
The first association between uremia and bone disease was made by Lucas and reported in Lancet in 1883 [1]. However, it was not until nearly 40 years later that the major clinical and radiological manifestations of the skeletal changes were accurately defined [2, 3]. In 1943, the histopathology of osteitis fibrosa and osteomalacia was described [4], and in the same year the term “renal osteodystrophy” was coined by Liu and Chu [5]. Subsequently, the abnormalities of bone mass that occur in osteopenia and osteosclerosis were also described [6]. Following the research of Stanbury and Lumb [7, 8], there began a period of rapid advance in the understanding of the processes behind altered divalent ion metabolism, and the abnormalities of parathyroid hormone and vitamin D3 production that are seen in end-stage renal disease. Despite these advances with the introduction of vitamin D3 replacement therapy, new oral phosphate binders and, most recently, calcimimetic therapy, osteodystrophy remains a common complication of end-stage renal failure, and continues to pose diagnostic and therapeutic dilemmas for clinical nephrologists.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Peritoneal Dialysis
- Phosphate Binder
- Continuous Ambulatory Peritoneal Dialysis
- Peritoneal Dialysis Patient
- Renal Osteodystrophy
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
The first association between uremia and bone disease was made by Lucas and reported in Lancet in 1883 [1]. However, it was not until nearly 40 years later that the major clinical and radiological manifestations of the skeletal changes were accurately defined [2, 3]. In 1943, the histopathology of osteitis fibrosa and osteomalacia was described [4], and in the same year the term “renal osteodystrophy” was coined by Liu and Chu [5]. Subsequently, the abnormalities of bone mass that occur in osteopenia and osteosclerosis were also described [6]. Following the research of Stanbury and Lumb [7, 8], there began a period of rapid advance in the understanding of the processes behind altered divalent ion metabolism, and the abnormalities of parathyroid hormone and vitamin D3 production that are seen in end-stage renal disease. Despite these advances with the introduction of vitamin D3 replacement therapy, new oral phosphate binders and, most recently, calcimimetic therapy, osteodystrophy remains a common complication of end-stage renal failure, and continues to pose diagnostic and therapeutic dilemmas for clinical nephrologists.
It has become apparent that the spectrum of bone lesions seen in dialysis patients has changed over the decades. In the first two decades of dialysis, hyperparathyroid disease was usual but then became much less common with the introduction of calcium-based phosphate binders and oral vitamin D therapy [9, 10]. Furthermore, a different pattern of bone lesions is found in peritoneal dialysis (PD) and hemodialysis patients [9–12]. In a histological study of 259 chronic dialysis patients in Canada in the early 1990s, the commonest bone lesion found was high turnover hyperparathyroid disease (50%) in hemodialysis patients, and low turnover, adynamic bone (61%) in PD patients [10]. In contrast, Malluche and Monier-Faugere (Kentucky, USA) reported that in a retrospective survey of 602 patients from 1982 to 1991 the mixed lesion was the commonest diagnosis [9], regardless of mode of dialysis (56% in CAPD and 49% in hemodialysis). A more recent study of 96 hemodialysis patients demonstrated 60% had low turnover lesions [13]. The difference between these reports is noteworthy in itself, since they are large and reliable studies, but from centers thousands of miles apart. While varying diagnostic criteria may account for some of the difference, it emphasizes the fact that, in dialysis patients, histomorphometric data represent the result of pathological processes, treatment regimes, and environmental effects that have been on-going for many years.
Over the past decade, interest in osteodystrophy has exploded as the connections between calcium, phosphate, parathyroid hormone, and cardiovascular disease have emerged. Unfortunately, most of the reports to date are from retrospective analyses and are yet to be confirmed in interventional randomized, prospective studies.
Classification of Renal Osteodystrophy
In this chapter, the term renal osteodystrophy is used to encompass all its skeletal manifestations such as osteitis fibrosa, osteomalacia, mixed lesions, the adynamic lesion, osteoporosis, osteosclerosis, and (in children) retardation of growth. However, renal osteodystrophy also includes a variety of extraskeletal problems including myopathy, peripheral ischemic necrosis, visceral calcification, and, perhaps most importantly, vascular and valvular calcification.
Since the introduction of the undecalcified bone biopsy, significant advances have been made in the understanding of the histological changes underlying all forms of renal osteodystrophy. Renal osteodystrophy has its origins early in the course of renal failure [14, 15], so that by the time GFR has fallen to 50% of normal, at least 50% of the patients exhibit abnormal bone histology [16]. In a study of 16 patients with creatinine clearances between 20 and 59 mL/min, Baker et al. found all of them to have abnormal bone histology [17]. By the time patients start peritoneal dialysis the majority have an identifiable histological abnormality [18], the nature of which depends to some extent on medical management up to that time.
The classification of renal osteodystrophy is simplified by the recognition that there are essentially two groups of histological lesions – high and low turnover bone lesions plus a third intermediate category referred to as a “mixed” lesion. There may also be osteopenia or osteoporosis superimposed on these lesions, particularly in elderly female patients.
High Turnover Bone Lesions
In osteitis fibrosa cystica, the characteristic findings include a marked increase in bone resorption, osteoblastic and osteoclastic activity, and endosteal fibrosis (Fig. 22.1). In particular, the number of osteoclasts is markedly increased, and they may be larger than normal with multiple nuclei. There may be a large increase in surface resorption with dissecting cavities where the osteoclasts have tunneled through the trabecular bone. This results in deposition of fibrous tissue in the marrow spaces (peritrabecular fibrosis), and the formation of so-called “woven bone,” new bone matrix that is not lamellar but disorganized in structure. Although the bone may show increased osteoid, the use of tetracycline labeling prior to biopsy demonstrates that mineralization proceeds relatively normally. Skeletal mass may diminish as the rate of resorption exceeds that of formation. The term osteitis implies inflammation of bone, which is not present, so that is it preferable to refer to this lesion as severe or predominant hyperparathyroid bone disease.

Bone histology in severe hyperparathyroid disease. Numerous large, multinucleate osteoclasts can be seen tunnelling into mineralized trabecular bone. Osteoblasts are also numerous, and peritrabecular fibrous tissue has been deposited in the marrow cavity (toluidine blue stain; original magnification × 100)
In mild hyperparathyroidism, elevated parathyroid hormone levels increase bone turnover but peritrabecular fibrosis is minimal or absent.
Low Turnover Bone Lesions
In osteomalacia, defective mineralization of bone, due to deficiency of 1,25-dihydroxyvitamin D3, results in a relative increase in the amount of osteoid or unmineralized bone matrix (Fig. 22.2).

Bone histology in osteomalacia (not aluminium-related). Broad lamellar osteoid seams surround the calcified trabecular bone. In some areas the failure of mineralization has resulted in ‘islands' of calcified bone so that the mechanical strength of the trabeculum is greatly reduced (toluidine blue stain; original magnification × 100)
Osteitis fibrosa can also increase osteoid mass, simply as a result of increased bone turnover, but bone biopsy with dual tetracycline labeling will reliably distinguish these diseases. Use of oral aluminium for phosphate binding is decreasing, but accumulation can also lead to an osteomalacic-type osteodystrophy even in the presence of adequate 1,25-dihydroxyvitamin D3 levels [19]. In bone, the site of aluminium deposition is at the interface between mineralized bone and unmineralized osteoid. Here it appears to reduce osteoblast numbers and delay the process of mineralization, as demonstrated by diminished uptake of tetracycline into trabecular bone [20]. Studies have established an inverse relationship between bone aluminium accumulation and the rate of bone formation [21] and, even in cell-free laboratory studies, aluminium has been shown to reduce both the formation and growth of hydroxyapatite crystals [22].
Adynamic bone lesions, previously thought to be a result of aluminium accumulation in bone, are now well recognized to occur in the absence of stainable bone aluminium (Fig. 22.3 ), and are characterized by an abnormally low bone formation rate, a defect of bone mineralization, normal or decreased osteoid thickness, decreased osteoblastic surfaces, and normal or decreased osteoclastic surfaces [16, 23, 23–26]. This appearance was sometimes referred to as aplastic, a term usually reserved for structures that are congenitally absent, whereas adynamic more accurately conveys the inactivity of bone cells in this lesion [27]. Little is known about its etiology, and even less about its natural history, although there is evidence to suggest that it is commoner in patients with diabetes, elderly dialysis patients, and those on continuous ambulatory peritoneal dialysis (CAPD) [23–25, 28]. Overtreatment with vitamin D, and use of corticosteroids, as well as low sexual and thyroid hormone levels, are other causes that have been considered [25, 29]. Recently, a small study of PD patients implicated albumin and the malnutrition-inflammation-cachexia syndrome in the pathophysiology of the adynamic lesion [27].

Bone histology in the adynamic lesion. Osteoid seams are very thin or almost absent. Numbers of osteoclasts and osteoblasts are greatly reduced and unrepaired microfractures can be seen within the mineralized bone (hematoxylin and eosin stain; original magnification, × 100)
Mixed Bone Lesions
In many patients, hyperparathyroidism and defective mineralization coexist with variable bone volume and rates of bone turnover. This probably reflects the fact that changes to rates of bone turnover occur much more slowly than changes to biochemical parameters. These changes will also be influenced by local structural stresses and may therefore vary in space as well as time within the skeleton.
Osteoporosis
In osteoporosis, the bone mineral density is reduced, bone microarchitecture is disrupted, and the amount and variety of noncollagenous proteins in bone is altered and are therefore more at risk of fracture. Osteoporosis is defined by the World Health Organization as a bone mineral density 2.5 SD below peak bone mass in women (20-year-old sex-matched healthy person average) as measured by DEXA. Making a diagnosis in patients with chronic kidney disease (CKD) is more difficult since any type of osteodystrophy can be associated with reduced bone density [18], with no apparent correlation. Nevertheless, peritoneal dialysis patients are at greater risk of osteoporosis than the general population because of numerous risk factors found more commonly in CKD 5. These include poor nutrition, decreased physical activity, smoking and peripheral vascular disease, estrogen or androgen deficiency, low body mass index, reduced mobility, previous steroid and heparin usage, low parathyroid hormone levels, β-2-microglobulin amyloidosis, and chronic acidosis [30].
Pathogenesis of Renal Osteodystrophy
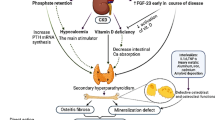
Renal osteodystrophy is recognized to be a common complication of end-stage renal failure and is believed to have its origins early in the onset of renal impairment [31]. The mechanism of its development is both multifactorial and controversial, but since normal kidneys maintain calcium, phosphate, magnesium and bicarbonate balance, synthesise 1.25- and 24,25-dihydroxyvitamin D3, act as a major target organ and excretory organ for parathyroid hormone, and also excrete aluminium, it is self-evident that renal failure will have numerous profound effects on mineral metabolism. These various factors all interact to a greater or lesser extent, but for simplicity are considered separately in the following sections.
Parathyroid Hormone and Calcium Metabolism
Bone is continually being remodeled, and in health a balance is maintained between synthesis of bone matrix (osteoid formation), its mineralization, and subsequent resorption. This balance is governed by the relative activity of osteoblasts, osteoclasts, and osteocytes. Increased secretion of parathyroid hormone (PTH) increases both the activity and numbers of these bone cells, causing an overall increase in bone turnover. Excessive production may result in deposition of fibrous tissue in the marrow spaces (osteitis fibrosa), endosteal fibrosis, and the formation of so-called “woven bone,” new bone matrix that is not lamellar but disorganized in structure. Skeletal mass may diminish as the rate of resorption exceeds that of formation. Studies from both Europe and the United States suggest that parathyroidectomy is still required in a significant number of patients. A study of 14,180 patients undergoing dialysis in Lombardy, Italy, from 1983 to 1996, reported parathyroidectomy rates of 3.3 cases per 1,000 patient-years for patients receiving renal replacement therapy for less than 5 years, and 30 cases per 1,000 patient-years for those receiving renal replacement therapy for more than 10 years [32]. In the United States, parathyroidectomy rates among prevalent hemodialysis patients declined between 1988 and 1998 but increased progressively after 1998 despite introduction of better therapies for preventing severe hyperparathyroidism [33]. However, it is generally accepted that, in the uremic patient, low/normal levels of PTH result in excessively low bone cell activity and bone turnover, or adynamic bone [9, 10, 25, 29, 34–39], and this is associated with increased rates of vascular calcification [40].
PTH is a single-chain protein of 84 amino acids, the sequence of which was established by Keutmann et al. in 1978 [41]. It is synthesized in the parathyroid chief cell via two precursors, pre-pro-PTH and pro-PTH (115 and 90 amino acids, respectively). PTH secretion occurs approximately 20 min after synthesis of the original pre-pro-PTH [42].
Significant elevations of serum parathyroid hormone have been reported in patients with only moderately abnormal glomerular filtration rates of 60–80 mL/min [17, 43, 44]. The secretion of PTH is controlled by many factors, but in renal impairment the most important stimulus is thought to be reduction in the level of serum-ionized calcium (Fig. 22.4 ). Factors which contribute to hypocalcemia and elevation of serum PTH are phosphate retention, defective vitamin D metabolism, skeletal resistance to the calcemic action of PTH, elevation of the “set point” at which serum calcium suppresses PTH release and impaired degradation of circulating PTH [44].

Factors contributing to the production of excess parathyroid hormone in dialysis patients
Secretion of PTH is primarily controlled by the concentration of ionized calcium in the extracellular space so that hypocalcemia stimulates, and hypercalcemia suppresses PTH release [45]. This relationship between PTH and serum calcium can be represented as a sigmoidal curve, with a basal rate of secretion persisting even during hypercalcemia [46]. In normal individuals, the basal PTH level is approximately 20–25% of the maximally stimulated PTH level and is positioned in the initial part of the steep ascent of the sigmoidal curve. Therefore, a small decrease in serum calcium produces a large increase in serum PTH secretion. Felsenfeld observed that for the same ionized calcium level serum PTH was higher during the induction of hypocalcemia than during the recovery from hypocalcemia [47]. Conversely, the PTH level was greater when hypercalcemia was induced from the nadir of hypocalcemia than when hypercalcemia was induced from basal serum calcium. Furthermore, the set point of calcium was greater during the induction of hypocalcemia than during the recovery from hypocalcemia. This differential response of PTH to the direction of change of serum calcium is known as “hysteresis,” and there is evidence that the PTH–calcium curves may differ in different forms of renal osteodystrophy or after a specific form of therapy such as desferrioxamine or calcitriol [47–49].
As well as suppressing PTH secretion, hypercalcemia is also known to decrease parathyroid cell cyclic adenosine monophosphate (cAMP) but it is not clear whether this is the means by which PTH is controlled or whether it is a secondary phenomenon [50].
Hyperplastic parathyroid glands are less sensitive to ionized calcium levels than are normal glands, suggesting that one cause of elevated PTH in chronic renal failure may be a shift in the “set point” for calcium-regulated PTH secretion, in addition to the increase in parathyroid mass. The set point is defined as a calcium ion concentration necessary to suppress the secretion of PTH by 50%. Furthermore the degree of responsiveness across the calcium-sensitive range is altered so that hyperparathyroidism is a product of the increase in tissue mass (because the nonsuppressible, basal secretion is increased) and the lack of suppression by calcium in the normocalcemic range. Thus normal concentrations of ionized calcium may not be sufficient to suppress hyperplastic parathyroid glands, and serum levels have to be increased to the upper limits of normal to control the release of PTH in patients with secondary hyperparathyroidism [46]. However, there is evidence that once the parathyroid gland size exceeds a certain limit, the nonsuppressible basal secretion alone becomes sufficient to increase serum parathyroid hormone to hyperparathyroid levels [51]. When this occurs parathyroidectomy was previously the only remaining option, but with the introduction of cinacalcet many patients may now avoid this procedure.
Firm evidence now exists that parathyroid cells possess specific nuclear receptors for 1,25-dihydroxyvitamin D3 [51–53]. When given intravenously to a group of 20 hemodialysis patients calcitriol produced a marked suppression (70.1 ± 3.2%) of PTH levels without a significant change in serum calcium, confirming that it is an important regulator of PTH secretion at least in states of calcitriol depletion [53]. Substantial degradation of calcitriol occurs in the intestine so that oral vitamin D increases intestinal calcium absorption but the delivery of calcitriol to peripheral target organs is limited [54, 55]. This could explain the greater effect of intravenous, compared to oral, calcitriol. PTH secretion is also affected by ionized magnesium, however, only severe hypomagnesemia seems to have any clinical relevance in that it has been shown to inhibit PTH secretion [56].
In addition to the abnormalities of secretion that occur in chronic renal failure, the process of degradation is incomplete. Normally, intact PTH is degraded by the liver and kidneys, resulting in the production of amino (N)- and carboxy (C)-terminal fragments. The fragments are further metabolized by the kidney, so that in the absence of renal function they will accumulate. C-terminal fragments are detectable up to 2 weeks after parathyroidectomy in chronic renal failure, yet decrease by 80% within 24 h of a successful renal transplant. So-called “second-generation” PTH assays, called “intact” PTH assays, have been developed since 1987. They use two different antibodies, the first, directed against the C-terminal region of PTH, and the second directed against the N-terminal region. These “sandwich” assays were the first radioimmunometric assays, and were thought to measure only the full-length 1–84 PTH, however, they also measure large PTH fragments (namely 7–84 PTH). These N-terminal truncated PTH fragments inhibit the action of PTH by blocking its binding to its normal receptor. They may also bind to a specific C-terminal PTH receptor and may have biological functions in the skin, bone, hematopoietic system, and placenta [57]. The third generation of PTH assays have been developed since 2000 and use the same sandwich and radioimmunometric techniques, with the first antibody directed against amino acids 39–84, but the second antibody has been restricted and directed against the first six N-terminal residues of 1–84 PTH. They have been demonstrated to be most sensitive, and more specific, when measuring bioactive intact 1–84 PTH. It is now clear that different assays can give significantly different results as demonstrated by a study of 15 different methods [58]. In terms of correlation with bone histology, most experience has been gained with sandwich assays specific to the whole, or intact, 1–84 PTH molecule. These appear to correlate well with the biological effects of PTH on bone in chronic renal failure [18, 59], and whether “third-generation” assays have any advantages remains to be seen [60].
Vitamin D Metabolism
The similarity between the bone disease caused by simple vitamin D deficiency and that which occurs in chronic renal failure has been recognized for many years, both at clinical [1] and histological levels [4]. It was also known that renal failure was associated with impaired intestinal absorption of calcium [5]. In renal failure both the bone lesions and the defect in calcium absorption were shown to be correctable by oral calciferol, but the amount required to have an effect is much larger than in simple deficiency states. The disease is “vitamin D resistant.” Vitamin D3 (calciferol) circulates in the blood bound to vitamin D-binding protein, after having been synthesized in the skin or absorbed from the diet. In the liver it is metabolized by the enzyme vitamin-D-25-hydroxylase to form 25-hydroxyvitamin D (calcidiol, 25-OH-D). In most patients with chronic renal failure, 25-OH-D levels are normal if they eat a balanced diet and their skin is not completely covered from the sun.
In the kidney 25-OH–D is further metabolized by a mitochondrial cytochrome P–450 oxidase, 25-OH–D-1α-hydroxylase, to form 1,25-dihydroxyvitamin D3 (calcitriol), the biologically active form of vitamin D. Various studies have indicated that the proximal convoluted tubules (PCT) are the principal site of 1,25(OH)2D3 production [61]. In 1973, Mawer et a1. showed that calcitriol could not be detected in the serum of patients with chronic renal failure, after injection of radioactive cholecalciferol, and suggested that it was the inability to form this metabolite that was the cause of the vitamin D resistance[62]. This is now known to be the case, and is a result of reduced renal 1α-hydroxylase activity caused by loss of renal mass, hyperphosphatemia and possibly by uremic toxins [63]. In addition to renal production of 1,25-OH–D3, humans can, in certain pathological states, produce it extrarenally. In sarcoidosis cultured alveolar macrophages and lymph node homogenates can convert 25-OH–D to 1,25-OH–D3 and a similar process probably accounts for the hypercalcemia and hypercalciuria sometimes seen in other granulomatous diseases such as tuberculosis, silicosis, berylliosis, and fungal diseases. In CAPD patients who have had one or more episodes of peritonitis cultured peritoneal macrophages are also able to convert 25-OH–D3 to 1,25-OH–D3 [64].
Once synthesized in the kidney, 1,25-OH–D3 is transported by vitamin D-binding protein to its target cells. It enters the cell by a mechanism that is poorly understood and is then transported to the nucleus. Here it interacts with its nuclear receptor, phosphorylating it to bring about interaction with chromatin and transcription of specific genes. In the small intestine this results in expression of the gene coding for calbindin, the calcium-binding protein. The activity of other proteins is also affected, with the net result that calcium and phosphate absorption from the intestine is stimulated. The effect of 1,25-OH–D3 on bone is to increase removal of calcium. A small decrease in serum ionized calcium stimulates PTH production which in turn stimulates the kidney to produce 1,25-OH–D3, 1,25-OH–D3 in conjunction with PTH increases osteoclastic activity and release of calcium, returning the ionized calcium level to normal.
1,25-OH–D3 has been conclusively shown to suppress PTH secretion in dialysis patients when administered orally or intravenously [53, 65–71]. It undoubtedly also has important immunoregulatory functions and is able to decrease the rate of proliferation of certain tumour cells, such as the HL-60 (human promyelocytic) cell, and even transform them into mature macrophages. 1,25-OH–D3 can also inhibit the proliferation of cultured human keratinocytes, an ability that has important clinical implications in that it has been shown to dramatically improve psoriasis in 75% of a group of patients given up to 2 μg/day [72]. It has recently been suggested that vitamin D deficiency may be an important factor in the pathogenesis of hypertension and insulin resistance in end-stage renal failure [73].
Phosphate Metabolism
In the past, overactivity of the parathyroid glands in renal failure was explained by the trade-off hypothesis of Bricker [74, 75]. He postulated that as renal failure progresses there is a tendency for serum phosphate levels to rise and ionized calcium levels to fall, resulting in a compensatory rise in PTH. The increase in PTH reduces tubular reabsorption of phosphate in the remaining nephrons and increases serum calcium. Thus serum values of phosphate and calcium may be kept within, or near, the normal range at the expense of rising PTH levels and its resultant effects on the skeleton. However, more recent work has cast doubt on this, with the observations that hyperparathyroidism can develop even in the presence of a high serum calcium [75], and that hyperphosphatemia stimulates PTH secretion independent of serum calcium concentration [76–78]. However, increased phosphate excretion results in decreased activity of 25-OH–D-1α-hydroxylase, and consequently decreased production of 1,25-dihydroxyvitamin D3 [72]. This in turn stimulates increased synthesis and secretion of PTH in an attempt to stimulate renal production of 1,25-dihydroxyvitamin D3. As renal failure progresses the compensatory effect of PTH on 1,25-dihydroxyvitamin D3 deficiency is overcome and an absolute deficiency develops [16]. This further stimulates PTH levels and decreases gastrointestinal calcium absorption [79]. With further progression of renal failure, to a glomerular filtration rate of around 10 mL/min, phosphate excretion can no longer be increased and hyperphosphatemia occurs, exacerbating the hypocalcemia. At this stage, hypocalcemia further stimulates PTH secretion, although doubt remains as to its role in the earlier stages of renal failure. In addition hyperphosphatemia is associated with extraskeletal calcification in soft tissues and, perhaps more worryingly, in blood vessels.
Magnesium Metabolism
Although magnesium metabolism is affected by decreasing renal function, the clinical relevance of this is unknown. In normal subjects magnesium is absorbed from the small intestine and excreted in the urine, so that elevated serum levels are seen in renal failure [80]. In vitro, magnesium is an inhibitor of crystallization and may increase bone levels of pyrophosphate – another inhibitor of mineralization [81]. In uremia, bone magnesium is correlated with serum magnesium, and serum pyrophosphate is increased [82]. Hence, it is theoretically possible that elevated serum magnesium could play a role in the development of osteomalacia, but there is no evidence in the literature that this is the case.
Moderate hypomagnesemia can contribute to elevation of serum PTH [83], whereas severe hypomagnesemia has been shown to inhibit PTH secretion [56].
Aluminium and Osteodystrophy
In 1978, Ward et al. made the association between aluminium and bone disease in dialysis patients [19]. Since then the importance attached to reducing exposure to aluminium has increased, but aluminium hydroxide is still in use as a “rescue therapy” in cases of severe hyperphosphatemia.
A normal human daily intake of aluminium ranges from 2 to 20 mg but gastrointestinal uptake is estimated to be only 0.5–1% of this. Normal urinary aluminium excretion varies between 20 and 50 mg/day but was shown to increase to 200–400 mg/day when normal individuals were given aluminium hydroxide in amounts commonly given to dialysis patients [84]. In end-stage renal failure the loss of urinary excretion plus exposure to aluminium in dialysate solutions, phosphate binders, and volume replacement fluids can result in the total body content rising by a factor of up to 20. Serum levels are a poor guide to total body load, since it is strongly protein-bound and largely deposited quickly in tissues such as bone, liver and spleen.
Aluminium accumulates at the interface between mineralized bone and unmineralized osteoid, where it delays the process of mineralization. Aluminium also accumulates in parathyroid glands and suppresses their secretion of PTH. In patients with aluminium-related bone disease and renal failure, PTH levels are commonly lower than would be expected and may be suppressed to normal levels, giving some degree of protection from hyperparathyroid bone disease [85], but possibly resulting in development of the adynamic bone lesion [59] and other toxic effects.
It is likely that aluminium-related bone disease will gradually disappear as fewer physicians use aluminium-based phosphate binders, and exposure from other sources is sought out and reduced to minimal levels.
Acid-Base Balance
The role of acidosis in the pathogenesis of renal osteodystrophy is unclear. However, acidosis is involved in both calcium balance and PTH release. Acidotic azotemic patients show increased losses of urinary and faecal calcium which can be reduced by alkali treatment, resulting in restoration of a neutral calcium balance [86]. In a study of 54 uremic patients, infusion of sodium bicarbonate produced a rise in arterialized capillary blood pH and a proportional fall of around 20% in serum PTH [87, 88]. No significant change in serum ionized calcium was observed during the study. The clinical significance of these findings remains to be elucidated but it seems likely that, as with changes in magnesium metabolism, the effects are of much less importance than those associated with PTH and vitamin D.
Calcitonin
Calcitonin is a 32-amino acid single-chain peptide, and the major stimulus for its secretion is hypercalcemia. Circulating calcitonin has a short half-life (around 10 min) and depends on renal function for degradation and excretion, so that high circulating levels are found in patients with renal failure [89–91]. Its role in normal human subjects is debated, since neither the absence of this hormone (as in completely thyroidectomized patients) nor its thousand-fold excess (as in patients with thyroid medullary carcinoma) is generally associated with any abnormality of calcium homeostasis or skeletal integrity [92]. However, there is around 40% structural homology between PTH and calcitonin receptors, the latter being found in bone, kidney, central nervous system, testis, placenta and on some tumour cells.
Clinical and Radiological Features of Renal Osteodystrophy
Clinical features of the altered mineral and skeletal metabolism that occurs in renal failure may be considered under two broad headings: extraskeletal and skeletal manifestations (Table 22.1 ).
Extraskeletal Manifestations
Extraskeletal manifestations are a result of deposition of phosphate and calcium in soft tissues. Calcium deposition in the skin may contribute to the pruritus that in severe cases can be quite disabling for dialysis patients, preventing sleep and resulting in widespread excoriations with skin sepsis [93]. Calcification in the conjunctiva is another common problem leading to the intensely painful “red eye,” with flecks of calcium often clearly visible on examination.
Perhaps the most important extraskeletal manifestation is calcification of the vascular tree that frequently becomes visible on plain X-rays. Most commonly the calcification is localized to the medial layer of small and medium-sized arteries (Monckeberg’s sclerosis) [40, 94–98]. However, the abdominal aorta, femoral and digital arteries are often clearly outlined on films taken for skeletal survey, but the same process is undoubtedly occurring in the mesenteric, cerebral and coronary vasculature, resulting in considerable morbidity and mortality in dialysis and transplant patients [36, 99]. In severe cases, vascular calcification in the iliac and femoral vessels may render a patient untransplantable because anastomosis of the vessels becomes impossible. Furthermore the risk of preoperative myocardial infarction is greatly increased, and heart failure, controlled by strict attention to fluid balance while on dialysis, may be unmasked by renal transplantation.
Although hyperphosphatemia can cause an increase in the calcium–phosphate product, to a point where its solubility product is exceeded and precipitation may occur, analysis of vascular tissue has shown that calcification is not simply a passive process. Rather, under the right conditions, vascular cells can express bone cell surface markers and lay down hydroxyapatite, suggesting a much more active and regulated process. Recent studies have shown that arterial calcification is an active process that is regulated by a variety of genes and proteins [100]. Arterial calcification appears to be a process similar to bone formation implicating a variety of proteins involved in bone and mineral metabolism detected in atherosclerotic plaques and/or medial calcifications Since elevations of phosphate, calcium, and calcium–phosphate product have been associated with mortality and morbidity, it is now widely believed that vascular calcification could be the link.
A study of the calcium–phosphate product in a large number of hemodialysis patients demonstrated that the relative risk of death for those with a serum phosphate level greater than 6.5 mg/dL was 1.27 relative to those with a lower level [101]. The increased risk was not reduced by statistical adjustment for coexisting medical conditions, delivered dose of dialysis, PTH level, nutritional parameters or markers of noncompliance. The calcium–phosphate product showed a mortality trend similar to phosphate alone, with those patients having products greater than 72 mg2/dL2 showing a relative mortality risk of 1.34 compared to those with products less than 52 mg2/dL2. Similar findings have been reported from other studies of dialysis patients [102] as well as those with earlier CKD [103]. There seems little doubt that tight control of serum phosphate is vitally important for any dialysis patient, although it must be remembered that these studies are retrospective and not interventional.
Skeletal Manifestations
Skeletal signs and symptoms of renal osteodystrophy include bone pain, bone tenderness, spontaneous fractures, retardation of growth, and joint disease. With the exception of adolescents with tubulo-interstitial pathology, symptoms are unusual in patients with end-stage renal disease, unless the decline in renal function has been particularly slow. However, those with tubulo-interstitial disease and adolescent patients are more prone to overt bone disease. The prevalence of symptoms among dialysis patients varies greatly from unit to unit, which may reflect differences in reporting or true differences due to a dialysis-induced cause such as aluminium intake [104, 105]. Both osteomalacia and osteitis fibrosa may be associated with bone pain, tenderness, fatigue, and proximal muscle weakness. In addition, lower back and lower limb pain contribute to the reduced exercise ability that is common in dialysis patients. This in turn worsens muscle weakness and loss of skeletal mass. Periosteal new bone growth and osteosclerosis are usually asymptomatic but may often be seen on skeletal radiography.
Radiological Features
Regular radiological assessment using plain radiographs – the traditional annual “skeletal survey” – is now rarely carried out in order to monitor renal osteodystrophy since radiological signs of hyperparathyroid disease detected by such radiographs are relatively late features.
The earliest radiological feature of hyperparathyroidism is subperiosteal erosion occurring at the tufts of the terminal phalanges, the radial aspect of the middle phalanges and the distal ends of the clavicles. In a study of 30 end-stage renal failure patients, performed immediately prior to commencing dialysis, erosion of the terminal phalanges was only seen in five of the eight patients who had severe hyperparathyroid disease on bone biopsy and serum PTH values [18]. However, plain radiographs did not identify patients with mild hyperparathyroid disease, and the majority of patients were judged to have essentially normal skeletal surveys. These findings are in agreement with those of Owen et al., who compared plain skeletal radiology with bone histology in 82 patients with renal failure, and found no correlation between radiological and histological indices [106] Malluche and Faugere agree that information obtained from skeletal X-rays is limited and often misleading, and that most radiological signs considered to be pathognomonic of severe osteitis fibrosa can be found in any of the three histological types of renal osteodystrophy [16]. In addition, a skeletal survey provides a relatively high dose of ionizing radiation for such inconclusive information.
Low turnover adynamic bone may have no specific radiological features [36], and the introduction of oral vitamin D metabolites has largely abolished osteomalacia so that it is rarely found, even in histological studies [107].
In recent years, other radiological techniques have been developed for examining bone in a more quantitative fashion, including skeletal scintigraphy, measurement of bone density and mineral content by single or dual photon densitometry, plus single and dual energy quantitative computed tomography (QCT) scan [108, 109]. These techniques are discussed later in the chapter.
Renal Osteodystrophy and PD
The introduction of CAPD in the 1970s provided new opportunities for the investigation and management of renal osteodystrophy. However, reports of its management in CAPD remain confusing, with some showing improvement [55, 104, 110] and others showing deterioration [111–113].
The different pattern of bone lesions seen in PD and hemodialysis is now well described [9–12, 114]. There are several differences between the dialysis modalities which may affect mineral metabolism [11]. PD is associated with far greater losses of middle- and large-molecular-weight protein fractions, thereby removing more transferrin-bound aluminium, as well as 25-hydroxyvitamin D3. With PD, weekly phosphate removal is greater than hemodialysis, and it provides a steady-state biochemical profile unlike the “saw-tooth” pattern of hemodialysis. Furthermore, the high calcium concentration in standard peritoneal dialysis fluids of the past may have significantly suppressed PTH levels, contributing to the higher incidence of low-turnover bone disease seen in PD. In contrast, a hemodialysis patient may experience episodes of relative hypocalcemia two or three times each week, which may well stimulate PTH production.
Calcium and Phosphate Balance in PD
In end-stage renal disease serum phosphate levels begin to rise and ionized calcium levels begin to fall once the GFR is less than 20 mL/min. These abnormalities can be at least partially corrected by the administration of oral calcium carbonate, although some concern exists about its use in CKD 5. However, ionized calcium levels may still be low (0.9–1.1 mmol/L) when patients start PD, even though total serum calcium levels are normal [115]. Serum levels usually rise once PD has begun, despite the majority of patients now using dialysis fluids with a calcium concentration of 1.25 mmol/L. Gastrointestinal absorption and peritoneal flux of calcium during dialysis are the two major determinants of overall calcium mass balance in peritoneal dialysis patients.
Gastrointestinal Absorption
The gastrointestinal absorption of calcium has been studied by several groups and is known to be dependent on many factors including the degree of uremia, serum phosphate level, PTH level, 1,25-dihydroxyvitamin D3 level, and total calcium intake. In uremic subjects, Recker and Saville [116] found that calcium absorption ranged from 5 to 59%, while Clarkson and colleagues [117] and Ramirez et al. [118] reported figures of 8% and 28%, respectively. In a study of CAPD patients in our own unit, calcium absorption rate was subnormal in 18 of 19 subjects, although significant variation existed between patients. Percentage absorption ranged from 3.2 to 23.9%, results not dissimilar from those in uremics [119]. Blumenkrantz examined absorption of dietary calcium in CAPD patients over the range 500–2,500 mg/day [120] and suggested that it can be represented by the empirical relationship Y = 0.42X – 277 (where Y = amount absorbed, X = intake in mg/day). Therefore if the intake is around 730 mg/day, approximately 30 mg of calcium is absorbed by the patient.
If calcium salts are to be used as first-line therapy for hyperphosphatemia then oral intake and gastrointestinal absorption will be necessarily high. Once a patient is established on dialysis, control of hyperphosphatemia is very important, not only to minimize further stimulation of PTH secretion, but also to keep the calcium–phosphate product within the normal range. Failure to do this can result in rapid progression of vascular and soft-tissue calcification. The PD patient’s high-protein diet (recommended minimum protein intake 1.2 g/kg/day) provides an obligatory phosphate intake of up to 1,200 mg daily [120, 121]. Although peritoneal dialysis controls the hyperphosphatemia of end-stage renal disease more effectively than does hemodialysis [113, 122, 123], it removes only 310–320 mg/day [120, 124] – rather less than one-third of the amount required to bring phosphate levels into the normal range. Therefore, if a neutral phosphate balance is to be achieved, the gastrointestinal elimination of phosphorus needs to be around 700 mg/day [125]. Since 40–80% of dietary phosphorus is absorbed by patients with renal failure [123], the fraction of phosphate absorbed must be reduced, and hence gastrointestinal elimination increased, by oral phosphate-binding agents.
The Role of Calcium Salts in Renal Osteodystrophy
Established phosphate binders, available for clinical use, include aluminium hydroxide and carbonate, calcium carbonate and acetate, magnesium carbonate, keto-analogues of amino acids, sevelamer hydrochloride, and lanthanum carbonate (Table 22.2 ). Each of these binders has advantages and disadvantages, but only four are in widespread use – calcium carbonate, calcium acetate, sevelamer hydrochloride, and lanthanum carbonate. Calcium carbonate and acetate remain in regular use in many countries as first-line phosphate binders in PD patients, where maintenance of optimal serum calcium and phosphate levels is central to the treatment of hyperparathyroidism. However, concern has arisen that calcium overload may contribute to adynamic bone and vascular calcification [94, 97]. Therefore, the U.S. National Kidney Foundation Kidney Disease Outcomes Quality Initiative (K/DOQI) guidelines suggest limiting the dose to a maximum of 1.5 g elemental calcium per day [126]. Calcium ketoglutarate is also in use, particularly in certain European countries [127].
When CAPD was first introduced aluminium gels were the standard phosphate binders, and a high calcium concentration in the dialysis fluid (1.75 mmol/L–3.5 mEq/L) was therefore beneficial, rapidly bringing serum calcium levels into the normal range. As the dangers of aluminium accumulation became apparent [85, 128], aluminium-containing phosphate binders were replaced by calcium salts as first-line therapy for hyperphosphatemia in most renal units. Unfortunately calcium salts frequently result in hypercalcemia when given in sufficiently large oral doses to control serum phosphate [129–135], so that aluminium-containing phosphate binders continued to be used. It has been suggested that, when used in low doses, with careful monitoring of serum aluminium, these binders are safe [85]. However, although only about 1% of the oral dose of aluminium is absorbed [136], even on modest doses this represents between 5 and 10 mg of elemental aluminium daily. Since PD removes only 40–50 mg daily [137] it is evident that tissue accumulation is inevitable and significant. Reduction of dialysis fluid calcium concentration was advocated as a means of extending the use of calcium salts and preventing hypercalcemia and was studied both in hemodialysis [138, 139] and peritoneal dialysis patients [140–146].
The demonstration of an association between mortality and high/normal calcium levels by United States Renal Data System (USRDS) and Dialysis Outcomes and Practice Patterns Study (DOPPS) data [101, 102, 147] has persuaded many physicians that utilizing high/normal serum calcium levels to suppress PTH is no longer tenable. However, no prospective randomized trials have been undertaken so far to validate current guidelines. In order to maintain serum calcium below 2.37 mmol/L while controlling hyperphosphatemia use of the more expensive non-calcium-containing phosphate binders is required. This is impossible in many countries around the world on grounds of cost, and is even causing concern the United States [148–150].
Control of serum phosphate is not only important in terms of its effect on PTH, but also for prevention, and sometimes treatment, of extraskeletal calcification. This potentially lethal aspect of renal osteodystrophy is a particular hazard in patients who have persistent hypercalcemia and hyperphosphatemia. It was traditional to prescribe aluminium-containing binders for such patients on the grounds that the aluminium absorbed is likely to be less harmful than allowing the process of vascular calcification to continue unchecked. Utilization of noncalcemic phosphate binders is now a more appropriate (and expensive) approach.
Noncalcemic Phosphate Binders
A more efficient dialysis fluid is an attractive way of improving phosphate clearance, and this was tried in the early 1980s in an experimental rat model [151]. Polyethylenimine was utilized as an osmotic agent, and demonstrated measurable binding to phosphate. Unfortunately it was toxic to the rats and produced gross morphological changes in the visceral mesothelium and associated organs. A variety of other oral phosphate-binding agents have been tested and some are in clinical use [152]. However, none has completely abolished the problem of hyperphosphatemia because of problems with efficacy, potency or palatability.
Two noncalcemic compounds now generally available are poly[allylamine hydrochloride] (RenaGel, Geltex Pharmaceuticals, Waltham, Mass., USA) and lanthanum carbonate (Fosrenol, Shire Pharmaceuticals, Andover, UK). Poly[allylamine hydrochloride] is a nonabsorbable calcium- and aluminium-free compound that is as effective as calcium carbonate [153]. It also has a beneficial effect on the patients' lipid profile with a reduction in serum total and LDL cholesterol.
Lanthanum was first reported as a potential oral phosphate binder by Graff and Burnel [154]. Phosphate binding was estimated by the reduction of urinary excretion and increase in faecal excretion, and lanthanum citrate was found to be as effective as aluminium chloride, with less (but not zero) systemic absorption. Human studies have confirmed its phosphate binding properties in vivo [155–159] and it has been studied for up to 3 and 6 years, and has been commercially available since 2005. Although concerns exist in some quarters about its long-term safety, no clinical data has emerged to justify this [160]. The renewed interest in control of serum phosphate has reinvigorated the search for other more effective binders and several are in various stages of development [161].
Peritoneal Flux and Reduced Calcium Dialysis Fluid
During an exchange of 2 L of 1.36% glucose, 1.75 mmol/L calcium PD solution there is a net influx of calcium to the patient, although the amount varies from one study to the next (84 ± 18 mg/day [120], 300 mg/day [162]). The transfer of calcium is also influenced by ultrafiltration rate and volume [163], so that a 1.36% glucose solution results in a 10 mg calcium uptake by the patient but the greater ultrafiltration from a 3.86% solution leads to a loss of 20 mg. This gives a net daily absorption of 10 mg if one hypertonic bag is used per day. In another study, Kwong et al. [164] found an uptake of 29 mg per 1.36% exchange and a loss of 6 mg per 3.86% exchange, suggesting a larger net gain of around 80 mg daily. However, a lower PD fluid calcium concentration of 1.5 mmol/L causes the balance to become negative with a loss of 50 ± 36 mg/day [105]. These findings suggest that patients using dialysis solutions containing 1.75 mmol/L of calcium are in a significantly positive calcium balance even before considering the additional gut absorption from oral calcium carbonate and vitamin D therapy.
The theoretical work by Martis et al. [125] formed the basis for the commercial production of a peritoneal dialysis fluid with a calcium concentration of 1.25 mmol/L, in an attempt to decrease the incidence of hypercalcemia in CAPD patients taking oral calcium salt phosphate binders. Clinical studies have now confirmed this theoretical work [145, 165] and it is now the commonest concentration in general use. Although dialysis fluids with other concentrations of calcium can be obtained (0, 0.60, 1.00, 1.45 mmol/L), 1.25 mmol/L would appear to be the logical choice for a standard PD fluid because it is so close to normal serum ionized calcium levels. This results in a homeostatic effect, with calcium being lost into the peritoneum when serum levels are above 1.25 mmol/L, but being absorbed from the peritoneum during times of relative hypocalcemia. All other proposed calcium concentrations are outside the normal range of serum ionized calcium and therefore cannot exert this homeostatic effect.
Convective effects of ultrafiltration increase the removal of calcium from the peritoneum so that patients using one or more 3.86% glucose exchanges per day will have a significantly greater negative peritoneal calcium balance than patients using only 1.36% exchanges [166]. While in theory this could result in some degree of hypocalcemia, in practice it rarely occurs, as calcium absorption from oral phosphate binders is usually sufficient to compensate.
A 2-year prospective biochemical, radiological, and histological study of 1.25 mmol/L calcium PD fluid showed it to be safe in compliant, well-monitored patients [141, 144]. It allowed administration of larger doses of calcium carbonate (now considered less desirable) and achievement of good control of serum phosphate and calcium–phosphate product. Parathyroid hormone levels were suppressed in the majority of patients, and bone histology and density did not deteriorate.
Cunningham et al. [146] used 1.25 mmol/L calcium dialysis fluid to enable the use of calcium carbonate plus alphacalcidol in a group of CAPD patients. In 17 CAPD patients taking oral calcium carbonate, reductions in dialysis fluid calcium concentration to 1.45 mmol/L or 1.00 mmol/L enabled most of the patients to also take oral alphacalcidol. Parathyroid hormone, serum aluminium, and alkaline phosphatase levels were all decreased during the 11 months of the study, with the authors concluding that a dialysate calcium concentration of 1.75 mmol/L is too high for the majority of calcium carbonate–treated patients, and that substantial reductions of the dialysate calcium concentration are required. Other workers have used 1.0 mmol/L calcium fluid in PD patients, again with similar results [39, 167]. Utilization of solutions with calcium concentrations of 1.00 mmol/L and below put the patient into a permanent negative calcium balance, so that very close attention must be given to PTH levels and compliance with oral calcium and vitamin D therapy, but this approach has been reported to increase PTH levels and bone turnover in patients with adynamic bone [168].
Serum Magnesium in PD
Magnesium levels are consistently elevated in PD patients managed with standard dialysate containing 0.75 mmol/L of magnesium [140]. No toxicity has been reported at these levels, indeed hypermagnesemia may have a suppressive effect on PTH release and retard the development of arterial calcification in PD patients [169]. Hypermagnesemia may therefore be beneficial, but it has also been shown that normalization of serum magnesium is associated with an improvement in bone histology in hemodialysis patients [170]. Reducing the magnesium content to 0.25 mmol/L normalizes serum magnesium levels in CAPD patients [122, 165]. Parsons et al. [171, 172] have described the use of a low-calcium/zero magnesium PD fluid with a combination of calcium carbonate and magnesium carbonate in liquid form as the phosphate binder. Using this approach, mean serum phosphate levels of 1.4–1.5 mmol/L were obtained without causing hypermagnesemia, although hypomagnesemia was seen in two of 32 patients. Zero magnesium fluids have also been studied by Shah et al. [173] and offer the advantage of permitting larger doses of magnesium salt phosphate binders, but there are two disadvantages. First, patients may experience gastrointestinal upset, since magnesium salts have a laxative effect [174], and secondly monitoring of compliance and serum magnesium levels becomes obligatory, as hypomagnesemia has been associated with cardiac rhythm disturbances [175–177] and electrocardiographic abnormalities [178, 179].
Acid–Base Balance and 40 mmol/L Lactate PD Fluid
There is considerable evidence that as renal mechanisms for acid excretion fail, bone mineral stores become an important source of buffer [180, 181]. Acetazolamide produces a metabolic acidosis in normal subjects by inhibiting carbonic anhydrase in the proximal tubular epithelium, resulting in a bicarbonate diuresis. In virtually anuric hemodialysis patients it might therefore be expected to have little effect, but in fact produces a severe metabolic acidosis [182], suggesting that it is interfering with extrarenal buffering. Carbonic anhydrase is present in osteoclasts [183], and may be activated by PTH to promote bone resorption by release of H+ ions [184]. The availability of bone buffers and bicarbonate would therefore depend on the activity of PTH, and could be inhibited by acetazolamide. It can therefore be seen that during a time of prolonged metabolic acidosis, such as exists in many PD patients using PD fluid with only 35 mmol/L lactate [185], buffering by bone would be linked to bone resorption and increased PTH levels.
The use of PD fluids containing 40 mmol/L lactate, or newer so-called biocompatible fluids containing only bicarbonate, or bicarbonate plus lactate, correct the mild acidosis experienced by most PD patients using the older lower lactate concentration [186]. Optimal correction of acidosis has been shown to change the progression of osteodystrophy in hemodialysis patients by Lefebvre et al., who, over 18 months, prospectively studied two groups of patients, dialyzed against either standard dialysis fluid (32–24 mmol/L), or against fluid supplemented with 7–15 mmol/L of bicarbonate to achieve a predialysis plasma bicarbonate of 24 mmol/L. The supplemented group had a decreased rate of progression of secondary hyperparathyroidism in patients with high bone turnover, and stimulated bone turnover in those with low bone formation rates [187].
Parathyroid Hormone in PD
Although the prevalence of symptomatic bone disease has decreased in recent years, the 1989 EDTA Registry report showed that around 40% of all patients dialyzed for up to 15 years still required parathyroidectomy [188]. This partly reflects the poor understanding of the pathogenesis of secondary hyperparathyroidism that existed in the 1970s and 1980s, and partly the difficulty of monitoring vitamin D and PTH levels. However, only slightly better rates have been reported more recently from the Lombardy registry in Italy, where it was noted that PD patients had a higher likelihood of parathyroidectomy than HD patients [32]. In the multicenter DOPPS study, the adjusted rate of parathyroidectomy varied four-fold across the DOPPS countries, and was significantly associated with baseline concentrations of phosphorus, calcium, calcium-phosphorus product, PTH, and dialysate calcium concentration [102].
PD has been shown to clear significant amounts of PTH from the serum. Using a C-terminal assay Delmez et al. [163] found a clearance rate of 1.5 mL/min or 13.6 ± 3.2% of the estimated total extracellular iPTH. Despite this, there is no clearcut consensus on the effect of PD on PTH levels, although the weight of evidence is in favor of a steady decline with time [104, 110, 189, 190]. However, other reports show no change [163], an increase in the levels [111] or a variable response [112]. The reason for these differences probably lies in the widely varying practices between centres with regard to the use of phosphate binders, vitamin D3 treatment and also the different radioimmunoassays used for measurement of iPTH and its fragments.
Until the 1990s, CAPD tended to be seen as a prescription in itself, with a standard set of guidelines that were suitable for every patient. As a result, the majority were treated with four 2-L exchanges, a phosphate binder, vitamin supplements and a small oral dose of 1,25-dihydroxyvitamin D3. PTH levels were rarely measured, and the dosage of calcitriol was changed only if hypercalcemia occurred or evidence of osteitis fibrosa appeared on plain radiology of the hands.
Maintenance of a high serum ionized calcium (1.2–1.3 mmol/L) and strict control of serum phosphate, from the time of first starting dialysis, has been shown to decrease PTH levels in CAPD patients without the addition of vitamin D3 therapy [142, 143]. However, the increased incidence of adynamic bone, its association with vascular calcification and studies of bone turnover rate, have resulted in a plethora of guidelines suggesting that the appropriate target range for PTH is somewhere between 2 and 5 × ULN (upper limit of normal) [126]. Although these opinion-based guidelines seem sensible given the current state of our knowledge, they have never been tested in a randomized controlled, outcome trial, and it is not known whether targets for PD patients should be different.
In a large and long-term study of PTH and all-cause mortality in 345 HD and 277 PD patients, over 14 years, survival after adjustment for age, race, gender, months on dialysis at enrollment, diabetic status, and nutritional markers were significantly better for patients with enrollment PTH greater than 200 pg/mL than for patients with PTH 65 to 199 pg/mL and patients with PTH less than 65 pg/mL. For PD patients, age, diabetes, and months on PD at enrollment were inversely associated with PTH, whereas black race, albumin, creatinine, and phosphate were associated positively [191]. However, in a very similar size study of 251 PD patients, Oreopolous’s group found no such association [192] emphasizing the difficulty of interpreting observational, retrospective chart studies.
Vitamin D in PD
Vitamin D metabolism is well known to be abnormal in uremia, with very low levels of 1,25-dihydroxyvitamin D3 [62]. However, there are additional factors affecting vitamin D levels relating to the PD itself. Levels of 1,25-dihydroxyvitamin D3 are known to be very low, and sometimes undetectable, at the start of PD if prior treatment has not been given [18]. 25-Hydroxyvitamin D3 levels are usually within the normal range at the start of CAPD but begin to decline thereafter [104, 124]. This is not unexpected since peritoneal dialysis effluent contains significant amounts of vitamin D binding protein, an α2-globulin of molecular weight 59 kDa, which binds all three vitamin D metabolites (1,25-dihydroxyvitamin D3, 25-hydroxyvitamin D3, and 24,25-hydroxyvitamin D3). Losses of 1,25-dihydroxyvitamin D3 and 24,25-dihydroxyvitamin D3 have been shown to average approximately 6–8% of the plasma pool per day [193]. Thus, PD patients may require 2–3 times the maintenance doses used in hemodialysis patients if it is thought necessary to bring serum levels of 1,25-dihydroxyvitamin D3 into the normal range, These doses frequently produce the problem of hypercalcemia.
In England a seasonal variation in 25-hydroxy-vitamin D3 levels was found by Cassidy et al. [194], and in sunnier climates patients may be able to maintain 25-hydroxyvitamin D3 levels within the normal range throughout the year. Whether 24,25-dihydroxyvitamin D3 plays an important role in bone mineralization remains to be proved, but Dunstan et al. [195] have shown that the combination of 1,25-dihydroxyvitamin D3 and 24,25-dihydiroxy-vitamin D3 given orally did not appear to confer any additional benefit compared with 1,25-dihydrox-yvitamin D3 alone. This result is challenged by the work of Chaimovitz, Gal-Moscovitzer, and others, who suggest that 24,25-dihydroxyvitamin D3 plays a role in the regulation of PTH levels [196–199], and when given in conjunction with 1,25-dihydroxyvitamin D3 suppressed osteoclastic parameters without causing hypercalcemia.
The Role of Vitamin D Analogue Therapy in PD
Vitamin D therapy in PD has traditionally been used for the treatment of elevated PTH levels (greater than 5–6 × ULN) in one of two ways. If serum calcium levels are low, then a daily oral dose of calcitriol or 1-alphacalcidol will raise serum calcium and thereby suppress PTH secretion. Alternatively if serum calcium is already towards the top end of the normal range then twice weekly, “pulse therapy” will produce larger peak serum vitamin D levels, which, in theory, should also suppress PTH secretion, but with less stimulation of gastrointestinal calcium uptake. Small-scale studies have provided some evidence for this approach [67, 70, 71, 200, 201].
Oral Pulse Calcitriol Therapy
The idea of pulse therapy was initially investigated in hemodialysis patients, where thrice-weekly intravenous pulses of 1,25-(OH)2D3 were administered at the end of a hemodialysis session [53]. This regime resulted in marked suppression of iPTH levels with a mean decrement of around 70%, although Slatopolsky surmised that this was largely as a result of the rise in serum ionized calcium that occurred during the study. Furthermore, this study also demonstrated that when equal doses of intravenous or oral calcitriol were given, the serum concentration of calcitriol was 6–8 times higher with the intravenous preparation, resulting in a greater delivery to nonintestinal target tissues and allowing greater expression of its biological effect on the parathyroid glands. However, even this degree of suppression was insufficient to restore iPTH to satisfactory levels, because of the large nonsuppressible basal secretion rate of hypertrophied glands. Interestingly, it has also been shown that administering calcitriol at night reduces both the incidence and severity of hypercalcemia in hemodialysis patients [202].
Korkor demonstrated that the parathyroid glands from patients with chronic renal failure contained only one-third as many calcitriol receptors as are found in parathyroid adenomas [203], and in animal studies it is known that uremia results in a two- to four-fold decrease in receptor numbers as compared to normal values [204, 205]. Thus it is likely that reduced receptor numbers in the parathyroid glands of uremic patients render them less responsive to the inhibitory effects of calcitriol, so that suppression requires high peak serum levels.
Since the initial work of Slatopolsky several other workers have confirmed that both pulse intravenous 1,25-dihydroxyvitamin D3 and α-calcidol are effective in reducing serum PTH levels in hemodialysis patients [65, 66, 70], although all found difficulty in distinguishing direct effects on parathyroid secretion from indirect effects mediated by raising serum calcium. Subsequent studies in CAPD patients have tended to confirm this work, but some controversy still exists over the ideal regime of administration [206–208]. It is, however, clear that 1,25-dihydroxyvitamin D3 does have a direct suppressive effect on parathyroid cells by influencing transcription of the parathyroid gene [52, 205, 209].
With current concern about vascular calcification and calcium loading, oral pulse therapy would appear to be the most appropriate method of administration for PD patients, but outcome studies are once again lacking, and cinacalcet may have obviated the need to study this further. However, vitamin D manufacturers are “fighting back” with retrospective, observational data to suggest that perhaps vitamin D therapy confers a small survival benefit in HD patients [210], and its immunological effects may yet turn out to be important to dialysis patients.
Intraperitoneal Vitamin D Therapy
Delmez et al. demonstrated that calcitriol could also be given intraperitoneally in CAPD patients where again it produced a rise in serum ionized calcium levels and a significant fall in serum PTH [55]. However, continued control of serum phosphate is also required, since hyperphosphatemia will significantly blunt the effect of calcitriol therapy [211]. It has been shown that 22-oxa-calcitriol can be given effectively by the intraperitoneal route, but adherence to PD vinyl bags varies and may result in uncertain serum levels [212, 213]. However, the risk of introducing infection while injecting vitamin D, and the fact that such a route has additional practical difficulties in automated PD has prevented this technique from gaining general acceptance.
Calcitriol Analogues
In rats with normal renal function 22-oxa-calcitriol has been shown to have very little calcemic activity [214], yet it suppresses PTH mRNA levels equally as effectively as 1,25-(OH)2D3 [215]. A similar effect is reported in dogs with over 12 months of renal failure, where administration of a single intravenous dose of 5 mg of 22-oxa-calcitriol decreased PTH by 80% over 24 h without any change in serum calcium or phosphate levels [215]. However, Drueke and co-workers found similar degrees of hypercalcemia in rats with chronic renal failure treated with either 1,25-(OH)2D3 or 22-oxa-calcitriol [216], and this observation has been confirmed in other studies of hyperparathyroid hemodialysis patients, where PTH suppression was associated with a rise in serum calcium [217–219]. Other analogues, such as 2b-3-hydroxypropoxy calcitriol, EB 1089, and KH 1060, are under investigation as immunosuppressive agents, as well as for their effects on divalent ion metabolism [220].
Although prospective comparisons against calcitriol are very limited, current evidence suggests that new active vitamin D analogues, such as 19-nor-paracalcitol and doxercalciferol, adequately control PTH levels with minimal changes in serum calcium and phosphate during treatment with calcium-containing phosphate binders. In the past, the development of adynamic bone occurred in a substantial proportion of dialyzed patients treated with calcitriol therapy and calcium containing binders. Therefore, the long-term skeletal response to the new active vitamin D sterols remains to be established and whether patients will develop adynamic bone is not known at the present time. Furthermore, the impact of the addition of calcium-free phosphate binders on the control of secondary hyperparathyroidism in conjunction with the new active vitamin D analogues remains to be studied. A retrospective, observational study has suggested a small improvement in survival with intravenous paricalcitol but such data cannot be regarded as scientifically robust at this time [210]. Whether the cost of these new analogues can be justified by improved patient outcomes remains to be proven.
Calcimimetics
Compounds that act as calcium receptor agonists are called calcimimetics because they mimic or potentiate the effects of extracellular calcium on parathyroid cell function. By targeting the calcium sensing receptor, cinacalcet provides a new means of regulating PTH secretion by amplifying the receptor’s sensitivity to extracellular calcium and reducing PTH concentrations [221]. The discovery of such compounds with potent and selective activity enables a pharmacological approach to regulating plasma levels of PTH.
Results from clinical trials examining single and multiple doses up to 180 mg once daily suggest that treatment with cinacalcet not only reduces plasma PTH concentrations but also leads to a concomitant decrease of serum calcium and phosphate in patients with secondary hyperparathyroidism receiving hemodialysis [222–225]. It reduces PTH to within K/DOQI targets in 44–56% of patients with a greater number (∼60%) achieving at least a 30% reduction in serum PTH from baseline [226]. Analyses of combined data from randomized, blinded, placebo-controlled, 6- to 12-month studies of cinacalcet versus standard care for secondary HPT showed statistically significant and clinically meaningful reductions in the risks of parathyroidectomy, fracture, and cardiovascular hospitalization. Although the individual clinical studies were designed to assess changes in biochemical parameters and not a priori clinical end points, the prospective and interventional nature of the combined data lends credibility to the clinical relevance of biochemical control in secondary HPT and suggests that therapy with cinacalcet may lead to beneficial effects on clinical outcomes [227]. Long-term treatment has been shown to effectively sustain reductions in PTH for up to 3 years [228]. Cinacalcet’s efficacy appears to be similar in both hemodialysis and peritoneal dialysis patients [229].
Side effects reported in a trial of cinacalcet [230] compared to placebo include nausea (32 versus 19%) and vomiting (30 versus 16%). The frequency of nausea was unrelated to dose, but vomiting occurred more frequently at higher doses. Hypocalcemia (<1.90 mmol/L) occurred significantly more frequently in cinacalcet-treated than placebo-treated patients (5 versus 1%). In the titration phase of this study more cinacalcet-treated patients than placebo-treated patients withdrew (15 versus 7%), and just less than another 5% of patients later withdrew from treatment as a result of these adverse effects.
Renal Osteodystrophy in Diabetic PD Patients
A number of reports over the past 12 years have suggested that insulin-dependent diabetes mellitus is associated with lower serum levels of PTH [114, 231, 232], and decreased responsiveness to acute hypocalcemia [233]. Therefore, it has been suggested that diabetic patients may be more prone to low-turnover bone disease, and relatively protected from severe hyperparathyroidism. However, until recently it has been difficult to separate the effects of diabetes from those of aluminium accumulation, but Pei et al. [114] have now shown that diabetes mellitus is an important risk factor for both aluminium-related bone disease and the adynamic bone lesion. These authors also noted that diabetes appears to enhance the risk of developing aluminium bone disease, possibly by increasing gastrointestinal absorption and bone surface accumulation of aluminium. This finding adds further impetus to the drive to eliminate use of aluminium-based phosphate binders wherever possible.
The Idiopathic Adynamic Bone Lesion in PD
The spectrum of bone disease has changed over the past 15 years with the emergence of the adynamic bone lesion, now present in 20–60% of dialysis patients according to various histological studies [9, 23, 25, 35, 36, 234, 235], and the welcome decline in the usage of aluminium-containing phosphate binders. The association of adynamic bone with low PTH levels has resulted in a reassessment of attempts to suppress serum PTH to “normal” levels in both predialysis and dialysis patients [34, 35] and acknowledgement that blanket prescription of continuous low-dose oral calcitriol therapy for all dialysis patients is unwise [236].
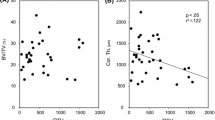
This lesion occurs more commonly in patients aged over 50 years at start of dialysis (Table 22.3 ), in those with a longer duration of predialysis renal failure [36], in diabetic patients [114], and in PD patients compared to HD patients [28]. In a longitudinal histological study of bone disease in CAPD [142], five of eight patients who completed a full 2 years of follow-up were found to have developed the adynamic lesion (Fig. 22.5 ). None of the patients reported symptoms attributable to the lesion, and it has no characteristic radiological findings. Bone mineral density did not decline over the 2 years, but even longer follow-up may be required for signs and symptoms to appear. It is associated with normal or suppressed levels of parathyroid hormone [9, 28, 36, 237, 238], and high or high normal levels of ionized calcium. Sherrard et al. noted that the adynamic bone lesion was the commonest histological diagnosis in their study of 267 dialysis patients [10], and that it occurred more frequently in PD (61%) than in hemodialysis patients (36%). They suggest that this may be due to the more sustained and higher calcium levels associated with PD, which may result in more effective suppression of PTH than the intermittent calcium load of hemodialysis.

Changes in bone histology in eight patients over 24 months of continuous ambulatory peritoneal dialysis. OM, Osteomalacia; HPTH, hyperparathyroidism
Perhaps the most significant feature of the adynamic bone lesion is its association with vascular calcification, which may be related to the higher serum calcium levels found in patients with this lesion. This is a worrying association since many of these patients will be hoping for renal transplantation, which may become impossible when vascular calcification is severe. Evidence has accumulated pointing to the active and regulated nature of the calcification process. Elevated phosphate and calcium, common in patients with adynamic bone, may stimulate sodium-dependent phosphate co-transport involving osteoblast-like changes in cellular gene expression. Arterial calcification is responsible for stiffening of the arteries with increased left-ventricular afterload and abnormal coronary perfusion as the principal clinical consequences [239]. It also seems likely that adynamic bone would be significantly more prone to the osteoporotic effects of high-dose steroid immunosuppression, as well as avascular necrosis of the femoral head [240]. Treatment of osteoporosis remains uncertain, since the long-term effects of bisphosphonates in CKH are unknown [241–243].
Under normal circumstances the skeleton provides a large buffering capacity for both serum calcium and phosphate. If the bone is adynamic its buffering ability may be significantly reduced, and serum levels are therefore much more easily influenced by dietary intake or absorption from the dialysis fluid. Under these conditions there is a greater likelihood of the calcium–phosphate product being exceeded, and the process of metastatic calcification beginning. If this is the case then it would be very important to allow PTH levels to rise slightly and stimulate bone turnover, while maintaining strict control of serum phosphate. In this way one would also hope to induce resorption of vascular calcium deposits [168]. It is evident that the use of calcitriol, a powerful modulator of calcium and PTH, should be tailored to the individual patient’s clinical situation, not merely prescribed in an unthinking fashion as a “vitamin supplement.” Tailoring of any therapy requires the clinician to gather certain data in order to determine appropriate management. In this case one needs to examine the patient’s serum calcium, phosphate, and parathyroid hormone levels, in conjunction with bone histology data in certain cases. On the basis of these findings one can plan individual treatment along the lines of the clinical algorithm shown in Fig. 22.6 .

Clinical algorithm for the monitoring and management of renal osteodystrophy in PD patients
Recommendations for Management of Osteodystrophy in PD
Techniques for monitoring renal osteodystrophy in PD patients are still evolving. In the past, different units have approached the problem in widely differing ways, with some only monitoring serum calcium, phosphate and alkaline phosphatase plus annual skeletal surveys, and others performing much more detailed (and expensive) investigations, sometimes including bone biopsy. If we consider the available techniques under three broad headings – biochemical, radiological and histological – certain recommendations can be made, given the present state of understanding (Table 22.4 ).
Biochemical Monitoring
In dialysis patients, changes in serum calcium and phosphate do not reflect disease processes within the skeleton as they may do in patients with normal renal function. They are primarily influenced by the patient’s diet and oral intake of phosphate binders plus the amount of dialysis that patient is receiving. A rise in serum alkaline phosphatase is more indicative of increased bone turnover, but is seen only in advanced hyperparathyroidism and levels are influenced by liver and intestinal production. A rising level is generally associated with histologically severe osteitis fibrosa. The bone isoenzyme of alkaline phosphatase is a more sensitive indicator and low levels are associated with a greater likelihood of adynamic bone histology [36]. Measurement of serum osteocalcin is possible, providing a marker of osteoblast activity, but the very short half-life of the molecule means that unless the serum is spun and frozen within 20 min any subsequent assay is likely to be invalid. For this reason it seems unlikely to be helpful in clinical practice, and in any case reports show a close correlation with intact PTH levels in dialysis patients [244]. Another marker of hyperparathyroid bone disease, tartrate-resistant acid phosphatase (TRAP), has recently been reported to be a sensitive indicator of high- and low-turnover bone states [245–247]. This enzyme is produced by actively resorbing osteoclasts, and can be measured in serum by immunoassay or colorimetrically. Experience with this marker is limited at present [247].
The most widely available, and generally useful, marker at present, is intact PTH, which should be measured every 3 or 4 months in all PD patients. Regular measurement of total alkaline phosphatase is unnecessary, although it will continue to be performed as part of the “liver screen.”
Radiological Monitoring
Routine, full skeletal surveys are unnecessary, and have largely been replaced by regular monitoring of serum PTH with plain X-rays of the hands plus a single lateral view of the lumbar spine [106, 248, 249]. This greatly reduces the dose of radiation previously received by PD patients without detriment to patient care. Ideally, some measurement of skeletal bone density should be made at the time of starting dialysis in all patients, so that those with subnormal bone density can be identified and special efforts made to improve the situation. Whether this measurement is best performed by QCT, SPA, DPA or DEXA scan would depend on local facilities, finance, and expertise.
Skeletal scintigraphy may be performed using technetium labeled 99m-methylene diphosphonate, but correlation of scan results with bone histology is poor, although increased uptake has been demonstrated in severe hyperparathyroid bone disease [250–252], and it has been demonstrated to be a method of monitoring response to treatment [253, 254].
Single-photon absorptiometry (SPA): A highly collimated beam of photons is used with 125I as a source. However, the single-energy beam requires a constant thickness of soft tissue around the bone to produce reliable results, and it cannot distinguish between trabecular and cortical bone. Studies have found that mineral content tends to be lower than normal in patients with renal failure [18, 255–257], but it does not allow differentiation between different types of osteodystrophy.
Dual photon absorptiometry (DPA): DPA utilizes an isotope with two energies (153 Gd), thereby overcoming the requirement of SPA for a constant thickness of soft tissue around the bone. It can therefore be used to measure bone mineral density in any skeletal site, but normal scan results have been seen in hemodialysis patients with histologically proven osteodystrophy [249].
Quantitative computed tomography (QCT): the principle of this technique is the same as for standard CT scanning, and a calibration phantom placed under the patient is used to relate bone mineral density to Hounsfield units. True bone density is measured in g/cm3 and it is less affected by artefacts such as soft tissue calcification. However, it delivers a relatively high radiation dose.
In our own experience, bone density measurements are, if anything, less helpful than plain radiographs, since there is no correlation with histological diagnosis. Although end-stage renal failure patients were found to have reduced bone density, compared to age- and sex-matched normal values, the majority of patients had both QCT and SPA results within 2 SD of normal. Piraino et al. used QCT to measure bone mineral density in a group of 31 patients who had been on dialysis (19 peritoneal dialysis, 10 hemodialysis) for a mean of 5.3 years [258]. It is now known that the type of dialysis influences bone histology [11], therefore it may be misleading to consider peritoneal dialysis and hemodialysis patients together. However, as in our study, Piraino et al. found that serum PTH was a better indicator of the type of bone disease present than was QCT-determined bone density, and concluded that the usefulness of this technique in patients with renal failure is limited.
Dual-energy X-ray absorptiometry (DEXA): this has become the most widely used method of quantifying bone mineral density in renal osteodystrophy and osteoporosis. It has several theoretical advantages over the other techniques, including low radiation dose, greater precision and rapidity of scanning. Despite this, measurements can be distorted by several artefacts such as osteophytes, calcification of the aorta or other soft tissues, and previous vertebral collapse. In 25 dialysis patients no correlation was found between QCT and DEXA measurements of vertebral bone mineral density [259]. Results can be difficult to interpret because low density can be associated with almost any type of osteodystrophy [18, 36, 260–262].
While single bone density measurements may be unhelpful, sequential studies might be more helpful, especially if one has initially established the histological diagnosis by biopsy. Decreasing bone density in a patient known to previously have osteitis fibrosa would suggest deterioration, especially if associated with a high iPTH.
Transiliac Bone Biopsy
Bone biopsy with tetracycline labeling is undoubtedly the only reliable method of diagnosing the type and severity of renal osteodystrophy. This is rarely performed in clinical practice because of patients' and doctors' perceptions of its painfulness and invasiveness. In our experience it is no more painful than Jamshidi marrow biopsies, which are routinely performed in many clinical situations if the blood film suggests it is necessary.
Local expertise permitting, PD patients could undergo a tetracycline-labeled bone biopsy at the time of starting dialysis, to establish the exact histology of their bone plus the degree of aluminium accumulation. In many cases this could be performed under general anesthetic by the surgeon as the Tenckhoff catheter is inserted. Thereafter, one can probably surmise what is happening to the histology on the basis of regular PTH measurements once the initial histology is known. However, few centers have the expertise to either perform a biopsy or to interpret them histologically.
Most countries have created guidelines for management of bone and mineral metabolism, or else they follow European Best Practice or K/DOQI. However, all these guidelines have similar underlying principles. They all depend first on maximal restriction of dietary phosphate, within the limitations of a diet providing 1.2 g protein/kg body weight per day, and usually on the routine use of PD fluid with a calcium concentration of 1.25 mmol/L, magnesium of 0.25 mmol/L, and lactate of 40 mmol/L or bicarbonate buffer. Appropriate phosphate binders should be given in doses adequate to maintain serum phosphate below 1.8 mmol/L and aluminium-containing binders should be avoided. If hypercalcemia occurs, calcium carbonate could be partially or completely replaced by lanthanum carbonate [157] or sevelamer hydrochloride [153]. Serum ionized calcium and iPTH should be measured every 3 months to provide a guide for treatment with oral calcitriol. If iPTH is less than 300 pg/mL (or 5 × ULN) then vitamin D3 is not required. If iPTH exceeds 300 pg/mL then attention should be paid to maintenance of a mid-range serum calcium level and tighter dietary control of phosphate where possible. Calcitriol should be given as intermittent-pulse doses [71], and iPTH remeasured after 3 months. If the level remains significantly elevated, or is rising, then introduction of cinacalcet may be appropriate if it is financially affordable. If PTH continues to rise appropriate scans should be ordered to search for a parathyroid adenoma with a view to possible parathyroidectomy (provided the patient does not have significant aluminium deposition).
Serum magnesium levels should be measured once every 6 months, especially in those patients whose dietary intake may be low, to check for hypomagnesemia. Plasma aluminium levels should also be monitored every 6 months depending on local circumstances, although some units now reserve this only for patients taking aluminium-based phosphate binders. It must be remembered that aluminium uptake can be significantly enhanced by citrate ingestion and oral vitamin D3.
Utilization of any carefully thought out guidelines should enable reasonable control of divalent ion metabolism, and prevention or amelioration of osteodystrophy in the majority of PD patients. However, while the targets for calcium, phosphate, calcium-phosphate product, and PTH are more easily achieved in PD patients than HD [263], some may be mutually exclusive. Further advances will hinge on a greater understanding of the vitamin D/parathyroid axis in renal failure, along with the development of safe, noncalcemic analogues of vitamin D3 plus more efficient phosphate binders and calcimimetics, and most importantly prospective, randomized, controlled, interventional studies to examine patient outcomes.
Summary
Disorders of calcium, magnesium, phosphate, vitamin D3, and parathyroid hormone can combine to produce a variety of skeletal and extraskeletal pathologies in PD patients. The recognition that osteodystrophy is linked to vascular disease and patient mortality has reinvigorated research into the underlying mechanisms and the creation of new pharmaceutical agents.
New noncalcemic phosphate binders have now entered clinical use, and further increase the options available to clinicians. Calcimimetic agents are also available and provide an effective means of controlling PTH independently of serum calcium levels. Vitamin D analogues are currently perhaps of least certain value in PD, especially since they are largely only available for intravenous use. However, new pharmaceuticals come with significant expense, to the point that many patients, even in wealthy countries, will never be able to receive them, and at present their effect on patient outcome remains unknown. It is imperative that both pharma and academia combine to demonstrate these agents' benefit, or lack of benefit, in order not to waste large amounts of money which might be better invested in the dialysis process itself.
The judicious use of a variety of biochemical and radiological investigations, plus bone histomorphometry where appropriate, can provide a rational basis for the monitoring and management of divalent ion metabolism and renal osteodystrophy in PD patients. Sadly, most guidelines can at present only be based on opinion, and therefore we must heed the words of the K/DOQI authors: Since the majority of the recommendations made in this set of guidelines are based on opinion, it is imperative that evaluation of their clinical outcomes be made a component of their implementation. In addition, the paucity of evidence based information in this field requires that a more integrated approach to research efforts be planned and conducted to provide answers to the many issues that remain to be elucidated [126].
References
Lucas R. On a form of late rickets associated with albuminuria, rickets of adolescence. Lancet 1883; i: 993–994.
Fletcher H. Case of infantilism with polyuria and chronic renal disease. Proc R Soc Med Lond 1911; 4(Sect Study Dis Child): 95–97 .
Barber H. The bone deformities of renal dwarfism. Lancet 1920; i: 18–20.
Follis R, Jackson D. Renal osteomalacia and osteitis fibrosa. Bull Johns Hopkins Hosp 1943; 72: 232–234.
Liu S, Chu H. Studies of calcium and phosphorus metabolism with special reference to pathogenesis and effects of dihydrotachysterol (A.T. 10) and iron. Medicine 1943; 22: 103–107.
Garner A, Ball J. Quantitative observations on mineralised and unmineralised bone in chronic renal azotaemia and intestinal malabsorption syndrome. J Pathol Bacteriol 1966; 91: 545–549.
Stanbury SW, Lumb GA. Metabolic studies of renal osteodystrophy. I. Calcium, phosphorus and nitrogen metabolism in rickets, osteomalacia and hyperparathyroidism complicating chronic uremia and in the osteomalacia of the adult Fanconi syndrome. Medicine (Baltimore) 1962; 41: 1–34.
Stanbury SW, Lumb GA. Parathyroid function in chronic renal failure. A statistical survey of the plasma biochemistry in azotaemic renal osteodystrophy. Q J Med 1966; 35: 1–23.
Malluche HH, Monier-Faugere MC. Risk of adynamic bone disease in dialyzed patients. Kidney Int Suppl 1992; 38: S62–S67.
Sherrard DJ, Hercz G, Pei Y et al. The spectrum of bone disease in end-stage renal failure – an evolving disorder. Kidney Int 1993; 43: 436–442.
Coburn JW. Mineral metabolism and renal bone disease: effects of CAPD versus hemodialysis. Kidney Int Suppl 1993; 40: S92–S100.
Anwar N, Hutchison AJ, Gokal R. Comparison of renal osteodystrophy in patients undergoing continuous ambulatory peritoneal dialysis and hemodialysis. Perit Dial Int 1993; 13 Suppl 2: S451–S453.
Gal-Moscovici A, Popovtzer MM. New worldwide trends in presentation of renal osteodystrophy and its relationship to parathyroid hormone levels. Clin Nephrol 2005; 63: 284–289.
Llach F, Massry SG, Singer FR, Kurokawa K, Kaye JH, Coburn JW. Skeletal resistance to endogenous parathyroid hormone in pateints with early renal failure. A possible cause for secondary hyperparathyroidism. J Clin Endocrinol Metab 1975; 41: 339–345.
Avioli LV. The Renal Osteodystrophies. In: Brenner B, Rector FC, eds. The Kidney. Saunders, Philadelphia, USA, 1986; 1542–1580.
Malluche H, Faugere MC. Renal bone disease 1990: an unmet challenge for the nephrologist. Kidney Int 1990; 38: 193–211.
Baker LR, Abrams L, Roe CJ et al. 1,25(OH)2D3 administration in moderate renal failure: a prospective double-blind trial. Kidney Int 1989; 35: 661–669.
Hutchison AJ, Whitehouse RW, Boulton HF et al. Correlation of bone histology with parathyroid hormone, vitamin D3, and radiology in end-stage renal disease. Kidney Int 1993; 44: 1071–1077.
Ward MK, Feest TG, Ellis HA, Parkinson IS, Kerr DN. Osteomalacic dialysis osteodystrophy: evidence for a water-borne aetiological agent, probably aluminium. Lancet 1978; 1: 841–845.
Cournot-Witmer G, Plachot JJ, Bourdeau A et al. Effect of aluminum on bone and cell localization. Kidney Int Suppl 1986; 18: S37–S40.
Ott SM, Maloney NA, Coburn JW, Alfrey AC, Sherrard DJ. The prevalence of bone aluminum deposition in renal osteodystrophy and its relation to the response to calcitriol therapy. N Engl J Med 1982; 307: 709–713.
Posner AS, Blumenthal NC, Boskey AL. Model of aluminum-induced osteomalacia: inhibition of apatite formation and growth. Kidney Int Suppl 1986; 18: S17–S19.
Cannata Andia JB. Adynamic bone and chronic renal failure: an overview. Am J Med Sci 2000; 320: 81–84.
Heaf J. Adynamic bone disease and malnutrition-inflammation-cachexia syndrome. Kidney Int 2007; 71: 1326.
Heaf J. Causes and consequences of adynamic bone disease. Nephron 2001; 88: 97–106.
Weinreich T, Zapf J, Schmidt-Gayk H, Ritzel H, Delling G, Reichel H. Insulin-like growth factor 1 and 2 serum concentrations in dialysis patients with secondary hyperparathyroidism and adynamic bone disease. Clin Nephrol 1999; 51: 27–33.
Sanchez CP. Adynamic bone revisited: is there progress? Perit Dial Int 2006; 26: 43–48.
Mucsi I, Hercz G. Adynamic bone disease: pathogenesis, diagnosis and clinical relevance. Curr Opin Nephrol Hypertens 1997; 6: 356–361.
Rocha LA, Higa A, Barreto FC et al. Variant of adynamic bone disease in hemodialysis patients: fact or fiction? Am J Kidney Dis 2006; 48: 430–436.
Stehman-Breen C. Osteoporosis and chronic kidney disease. Semin Nephrol 2004; 24: 78–81.
Coburn JW. Renal osteodystrophy. Kidney Int 1980; 17: 677–623.
Malberti F, Marcelli D, Conte F, Limido A, Spotti D, Locatelli F. Parathyroidectomy in patients on renal replacement therapy: an epidemiologic study. J Am Soc Nephrol 2001; 12: 1242–1248.
Kestenbaum B, Andress DL, Schwartz SM et al. Survival following parathyroidectomy among United States dialysis patients. Kidney Int 2004; 66: 2010–2016.
Cohen-Solal ME, Sebert JL, Boudailliez B et al. Non-aluminic adynamic bone disease in non-dialyzed uremic patients: a new type of osteopathy due to overtreatment? Bone 1992; 13: 1–5.
Fournier A, Moriniere P, Cohen Solal ME et al. Adynamic bone disease in uremia: may it be idiopathic? Is it an actual disease? Nephron 1991; 58: 1–12.
Hutchison AJ, Whitehouse RW, Freemont AJ, Adams JE, Mawer EB, Gokal R. Histological, radiological, and biochemical features of the adynamic bone lesion in continuous ambulatory peritoneal dialysis patients. Am J Nephrol 1994; 14: 19–29.
Dunstan CR, Evans RA, Hills E, Wong SY, Alfrey AC. Effect of aluminum and parathyroid hormone on osteoblasts and bone mineralization in chronic renal failure. Calcif Tissue Int 1984; 36: 133–138.
Charhon SA, Chavassieux PM, Chapuy MC, Boivin GY, Meunier PJ. Low rate of bone formation with or without histologic appearance of osteomalacia in patients with aluminum intoxication. J Lab Clin Med 1985; 106: 123–131.
Hercz G, Pei Y, Greenwood C et al. Aplastic osteodystrophy without aluminum: the role of ‘suppressed' parathyroid function. Kidney Int 1993; 44: 860–866.
London GM, Marty C, Marchais SJ, Guerin AP, Metivier F, de Vernejoul MC. Arterial calcifications and bone histomorphometry in end-stage renal disease. J Am Soc Nephrol 2004; 15: 1943–1951.
Keutmann HT, Sauer MM, Hendy GN, O'Riordan LH, Potts JT, Jr. Complete amino acid sequence of human parathyroid hormone. Biochemistry 1978; 17: 5723–5729.
Chu LL, MacGregor RR, Hamilton JW, Cohn DV. Conversion of proparathyroid hormone to parathyroid hormone: the use of amines as specific inhibitors. Endocrinology 1974; 95: 1431–1438.
Arnaud CD. Hyperparathyroidism and renal failure. Kidney Int 1973; 4: 89–95.
Slatopolsky E, Lopez-Hilker S, Dusso A, Morrissey JJ, Martin KJ. Parathyroid hormone secretion: perturbations in chronic renal failure. Contrib Nephrol 1988; 64: 16–24.
Sherwood LM, Mayer GP, Ramberg CF, Jr., Kronfeld DS, Aurbach GD, Potts JT, Jr. Regulation of parathyroid hormone secretion: proportional control by calcium, lack of effect of phosphate. Endocrinology 1968; 83: 1043–1051.
Slatopolsky E, Lopez-Hilker S, Dusso A. The interrelationship between vitamin D and parathyroid hormone secretion in health and disease. In: Davidson AM, ed. Nephrology. Proceedings of the International Congress of Nephrology. Balliere Tindall, London: 1988; 1067–1075.
Felsenfeld AJ, Ross D, Rodriguez M. Hysteresis of the parathyroid hormone response to hypocalcemia in hemodialysis patients with low turnover aluminum bone disease. J Am Soc Nephrol 1991; 2: 1136–1143.
Dunlay R, Rodriguez M, Felsenfeld AJ, Llach F. Direct inhibitory effect of calcitriol on parathyroid function (sigmoidal curve) in dialysis. Kidney Int 1989; 36: 1093–1098.
Cunningham J, Altmann P, Gleed JH, Butter KC, Marsh FP, O'Riordan JL. Effect of direction and rate of change of calcium on parathyroid hormone secretion in uraemia. Nephrol Dial Transplant 1989; 4: 339–344.
Brown EM, Gardner DG, Windeck RA, Hurwitz S, Brennan MF, Aurbach GD. Beta-adrenergically stimulated adenosine 3′,5′-monophosphate accumulation in and parathyroid hormone release from dispersed human parathyroid cells. J Clin Endocrinol Metab 1979; 48: 618–626.
Fukagawa M, Kitaoka M, Yi H et al. Serial evaluation of parathyroid size by ultrasonography is another useful marker for the long-term prognosis of calcitriol pulse therapy in chronic dialysis patients. Nephron 1994; 68: 221–228.
Fukagawa M, Kaname S, Igarashi T, Ogata E, Kurokawa K. Regulation of parathyroid hormone synthesis in chronic renal failure in rats. Kidney Int 1991; 39: 874–881.
Slatopolsky E, Weerts C, Thielan J, Horst R, Harter H, Martin KJ. Marked suppression of secondary hyperparathyroidism by intravenous administration of 1,25-dihydroxy-cholecalciferol in uremic patients. J Clin Invest 1984; 74: 2136–2143.
Jones CL, Vieth R, Spino M et al. Comparisons between oral and intraperitoneal 1,25-dihydroxyvitamin D3 therapy in children treated with peritoneal dialysis. Clin Nephrol 1994; 42: 44–49.
Delmez JA, Dougan CS, Gearing BK et al. The effects of intraperitoneal calcitriol on calcium and parathyroid hormone. Kidney Int 1987; 31: 795–799.
Chase LR, Slatopolsky E. Secretion and metabolic efficacy of parathyroid hormone in patients with severe hypomagnesemia. J Clin Endocrinol Metab 1974; 38: 363–371.
Torres PU. The need for reliable serum parathyroid hormone measurements. Kidney Int 2006; 70: 240–243.
Souberbielle JC, Boutten A, Carlier MC et al. Inter-method variability in PTH measurement: implication for the care of CKD patients. Kidney Int 2006; 70: 345–350.
Cohen-Solal ME, Sebert JL, Boudailliez B et al. Comparison of intact, midregion, and carboxy terminal assays of parathyroid hormone for the diagnosis of bone disease in hemodialyzed patients. J Clin Endocrinol Metab 1991; 73: 516–524.
Boudou P, Ibrahim F, Cormier C, Chabas A, Sarfati E, Souberbielle JC. Third- or second-generation parathyroid hormone assays: a remaining debate in the diagnosis of primary hyperparathyroidism. J Clin Endocrinol Metab 2005; 90: 6370–6372.
Brunette MG, Chan M, Ferriere C, Roberts KD. Site of 1,25(OH)2 vitamin D3 synthesis in the kidney. Nature 1978; 276: 287–289.
Mawer EB, Taylor CM, Backhouse J, Lumb GA, Stanbury SW. Failure of formation of 1,25-dihydroxycholecalciferol in chronic renal insufficiency. Lancet 1973; 1: 626–628.
Rapoport J, Shany S, Chaimovitz C. Continuous ambulatory peritoneal dialysis and vitamin D. Nephron 1988; 48: 1–3.
Hayes ME, O'Donoghue DJ, Ballardie FW, Mawer EB. Peritonitis induces the synthesis of 1 alpha,25-dihydroxyvitamin D3 in macrophages from CAPD patients. FEBS Lett 1987; 220: 307–310.
Lind L, Wengle B, Wide L, Wrege U, Ljunghall S. Suppression of serum parathyroid hormone levels by intravenous alphacalcidol in uremic patients on maintenance hemodialysis. A pilot study. Nephron 1988; 48: 296–299.
Ljunghall S, Althoff P, Fellstrom B et al. Effects on serum parathyroid hormone of intravenous treatment with alphacalcidol in patients on chronic hemodialysis. Nephron 1990; 55: 380–385.
Andress DL, Norris KC, Coburn JW, Slatopolsky EA, Sherrard DJ. Intravenous calcitriol in the treatment of refractory osteitis fibrosa of chronic renal failure. N Engl J Med 1989; 321: 274–279.
Tsukamoto Y, Nomura M, Takahashi Y et al. The ‘oral 1,25-dihydroxyvitamin D3 pulse therapy' in hemodialysis patients with severe secondary hyperparathyroidism. Nephron 1991; 57: 23–28.
Gallieni M, Brancaccio D, Padovese P et al. Low-dose intravenous calcitriol treatment of secondary hyperparathyroidism in hemodialysis patients. Italian Group for the Study of Intravenous Calcitriol. Kidney Int 1992; 42: 1191–1198.
Moriniere P, Maurouard C, Boudailliez B et al. Prevention of hyperparathyroidism in patients on maintenance dialysis by intravenous 1-alpha-hydroxyvitamin D3 in association with Mg(OH)2 as sole phosphate binder. A randomized comparative study with the association CaCO3 +/− Mg(OH)2. Nephron 1992; 60: 154–163.
Martin KJ, Ballal HS, Domoto DT, Blalock S, Weindel M. Pulse oral calcitriol for the treatment of hyperparathyroidism in patients on continuous ambulatory peritoneal dialysis: preliminary observations. Am J Kidney Dis 1992; 19: 540–545.
Holick MF. Vitamin D and the kidney. Kidney Int 1987; 32: 912–929.
Mak RH, Wong JH. The vitamin D/parathyroid hormone axis in the pathogenesis of hypertension and insulin resistance in uremia. Miner Electrolyte Metab 1992; 18: 156–159.
Bricker NS. On the pathogenesis of the uremic state. An exposition of the ‘trade-off hypothesis’. N Engl J Med 1972; 286: 1093–1099.
Faugere MC, Friedler R, Fanti P, Malluche H. Lack of histologic signs of Vit D deficiency in early development of renal osteodystrophy. J Bone Miner Res 1988; 3 suppl 1: s95.
Lopez-Hilker S, Galceran T, Chan YL, Rapp N, Martin KJ, Slatopolsky E. Hypocalcemia may not be essential for the development of secondary hyperparathyroidism in chronic renal failure. J Clin Invest 1986; 78: 1097–1102.
Nielsen PK, Feldt-Rasmussen U, Olgaard K. A direct effect in vitro of phosphate on PTH release from bovine parathyroid tissue slices but not from dispersed parathyroid cells. Nephrol Dial Transplant 1996; 11: 1762–1768.
Ritz E, Matthias S, Seidel A, Reichel H, Szabo A, Horl WH. Disturbed calcium metabolism in renal failure – pathogenesis and therapeutic strategies. Kidney Int Suppl 1992; 38: S37–S42.
Massry SG, Tuma S, Dua S, Goldstein DA. Reversal of skeletal resistance to parathyroid hormone in uremia by vitamin D metabolites: evidence for the requirement of 1,25(OH)2D3 and 24,25(OH)2D3. J Lab Clin Med 1979; 94: 152–157.
Randall RE, Jr., Cohen MD, Spray CC, Jr., Rossmeisl EC. Hypermagnesemia in renal failure. Etiology and toxic manifestations. Ann Intern Med 1964; 61: 73–88.
Moriniere P, Vinatier I, Westeel PF et al. Magnesium hydroxide as a complementary aluminium-free phosphate binder to moderate doses of oral calcium in uraemic patients on chronic haemodialysis: lack of deleterious effect on bone mineralisation. Nephrol Dial Transplant 1988; 3: 651–656.
Alfrey AC, Solomons CC. Bone pyrophosphate in uremia and its association with extraosseous calcification. J Clin Invest 1976; 57: 700–705.
Macintyre I, Davidsson D. The production of secondary potassium depletion, sodium retention, nephrocalcinosis and hypercalcaemia by magnesium deficiency. Biochem J 1958; 70: 456–462.
Kaehny WD, Hegg AP, Alfrey AC. Gastrointestinal absorption of aluminum from aluminum-containing antacids. N Engl J Med 1977; 296: 1389–1390.
Malluche HH. Aluminium and bone disease in chronic renal failure. Nephrol Dial Transplant 2002; 17 Suppl 2: 21–24.
Litzow JR, Lemann J, Jr., Lennon EJ. The effect of treatment of acidosis on calcium balance in patients with chronic azotemic renal disease. J Clin Invest 1967; 46: 280–286.
Kraut JA. The role of metabolic acidosis in the pathogenesis of renal osteodystrophy. Adv Ren Replace Ther 1995; 2: 40–51.
Kraut JA, Kurtz I. Metabolic acidosis of CKD: diagnosis, clinical characteristics, and treatment. Am J Kidney Dis 2005; 45: 978–993.
Chittal SM, Oreopoulos DG, DeVeber GA et al. Plasma calcitonin in renal osteodystrophy. Can Med Assoc J 1971; 104: 1098–1100.
Opatrna S, Klaboch J, Opatrny K, Jr. et al. Procalcitonin levels in peritoneal dialysis patients. Perit Dial Int 2005; 25: 470–472.
Silva OL, Becker KL, Shalhoub RJ, Snider RH, Bivins LE, Moore CF. Calcitonin levels in chronic renal disease. Nephron 1977; 19: 12–18.
Hirsch PF, Baruch H. Is calcitonin an important physiological substance? Endocrine 2003; 21: 201–208.
Patel TS, Freedman BI, Yosipovitch G. An update on pruritus associated with CKD. Am J Kidney Dis 2007; 50: 11–20.
Kalpakian MA, Mehrotra R. Vascular calcification and disordered mineral metabolism in dialysis patients. Semin Dial 2007; 20: 139–143.
Caplin B, Wheeler DC. Arterial calcification in dialysis patients and transplant recipients. Semin Dial 2007; 20: 144–149.
Stompor T, Rajzer M, Pasowicz M et al. Coronary artery calcification, common carotid artery intima-media thickness and aortic pulse wave velocity in patients on peritoneal dialysis. Int J Artif Organs 2006; 29: 736–744.
Moe SM. Vascular calcification and renal osteodystrophy relationship in chronic kidney disease. Eur J Clin Invest 2006; 36 Suppl 2: 51–62.
Mehrotra R. Disordered mineral metabolism and vascular calcification in nondialyzed chronic kidney disease patients. J Ren Nutr 2006; 16: 100–118.
Goldsmith DJ, Covic A, Sambrook PA, Ackrill P. Vascular calcification in long-term haemodialysis patients in a single unit: a retrospective analysis. Nephron 1997; 77: 37–43.
Davies MR, Hruska KA. Pathophysiological mechanisms of vascular calcification in end-stage renal disease. Kidney Int 2001; 60: 472–479.
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31: 607–617.
Young EW, Albert JM, Satayathum S et al. Predictors and consequences of altered mineral metabolism: the Dialysis Outcomes and Practice Patterns Study. Kidney Int 2005; 67: 1179–1187.
Kestenbaum B, Sampson JN, Rudser KD et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol 2005; 16: 520–528.
Gokal R, Ramos JM, Ellis HA et al. Histological renal osteodystrophy, and 25 hydroxycholecalciferol and aluminum levels in patients on continuous ambulatory peritoneal dialysis. Kidney Int 1983; 23: 15–21.
Calderaro V, Oreopoulos DG, Meema HE et al. The evolution of renal osteodystrophy in patients undergoing continuous ambulatory peritoneal dialysis (CAPD). Proc Eur Dial Transplant Assoc 1980; 17: 533–542.
Owen JP, Parnell AP, Keir MJ et al. Critical analysis of the use of skeletal surveys in patients with chronic renal failure. Clin Radiol 1988; 39: 578–582.
Couttenye MM, D'Haese PC, Verschoren WJ, Behets GJ, Schrooten I, De Broe ME. Low bone turnover in patients with renal failure. Kidney Int Suppl 1999; 73: S70–S76.
Adams JE, Chen SZ, Adams PH, Isherwood I. Measurement of trabecular bone mineral by dual energy computed tomography. J Comput Assist Tomogr 1982; 6: 601–607.
Genant HK, Block JE, Steiger P, Glueer CC, Ettinger B, Harris ST. Appropriate use of bone densitometry. Radiology 1989; 170: 817–822.
Rahman R, Heaton A, Goodship THJ et al. Renal osteodystrophy in patients on CAPD: A five year study. Perit Dial Int 1987; 7: 20–26.
Digenis G, Khanna R, Pierratos A et al. Renal osteodystrophy in patients maintained on CAPD for more than three years. Perit Dial Int 1983; 3: 81–86.
Kurtz SB. Clinical parameters of renal bone disease: A comparison of CAPD and HD. Dial Transplant 1985; 14: 30–37.
Buccianti G, Bianchi ML, Valenti G. Progress of renal osteodystrophy during continuous ambulatory peritoneal dialysis. Clin Nephrol 1984; 22: 279–283.
Pei Y, Hercz G, Greenwood C et al. Renal osteodystrophy in diabetic patients. Kidney Int 1993; 44: 159–164.
Cozzolino M, Gallieni M, Chiarelli G, Brancaccio D. Calcium and phosphate handling in peritoneal dialysis. Contrib Nephrol 2006; 150: 214–225.
Recker RR, Saville PD. Calcium absorption in renal failure: its relationship to blood urea nitrogen, dietary calcium intake, time on dialysis, and other variables. J Lab Clin Med 1971; 78: 380–388.
Clarkson EM, Eastwood JB, Koutsaimanis KG, de Wardener HE. Net intestinal absorption of calcium in patients with chronic renal failure. Kidney Int 1973; 3: 258–263.
Ramirez JA, Emmett M, White MG et al. The absorption of dietary phosphorus and calcium in hemodialysis patients. Kidney Int 1986; 30: 753–759.
Hutchison AJ, Boulton HF, Herman K, Day JP, Prescott M, Gokal R. Use of oral stable strontium to provide an index of intestinal calcium absorption in chronic ambulatory peritoneal dialysis patients. Miner Electrolyte Metab 1992; 18: 160–165.
Blumenkrantz MJ, Kopple JD, Moran JK, Coburn JW. Metabolic balance studies and dietary protein requirements in patients undergoing continuous ambulatory peritoneal dialysis. Kidney Int 1982; 21: 849–861.
Lindholm B, Bergstrom J. Nutritional aspects on peritoneal dialysis. Kidney Int Suppl 1992; 38: S165–S171.
Nolph KD, Prowant B, Serkes KD et al. Multicenter evaluation of a new peritoneal dialysis solution with a high lactate and a low magnesium concentration. Perit Dial Int 1983; 3: 63–65.
Hercz G, Coburn JW. Prevention of phosphate retention and hyperphosphatemia in uremia. Kidney Int Suppl 1987; 22: S215–S220.
Delmez JA, Fallon MD, Bergfeld MA, Gearing BK, Dougan CS, Teitelbaum SL. Continuous ambulatory peritoneal dialysis and bone. Kidney Int 1986; 30: 379–384.
Martis L, Serkes KD, Nolph KD. Calcium carbonate as a phosphate binder: is there a need to adjust peritoneal dialysate calcium concentrations for patients using CaCO3? Perit Dial Int 1989; 9: 325–328.
K/DOQIClinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42: 1–202.
Bro S, Rasmussen RA, Handberg J, Olgaard K, Feldt-Rasmussen B. Randomized crossover study comparing the phosphate-binding efficacy of calcium ketoglutarate versus calcium carbonate in patients on chronic hemodialysis. Am J Kidney Dis 1998; 31: 257–262.
Andreoli SP, Bergstein JM, Sherrard DJ. Aluminum intoxication from aluminum-containing phosphate binders in children with azotemia not undergoing dialysis. N Engl J Med 1984; 310: 1079–1084.
Salusky IB, Coburn JW, Foley J, Nelson P, Fine RN. Effects of oral calcium carbonate on control of serum phosphorus and changes in plasma aluminum levels after discontinuation of aluminum-containing gels in children receiving dialysis. J Pediatr 1986; 108: 767–770.
Slatopolsky E, Weerts C, Lopez-Hilker S et al. Calcium carbonate as a phosphate binder in patients with chronic renal failure undergoing dialysis. N Engl J Med 1986; 315: 157–161.
Pflanz S, Henderson IS, McElduff N, Jones MC. Calcium acetate versus calcium carbonate as phosphate-binding agents in chronic haemodialysis. Nephrol Dial Transplant 1994; 9: 1121–1124.
Taylor JE, Henderson IS, Stewart WK, Mactier RA. Calcium carbonate 1250 mg/1260 mg: an effective phosphate binder. Scott Med J 1990; 35: 45–47.
Lerner A, Kramer M, Goldstein S, Caruana R, Epstein S, Raja R. Calcium carbonate. A better phosphate binder than aluminum hydroxide. ASAIO Trans 1986; 32: 315–318.
Kobrin SM, Epstein SE, Goldstein SJ, Kramer MS, Raja RM. Calcium carbonate as a phosphate binder. One year's experience. ASAIO Trans 1987; 33: 518–520.
Fournier A, Moriniere P, Hamida FB, Ghazali A. CaCO3: an inefficient and unsafe phosphate binder? Nephrol Dial Transplant 1994; 9: 335–336.
Ott SM. Aluminum accumulation in individuals with normal renal function. Am J Kidney Dis 1985; 6: 297–301.
Joffe P, Olsen F, Heaf JG, Gammelgaard B, Podenphant J. Aluminum concentrations in serum, dialysate, urine and bone among patients undergoing continuous ambulatory peritoneal dialysis (CAPD). Clin Nephrol 1989; 32: 133–138.
Slatopolsky E, Weerts C, Norwood K et al. Long-term effects of calcium carbonate and 2.5 mEq/L calcium dialysate on mineral metabolism. Kidney Int 1989; 36: 897–903.
Van der Merwe WM, Rodger RS, Grant AC et al. Low calcium dialysate and high-dose oral calcitriol in the treatment of secondary hyperparathyroidism in haemodialysis patients. Nephrol Dial Transplant 1990; 5: 874–877.
Gokal R, Hutchison A. Calcium, phosphorus, aluminium and bone disease in continuous ambulatory peritoneal dialysis patients. In: Hatano M, ed. Nephrology. Springer, Tokyo: 1991; 1602–1609.
Hutchison AJ, Gokal R. Towards tailored dialysis fluids in CAPD – the role of reduced calcium and magnesium in dialysis fluids. Perit Dial Int 1992; 12: 199–203.
Hutchison AJ, Were AJ, Boulton HF, Mawer EB, Laing I, Gokal R. Hypercalcaemia, hypermagnesaemia, hyperphosphataemia and hyperaluminaemia in CAPD: improvement in serum biochemistry by reduction in dialysate calcium and magnesium concentrations. Nephron 1996; 72: 52–58.
Hutchison AJ, Freemont AJ, Boulton HF, Gokal R. Low-calcium dialysis fluid and oral calcium carbonate in CAPD. A method of controlling hyperphosphataemia whilst minimizing aluminium exposure and hypercalcaemia. Nephrol Dial Transplant 1992; 7: 1219–1225.
Hutchison AJ, Gokal R. Improved solutions for peritoneal dialysis: physiological calcium solutions, osmotic agents and buffers. Kidney Int Suppl 1992; 38: S153–S159.
Piraino B, Perlmutter JA, Holley JL, Johnston JR, Bernardini J. The use of dialysate containing 2.5 mEq/L calcium in peritoneal dialysis patients. Perit Dial Int 1992; 12: 75–76.
Cunningham J, Beer J, Coldwell RD, Noonan K, Sawyer N, Makin HL. Dialysate calcium reduction in CAPD patients treated with calcium carbonate and alfacalcidol. Nephrol Dial Transplant 1992; 7: 63–68.
Kimata N, Albert JM, Akiba T et al. Association of mineral metabolism factors with all-cause and cardiovascular mortality in hemodialysis patients: the Japan dialysis outcomes and practice patterns study. Hemodial Int 2007; 11: 340–348.
White CA, Jaffey J, Magner P. Cost of applying the K/DOQI guidelines for bone metabolism and disease to a cohort of chronic hemodialysis patients. Kidney Int 2007; 71: 312–317.
Lorenzo V, Martin-Malo A, Perez-Garcia R et al. Prevalence, clinical correlates and therapy cost of mineral abnormalities among haemodialysis patients: a cross-sectional multicentre study. Nephrol Dial Transplant 2006; 21: 459–465.
Manns B, Stevens L, Miskulin D, Owen WF, Jr., Winkelmayer WC, Tonelli M. A systematic review of sevelamer in ESRD and an analysis of its potential economic impact in Canada and the United States. Kidney Int 2004; 66: 1239–1247.
McGary TJ, Nolph KD, Moore HL, Kartinos NJ. Polycation as an alternative osmotic agent and phosphate binder in peritoneal dialysis. Uremia Invest 1984; 8: 79–84.
Hutchison AJ. Calcitriol, lanthanum carbonate, and other new phosphate binders in the management of renal osteodystrophy. Perit Dial Int 1999; 19 (suppl 2): S408–S412.
Chertow GM, Burke SK, Lazarus JM et al. Poly[allylamine hydrochloride] (RenaGel): a noncalcemic phosphate binder for the treatment of hyperphosphatemia in chronic renal failure. Am J Kidney Dis 1997; 29: 66–71.
Graff L, Burnel D. A possible non-aluminum oral phosphate binder? A comparative study on dietary phosphorus absorption. Res Commun Mol Pathol Pharmacol 1995; 89: 373–388.
Albaaj F, Hutchison AJ. Lanthanum carbonate (Fosrenol): a novel agent for the treatment of hyperphosphataemia in renal failure and dialysis patients. Int J Clin Pract 2005; 59: 1091–1096.
Finn WF, Joy MS. A long-term, open-label extension study on the safety of treatment with lanthanum carbonate, a new phosphate binder, in patients receiving hemodialysis. Curr Med Res Opin 2005; 21: 657–664.
Hutchison AJ, Maes B, Vanwalleghem J et al. Long-term efficacy and tolerability of lanthanum carbonate: results from a 3-year study. Nephron Clin Pract 2006; 102: c61–c71.
Persy VP, Behets GJ, Bervoets AR, De Broe ME, D'Haese PC. Lanthanum: a safe phosphate binder. Semin Dial 2006; 19: 195–199.
Hutchison AJ. Improving phosphate-binder therapy as a way forward. Nephrol Dial Transplant 2004; 19 (suppl 1): i19–i24.
Drueke TB. Lanthanum carbonate as a first-line phosphate binder: the ‘cons’. Semin Dial 2007; 20: 329–332.
McIntyre CW. New developments in the management of hyperphosphatemia in chronic kidney disease. Semin Dial 2007; 20: 337–341.
Parker A, Nolph KD. Magnesium and calcium mass transfer during continuous ambulatory peritoneal dialysis. Trans Am Soc Artif Intern Organs 1980; 26: 194–196.
Delmez JA, Slatopolsky E, Martin KJ, Gearing BN, Harter HR. Minerals, vitamin D, and parathyroid hormone in continuous ambulatory peritoneal dialysis. Kidney Int 1982; 21: 862–867.
Kwong MBL, Lee JSK, Chan MK. Transperitoneal calcium and magnesium transfer during an 8-hour dialysis. Perit Dial Int 1987; 7: 85–89.
Hutchison AJ, Merchant M, Boulton HF, Hinchcliffe R, Gokal R. Calcium and magnesium mass transfer in peritoneal dialysis patients using 1.25 mmol/L calcium, 0.25 mmol/L magnesium dialysis fluid. Perit Dial Int 1993; 13: 219–223.
Simonsen O, Venturoli D, Wieslander A, Carlsson O, Rippe B. Mass transfer of calcium across the peritoneum at three different peritoneal dialysis fluid Ca2+ and glucose concentrations. Kidney Int 2003; 64: 208–215.
Hamdy NA, Brown CB, Boletis J et al. Mineral metabolism in CAPD. Contrib Nephrol 1990; 85: 100–110.
Haris A, Sherrard DJ, Hercz G. Reversal of adynamic bone disease by lowering of dialysate calcium. Kidney Int 2006; 70: 931–937.
Meema HE, Oreopoulos DG, Rapoport A. Serum magnesium level and arterial calcification in end-stage renal disease. Kidney Int 1987; 32: 388–394.
Gonella M, Ballanti P, Della RC et al. Improved bone morphology by normalizing serum magnesium in chronically hemodialyzed patients. Miner Electrolyte Metab 1988; 14: 240–245.
Breuer J, Moniz C, Baldwin D, Parsons V. The effects of zero magnesium dialysate and magnesium supplements on ionised calcium concentration in patients on regular dialysis treatment. Nephrol Dial Transplant 1987; 2: 347–350.
Parsons V, Baldwin D, Moniz C, Marsden J, Ball E, Rifkin I. Successful control of hyperparathyroidism in patients on continuous ambulatory peritoneal dialysis using magnesium carbonate and calcium carbonate as phosphate binders. Nephron 1993; 63: 379–383.
Shah GM, Winer RL, Cutler RE et al. Effects of a magnesium-free dialysate on magnesium metabolism during continuous ambulatory peritoneal dialysis. Am J Kidney Dis 1987; 10: 268–275.
Gastro-intestinal system; Antacids and simeticone. In: Martin J, ed. British National Formulary. BMJ Publishing Group & RPS Publishing, London: 2007; 37–39.
Dyckner T, Wester PO. Relation between potassium, magnesium and cardiac arrhythmias. Acta Med Scand Suppl 1981; 647: 163–169.
Whang R. Clinical disorders of magnesium metabolism. Compr Ther 1997; 23: 168–173.
Hollifield JW. Thiazide treatment of systemic hypertension: effects on serum magnesium and ventricular ectopic activity. Am J Cardiol 1989; 63: 22G–25G.
Seelig M. Cardiovascular consequences of magnesium deficiency and loss: pathogenesis, prevalence and manifestations–magnesium and chloride loss in refractory potassium repletion. Am J Cardiol 1989; 63: 4G–21G.
Seelig MS. Interrelationship of magnesium and congestive heart failure. Wien Med Wochenschr 2000; 150: 335–341.
Lemann J, Jr., Lennon EJ. Role of diet, gastrointestinal tract and bone in acid-base homeostasis. Kidney Int 1972; 1: 275–279.
Kaye M, Frueh AJ, Silverman M, Henderson J, Thibault T. A study of vertebral bone powder from patients with chronic renal failure. J Clin Invest 1970; 49: 442–453.
De MS, Cecchin E. Severe metabolic acidosis and disturbances of calcium metabolism induced by acetazolamide in patients on haemodialysis. Clin Sci (Lond) 1990; 78: 295–302.
Maren TH. Carbonic anhydrase. N Engl J Med 1985; 313: 179–181.
Waite LC. Carbonic anhydrase inhibitors, parathyroid hormone and calcium metabolism. Endocrinology 1972; 91: 1160–1165.
Nolph KD, Sorkin M, Rubin J et al. Continuous ambulatory peritoneal dialysis: three-year experience at one center. Ann Intern Med 1980; 92: 609–613.
Fourtounas C, Savidaki E, Roumelioti M et al. Acid-base profile and predictors of metabolic acidosis in patients undergoing peritoneal dialysis with lactate- and bicarbonate-buffered peritoneal dialysis solutions. Adv Perit Dial 2006; 22: 187–191.
Lefebvre A, de Vernejoul MC, Gueris J, Goldfarb B, Graulet AM, Morieux C. Optimal correction of acidosis changes progression of dialysis osteodystrophy. Kidney Int 1989; 36: 1112–1118.
Fassbinder W, Brunner FP, Brynger H et al. Combined report on regular dialysis and transplantation in Europe, XX, 1989. Nephrol Dial Transplant 1991; 6 (suppl 1): 5–35.
Loschiavo C, Fabris A, Adami S et al. Effects of continuous ambulatory peritoneal dialysis (CAPD) on renal osteodystrophy. Perit Dial Int 1985; 5: 53–55.
Heaf JG, Lokkegard H. Parathyroid hormone during maintenance dialysis: influence of low calcium dialysate, plasma albumin and age. J Nephrol 1998; 11: 203–210.
Avram MM, Mittman N, Myint MM, Fein P. Importance of low serum intact parathyroid hormone as a predictor of mortality in hemodialysis and peritoneal dialysis patients: 14 years of prospective observation. Am J Kidney Dis 2001; 38: 1351–1357.
Dimkovic NB, Bargman J, Vas S, Oreopoulos DG. Normal or low initial PTH levels are not a predictor of morbidity/mortality in patients undergoing chronic peritoneal dialysis. Perit Dial Int 2002; 22: 204–210.
Aloni Y, Shany S, Chaimovitz C. Losses of 25-hydroxyvitamin D in peritoneal fluid: possible mechanism for bone disease in uremic patients treated with chronic ambulatory peritoneal dialysis. Miner Electrolyte Metab 1983; 9: 82–86.
Cassidy MJ, Owen JP, Ellis HA et al. Renal osteodystrophy and metastatic calcification in long-term continuous ambulatory peritoneal dialysis. Q J Med 1985; 54: 29–48.
Dunstan CR, Hills E, Norman AW et al. Treatment of hemodialysis bone disease with 24,25-(OH)2D3 and 1,25-(OH)2D3 alone or in combination. Miner Electrolyte Metab 1985; 11: 358–368.
Ben-Ezer D, Shany S, Conforty A et al. Oral administration of 24,25(OH)2D3 suppresses the serum parathyroid hormone levels of dialysis patients. Nephron 1991; 58: 283–287.
Gal-Moscovici A, Rubinger D, Popovtzer MM. 24,25-dihydroxyvitamin D3 in combination with 1,25-dihydroxyvitamin D3 ameliorates renal osteodystrophy in rats with chronic renal failure. Clin Nephrol 2000; 53: 362–371.
Mortensen BM, Aarseth HP, Ganss R, Haug E, Gautvik KM, Gordeladze JO. 24,25-dihydroxy vitamin D3 treatment inhibits parathyroid-stimulated adenylate cyclase in iliac crest biopsies from uremic patients. Bone 1993; 14: 125–131.
Muirhead N, Adami S, Sandler LM et al. Long-term effects of 1,25–dihydroxy vitamin D3 and 24,25-dihydroxy vitamin D3 in renal osteodystrophy. Q J Med 1982; 51: 427–444.
Baskin E, Ozen S, Karcaaltincaba M et al. Beneficial role of intravenous calcitriol on bone mineral density in children with severe secondary hyperparathyroidism. Int Urol Nephrol 2004; 36: 113–118.
Greenbaum LA, Grenda R, Qiu P et al. Intravenous calcitriol for treatment of hyperparathyroidism in children on hemodialysis. Pediatr Nephrol 2005; 20: 622–630.
Schaefer K, Umlauf E, von HD. Reduced risk of hypercalcemia for hemodialysis patients by administering calcitriol at night. Am J Kidney Dis 1992; 19: 460–464.
Korkor AB. Reduced binding of [3H]1,25-dihydroxyvitamin D3 in the parathyroid glands of patients with renal failure. N Engl J Med 1987; 316: 1573–1577.
Merke J, Hugel U, Zlotkowski A et al. Diminished parathyroid 1,25(OH)2D3 receptors in experimental uremia. Kidney Int 1987; 32: 350–353.
Brown AJ, Dusso A, Lopez-Hilker S, Lewis-Finch J, Grooms P, Slatopolsky E. 1,25-(OH)2D receptors are decreased in parathyroid glands from chronically uremic dogs. Kidney Int 1989; 35: 19–23.
Scanziani R, Dozio B, Bonforte G, Surian M. Effects of calcitriol pulse therapy per os in CAPD patients. Adv Perit Dial 1994; 10: 270–274.
Bechtel U, Mucke C, Feucht HE, Schiffl H, Sitter T, Held E. Limitations of pulse oral calcitriol therapy in continuous ambulatory peritoneal dialysis patients. Am J Kidney Dis 1995; 25: 291–296.
Romanini D, Gazo A, Bellazzi R, de VA, Nai M, Santagostino M. Long-term effect of oral calcitriol single weekly pulse in CAPD and in HD. Adv Perit Dial 1994; 10: 267–269.
Pedrozo HA, Schwartz Z, Rimes S et al. Physiological importance of the 1,25(OH)2D3 membrane receptor and evidence for a membrane receptor specific for 24,25(OH)2D3. J Bone Miner Res 1999; 14: 856–867.
Teng M, Wolf M, Lowrie E, Ofsthun N, Lazarus JM, Thadhani R. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med 2003; 349: 446–456.
Shoji S, Nishizawa Y, Tabata T et al. Influence of serum phosphate on the efficacy of oral 1,25-dihydroxyvitamin D3 pulse therapy. Miner Electrolyte Metab 1995; 21: 223–228.
Kubota M, Iwanaga Y, Ishiguro N. The effect of intraperitoneal 22-oxacalcitriol on secondary hyperparathyroidism in continuous ambulatory peritoneal dialysis patients (IPOX study). Adv Perit Dial 2003; 19: 227–230.
Murakami K, Miyachi H, Watanabe A et al. Suppression of parathyroid hormone secretion in CAPD patients by intraperitoneal administration of Maxacalcitol. Clin Exp Nephrol 2004; 8: 134–138.
Brown AJ, Ritter CR, Finch JL et al. The noncalcemic analogue of vitamin D, 22-oxacalcitriol, suppresses parathyroid hormone synthesis and secretion. J Clin Invest 1989; 84: 728–732.
Brown AJ, Finch JL, Lopez-Hilker S et al. New active analogues of vitamin D with low calcemic activity. Kidney Int Suppl 1990; 29: S22–S27.
Kubrusly M, Gagne ER, Urena P et al. Effect of 22-oxa-calcitriol on calcium metabolism in rats with severe secondary hyperparathyroidism. Kidney Int 1993; 44: 551–556.
Hirata M, Katsumata K, Masaki T et al. 22-Oxacalcitriol ameliorates high-turnover bone and marked osteitis fibrosa in rats with slowly progressive nephritis. Kidney Int 1999; 56: 2040–2047.
Kurokawa K, Akizawa T, Suzuki M, Akiba T, Ogata E, Slatopolsky E. Effect of 22-oxacalcitriol on hyperparathyroidism of dialysis patients: results of a preliminary study. Nephrol Dial Transplant 1996; 11 (suppl 3): 121–124.
Yasuda M, Akiba T, Nihei H. Multicenter clinical trial of 22-oxa-1,25-dihydroxyvitamin D3 for chronic dialysis patients. Am J Kidney Dis 2003; 41: S108–S111.
Posner GH. New vitamin D analogues. Nephrol Dial Transplant 1996; 11 (suppl 3): 32–36.
Nemeth EF, Bennett SA. Tricking the parathyroid gland with novel calcimimetic agents. Nephrol Dial Transplant 1998; 13: 1923–1925.
Lindberg JS. Calcimimetics: a new tool for management of hyperparathyroidism and renal osteodystrophy in patients with chronic kidney disease. Kidney Int Suppl 2005; S33–S36.
Lindberg JS, Moe SM, Goodman WG et al. The calcimimetic AMG 073 reduces parathyroid hormone and calcium x phosphorus in secondary hyperparathyroidism. Kidney Int 2003; 63: 248–254.
Quarles LD, Sherrard DJ, Adler S et al. The calcimimetic AMG 073 as a potential treatment for secondary hyperparathyroidism of end-stage renal disease. J Am Soc Nephrol 2003; 14: 575–583.
Block GA. The impact of calcimimetics on mineral metabolism and secondary hyperparathyroidism in end-stage renal disease. Kidney Int Suppl 2003; S131–S136.
Moe SM, Chertow GM, Coburn JW et al. Achieving NKF-K/DOQI bone metabolism and disease treatment goals with cinacalcet HCl. Kidney Int 2005; 67: 760–771.
Cunningham J, Danese M, Olson K, Klassen P, Chertow GM. Effects of the calcimimetic cinacalcet HCl on cardiovascular disease, fracture, and health-related quality of life in secondary hyperparathyroidism. Kidney Int 2005; 68: 1793–1800.
Moe SM, Cunningham J, Bommer J et al. Long-term treatment of secondary hyperparathyroidism with the calcimimetic cinacalcet HCl. Nephrol Dial Transplant 2005; 20: 2186–2193.
Lindberg JS, Culleton B, Wong G et al. Cinacalcet HCl, an oral calcimimetic agent for the treatment of secondary hyperparathyroidism in hemodialysis and peritoneal dialysis: a randomized, double-blind, multicenter study. J Am Soc Nephrol 2005; 16: 800–807.
Block GA, Martin KJ, de Francisco AL et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med 2004; 350: 1516–1525.
Andress DL, Hercz G, Kopp JB et al. Bone histomorphometry of renal osteodystrophy in diabetic patients. J Bone Miner Res 1987; 2: 525–531.
McNair P, Christensen MS, Madsbad S, Christiansen C, Transbol I. Hypoparathyroidism in diabetes mellitus. Acta Endocrinol (Copenh) 1981; 96: 81–86.
Heidbreder E, Gotz R, Schafferhans K, Heidland A. Diminished parathyroid gland responsiveness to hypocalcemia in diabetic patients with uremia. Nephron 1986; 42: 285–289.
Ballanti P, Wedard BM, Bonucci E. Frequency of adynamic bone disease and aluminum storage in Italian uraemic patients – retrospective analysis of 1429 iliac crest biopsies. Nephrol Dial Transplant 1996; 11: 663–667.
Torres A, Lorenzo V, Hernandez D et al. Bone disease in predialysis, hemodialysis, and CAPD patients: evidence of a better bone response to PTH. Kidney Int 1995; 47: 1434–1442.
Fournier A, Moriniere P, Ben HF et al. Use of alkaline calcium salts as phosphate binder in uremic patients. Kidney Int Suppl 1992; 38: S50–S61.
Kurz P, Monier-Faugere MC, Bognar B et al. Evidence for abnormal calcium homeostasis in patients with adynamic bone disease. Kidney Int 1994; 46: 855–861.
Mucsi I, Hercz G. Relative hypoparathyroidism and adynamic bone disease. Am J Med Sci 1999; 317: 405–409.
London GM, Marty C, Marchais SJ, Guerin AP, Metivier F, de Vernejoul MC. Arterial calcifications and bone histomorphometry in end-stage renal disease. J Am Soc Nephrol 2004; 15: 1943–1951.
Miller PD. Treatment of osteoporosis in chronic kidney disease and end-stage renal disease. Curr Osteoporos Rep 2005; 3: 5–12.
Ott SM. Long-term safety of bisphosphonates. J Clin Endocrinol Metab 2005; 90: 1897–1899.
Fan SL, Cunningham J. Bisphosphonates in renal osteodystrophy. Curr Opin Nephrol Hypertens 2001; 10: 581–588.
Geng Z, Monier-Faugere MC, Bauss F, Malluche HH. Short-term administration of the bisphosphonate ibandronate increases bone volume and prevents hyperparathyroid bone changes in mild experimental renal failure. Clin Nephrol 2000; 54: 45–53.
Mazzaferro S, Coen G, Ballanti P et al. Osteocalcin, iPTH, alkaline phosphatase and hand X-ray scores as predictive indices of histomorphometric parameters in renal osteodystrophy. Nephron 1990; 56: 261–266.
Maruyama Y, Arai K, Yoshida K et al. Study of tartrate resistant acid phosphatase in patients with chronic renal failure on maintenance hemodialysis. Nippon Jinzo Gakkai Shi 1991; 33: 397–402.
Kaneko Y, Maruyama Y, Tunemi K et al. Studies of serum bone Al-P isoenzyme and serum osteocalcin in patients on maintenance hemodialysis. Nippon Jinzo Gakkai Shi 1990; 32: 345–351.
Chu P, Chao TY, Lin YF, Janckila AJ, Yam LT. Correlation between histomorphometric parameters of bone resorption and serum type 5b tartrate-resistant acid phosphatase in uremic patients on maintenance hemodialysis. Am J Kidney Dis 2003; 41: 1052–1059.
Mohini R, Dumler F, Rao DS. Skeletal surveys in renal osteodystrophy. ASAIO Trans 1991; 37: 635–637.
DeVita MV, Rasenas LL, Bansal M et al. Assessment of renal osteodystrophy in hemodialysis patients. Medicine (Baltimore) 1992; 71: 284–290.
Olgaard K, Madsen S, Heerfordt J, Hammer M, Jensen H. Scintigraphic skeletal changes in non-dialyzed patients with advanced renal failure. Clin Nephrol 1979; 12: 273–278.
Hodson EM, Howman-Giles RB, Evans RA et al. The diagnosis of renal osteodystrophy: a comparison of Technetium-99m-pyrophosphate bone scintigraphy with other techniques. Clin Nephrol 1981; 16: 24–28.
Karsenty G, Vigneron N, Jorgetti V et al. Value of the 99mTc-methylene diphosphonate bone scan in renal osteodystrophy. Kidney Int 1986; 29: 1058–1065.
Kaida H, Ishibashi M, Nishida H et al. Assessment of therapeutic effect in patients with secondary hyperparathyroidism using bone scintigraphy. Ann Nucl Med 2005; 19: 367–372.
Kurata S, Ishibashi M, Nishida H, Hiromatsu Y, Hayabuchi N. A clinical assessment of the relationship between bone scintigraphy and serum biochemical markers in hemodialysis patients. Ann Nucl Med 2004; 18: 513–518.
Heaf JG, Nielsen LP, Mogensen NB. Use of bone mineral content determination in the evaluation of osteodystrophy among hemodialysis patients. Nephron 1983; 35: 103–107.
Rickers H, Christensen M, Rodbro P. Bone mineral content in patients on prolonged maintenance hemodialysis: a three year follow-up study. Clin Nephrol 1983; 20: 302–307.
Lindergard B, Johnell O, Nilsson BE, Wiklund PE. Studies of bone morphology, bone densitometry and laboratory data in patients on maintenance hemodialysis treatment. Nephron 1985; 39: 122–129.
Piraino B, Chen T, Cooperstein L, Segre G, Puschett J. Fractures and vertebral bone mineral density in patients with renal osteodystrophy. Clin Nephrol 1988; 30: 57–62.
Funke M, Maurer J, Grabbe E, Scheler F. Comparative studies with quantitative computed tomography and dual-energy x-ray absorptiometry on bone density in renal osteopathy. Rofo 1992; 157: 145–149.
Johnson DW, McIntyre HD, Brown A, Freeman J, Rigby RJ. The role of DEXA bone densitometry in evaluating renal osteodystrophy in continuous ambulatory peritoneal dialysis patients. Perit Dial Int 1996; 16: 34–40.
Chesney RW. Bone mineral density in chronic renal insufficiency and end-stage renal disease: how to interpret the scans. J Pediatr Endocrinol Metab 2004; 17 (suppl 4): 1327–1332.
Gerakis A, Hadjidakis D, Kokkinakis E, Apostolou T, Raptis S, Billis A. Correlation of bone mineral density with the histological findings of renal osteodystrophy in patients on hemodialysis. J Nephrol 2000; 13: 437–443.
Wei M, Taskapan H, Esbaei K, Jassal SV, Bargman JM, Oreopoulos DG. K/DOQI guideline requirements for calcium, phosphate, calcium phosphate product, and parathyroid hormone control in dialysis patients: can we achieve them? Int Urol Nephrol 2006; 38: 739–743.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Vardhan, A., Hutchison, A.J. (2009). Calcium, Phosphate, and Renal Osteodystrophy. In: Khanna, R., Krediet, R.T. (eds) Nolph and Gokal's Textbook of Peritoneal Dialysis. Springer, Boston, MA. https://doi.org/10.1007/978-0-387-78940-8_22
Download citation
DOI: https://doi.org/10.1007/978-0-387-78940-8_22
Publisher Name: Springer, Boston, MA
Print ISBN: 978-0-387-78939-2
Online ISBN: 978-0-387-78940-8
eBook Packages: MedicineMedicine (R0)