneal dialysis. FEBS Lett 1999; 463 (3): 260–264.
Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J 1999; 344 (Pt 1): 109–116.
Morgan LW, Wieslander A, Davies M, et al. Glucose degradation products (GDP) retard remesothelialization independently of D-glucose concentration. Kidney Int 2003; 64 (5): 1854–1866.
Witowski J, Wisniewska J, Korybalska K, et al. Prolonged exposure to glucose degradation products impairs viability and function of human peritoneal mesothelial cells. J Am Soc Nephrol 2001; 12 (11): 2434–2441.
Devuyst O. New insights in the molecular mechanisms regulating peritoneal permeability. Nephrol Dial Transplant 2002; 17 (4): 548–551.
Witowski J, Korybalska K, Wisniewska J, et al. Effect of glucose degradation products on human peritoneal mesothelial cell function. J Am Soc Nephrol 2000; 11 (4): 729–739.
Lai KN, Leung JC, Chan LY, et al. Differential expression of receptors for advanced glycation end-products in peritoneal mesothelial cells exposed to glucose degradation products. Clin Exp Immunol 2004; 138 (3): 466–475.
Boulanger E, Grossin N, Wautier MP, Taamma R, Wautier JL. Mesothelial RAGE activation by AGEs enhances VEGF release and potentiates capillary tube formation. Kidney Int 2007; 71 (2): 126–133.
Bredt DS, Snyder SH. Nitric oxide: a physiologic messenger molecule. Annu Rev Biochem 1994; 63: 175–195.
Bikfalvi A, Klein S, Pintucci G, Rifkin DB. Biological roles of fibroblast growth factor-2. Endocr Rev 1997; 18 (1): 26–45.
Pecoits-Filho R, Araujo MR, Lindholm B, et al. Plasma and dialysate IL-6 and VEGF concentrations are associated with high peritoneal solute transport rate. Nephrol Dial Transplant 2002; 17 (8): 1480–1486.
Kubes P. Nitric oxide affects microvascular permeability in the intact and inflamed vasculature. Microcirculation 1995; 2 (3): 235–244.
Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J 1999; 13 (1): 9–22.
Fukumura D, Gohongi T, Kadambi A, et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci USA 2001; 98 (5): 2604–2609.
Hood JD, Meininger CJ, Ziche M, Granger HJ. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am J Physiol 1998; 274 (3 Pt 2): H1054–H1058.
Lee HB, Yu MR, Song JS, Ha H. Reactive oxygen species amplify protein kinase C signaling in high glucose-induced fibronectin expression by human peritoneal mesothelial cells. Kidney Int 2004; 65 (4): 1170–1179.
Noh H, Kim JS, Han KH, et al. Oxidative stress during peritoneal dialysis: implications in functional and structural changes in the membrane. Kidney Int 2006; 69 (11): 2022–2028.
Rubin J, Rust P, Brown P, Popovich RP, Nolph KD. A comparison of peritoneal transport in patients with psoriasis and uremia. Nephron 1981; 29 (3–4): 185–189.
Lamb EJ, Worrall J, Buhler R, Harwood S, Cattell WR, Dawnay AB. Effect of diabetes and peritonitis on the peritoneal equilibration test. Kidney Int 1995; 47 (6): 1760–1767.
Topley N, Liberek T, Davenport A, Li FK, Fear H, Williams JD. Activation of inflammation and leukocyte recruitment into the peritoneal cavity. Kidney Int (suppl. 56)1996; S17–S21.
Topley N, Mackenzie RK, Williams JD. Macrophages and mesothelial cells in bacterial peritonitis. Immunobiology 1996; 195 (4–5): 563–573.
Topley N, Mackenzie R, Jörres A, Coles GA, Williams JD. Cytokine networks in continuous ambulatory peritoneal dialysis: interactions of resident cells during inflammation in the peritoneal cavity. Perit Dial Int 1993; 13 (suppl. 2): S282–S285.
Li FK, Davenport A, Robson RL, et al. Leukocyte migration across human peritoneal mesothelial cells is dependent on directed chemokine secretion and ICAM-1 expression. Kidney Int 1998; 54 (6): 2170–2183.
Robson RL, McLoughlin RM, Witowski J, et al. Differential regulation of chemokine production in human peritoneal mesothelial cells: IFN-gamma controls neutrophil migration across the mesothelium in vitro and in vivo. J Immunol 2001; 167 (2): 1028–1038.
McLoughlin RM, Hurst SM, Nowell MA, et al. Differential regulation of neutrophil-activating chemokines by IL-6 and its soluble receptor isoforms. J Immunol 2004; 172 (9): 5676–5683.
McLoughlin RM, Jenkins BJ, Grail D, et al. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc Natl Acad Sci U S A 2005; 102 (27): 9589–9594.
Cannistra SA, Ottensmeier C, Tidy J, DeFranzo B. Vascular cell adhesion molecule-1 expressed by peritoneal mesothelium partly mediates the binding of activated human T lymphocytes. Exp Hematol 1994; 22: 996–1002.
Liberek T, Topley N, Luttmann W, Williams JD. Adherence of neutrophils to human peritoneal mesothelial cells: role of intercellular adhesion molecule-1. J Am Soc Nephrol 1996; 7: 208–217.
Jonjic N, Peri G, Bernasconi S, et al. Expression of adhesion molecules and chemotactic cytokines in cultured human mesothelial cells. J Exp Med 1992; 176: 1165–1174.
Basok A, Shnaider A, Man L, Chaimovitz C, Douvdevani A. CD40 is expressed on human peritoneal mesothelial cells and upregulates the production of interleukin-15 and RANTES. J Am Soc Nephrol 2001; 12 (4): 695–702.
Glik A, Douvdevani A. T lymphocytes: the “cellular” arm of acquired immunity in the peritoneum. Perit Dial Int 2006; 26 (4): 438–448.
Glik A, Mazar J, Rogachev B, Zlotnik M, Douvdevani A. CD40 ligand expression correlates with resolution of peritonitis and mononuclear cell recruitment. Perit Dial Int 2005; 25 (3): 240–247.
Man L, Lewis E, Einbinder T, Rogachev B, Chaimovitz C, Douvdevani A. Major involvement of CD40 in the regulation of chemokine secretion from human peritoneal mesothelial cells. Kidney Int 2003; 64 (6): 2064–2071.
Mazar J, Agur T, Rogachev B, et al. CD40 ligand (CD154) takes part in regulation of the transition to mononuclear cell dominance during peritonitis. Kidney Int 2005; 67 (4): 1340–1349.
Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol 2005; 175 (6): 3463–3468.
Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM. The soluble interleukin 6 receptor: mechanisms of production and implications in disease. FASEB J 2001; 15 (1): 43–58.
Fricke H. Expression of CD44v9 on peritoneal T cells from patients on continuous ambulatory peritoneal dialysis. Nephron 1996; 73: 373–374.
Fricke H, Hartmann J, Sitter T, Steldinger R, Rieber P, Schiffl H. Continuous ambulatory peritoneal dialysis impairs T lymphocyte selection in the peritoneum. Kidney Int 1996; 49: 1386–1395.
Williams JD, Topley N, Craig KJ, et al. The Euro-Balance Trial: the effect of a new biocompatible peritoneal dialysis fluid (balance) on the peritoneal membrane. Kidney Int 2004; 66 (1): 408–418.
Cooker LA, Luneburg P, Holmes CJ, Jones S, Topley N. Interleukin-6 levels decrease in effluent from patients dialyzed with bicarbonate/lactate-based peritoneal dialysis solutions. Perit Dial Int 2001; 21 (suppl. 3): S102–S107.
Martikainen TA, Ekstrand AV, Honkanen EO, Teppo AM, Gronhagen-Riska C. Dialysate leukocytes, sICAM-1, hyaluronan and IL-6: predictors of outcome of peritonitis? Blood Purif 2004; 22 (4): 360–366.
Pecoits-Filho R, Barany P, Lindholm B, Heimburger O, Stenvinkel P. Interleukin-6 is an independent predictor of mortality in patients starting dialysis treatment. Nephrol Dial Transplant 2002; 17 (9): 1684–1688.
Pecoits-Filho R, Carvalho MJ, Stenvinkel P, Lindholm B, Heimburger O. Systemic and intraperitoneal interleukin-6 system during the first year of peritoneal dialysis. Perit Dial Int 2006; 26 (1): 53–63.
Szeto CC, Chow KM, Lam CW, et al. Clinical biocompatibility of a neutral peritoneal dialysis solution with minimal glucose-degradation products--a 1-year randomized control trial. Nephrol Dial Transplant 2007; 22 (2): 552–559.
Holmes CJ. Glucotoxicity in peritoneal dialysis – solutions for the solution! Adv Chronic Kidney Dis 2007; 14 (3): 269–278.
Rodrigues A, Cabrita A, Maia P, Guimaraes S. Peritoneal rest may successfully recover ultrafiltration in patients who develop peritoneal hyperpermeability with time on continuous ambulatory peritoneal dialysis. Adv Perit Dial 2002; 18: 78–80.
de Alvaro F, Castro MJ, Dapena F, et al. Peritoneal resting is beneficial in peritoneal hyperpermeability and ultrafiltration failure. Adv Perit Dial 1993; 9: 56–61.
Zhe XW, Tian XK, Cheng L, Wang T. Effects of peritoneal resting on peritoneal fluid transport kinetics. Perit Dial Int 2007; 27 (5): 575–579.
Kim YL, Kim SH, Kim JH, et al. Effects of peritoneal rest on peritoneal transport and peritoneal membrane thickening in continuous ambulatory peritoneal dialysis rats. Perit Dial Int 1999; 19 (suppl. 2): S384–S387.
Davies SJ. Monitoring of long-term peritoneal membrane function. Perit Dial Int 2001; 21 (2): 225–230.
Twardowski ZJ, Nolph K, Khanna R. Peritoneal equilibration test. Perit Dial Bull 1987; 7: 138–147.
Rumpsfeld M, McDonald SP, Johnson DW. Higher peritoneal transport status is associated with higher mortality and technique failure in the Australian and New Zealand peritoneal dialysis patient populations. J Am Soc Nephrol 2006; 17 (1): 271–278.
Rumpsfeld M, McDonald SP, Purdie DM, Collins J, Johnson DW. Predictors of baseline peritoneal transport status in Australian and New Zealand peritoneal dialysis patients. Am J Kidney Dis 2004; 43 (3): 492–501.
Gillerot G, Goffin E, Michel C, et al. Genetic and clinical factors influence the baseline permeability of the peritoneal membrane. Kidney Int 2005; 67 (6): 2477–2487.
Selgas R, Bajo MA, Cirugeda A, et al. Ultrafiltration and small solute transport at initiation of PD: questioning the paradigm of peritoneal function. Perit Dial Int 2005; 25 (1): 68–76.
Goffin E, Devuyst O. Phenotype and genotype: perspectives for peritoneal dialysis patients. Nephrol Dial Transplant 2006; 21 (11): 3018–3022.
Wong TY, Szeto CC, Szeto CY, Lai KB, Chow KM, Li PK. Association of ENOS polymorphism with basal peritoneal membrane function in uremic patients. Am J Kidney Dis 2003; 42 (4): 781–786.
Szeto CC, Chow KM, Poon P, Szeto CY, Wong TY, Li PK. Genetic polymorphism of VEGF: Impact on longitudinal change of peritoneal transport and survival of peritoneal dialysis patients. Kidney Int 2004; 65 (5): 1947–1955.
Ioannidis JP. Genetic associations: false or true? Trends Mol Med 2003; 9 (4):135–138.
Cordell HJ, Clayton DG. Genetic association studies. Lancet 2005; 366 (9491): 1121–1131.
Passlick-Deetjen J, Pischetsrieder M, Witowski J, Bender TO, Jorres A, Lage C. In vitro superiority of dual-chambered peritoneal dialysis solution with possible clinical benefits. Perit Dial Int 2001; 21 (suppl. 3): S96–S101.
Erixon M, Wieslander A, Linden T, et al. Take care in how you store your PD fluids: actual temperature determines the balance between reactive and non-reactive GDPs. Perit Dial Int 2005; 25 (6): 583–590.
Erixon M, Wieslander A, Linden T, et al. How to avoid glucose degradation products in peritoneal dialysis fluids. Perit Dial Int 2006; 26 (4): 490–497.
Kjellstrand P, Erixon M, Wieslander A, Linden T, Martinson E. Temperature: the single most important factor for degradation of glucose fluids during storage. Perit Dial Int 2004; 24 (4): 385–391.
Linden T, Cohen A, Deppisch R, Kjellstrand P, Wieslander A. 3,4-Dideoxyglucosone-3-ene (3,4-DGE): a cytotoxic glucose degradation product in fluids for peritoneal dialysis. Kidney Int 2002; 62 (2): 697–703.
Jones S, Holmes CJ, Krediet RT, et al. Bicarbonate/lactate-based peritoneal dialysis solution increases cancer antigen 125 and decreases hyaluronic acid levels. Kidney Int 2001; 59 (4):1529–1538.
Jones S, Holmes CJ, Mackenzie RK, et al. Continuous dialysis with bicarbonate/lactate-buffered peritoneal dialysis fluids results in a long-term improvement in ex vivo peritoneal macrophage function. J Am Soc Nephrol 2002; 13 (suppl. 1): S97–S103.
Ueda Y, Miyata T, Goffin E, et al. Effect of dwell time on carbonyl stress using icodextrin and amino acid peritoneal dialysis fluids. Kidney Int 2000; 58 (6): 2518–2524.
Mortier S, De Vriese AS, McLoughlin RM, et al. Effects of conventional and new peritoneal dialysis fluids on leukocyte recruitment in the rat peritoneal membrane. J Am Soc Nephrol 2003; 14 (5): 1296–1306.
Mortier S, De Vriese AS, Van de Voorde J, Schaub TP, Passlick-Deetjen J, Lameire NH. Hemodynamic effects of peritoneal dialysis solutions on the rat peritoneal membrane: role of acidity, buffer choice, glucose concentration, and glucose degradation products. J Am Soc Nephrol 2002; 13 (2): 480–489.
Rippe B, Simonsen O, Heimburger O, et al. Long-term clinical effects of a peritoneal dialysis fluid with less glucose degradation products. Kidney Int 2001; 59 (1): 348–357.
Miyata T, Devuyst O, Kurokawa K, van Ypersele de Strihou C. Toward better dialysis compatibility: advances in the biochemistry and pathophysiology of the peritoneal membranes. Kidney Int 2002; 61 (2): 375–386.
Hobbs AJ, Higgs A, Moncada S. Inhibition of nitric oxide synthase as a potential therapeutic target. Annu Rev Pharmacol Toxicol 1999; 39: 191–220.
Duman S, Gunal AI, Sen S, et al. Does enalapril prevent peritoneal fibrosis induced by hypertonic (3.86%) peritoneal dialysis solution? Perit Dial Int 2001; 21 (2): 219–224.
Duman S, Sen S, Duman C, Oreopoulos DG. Effect of valsartan versus lisinopril on peritoneal sclerosis in rats. Int J Artif Organs 2005; 28 (2): 156–163.
Duman S, Sen S, Sozmen EY, Oreopoulos DG. Atorvastatin improves peritoneal sclerosis induced by hypertonic PD solution in rats. Int J Artif Organs 2005; 28 (2): 170–176.
Duman S, Wieczorowska-Tobis K, Styszynski A, Kwiatkowska B, Breborowicz A, Oreopoulos DG. Intraperitoneal enalapril ameliorates morphologic changes induced by hypertonic peritoneal dialysis solutions in rat peritoneum. Adv Perit Dial 2004; 20: 31–36.
Kyuden Y, Ito T, Masaki T, Yorioka N, Kohno N. Tgf-beta1 induced by high glucose is controlled by angiotensin-converting enzyme inhibitor and angiotensin II receptor blocker on cultured human peritoneal mesothelial cells. Perit Dial Int 2005; 25 (5): 483–491.
Sauter M, Cohen CD, Wornle M, Mussack T, Ladurner R, Sitter T. ACE inhibitor and AT1-receptor blocker attenuate the production of VEGF in mesothelial cells. Perit Dial Int 2007; 27 (2): 167–172.
Fang CC, Yen CJ, Chen YM, et al. Diltiazem suppresses collagen synthesis and IL-1beta-induced TGF-beta1 production on human peritoneal mesothelial cells. Nephrol Dial Transplant 2006; 21 (5): 1340–1347.
Griffioen AW, Molema G. Angiogenesis: potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol Rev 2000; 52 (2): 237–268.
Yoshio Y, Miyazaki M, Abe K, et al. TNP-470, an angiogenesis inhibitor, suppresses the progression of peritoneal fibrosis in mouse experimental model. Kidney Int 2004; 66 (4): 1677–1685.
Tanabe K, Maeshima Y, Ichinose K, et al. Endostatin peptide, an inhibitor of angiogenesis, prevents the progression of peritoneal sclerosis in a mouse experimental model. Kidney Int 2007; 71 (3): 227–238.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Devuyst, O., van Westrhenen, R., Topley, N. (2009). Long-Term Peritoneal Dialysis Patients. In: Khanna, R., Krediet, R.T. (eds) Nolph and Gokal's Textbook of Peritoneal Dialysis. Springer, Boston, MA. https://doi.org/10.1007/978-0-387-78940-8_27
Download citation
DOI: https://doi.org/10.1007/978-0-387-78940-8_27
Publisher Name: Springer, Boston, MA
Print ISBN: 978-0-387-78939-2
Online ISBN: 978-0-387-78940-8
eBook Packages: MedicineMedicine (R0)