
Abstract
This chapter focuses on insulin resistance and its role in diabetes, obesity, and cardiovascular (CV) disease. There is significant evidence that both CV disease and diabetes have common metabolic antecedents. The metabolic syndrome and its relation to insulin resistance, cardiovascular disease, and diabetes will be critically reviewed with a focus on current clinical controversies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Insulin Resistance
- Metabolic Syndrome
- Visceral Adipose Tissue
- Subcutaneous Adipose Tissue
- Fast Plasma Insulin
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
This chapter focuses on insulin resistance and its role in diabetes, obesity, and cardiovascular (CV) disease. There is significant evidence that both CV disease and diabetes have common metabolic antecedents.1,2 The metabolic syndrome and its relation to insulin resistance, cardiovascular disease, and diabetes will be critically reviewed with a focus on current clinical controversies.
2 Insulin Action and Insulin Secretion
To maintain normal glucose homeostasis, there must be adequate insulin secretion synchronized with target tissue insulin responsiveness. Following the ingestion of a meal, insulin, secreted from the pancreas, permits the circulating blood glucose to be transported to muscle and adipose cells for metabolism or storage and also suppresses the release of glucose from the liver.3
Insulin acts to increase glucose uptake for storage and metabolism in muscle and adipose cells via the tissue-specific glucose transporter glut-4 and in liver via glut-2. Insulin also decreases lipolysis and promotes lipogenesis. In liver, insulin decreases gluconeogenesis and glycolysis and promotes glycogen synthesis. Insulin also regulates amino acid uptake and protein synthesis, has important actions on the vasculature and endothelial cells and, among its least understood functions, acts on the brain to integrate fuel and energy homeostasis.
Insulin activates its receptor by binding to its two alpha subunits4,5 (Figure 34.1). This auto-phosphorylates and activates tyrosine kinases intrinsic to the beta subunits, to promote phosphorylation of other tyrosines on downstream molecules known as insulin receptor substrates (IRS) and Shc. The IRS family of molecules consists of tissue-specific subtypes, for example, IRS-1 in muscle and IRS-2 in liver. IRS and Shc in turn activate phosphatidylinositol-3 kinase (PI-3-kinase) and mitogen-activated protein kinase (MAP kinase). The PI-3-kinase path is involved with metabolic activity and glucose transport via Glut-4 transporters in muscle and the MAP kinase pathway regulates mitogenesis and growth. Other metabolic activities controlled by the PI-3-kinase pathway include glycogen synthesis, lipolysis, fatty acid, and protein syntheses. Defects in this pathway are likely to reflect a combination of genetic and acquired defects.6,7 One example of a defect in the pathways involves phosphorylation of serine or threonine residues of the IRS-1 complex instead of tyrosine which results in decreased downstream insulin receptor activity.

2.1 Physiological Effects of Insulin Resistance
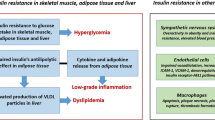
Resistance to insulin’s effects may occur in different tissues.8 In muscle, decreased insulin-mediated glucose uptake causes elevated blood glucose. Usually, a compensatory increase in insulin secretion develops which results in hyperinsulinemia sufficient to maintain normal glucose homeostasis. Without this, hyperglycemia and diabetes ensue. This compensatory increase in insulin or hyperinsulinemia while perfectly appropriate for maintaining the blood glucose level, may result in the enhancement of other actions of insulin.9,10
In the liver, a decreased insulin effect results in a lower suppression of hepatic glucose output and may lead to increased blood glucose. In adipose tissue, increased lipolysis and free fatty acids occur. Increased blood glucose and free fatty acid in turn promote further beta-cell dysfunction, decreased insulin secretion, and action. This negative amplification is also known as glucose toxicity and lipotoxicity.11 In the brain, insulin resistance may be involved in altered feeding and energy regulation. In endothelial cells, insulin action via the PI-3-kinase pathway promotes the release of nitric oxide from endothelial cells. Nitric oxide, a vasodilator, increases blood flow and aids in peripheral glucose uptake. In contrast, insulin, working through the MAP kinase pathway, also promotes the release of endothelin-1 which is a potent vasoconstrictor. Normally, the vasodilator and vasoconstrictor aspects of insulin are in balance. However, in insulin resistance, hyperinsulinemia sufficient to maintain normal blood glucose through the PI-3-kinase and glut-4 pathway overactivates the MAP kinase pathway resulting in vasoconstriction. Activation of MAP kinase also promotes proliferation of vascular smooth muscle cells, an early vascular abnormality.12,13
2.2 Measuring Insulin Action in Humans
Although insulin regulates many physiological functions, our understanding of defects in insulin action in humans centers around muscle glucose uptake which has become a defining feature. Dynamic measures of insulin action include the euglycemic insulin clamp,14,15 the insulin suppression test with steady-state plasma glucose determination,16 the frequently sampled intravenous glucose tolerance test,17–22 and the insulin tolerance test. Estimates of insulin sensitivity can also be obtained during an oral glucose tolerance test and include the Avignon, Matzuda, Gutt, and Stumvoll indices and the IS0,120.23–26 Static measures use fasting plasma insulin alone or, together with glucose values, result in indices such as the insulin/glucose ratio, HOMA-IR, and the QUICKI. All correlate to varying degrees with clamp-derived insulin sensitivity27–34 (Table 34.1).
The euglycemic insulin clamp, considered the gold standard, involves infusing a constant amount of insulin and a variable amount of glucose over time so that the plasma glucose concentration remains constant.14,15 By quantitating the amount of glucose required, the effect of insulin on whole-body glucose uptake is determined and reported as milligrams of glucose per kg body weight (or lean body mass) per minute. The higher the number of mg/kg/min of glucose infused, the greater the sensitivity at any particular insulin infusion. Adjustments may be made for plasma insulin concentration. Usually data from the last 30 min are used to calculate glucose disposal after a steady-state plasma glucose has been achieved. In its simplest form, the euglycemic insulin clamp method measures whole-body glucose uptake largely in muscle. In general, a glucose uptake above 5 mg/kg/min during a 1 mU/kg/min insulin infusion, which achieves a circulating insulin concentration of approximately 100 μU/ml, is usually considered normal insulin sensitivity, although this should be determined in individual populations. The choice of insulin dose infused during the clamp depends upon the hypothesis being tested. Liver glucose output is suppressed at low insulin concentrations while glucose uptake in muscle occurs at higher insulin concentrations. By combining this method with tracer techniques (labeled glucose or glycerol), the effect of insulin on the liver (suppression of hepatic glucose production, gluconeogenesis) and adipose tissue (lipolysis) can also be determined.35,36 The procedure is reproducible, time consuming, and requires a degree of experience. A variation on the euglycemic insulin clamp, popularized by Reaven, involves the infusion of a fixed dose of insulin and glucose and the resultant steady state plasma glucose is the measure of insulin sensitivity with suppressed insulin secretion.16
The frequently sampled intravenous glucose tolerance test (FSIVGTT) relies on a rise of endogenous insulin in response to a bolus infusion of intravenous glucose (0.3 g/kg of 50% dextrose) delivered over 1 min with plasma samples obtained at 0, 2, 3, 4, 5, 6, 8, 10, 12, 14, 16, 22, 25, 30, 40, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 min.17–22 The minimal model mathematical analysis of the kinetics of the resulting plasma glucose and insulin concentrations determines the fractional glucose disappearance rates per unit of insulin, termed S I. Another parameter describes the effect of glucose on its own disposal at basal insulin concentrations, termed S G. Parameters from the standard FSIVGTT correlate moderately well with clamp-derived insulin sensitivity.8 One limitation is its reliance on release of endogenous insulin which may not be robust in patients with diabetes. Thus, a modification of the technique evolved which uses a bolus of insulin (30 mU/kg) 20 min after the test begins. This improves the overall correlation of the FSIVGTT parameters with the clamp technique (r=0.62, p<0.0001). Subgroups with impaired glucose tolerance or diabetes may not correlate as well (r=0.48, p=0.016 and r=0.41, p=0.03, respectively).18,20,22
Measures based on the oral glucose tolerance test, the Avignon, Matsuda, Stromvoll, and Gutt indexes,23–26 are simpler to perform but may be affected by different and variable rates of gastric emptying and incretin effects. Measures which rely on a fasting glucose and/or insulin are simplest and lend themselves to large population studies.
Correlations of fasting plasma insulin with clamp-derived insulin sensitivity show coefficients of 0.56, p=0.01.33 The HOMA-IR or homeostatic model assessment is calculated using the following formula: fasting plasma glucose mmol/l × fasting plasma insulin μU/ml/22.5. The number 22.5 is a factor derived from the product of a normal glucose of 4.0 mmol/l and a normal insulin 5.0 μU/ml. Thus the value of 1.0 would be found in a “normal” individual and higher numbers indicate insulin resistance. HOMA-IR correlates well with clamp-derived insulin sensitivity, r= 0.88, p<0.0010.30 The HOMA-IR is less accurate in diabetes and, because fasting insulin and glucose reflect liver metabolism, this test assumes that hepatic and peripheral insulin resistance are comparable. The log of HOMA-IR provides more consistent results (correlation with clamp data produces in healthy controls r=–46, p=0.056 and in obese subjects r=–0.79, p<0.0001). Another variation is the QUICKI or the Quantitative Insulin Sensitivity Check Index =1/(log insulin (μU/ml) + log glucose (mg/dl)).31 It demonstrates a good correlation with clamp-derived data but appears to be less robust in non-obese subjects (overall r=0.78, p<0.00001; normal, r=0.49, p=0.01; obese, r=0.8, p<0.008; diabetics, r= 0.7, p<0.0001). Modifications of QUICKI include free fatty acid or glycerol determinations and improve the relationship with the euglycemic clamp-derived insulin sensitivity: QUICKI FFA = 1/(log fasting insulin [μl/ml] + log fasting glucose [mg/dl] + log fasting FFA [mmol/l]).32,34 Limitations may include decreased ability to measure change in insulin sensitivity and reduced accuracy in uncontrolled diabetes. Thus, there are numerous ways to determine insulin sensitivity, each with its own advantages and limitations. The key is choosing one that is both feasible and best addresses the hypothesis being tested.
2.3 Insulin Resistance, Type 2 Diabetes, and Metabolic Abnormalities: Physiological Studies
Following the discovery of the radioimmunoassay technique by Berson and Yalow,37,38 insulin resistance was identified as important in type 2 diabetes. It is present in the majority of type 2 patients with type 2 diabetes, their first-degree relatives, individuals with impaired glucose tolerance and obesity.39–44 For years debates raged about whether insulin resistance or insulin secretion was more important in the pathogenesis of diabetes.39 Most arguments supported insulin resistance since most patients and their relatives were insulin-resistant and nondiabetic individuals without diabetic relatives were not. Fewer arguments supported defective insulin secretion, although it was known that when matched for obesity, patients with type 2 diabetes had lower insulin responses than nondiabetic obese individuals. An important concept was the hyperbolic nature of the relationship between insulin resistance and insulin secretion in maintaining normal glucose tolerance: beta cell function varied reciprocally with the degree of insulin resistance as a constant, called a “disposition index” or DI. Thus mis-matches of beta cell function relative to insulin requirements were predicted to result in hyperglycemia and the development of diabetes.45
Longitudinal studies tracking the progression from normal to impaired to diabetic glucose tolerance provided clearer answers.46–49 Among insulin-resistant Pima Indians, who were followed for 5 years, individuals who remained with normal glucose tolerance developed a slight worsening of insulin resistance and a complementary increase in insulin secretion. In contrast, in those individuals who developed diabetes, there was a slight worsening of insulin resistance but a very dramatic decrease in insulin secretion. Thus, even in the presence of insulin resistance, the key and essential physiological abnormality leading to hyperglycemia was a relative or absolute decrease in insulin secretion relative to insulin requirements.
Studies in a different ethnic group further illustrate this concept of insulin deficiency in diabetes. Among African-Americans, type 2 diabetes is heterogeneous: there are insulin-resistant and insulin-sensitive variants.50 Nearly 30% of African-Americans with a BMI < 28.5 kg/m2 exhibit the unusual insulin-sensitive variant.51 Individuals with the insulin-sensitive variant had fasting plasma insulin levels markedly lower than that in those with the insulin-resistant variant. Postprandial insulin levels were also lower in the insulin-sensitive subgroup suggesting that insulin deficiency was a significant defect in this group. Insulin responses in the typical insulin-resistant variant were lower than that in controls leading to the conclusion that this group had at least two defects, insulin resistance and insulin deficiency, similar to the Pima Indians.
Several important metabolic features distinguish the variants. The insulin-resistant variant is associated with metabolic abnormalities including high plasma triglyceride and low plasma HDL cholesterol levels52 while the insulin-sensitive variant exhibits normal plasma triglycerides, HDL cholesterol, and free fatty acid levels. Body composition also distinguishes the variants. There is a greater amount of visceral adipose tissue volume in the insulin-resistant variant compared with the BMI and age-matched insulin-sensitive variant.53 Increased visceral adipose tissue is inversely associated with insulin-mediated glucose disposal while total subcutaneous adipose tissue is not (Figure 34.2). Increased visceral adipose tissue volume is also associated with increased plasma triglyceride levels, intramyocellular fat, and liver fat54–56. These and other data show that the insulin-resistant form of type 2 diabetes is characterized by increased cardiovascular risk factors with fat in abnormal locations.

Left Panel shows the inverse nonlinear relationship of insulin action to visceral adipose tissue (r = –0.58, Pp < 0.0001; men r = –0.60 (squares) and women r = –0.59 (triangles); the slope and intercept were not different in men and women). Right panel shows there is no significant relationship between insulin-mediated glucose disposal and total subcutaneous adipose tissue volume. Insets shown above the left and right panel, respectively, are a cross section of an abdominal CT image highlighted to show a small compared to a large visceral adipose tissue area.62
Insulin resistance and increased myocellular fat are also associated with abnormalities in muscle mitochondria number, size, and function.57,58 Myocellular ATP production is decreased somewhat in the fasting state and abolished during insulin stimulation in individuals with type 2 diabetes, in their relatives, in obesity and nutrient overload.59 Both increased FFA and glucose may decrease mitochondrial fitness and expression of genes for oxidative phosphorylation, including PGC-1α.60,61 It is difficult to determine whether the fundamental cellular decrease in energy production is a result or a cause of obesity or insulin resistance.
There are population differences in the prevalence of insulin resistance in type 2 diabetes. For example, among Japanese, the insulin-sensitive form has a high frequency (∼60%) with decreased non-traditional cardiovascular risk factors.63 In contrast, South Asian Indians have a high prevalence of insulin resistant diabetes.64 In the US NHANES data, 85% are insulin resistant and only 15% are insulin sensitive.65
Insulin resistance has been associated with multiple metabolic abnormalities (Table 34.2), several of which are traditional CV risk factors (high LDL cholesterol, hypertension, obesity, and diabetes), while the rest are known as nontraditional CV risk factors. Because of the link between insulin resistance and these CV risk factors,66,67 it has been attractive to consider that insulin resistance is the underlying pathophysiological cause of increased CV disease in diabetes and obesity. These metabolic abnormalities include increased inflammatory markers such as increased hsCRP (highly sensitive C-reactive protein), interleukin-6, increased plasminogen activator inhibitor-1 (PAI-1) and increased fibrinogen (predisposing to thrombosis), increased uric acid and endothelial dysfunction with increased microalbuminuria and homocysteine levels.63–77 The association between insulin resistance and hypertension appears to be population specific as it is not present in all ethnic groups78–80 and because among individuals with essential hypertension, only 50% are insulin resistant. Many of these metabolic changes found in insulin resistance are also observed with obesity and will be discussed below. It is difficult to know which is of primary importance, obesity or insulin resistance.
2.4 Insulin Resistance and Obesity
In the 1940s and 1950s, Jean Vague of France described two types of obesity, both occurring in men and women: android or central obesity and gynoid or peripheral obesity.81 The android form was associated with increased rates of diabetes, hypertension, and coronary artery disease while the gynoid form was not.
Obesity is excess of body fat and is most simply defined by a body mass index (BMI) or weight in kilograms divided by the height in meter squared [(weight (kg)/height (m)2]. By convention, an individual with a BMI of less than 25 kg/m2 is lean, with a BMI of 25–29.9 kg/m2 is overweight, and with a BMI greater than 30 kg/m2 is obese. Because of racial differences in body composition in South Asian Indians, an individual with a BMI < 23 is defined as normal, 23–25 is defined as overweight, and > 25 is defined as obese. The BMI is an imperfect measure and for a greater understanding of obesity and its relationship to metabolism, it is important to describe body composition. Total body fat can be estimated by body volume (using an underwater or an air displacement method), density, and weight or by using dual photon absorptiometry (DXA). Visceral and subcutaneous fat and muscle are measured using computed tomography or magnetic resonance imaging and in vivo measurement of metabolic activity of muscle can be obtained by NMR spectroscopy.
Obesity is related to insulin resistance.82,83,87 In studies of obese persons (BMI 30–34.9), McLaughlin84 showed that insulin-resistant individuals compared to insulin sensitive, had significantly higher systolic and diastolic blood pressures and serum triglyceride levels and low HDL cholesterol levels as well as a higher prevalence of impaired glucose tolerance (48% vs 2%). However, not all obese individuals are insulin resistant84,85 as highlighted in Ruderman’s concept of the metabolically “obese” normal-weight individual.86 The European Group for the Study of Insulin Resistance (EGIR) examined the relationship of insulin resistance measured by the euglycemic insulin clamp and hyperinsulinemia in healthy European individuals without hypertension over a wide range of body mass indices.85 Similarly, EGIR reported that only 25% of nondiabetic obese persons (BMI> 25 kg /m2; mean of 29 kg /m2) were insulin resistant based on the euglycemic insulin clamp measurements. This unexpected result suggests that 75% of obese (or non-lean) individuals are insulin sensitive. After correcting for age, sex, and BMI, the waist circumference and waist–hip ratio were no longer associated with insulin resistance. This is not surprising as the waist is highly correlated with BMI and reflects both the visceral and the subcutaneous adipose tissue compartments.
Whether visceral or the subcutaneous adipose tissue is most important relative to insulin resistance has been the subject of considerable debate. In a comprehensive review of studies in adults, correlating subcutaneous and visceral adipose tissue measurements made by CT or MRI to insulin resistance measured using either the euglycemic clamp or the steady-state plasma glucose by insulin sensitivity index, Reaven found that the majority (11 of 18 studies) showed that visceral adipose tissue was most highly correlated with insulin resistance in men and women, blacks, whites, and South Asians (with correlation coefficient ranging from –0.33 to –0.60). However five studies reported that subcutaneous adipose tissue had higher correlation coefficients and two had equivalent correlations between the two fat depots. In favor of subcutaneous adipose tissue is an observation that most circulating free fatty acids derive from this depot and have adverse metabolic effects on both insulin secretion and action and promote inflammation. Although, most of these cross-sectional studies suggest the importance of visceral adipose tissue, several well-constructed intervention studies and one longitudinal one provide the most convincing evidence that excess visceral fat has adverse effects.
An initial report from Goodpaster et al. noted that the subcutaneous but not visceral adipose tissue was most significantly correlated with insulin resistance (r= –0.61 vs –0.52).88 Subsequently, these authors reported that, after diet-induced weight loss, it was the decrease in visceral (not subcutaneous) adipose tissue which correlated most significantly with the improvement in insulin sensitivity.89 Thus, the visceral adipose tissue depot was clearly critical in determining insulin resistance. Lemieux followed a group of women for 7 years and reported on changes in body composition and insulin resistance.90 Comparisons of two subgroups with similar increases in visceral fat despite large differences in subcutaneous fat showed that there was no difference in metabolic parameters including glucose levels and insulin secretion. Moreover, individuals with the largest increase in visceral adipose tissue had significant deterioration in glucose tolerance and increase in insulin levels. A different approach – surgically removing subcutaneous adipose tissue - leads to the same conclusion. Klein studied the effects of subcutaneous adipose tissue liposuction on cardiovascular and metabolic risk factors and insulin action in obese diabetic and nondiabetic patients, before and 10 weeks after the procedure.91 The nondiabetic subjects had 10.5 kg of fat or 28% of the abdominal subcutaneous fat removed and the patients with type 2 diabetes had 9.1 kg of fat removed (44% of this depot); baseline BMIs were 39.9 and 35.1 kg/m2, respectively. Visceral adipose tissue volume did not change. Klein reported that despite a substantial weight loss there was no improvement in metabolic parameters including lipids, glucose, insulin, adiponectin, insulin resistance measured by the euglycemic insulin clamp method or other measures of inflammation including hsCRP, interleukin-6, and tumor necrosis factor alpha (TNF-α). A follow-up study, performed to evaluate long-term metabolic and CV benefit possibly overlooked in the earlier data, showed nearly identical results.92 Improved glycemic and metabolic control seen during the treatment of type 2 diabetes with thiazolidiendiones is frequently associated with increases in subcutaneous fat and decreases in liver, visceral and blood fat suggesting specific roles of different fat depots.95 Finally, a 10 year longitudinal study in Japanese Americans showed that visceral fat measured using the gold standard CT scanning was independently associated with the development of insulin resistance whereas total fat or subcutaneous fat was not.93 The importance of a longitudinal study in well characterized subjects cannot be over emphasized.
Since human studies cannot always provide definitive information, a study in an animal model serves to clarify the role of subcutaneous adipose tissue. In female Syrian hamsters, surgical removal of greater than 50% of the subcutaneous adipose tissue, followed by a high-fat diet, resulted in a marked increase in serum triglycerides and visceral fat as well as a worsening of glucose tolerance and increase in serum insulin levels.94 This demonstrates both the beneficial role of subcutaneous adipose tissue as a metabolic sink for excess calories and the adverse effects of storing calories in the viscera and in blood.95
Visceral adipose tissue has several unique metabolic properties. It demonstrates a high turnover, is more susceptible to catecholamine-induced lipolysis than subcutaneous adipose tissue,96,97 and is under different sex hormone regulation. 1-Beta hydroxysteroid dehydrogenase type 1 activity may also differ. This enzyme converts inactive cortisone to cortisol and thus may cause local tissue changes in hormonal milieu.98 Increased visceral adipose tissue is frequently associated with increased plasma triglyceride level, liver fat, and intramyocellular lipid.55–101 These data suggest that ectopic fat (or fat in the “wrong places”) may trigger inflammation with subsequent deleterious effects.
Adipose tissue is metabolically active and contributes to many factors which play a role in the adverse outcomes of obesity including insulin resistance, diabetes, and CV disease. These factors include increased resistin, increased visfatin, decreased adiponectin, increased inflammation, oxidative stress and increased reactive oxygen species102–113 as well as free fatty acids, plasminogen activator-1 (PAI-1), fibrinogen, and uric acid. While weight loss decreases many of these biomarkers and suggests their importance, the finding that the adipose tissue-derived pro-inflammatory cytokines such as tumor necrosis factor α (TNF-α) can directly trigger inflammation points to a mechanism.102 Several intracellular mediators of these inflammatory stimuli involve IKKβ/ NF-κB and the JNK pathways. Stimuli that activate the IKKβ/ NF-κB and JNK pathways include free fatty acid, glucose, reactive oxygen species, interleukin-6, ceramides, TNF-α, and advanced glycosylated end products (AGEs), as well as viral or bacterial elements. Activation of the pathways results in increased transcription of inflammatory moieties and the perpetuation of inflammation. Increased inflammation is associated with serine/threonine phosphorylation of IRS-1 and contributes to insulin resistance. Activation of macrophage can set in motion an inflammatory cascade of events leading to the vascular atheroma development and CV disease. In this context, two studies serve as proof of concept. Dandonna showed that treatment with insulin immediately after a myocardial infarction decreased inflammation (hsCRP and interleukin-6) and improved cardiac outcomes.114 Goldfine treated obese nondiabetic individuals with salsalate, an anti-inflammatory agent, and reported a decrease in inflammation as well as a decrease in C-peptide and glucose suggesting that decreasing inflammation improves insulin resistance.115 These data show some of the interrelationships of obesity, inflammation, insulin resistance, and CV disease.
2.5 Insulin Resistance, Obesity: Metabolic Heterogeneity
Insulin resistance and obesity are associated and are frequently assessed using the surrogate, hyperinsulinemia, especially in population studies. The European Group for the Study of Insulin Resistance (EGIR),85 which reported on insulin resistance measured by the euglycemic insulin clamp and hyperinsulinemia in healthy European individuals as mentioned earlier, defined insulin resistance as the bottom 10% of the insulin lean group and hyperinsulinemia as the top 10% of fasting plasma insulin. Obesity was defined as a BMI > 25 kg/m2. Insulin resistance was found in only 26% of obese subjects (mean BMI 29 kg/m2), far fewer than anticipated. Hyperinsulinemia was observed in 41% of the obese subjects and both hyperinsulinemia and insulin resistance were present only among 14% of the obese subjects compared to 1.6% of the lean. The frequency of insulin resistance was low in obese individuals and was exceeded by hyperinsulinemia. Thus, hyperinsulinemia may result not only from obesity and insulin resistance but also through other possibly central nervous system signals as well. Hyperinsulinemia, therefore, is not a precise surrogate for insulin resistance and the obese phenotype is heterogeneous in terms of insulin resistance and its metabolic abnormalities.
The heterogeneity of the obese phenotype is further demonstrated in the NHANES data of 5440 participants without known CV disease. Metabolic parameters assessed included fasting plasma glucose and insulin, insulin resistance measured by HOMA-IR, inflammation measured by hsCRP, lipids and blood pressure. Several interesting observations emerged. In the age-standardized group with normal body weight (BMI< 25 kg/m2), 30%, were metabolically unhealthy with two or more abnormalities while in the groups which were overweight (BMI 25–29.9 kg/m2) or obese (BMI > 30 kg/m2), 48.8, and 29%, respectively, were metabolically normal with 0–1 abnormalities. Racial sub-analyses were similar. Correlates of 0–1 metabolic abnormalities were younger age, black race/ethnicity, higher physical activity levels, and smaller waist circumference (Figures 34.3 and 34.4,116). A separate report confirms that obese individuals with high percent body fat can have favorable metabolic profiles characterized by normal insulin sensitivity, lack of high blood pressure, normal lipids, and adiponectin levels.117 These subjects had less liver, visceral and muscle fat, as well as less intima–media thickness, a surrogate for CV disease. Finally, an intervention study of diet-induced weight loss suggested that the two phenotypes might respond differently: the metabolically adverse group improved insulin sensitivity by 26% while in contrast, the metabolically normal group’s sensitivity deteriorated by 11%.118 Whether similarly divergent responses accompany exercise is not known.

Age-standardized prevalence of cardiometabolic abnormalities by body size and sex (A, men; B, women). *p <.001 for proportion metabolically abnormal vs normal weight. (modified from ref116)

Age- and sex-standardized prevalence of cardiometabolic abnormalities by body size and race/ethnicity. A, non-Hispanic whites; B, non-Hispanic blacks; and C, Mexican Americans. *p <.001 for proportion metabolically abnormal vs normal weight. (modified from ref116)
2.6 Insulin Resistance and Cardiovascular Disease – Population Studies
A prodigious number of population studies have tested the hypothesis that insulin resistance is a risk factor for cardiovascular disease in an attempt to understand the 2- to 4-fold increase in CV disease mortality with diabetes.119–121 Most used fasting plasma insulin or HOMA-IR to measure insulin resistance.
Small studies, using the euglycemic insulin clamp, showed a positive relationship between insulin resistance and CV disease. In a report of 208 persons followed for 6 years, those in the highest compared to the lowest tertile had increased CV disease.122 A report on the 6-year follow-up of 73 persons noted that CHD, hypertension, and microalbuminuria were increased in those with insulin resistance.123
Reports using the HOMA-IR showed variable results. The San Antonio Heart study which followed 2569 individuals for 7.5 years with 187 CV events. They reported an odds ratio (OR) of CV disease for the lowest versus the highest tertile of insulin resistance of 1.94 (95% CI 1.05–3.59) after adjustments for multiple confounders including sex, age, ethnicity, smoking alcohol use, physical activity, waist, blood pressure, HDL and LDL cholesterol, and triglycerides.124 The VA HT study125, The Study of Elderly Men127 and the DECODE126, the latter of which followed more than 10,000 individuals for 8.8 years also showed a positive association between insulin resistance and CVD.126,127 In contrast, the Strong Heart Study and the Framingham Offspring Study did not show a relationship of insulin resistance and CVD128,129
The relationship of insulin to CV disease is not as strong as it was initially hypothesized. In 1998, Ruige’s review showed an overall hazard ratio (HR) for insulin and CV disease of 1.18 (95% CI 1.08–1.29) for each 50 pmol/l of fasting plasma insulin and highlighted ethnic/racial heterogeneity.130 In whites the association of insulin and CV disease showed a HR of 1.4 (95% CI 1.23–1.65) compared to non-whites (Nauruans and Pima Indians) of 1.04 (95% CI 0.93–1.16). Whites were older, with clinical outcomes of death or myocardial infarction instead of ECG changes. Several specific studies are worthy of review. The ARIC study of 13,446 men and women with 305 events followed over 6 years showed no relationship of fasting plasma insulin to CV disease.131 Further follow-up revealed a relationship of fasting insulin to incident stroke with a HR of 1.54 (95% CI 1.01–1.3) for each 50 pmol/l of fasting insulin.132 In contrast, the Helsinki Policeman study of 970 men followed up to 22 years did not show a relationship of hyperinsulinemia to stroke after adjustment for age and other CV disease risk factors [HR 1.54 (95% CI 0.9–2.64)] while blood pressure, upper body obesity, and smoking were significantly predictive [HR 1.36 (95% CI 1.18–3.06), 1.59 (95% CI 1.26–2.00), 1.88 (95% CI 1.16–3.04), respectively]. This study highlights an interesting aspect of long-term follow-up. After adjustment for age and other CV disease risk factors, hyperinsulinemia (defined as the highest quintile of insulin area under the curve during an OGTT) was associated with a HR for major incident coronary heart disease at 5, 10, and 15 years [HR 2.36 (95% CI 1.00–5.57), 2.29 (95% CI 1.31–4.02), 1.76 (95% CI 1.09–2.82), respectively] but not at 22 years [HR 1.32 (95% CI 0.89–1.97)]. This attenuation of effect suggests a changing relationship of insulin to CV risk over time. The concept of a changing temporal relationship of a risk factor to a disease as the pathogenesis evolves may explain the varied findings in different studies.133,134 The Caerphilly study of 1056 subjects over 12 years with 127 events showed no relationship of fasting plasma insulin to CV disease.135 Among elderly men born between 1913 and 1923, the risk of coronary heart disease increased 2.4-fold in those in the highest compared to the lowest quintile of fasting plasma insulin after 13-year follow-up.136 The DECODE Study mentioned above,126 also reported that the risk of death was predicted by the highest compared to the lowest tertile of fasting insulin [HR 2.66 (95% CI 1.45–4.9) for women and 1.54 (95% CI 1.16–2.03) for men].The HOMA-IR results were significantly similar [HR 2.35 (95% CI 1.16–2.03) for women and 1.58 (95% CI 1.2–2.09) for men].
A recent meta-analysis of 19 studies in Western populations with 3600 events showed an overall modest predictive effect of insulin on incident coronary heart disease. For fasting insulin, the RR was 1.2 (95% CI 0.98–1.28) and for non-fasting insulin the RR was 1.35 (95% CI 1.14–1.6). Figure 34.5 shows the effect of insulin for all the studies.137

Prospective studies of concentrations of circulating insulin markers and coronary heart disease (CHD) (nonfatal MI or coronary death) in Western populations in 19 prospective studies involving a total of about 3600 incident CHD cases. It shows the risk ratio (RR) for CHD and confidence intervals (CI) for the top third vs the bottom third for fasting insulin (1.12 [95%C 0.98–1.28] for raised fasting insulin) and for non-fasting insulin (1.35 [95% CI 1.14–1.60] for raised non-fasting insulin).137 The left inset shows RR of various study characteristics. Specifically, top and bottom insets show RR of CHD: 1.12 [95% CI 0.98–1.28] for raised fasting insulin, 1.35 (1.14–1.60) for raised non-fasting insulin, respectively
2.7 Metabolic Syndrome: Risk for Type 2 Diabetes and Predictor of CV Disease
Although previously described, in 1988 Gerald Reaven’s Banting Lecture popularized the “metabolic syndrome,” linking insulin resistance as central to, if not the primary cause of a cluster of abnormalities including glucose intolerance, hypertension, and a distinct lipid profile of high triglyceride and low HDL cholesterol levels.8,136–145 The “syndrome” was a plausible explanation linking diabetes and cardiovascular disease. Diabetes, like obesity, is associated with a 2- to 4-fold increase in CV mortality and since most people with diabetes are obese and insulin resistant, clarifying the contribution of the components of each to cardiovascular disease is challenging. Reaven presented the metabolic syndrome as a testable hypothesis and ever since, the challenge has been met with an avalanche of articles.
2.8 Metabolic Syndrome – WHO, ATP III, and IDF Criteria
The central feature of the Reaven’s metabolic syndrome was insulin resistance measured by an insulin clamp. Since the insulin clamp was unwieldy, simpler measures were developed to assess if insulin resistance and metabolic syndrome were predictive of diabetes and increased CV. Various professional organizations devised clinically measurable but arbitrary sets of criteria as surrogates which were initially based on diabetes or insulin resistance. In chronological order these were the following: the World Health Organization (WHO, 1999), European Group for the Study of Insulin Resistance (EGIR, 1999), the National Cholesterol Education Panel (NCEP) of the American Heart Association (AHA), Adult Treatment Panel (ATP III) criteria (2001), the American Association of Clinical Endocrinologists (AACE) (2003), the revised AHA criteria (2005), and the International Diabetes Federation (IDF, 2005) (Tables 34.3 and 34.4 ).146–151
While the WHO and the EGIR were centered on diabetes, glucose intolerance, or insulin resistance, plus several other factors, the NCEP–ATP III version was developed by cardiologists with the express goal of targeting individuals at high risk for CV disease prevention. In NCEP–ATP III, three out of five equally weighted criteria defined the metabolic syndrome. Neither diabetes nor insulin resistance were necessary for the diagnosis of metabolic syndrome. In the IDF version, the central criterion was a high waist circumference (adjusted for differences in ethnic groups) plus two of four other elements. On the surface, these appear similar but the WHO, EGIR, AACE, and IDF are anchored hierarchically in diabetes/glucose intolerance/ insulin resistance or central obesity while in NCEP–ATP III all criteria are equal. Although conceptually NCEP–ATP III proposal appears simple, a large number of combinations of three out of five criteria raise the question of whether all combinations equally predict CV disease.152
2.9 Prevalence and Stability Over Time of the Metabolic Syndrome
The prevalence of metabolic syndrome varies by definition, population, and gender; reports suggest that its stability differs among populations. Its prevalence based on the NCEP–ATP III criteria was ∼24% in the US NHANES population of 1988–1994 and rose to ∼34% in 1999–2002 survey (Figure 34.6)153,154

Increase in metabolic syndrome over time in whites, African-Americans, and Mexican Americans
The San Antonio Heart study followed two cohorts over 10 and 7 years and reported an increased prevalence of the metabolic syndrome by the NCEP–ATP III criteria.155 Among nondiabetic individuals, percent rate increases were from 15.5 to 25.8% and 23.3 to 30.4% in men and 10.8 to 22.6% and 23.3 to 30.4 in women in the first and second cohorts. In diabetics these rates were significantly higher, ranging from 79 to 85% in the second cohort in men and 66 to 87% in both cohorts of women. In contrast, in Mexico, the rate in men was 38.9% without change over 7 years while in women there was no change over 3 years and a slight decrease (65–59%) over the final 4-year study interval.156 A Finnish cross-sectional study done 10 years apart (1992 and 2002) shows the increasing prevalence of NCEP–ATP III metabolic syndrome (1992 and 2002); men had a higher prevalence of metabolic syndrome compared to women at baseline but only women had a significant increase in incidence over time (32–38% for women and 48.8–52.6% for men).157 Among adolescents, the diagnosis of the metabolic syndrome is reported to fluctuate.158
Several important issues should be examined. (1) Is the metabolic syndrome useful in identifying individuals at high risk for diabetes and/or and increased CV disease in nondiabetic and diabetic populations? (2) Is insulin resistance the basis for the metabolic syndrome?
2.10 Does the Metabolic Syndrome Predict Diabetes in Nondiabetes Populations?
The metabolic syndrome strongly predicts diabetes. This may not be surprising because one component is an elevated blood glucose which alone has a similar predictive value.159,160 Indeed, metabolic syndrome accounts for up to half of the new cases of diabetes in the Framingham offspring study among those who did not have diabetes at baseline and were followed for 8 years.161 While the metabolic syndrome strongly predicts diabetes (HR ∼4–6), fasting plasma glucose is far more predictive (HR ∼18). The reader is referred to a recent review for more details.162
2.11 Does the Metabolic Syndrome Predict CV Disease in Nondiabetic Populations?
Early studies143 enthusiastically reported robust associations of insulin resistance and CV disease but subsequent more rigorous examinations adjusting for components of the metabolic syndrome found it less informative.
Ford summarized 17 prospective studies from 1998 to 2002, and after adjusting for confounders, the metabolic syndrome by WHO and NCEP–ATP III criteria modestly and similarly predicted CV disease (RR for WHO was 1.93 (95% CI 1.39–2.07) and NCEP was 1.65 (95% CI 1.38–1.00) and all-cause mortality (RR for WHO was 1.37 (95% CI 1.09–1.74) and for NCEP was 1.27 (95% CI 0.09–1.78)). In contrast, metabolic syndrome was a much more robust predictor of diabetes (RR 2.60–2.99).163 Among elderly Finns, followed for 13.5 years, Wang reported on the relationship of all the different definitions of metabolic syndrome and their ability to predict diabetes or CV mortality and disease.164 WHO and IDF criteria predicted CHD and CV disease mortality significantly in men [(HR 1.97 (95% CI 1.27–3.05) and 1.70 (95% CI 1.19–2.43) and 1.58 (95% CI 1.02–2.44) and 1.34 (0.94–1.93)], respectively. None predicted all-cause mortality. The individual components themselves significantly predicted disease by a similar magnitude as the metabolic syndrome: impaired glucose tolerance [HR 1.55 (95% CI 1.17–2.0)] in WHO, IDF, and ATP III metabolic syndrome; low HDL cholesterol (<1 mmol/l) [HR 1.50 (95% CI 1.12–2.01)] and microalbuminuria (albumin creatinine ratio greater than 3.39 mg/mmol) [HR 1.86 (95% CI 1.40–2.47)] in metabolic syndrome by WHO. In another publication of the same population followed for 14 years,165 metabolic syndrome predicted stroke with hazard ratios varying from 1.52 to 1.72 depending on the criteria used. This analysis also demonstrated that the individual components had nearly the same predictive value as the metabolic syndrome in predicting for outcomes and for CV disease risk.
The Malmo study followed over 5,000 Swedes for 11 years and showed a HR for a composite endpoint of MI and stroke of 1.11 (95% CI 0.86–1.44), 1.59 (1.25–2.03), and 1.35 (1.05–1.74), respectively, for the IDF, ATP III, and EGIR metabolic syndrome after adjusting for confounders.166 Only the NCEP–ATP III criteria were predictive for men but not for women. Individual components had significant hazard ratios of a similar order of magnitude: 2.97, 2.01, 1.81, and 1.75 for blood pressure, HDL cholesterol, obesity, and current smoking. NCEP–ATP III was most predictive of CV disease; however, other studies showed IDF to be equivalent to NCEP–ATP III167 and the DECODE study showed that WHO was better.168
Unlike the other studies, DECODE was a collaborative mortality analysis of 11 European studies of over 10,000 individuals, followed for 7–16 years.168 The prevalence rates of metabolic syndrome for men and women in the WHO were 27% and 19% NCEP, 25.9%, 23.4%; NCEP revised, 32.2% and 28.5%; IDF, 35.9% and 34.1%, respectively. In men the likelihood of CV mortality was 2.09 (95% CI 1.59–2.76), 1.74 (95% CI 1.31–2.3), 1.72 (95% CI 1.31–2.26), and 1.5 (95% CI 1.15–1.99) for WHO, NCEP, NCEP revised, and IDF metabolic syndrome, respectively; the corresponding values in women were weaker: 1.60 (95% CI 1.01–2.51), 1.39 (95% CI 0.89–2.18), 1.09 (95% CI 0.7–1.69), 1.53 (95% CI 0.99–2.39), respectively. Individual components were predictive of CV mortality in men and women with similar orders of magnitude (e.g., for blood pressure, HR was 2.3 for men, for triglyceride HR was 1.68).
Sattar reported on two studies in the elderly in Britain.169 One was the PROSPER study which randomized 4,812 nondiabetic individuals, age 70–82 years, to placebo or 40 mg pravastatin and followed them for 3.2 years; there were 774 incident cases of CV events and 287 of diabetes. The NCEP–ATP III criteria of metabolic syndrome did not predict CV events [HR 1.07 (95% CI 0.86–1.32)] nor did BMI > 30 g/m2, triglyceride levels > 1.69 mmol/l, fasting glucose > 6.1 mmol/l, or blood pressure > 130/80 (adjusted for confounders including treatment allocation). Positive predictors included age, male sex, prior CV disease, and low HDL cholesterol. A second study of 2,737 men, age 60–79, reported in the same paper, showed similar negative findings. In contrast, both studies showed that metabolic syndrome was a robust predictor of diabetes [HR 4.41 (95% CI 3.33–5.84) and 7.47 (95% CI 4.90–11.46), respectively]; however, the FPG was a nearly 4-fold better predictor [HR 18.0 (95% CI 13.9–24.5)]. Fasting plasma glucose and waist as dichotomized variables did not predict CV disease. Overall, metabolic syndrome was of no benefit in CV disease risk stratification for the elderly.
Similarly, Gami’s meta-analysis of 37 studies representing 172,572 middle age individuals without CV disease reported only a modest effect of metabolic syndrome in predicting CV risk with a HR of 1.49 (95% CI 1.37–1.61).170 Gami concluded that metabolic syndrome did not enhance risk prediction beyond that of the Framingham algorithm.
The prospective Framingham Heart Offspring Study161 followed 3323 individuals for 8 years and showed a more positive relationship of metabolic syndrome and CV disease. Of the 2649 with neither diabetes nor CV at baseline, there was a higher prevalence rate of NCEP–ATP III in men than women (26.8% vs 16.1%). Men had an age-adjusted HR of 2.88 (95% CI 1.96–4.16) for CV disease and 2.59 (95% CI 1.62–3.98) for CHD disease which was higher than that for women [HR 2.2 (95% CI 1.31–3.88) and 1.54 (95% CI 0.68–3.5)]. The population-attributable risk was 34 and 29% in men and 16 and 8% in women for CV disease and CHD, respectively, thus accounting for nearly a third of new CV disease cases in 8 years.
2.12 Does the Metabolic Syndrome Predict CV Disease in Diabetic Populations?
The evidence is mixed and suggests that metabolic syndrome does not uniquely predict incident CV disease or mortality over and above its components.
Three studies show a positive predictive value of the metabolic syndrome.171–173 Bonora171 studied over 900 individuals with diabetes and found that > 90% had the metabolic syndrome by the WHO criteria. Baseline CV disease was more common in patients with the metabolic syndrome than without (32.9% vs 17.8%); among a group without CV disease at baseline, metabolic syndrome was an independent predictor of incident CV disease with an OR of almost 5-fold over 4.5 years and 2-fold for prevalent CV disease. Guzder172 reported that in new onset type 2 diabetes, metabolic syndrome by ATP III criteria was present in 82% and predicted 2- and 4-fold increases in incident and prevalent CV disease. Tong173 reported on 4350 Chinese individuals followed for 7.1 years and found that metabolic syndrome by ATP III criteria (but not IDF criteria) predicted a 2.5-fold increase in CHD. This study also noted that micro- and macroalbuminuria, hypertension, and HDL cholesterol were all significant predictors of CV disease. The frequency of metabolic syndrome by NCEP–ATP III or IDF was 65%.
In contrast, three studies show that the metabolic syndrome has no clear predictive value for CV disease.174–176 Studies by Sone174 from Japan and Bruno175 from Italy showed similar results in approximately 1550 patient each followed for 8.9 and 11 years, respectively. Sone’s study in type 2 diabetes without baseline CV disease reported 51% with metabolic syndrome by WHO criteria and 45% with metabolic syndrome by ATP III criteria.174 ATP III was not predictive of CV disease in men or women. WHO was predictive only in women. In men, other factors such as triglycerides or HDL cholesterol were better or equivalent. Bruno’s study reported 75% with metabolic syndrome (WHO) but with no difference in mortality in patients with and without the metabolic syndrome (∼50% in each group) over 11 years.175 As with Sone’s study, metabolic syndrome with one or more components compared to zero components conferred an ∼2-fold increase in CV disease but the individual components were equally predictive. Finally, in 2007, Cull reported on the 10.3-year follow-up of 4,542 new onset type 2 diabetes patients in the UKPDS study.176 Metabolic syndrome was determined in four ways resulting in different prevalence rates: by ATP III 61%, by WHO 38%, by IDF 54%, and by EGIR 24%. The HR for CV disease was 1.3, 1.45, 1.23, and 1.0, respectively. Metabolic syndrome did not predict microvascular disease. The positive predictive value for CV disease was poor and ranged from 18 to 20%. Furthermore, in 47% of individuals without metabolic syndrome, there was a 10-year estimated CV disease risk of > 20% and in 37% of those with metabolic syndrome there was a 10-year estimated CV disease risk of < 20%.
Despite positive reports, overall, the metabolic syndrome is a rather poor predictor of CV disease in type 2 diabetes.
2.13 Why Does Not the Metabolic Syndrome Predict CV Disease in Type 2 Diabetes?
There are several potential explanations for the lack of predictive power of the MS for CV disease in patients with diabetes.176 The metabolic syndrome uses dichotomized variables while in fact these variables have a continuous relationship with CV disease (e.g., triglyceride and HDL). Not all the elements of the metabolic syndrome are equivalent in determining CV risk even though in the NCEP–ATP III criteria all are given equal weight. In fact, fasting plasma glucose of greater than 6.1 mmol/l is very strongly associated with CV disease risk.177,178 Diabetes is a greater risk factor for mortality and CV disease than metabolic syndrome (HR 5 and 3.6 vs 3.5 and 2.7, respectively),179 and the excess CV mortality in patients with known CV disease associated with metabolic syndrome is due mostly to diabetes; this excess disappears after controlling for diabetes.180,181
2.14 Does the Combination of Metabolic Syndrome and Insulin Resistance Predict CV Disease?
Although the elements do cluster, the metabolic syndrome does not improve the ability to identify a high CV risk cohort. In the current context, it is not clear whether this is because insulin resistance is not an antecedent CV risk factor or because metabolic syndrome does not capture insulin resistance. Another very real issue is that each component may have several causes besides insulin resistance. It is not clear if insulin resistance serves as an etiology for traditional or nontraditional CV disease risk factors.
Studies using the euglycemic insulin clamp (or FSIVGTT) showed that insulin resistance was present in only 33% of subjects with the metabolic syndrome182,183 and its sensitivity varied from 20 to 66%. Thus, metabolic syndrome may have a low sensitivity for identifying insulin resistance.
Several population studies bear on this question. The 11-year follow-up report of the Framingham Heart Offspring Study analyzed the impact of insulin resistance (measured using HOMA-IR) on CV disease and diabetes in a subset of people with and without the metabolic syndrome.184 Using ATP III criteria, approximately one quarter had metabolic syndrome (27.8%) and over half of these were insulin resistant, while in those without metabolic syndrome, 12.8% were insulin resistant. The study found that compared to those with neither the metabolic syndrome nor insulin resistance, metabolic syndrome alone or insulin resistance alone did not predict CV disease [HR 1.2 (95% CI 0.7–1.9)] and 1.3 (95% CI 0.9–1.19)] but both together doubled the risk [HR 2.3 (95% CI 1.7–3.1)] with a population-attributable risk for both men and women of 18% compared to neither metabolic syndrome nor insulin resistance (Figure 34.7). Thus, insulin resistance adds to the CV disease risk beyond just metabolic syndrome and confirms earlier reports that insulin resistance and metabolic syndrome are not identical but describe different subsets of population risk. In contrast, insulin resistance, metabolic syndrome, or both are increasingly predictive for diabetes.

Relative risk of incident CVD (left panel) or diabetes (right panel) and 8-year follow-up based on the presence of ATP III metabolic syndrome (MetS) or insulin resistance (IR) in the Framingham Heart Offspring Study. On both panels, open bars reflect data adjusted for age and sex. Hatched bars are adjusted for CV risks factors (left panel): age, sex, LDL cholesterol, and smoking and for diabetes risk factors (right panel): age, sex, family history of diabetes, BMI, and 2-h glucose during oral glucose tolerance testing. (modified from ref184)
To conclude, although the metabolic syndrome may add to the prediction of CV risk, overall its magnitude is not as great as was once thought. Nevertheless, the metabolic syndrome is likely to be not more than the sum of its parts after adjusting for standard CV risk factors.
3 Summary: Insulin
3.1 Resistance and the Metabolic Syndrome: The Debate Continues
From its popular inception in 1988, the metabolic syndrome, aka the insulin resistance syndrome, was presented as a testable hypothesis. Initially, insulin resistance was hypothesized to be the physiological basis for the observed clustering of metabolic variables including diabetes, lipid abnormalities, blood pressure, increased cardiovascular disease, and central obesity. It captured the imagination of thousands of scientific investigators and the lay public as the incidence of obesity and diabetes increased to epidemic proportions. Although a great deal of scientifically exciting and valid knowledge has been generated to understand how the variables are related, there is still no unifying consensus that links them.
In 2001, this scientific hypothesis was used as the basis to create a simple clinical screening tool to prevent cardiovascular disease. The presence of three of five variables defined the ATP III metabolic syndrome which was given the stamp of approval by the American Heart Association. It was a novel approach to the old problem of cardiovascular risk and set off a worldwide frenzy of investigations resulting in hundreds of papers, reviews and chapters. It was formally legitimized by an International Classification of Diseases (ICD-9) code. Many competing definitions of the “metabolic syndrome” developed. In 2005, the American Diabetes Association and the European Association for the Study of Diabetes concluded that the metabolic syndrome was an emperor with no clothes. A calculating look revealed that it did not provide sufficient predictive power for CV disease beyond conventional risk factors or the individual components of the metabolic syndrome. Although metabolic syndrome was strongly predictive of diabetes, the fasting plasma glucose was markedly better. The darling of two decades had lost favor.
In 2008, the Endocrine Society published a Clinical Practice Guideline for the Primary Prevention of CV disease or coronary heart disease.185 Expert opinion recommended that the metabolic syndrome could be used to identify an early likelihood of developing CV disease. Since the variables of the metabolic syndrome are CV risk factors and do cluster, the presence of any one should raise awareness to determine 10 years’ absolute risk for cardiovascular disease using the Framingham, PROCAM, or SCORE algorithms. Treatment should be initiated based on this with a focus on apoB-containing lipoproteins [LDL and VLDL cholesterol (triglyceride)], hypertension, increased blood glucose, enlarged waist, a prothrombotic, and a pro-inflammatory state. The guideline further advises that patients with metabolic risk initiate preventive measures with priority given to lifestyle modifications. The metabolic syndrome has risen again.
In the final analysis, what is needed is a way to identify individuals at high risk for cardiovascular disease who might benefit from prevention of the first CV event (primary prevention). Currently, the absolute prevalence rates of CV events are low, at ∼2–3% and in a recent trial of primary prevention of CV disease, only 1 individual benefited out of 100 treated with a statin.186 Given these data, most “healthy” individuals choose not to adhere to long-term preventive strategies, whether lifestyle or pharmacologic. It is obvious that the metabolic syndrome is not a robust marker and neither are the usual risk factors such as LDL cholesterol.
In order to identify individuals who are at risk before they have an event, future novel approaches might involve genomic analyses of CV risk. Another potential approach is to develop the concept of the vascular system phenotype or condition of the vascular using structural measures. This would enrich the target population for primary prevention of CV disease. With the advent of new technology such as ultrasound or CT, can we develop a better correlation between the structure of the vasculature and the risk for CV events? This would also identify a population enriched for risk of CV disease and an effective target for primary prevention. It is likely that these novel approaches would increase compliance, adherence, and cost-effectiveness of CV disease prevention.
References
Reaven G. The metabolic syndrome or the insulin resisvntance syndrome? Different names, different concepts, and different goals. Endocrinol Metab Clin North Am. 2004;33:283–303.
DeFronzo RA. Banting Lecture: from the triumvirate to the ominous Octet June 2008, American Diabetes Association. http://www.diabetesconnect.org/storetemplate/webcast_list.aspx?ses=902
Bergman RN, Finegood DT, Kahn SE. The evolution of beta-cell dysfunction and insulin resistance in type 2 diabetes. Eur J Clin Invest. 2002;32(Suppl 3):35–45.
Taguchi A, White MF. Insulin-like signaling, nutrient homeostasis and life span. Ann Rev Physiol. 2008;70:191–212.
Taniguchi CM, Emalnuelli B, Kahn CR. Critical nodes in signaling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96.
Chen Y, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435.
Muoio DM, Newgard CB. Molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:193–205.
Reaven G. Banting lecture. Role of insulin resistance in human disease. Diabetes. 1988;37:15.
Lebovitz HE. Insulin resistance – a common link between type 2 diabetes and cardiovascular disease. Diabetes Obes Metab. 2006;8:237–249. Review.
Cusi K, Maezono K, Osman A, et al. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–320.
Schwartz EA, Reaven PD. Molecular and signaling mechanisms of atherosclerosis in insulin resistance. Endocrinol Metab Clin North Am. 2006;35:525–549.
Fröjdő S, Vidal H, Pirola L. Alterations of insulin signaling in type 2 diabetes: a review of the current evidence from humans. Biochem Biophy Acta. 2009;1792(2):83–92. Review.
Leroith D, Accili D. Mechanisms of disease: using genetically altered mice to study concepts of type 2 diabetes. Nat Clin Pract Endocrinol Metab. 2008;4:164–172.
DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol Endocrinol Metab Gastrointest Physiol. 1979;237:E214–E223.
Sherwin RS, Kramer KJ, Tobin JD, et al. A model of the kinetics of insulin in man. J Clin Invest. 1974;53:1481–1492.
Shen SW, Reaven GM, Farquhar JW. Comparison of impedance to insulin-mediated glucose uptake in normal subjects and in subjects with latent diabetes. J Clin Invest. 1970;49:2151–2160.
Bergman RN, Phillips LS, Cobelli C. Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta cell glucose sensitivity from the response of intravenous glucose. J Clin Invest. 1981;68:1456–1467.
Bergman RN, Prager R, Volund A, Olefsky JM. Equivalence of the insulin sensitivity index in man derived by the minimal model method and the euglycemic glucose clamp. J Clin Invest. 1987;79:790–800.
Bergman RN, Ider YZ, Bowden CR, Cobelli C. Quantitative estimation of insulin sensitivity. Am J Physiol. 1979;236:E667–E677.
Yang YJ, Youn JH, Bergman RN. Modified protocols improve insulin sensitivity estimation using the minimal model. Am J Physiol Endocrinol Metab. 1987;253:E595–E602.
Beard JC, Bergman RN, Ward WK, Porte D Jr. The insulin sensitivity index in nondiabetic man. Correlation between clamp-derived and IVGTT-derived values. Diabetes. 1986;35:362–369.
Finegood DT, Hramiak IM, Dupre J. A modified protocol for estimation of insulin sensitivity with the minimal model of glucose kinetics in patients with insulin-dependent diabetes. J Clin Endocrinol Metab. 1990;70:1538–1549.
Stumvoll M, Mitrakou A, Pimenta W, et al. Use of the oral glucose tolerance test to assess insulin release and insulin sensitivity. Diabetes Care. 2000;23:295–330.
Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22:1462–1470.
Gutt M, Davis CL, Spitzer SB, et al. Validation of the insulin sensitivity index [ISI(0,120)]: comparison with other measures. Diabetes Res Clin Pract. 2000;47:177–184.
Avignon A, Boegner C, Mariano-Goulart D, Colette C, Monnier L. Assessment of insulin sensitivity from plasma insulin and glucose in the fasting or post oral glucose-load state. Int J Obes Relat Metab Disord. 1999;23:512–517.
Laakso M. How good a marker is insulin level for insulin resistance? Am J Epidemiol. 1993;137:959–965.
Yeni-Komshian H, Carantoni M, Abbasi F, Reaven GM. Relationship between several surrogate estimates of insulin resistance and quantification of insulin-mediated glucose disposal in 490 healthy, nondiabetic volunteers. Diabetes Care. 2000;23:71–175.
Hanson RL, Pratley RE, Bogardus C, et al. Evaluation of simple indices of insulin sensitivity and insulin secretion for use in epidemiologic studies. Am J Epidemiol. 2000;151:190–198.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419.
Katz A, Nambi SS, Mather K, et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab. 2000;85:2402–2410.
Rabasa-Lhoret R, Bastard JP, Jan V, et al. Modified quantitative insulin sensitivity check index is better correlated to hyperinsulinemic glucose clamp than other fasting-based index of insulin sensitivity in different insulin-resistant states. J Clin Endocrinol Metab. 2003;88:4917–4923.
Burén J, Lindmark S, Renström F, Eriksson JW. In vitro reversal of hyperglycemia normalizes insulin action in fat cells from type 2 diabetes patients: is cellular insulin resistance caused by glucotoxicity in vivo? Metabolism. 2003;52:239–245.
Perseghin G, Caumo A, Caloni M, Testolin G, Luzi L. Incorporation of the fasting plasma FFA concentration into QUICKI improves its association with insulin sensitivity in nonobese individuals. JCEM. 2001;86:4776–4781.
Radziuk J. Insulin sensitivity and its measurement: structural commonalities among the methods. J Clin Endocrinol Metab. 2000;85:4426–4433.
Finegood DT, Bergman RN, Vranic M. Estimation of endogenous glucose production during hyperinsulinemic-euglycemic glucose clamps. Comparison of labeled and unlabeled exogenous glucose infusates. Diabetes. 1987;36:914 –924.
Hales CN, Randle PJ. Immunoassay of insulin with insulin-antibody precipitate. Biochem J. 1963;88:137–146.
Kahn CR, Roth J. Berson, Yalow, and the JCI: the agony and the ecstasy. J Clin Invest. 2004;114:1051–1054.
Ferrannini E. Insulin resistance versus insulin deficiency in non-insulin-dependent diabetes mellitus: problems and prospects. Endocr Rev. 1998;19:477–490.
Ginsberg H, Kimmerling G, Olefsky JM, Reaven GM. Demonstration of insulin resistance in untreated adult onset diabetic subjects with fasting hyperglycemia. J Clin Invest. 1975;55:454 –461.
Gautier JF, Wilson C, Weyer C, et al. Low acute insulin secretory responses in adult offspring of people with early onset type 2 diabetes. Diabetes. 2001;50:1828–1833.
Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med. 1990;113:909–914.
Lillioja S, Mott DM, Spraul M, et al. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:1988–1992.
DeFronzo RA. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37:667–687.
Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW, Neifing JL, Ward WK, Beard JC, Palmer JP, et al. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes. 1993;42:1663–1672.
Cnop M, Vidal J, Hull RL, et al. Progressive loss of beta-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes. Diabetes Care. 2007;30:677–682.
Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787–794.
Weyer C, Tataranni PA, Bogardus C, Pratley RE. Insulin resistance and insulin secretory dysfunction are independent predictors of worsening of glucose tolerance during each stage of type 2 diabetes development. Diabetes Care. 2001;24:89–94.
Ferrannini E, Natali A, Bell P, Cavallo-Perin P, Lalic N, Mingrone G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). J Clin Invest. 1997;100:1166–1173.
Banerji MA, Lebovitz HE. Insulin-sensitive and insulin-resistant variants in NIDDM. Diabetes. 1989;38:784–792.
Chaiken RL, Banerji MA, Pasmantier RM, Huey H, Hirsch S, Lebovitz HE. Patterns of glucose and lipid abnormalities in Black NIDDM subjects. Diabetes Care. 1991;14:1036–1042.
Banerji MA, Lebovitz HE. Coronary heart disease risk factor profiles in Black patients with non-insulin dependent diabetes mellitus. Am J Med. 1991;91:51–58.
Banerji MA, Chaiken RL, Gorden D, Kral JG, Lebovitz HE. Does intra- abdominal adipose tissue in Black men determine whether NIDDM is insulin resistant or insulin-sensitive. Diabetes. 1995;44:141–146.
Petersen KF, Dufour S, Savage DB, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA. 2007;104:12587–12594.
Banerji MA, Buckley C, Chaiken RL, Gordon D, Lebovitz HE, Kral JG. The relationship of liver fat, triglycerides and visceral adipose tissue in insulin-resistant and insulin-sensitive black men with NIDDM. Int J Obesity. 1995;19:846–850.
Hwang JH, Stein DT, Barzilai N, et al. Increased intrahepatic triglyceride is associated with peripheral insulin resistance: in vivo MR imaging and spectroscopy studies. Am J Physiol Endocrinol Metab. 2007;293:E1663–E1669.
Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55(Suppl 2):S9–S15. Review.
Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14.
Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387.
Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471.
Szendoeli J, Roden M. Mitochondrial fitness and insulin sensitivity in humans. Review. Diabetelologia. 2008;51:2155–2167.
Banerji MA, Lebowitz J, Chaiken RL, Gordon D, Kral JG, Lebovitz HE. Relationship of visceral adipose tissue and glucose disposal is independent of sex in black NIDDM subjects. Am J Physiol. 1997;273:E425–432.
Taniguchi A, Fukushima M, Sakai M, et al. The role of the body mass index and triglyceride levels in identifying insulin-sensitive and insulin-resistant variants in Japanese non-insulin-dependent diabetic patients. Metabolism. 2000;49:1001–1005.
Banerji MA, Faridi N, Atluri R, Chaiken RL, Lebovitz HE. The relationship of body composition, insulin resistance and leptin in Asian Indian men. J Clin Endocrinol Metab. 1999;84:137–144.
Haffner SM, Mykkänen L, Festa A, Burke JP, Stern MP. Insulin-resistant prediabetic subjects have more atherogenic risk factors than insulin-sensitive prediabetic subjects: implications for preventing coronary heart disease during the prediabetic state. Circulation. 2000;101:975–980.
Pischon T, Boeing H, Hoffmann K, et al. General and abdominal adiposity and risk of death in Europe. N Engl J Med. 2008;359:2105–2120.
Flegal KM, Graubard BI, Williamson DF, Gail MH. Excess deaths associated with underweight, overweight and obesity. J Am Med Assoc. 2005;293:1861–1867.
Weyer C, Yudkin JS, Stehouwer CD, Schalkwijk CG, Pratley RE, Tataranni PA. Humoral markers of inflammation and endothelial dysfunction in relation to adiposity and in vivo insulin action in Pima Indians. Atherosclerosis. 2002;161:233–242.
Stefan N, Vozarova B, Funahashi T, et al. Plasma adiponectin concentration is associated with skeletal muscle insulin receptor tyrosine phosphorylation, and low plasma concentration precedes a decrease in whole-body insulin sensitivity in humans. Diabetes. 2002;51:1884–1888.
Vozarova B, Weyer C, Hanson K, Tataranni PA, Bogardus C, Pratley RE. Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obes Res. 2001;9:414–417.
Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. Review.
Salmenniemi U, Ruotsalainen E, Pihlajamäki J, et al. Multiple abnormalities in glucose and energy metabolism and coordinated changes in levels of adiponectin, cytokines, and adhesion molecules in subjects with metabolic syndrome. Circulation. 2004;110:3842–3848.
Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118:2992–3002. Review.
Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction: implications for the syndrome of insulin resistance. J Clin Invest. 1996;97:2601–2610.
Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904.
Qasim A, Mehta NN, Tadesse MG, et al. Adipokines, insulin resistance, and coronary artery. J Am Coll Cardiol. 2008;52:231–236.
Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950.
Reaven GM. Insulin resistance/compensatory hyperinsulinemia, essential hypertension, and cardiovascular disease. J Clin Endocrinol Metab. 2003;88:2399–2403.
Saad MF, Lillioja S, Nyomba BL, et al. Racial differences in the relation between blood pressure and insulin resistance. N Engl J Med. 1991;324:733–739.
Ferrannini E, Natali A, Capaldo B, Lehtovirta M, Jacob S, Yki-Järvinen H; for the European Group for the Study of Insulin Resistance (EGIR). Insulin resistance, hyperinsulinemia, and blood pressure. Role of age and obesity. Hypertension. 1992;30:1144–1149.
Vague J. The degree of masculine differentiation of obesities: a factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease. Am J Clin Nutr. 1956;4:20–34.
McLaughlin T, Allison G, Abbasi F, Lamendola C, Reaven G. Prevalence of insulin resistance and associated cardiovascular disease risk factors among normal weight, overweight, and obese individuals. Metabolism. 2004;53:495–499.
Bogardus C, Lillioja S, Mott DM, Hollenbeck C, Reaven G. Relationship between degree of obesity and in vivo insulin action in man. Am J Physiol. 1985;248(3 Pt 1):E286–E291.
McLaughlin T, Abbasi F, Lamendola C, Reaven G. Heterogeneity in the prevalence of risk factors for cardiovascular disease and type 2 diabetes mellitus in obese individuals: effect of differences in insulin sensitivity. Arch Int Med. 2007;167:642–648.
Ferrannini E, Natali A, Bell P, Cavallo-Perin P, Lalic N, Mingrone G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance, EGIR. J Clin Invest. 1997;100:1166–1173.
Ruderman N, Chisholm D, Pi-Sunyer X, Schneider S. The metabolically obese, normal-weight individual revisited. Diabetes. 1998;47:699–713. Review.
Reaven GM. Insulin resistance: the link between obesity and cardiovascular disease. Endocrinol Metab Clin North Am. 2008;37:581–601.
Goodpaster BH, Thaete FL, Simoneau J-A, Kelley DE. Subcutaneous abdominal fat and thigh muscle composition predict insulin sensitivity independently of visceral fat. Diabetes. 1997;46:1579–1585.
Effects of weight loss on regional fat distribution and insulin sensitivity in obesity. Diabetes. 1999;48:839–847.
Lemieux S, Després J-P, Moorjani S, et al. Are gender differences in cardiovascular disease risk factors explained by the level of visceral adipose tissue? Diabetologia. 1994;37:757–764.
Klein S, Fontana L, Young VL, et al. Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N Engl J Med. 2004;350:2549–2557.
Mohammed BS, Cohen S, Reeds D, Young VL, Klein S. Long-term effects of liposuction on metabolic risk factors for coronary heart disease. Obesity (Silver Spring). 2008;16:2648–51. Epub 2008 Sep 25.
Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359.
Weber RV, Buckley C, Fried SK, Kral JG. Subcutaneous lipectomy causes a metabolic syndrome in hamsters. Am J Physiol Regul Integr Comp Physiol. 2000;279:R936–R943.
Banerji M, Tiewala M, Catanzaro J, Lebovitz HE. Decreased plasma free fatty acids improve insulin sensitivity in African-American patients with type 2 diabetes treated with rosiglitazone. Presented in the 86th Annual Meeting of the Endocrine Society in New Orleans. 2004, Poster P1-330.
Lundgren M, Burén J, Lindgren P, Myrnäs T, Ruge T, Eriksson JW. Sex- and depot-specific lipolysis regulation in human adipocytes: interplay between adrenergic stimulation and glucocorticoids. Horm Metab Res. 2008;40:854–860.
Wajchenberg BL, Giannella-Neto D, da Silva ME, Santos RF. Depot-specific hormonal characteristics of subcutaneous and visceral adipose tissue and their relation to the metabolic syndrome. Horm Metab Res. 2002;34:616–621.
Tomlinson JW, Finney J, Gay C, Hughes BA, Hughes SV, Stewart PM. Impaired glucose tolerance and insulin resistance are associated with increased adipose 11β-hydroxysteroid dehydrogenase type 1 expression and elevated hepatic 5α-reductase activity. Diabetes. 2008;57:2652–2660.
Lettner A, Roden M. Ectopic fat and insulin resistance. Curr Diab Rep. 2008;8:185–191. Review.
Forouhi NG, Jenkinson G, Thomas EL, et al. Relation of triglyceride stores in skeletal muscle cells to central obesity and insulin sensitivity in European and South Asian men. Diabetologia. 1999;42:932–935.
Pan DA, Lillioja S, Kriketos AD, et al. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes. 1997;46:983–988.
Hotamisligil GS, Shargill NS, Speigelman BM. Adipose expression of tumor necrosis factor – alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91.
Arkan MC, Hevener AL, Greten FR, et al. IKK-β links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198.
Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336.
Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461.
Shimomura I, Funahashi T, Takahashi M, et al. Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat Med. 1996;2:800–803.
Itani SI, Ruderman NB, Schmieder F,, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011.
Tripathy D, Mohanty P, Dhindsa S, et al. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes. 2003;52:2882–2887.
Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72.
Pickup JC, Mattock MB, Chusney GD, Burt D. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia. 1997;40:1286–1292.
Yamauchi T, Kamon J, Waki H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946.
Trujillo ME, Scherer PE. Adipose tissue-derived factors: impact on health and disease. Endocr Rev. 2006;27:762–778.
Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119.
Chaudhuri A, Janicke D, Wilson MF, et al. Anti-inflammatory and profibrinolytic effect of insulin in acute ST-segment-elevation myocardial infarction. Circulation. 2004;109:849–854.
Fleischman A, Shoelson SE, Bernier R, Goldfine AB. Salsalate improves glycemia ad inflammatory parameters in obese young adults. Diabetes Care. 2008;3:289–294.
Wildman RP, Munter P, Reynolds K, et al. The obese without cardiometabolic risk factors clustering and the normal weight with cardiovascular risk factor clustering: prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch Int Med. 2008;168:1617–1724.
Stefan N, Kantartzis K, Maachan J, et al. Identification and charcterization of metabolically benign obesity in humans. Arch Int Med. 2008;168:1609–1616.
Karelis AD, Messier V, Brochu M, Rabasa-Lhoret R. Metabolically healthy but obese women: effect of an energy-restricted diet. Diabeteologia. 2008;51:1752–1754.
Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham Study. JAMA. 1979;241:2035–2038.
Hu FB, Stampfer MJ, Solomon CG, et al. The impact of diabetes mellitus on mortality from all causes and coronary heart disease in women: 20 years of follow-up. Arch Intern Med. 2001;23(161):1717–1723.
Huxley R, Barzi F, Woodward M. Excess risk of fatal coronary heart disease associated with diabetes in men and women: meta-analysis of 37 prospective cohort studies. BMJ. 2006;322:73–78. Review.
Facchini FS, Hua N, Abbasi F, Reaven GM. Insulin resistance as a predictor of age-related diseases. JCEM. 2001;86:3574–3578.
Nosadini R, Manzato E, Solini A, et al. Peripheral, rather than hepatic, insulin resistance and atherogenic lipoprotein phenotype predict cardiovascular complications in NIDDM. Eur J Clin Invest. 1994;24:258–266.
Hanley AJ, Williams K, Stern MP, Haffner SM. Homeostasis model assessment of insulin resistance in relation to the incidence of cardiovascular disease: the San Antonio Heart Study. Diabetes Care. 2002;25:1177–1184.
Robins SJ, Rubins HB, Faas FH, et al.; Veterans Affairs HDL Intervention Trial (VA-HIT). Insulin resistance and cardiovascular events with low HDL cholesterol: the Veterans Affairs HDL Intervention Trial (VA-HIT). Diabetes Care. 2003;26:1513–1517.
The DECODE Insulin Study Group. Plasma insulin and cardiovascular mortality in non diabetic European men and women: a metanalysis of data from eleven prospective studies. Diabeteologia. 2004;47:1245–1256.
Kuusisto J, Lempiäinen P, Mykkänen L, Laakso M. Insulin resistance syndrome predicts coronary heart disease events in elderly type 2 diabetic men. Diabetes Care. 2001;24:1629–1633.
Resnick HE, Jones K, Ruotolo G, et al. String Heart Study, Insulin resistance, the metabolic syndrome and risk of incident cardiobvascular disease in non-diabeteic American Indians: The Strong Heart Study. Diabetes Care. 2003;26:861–867.
Rutter MK, Meigs JB, Sullivan LM, D‘Agostino RB Sr, Wilson PW. Insulin resistance, the metabolic syndrome, and incident cardiovascular events in the Framingham Offspring Study. Diabetes. 2005;54:3252–3257.
Ruige JB, Assendelft WJ, Dekker JM, Kostense PJ, Heine RJ, Bouter LM. Insulin and risk of cardiovascular disease: a meta-analysis. Circulation. 1998;97:996–1001.
Folsom AR, Szklo M, Stevens J, Liao F, Smith R, Eckfeldt JH. A prospective study of coronary heart disease in relation to fasting insulin, glucose and diabetes. The Atherosclerosis Risk in Communities (ARIC) Study. Diabetes Care. 1997;20:935–942.
Folsom AR, Rasmussen ML, Chambless LE, et al. Prospective associations of fasting insulin, body fat distributuin, and diabetes with risk of ischemic stroke: The Atherosclerosis Risk in Communities (ARIC) Study. Diabetes Care. 1999;22:1077–1083.
Pyörälä M, Miettinen H, Laakso M, Pyörälä K. Hyperinsulinemia predicts coronary heart disease risk in healthy middle-aged men: the 22-year follow-up results of the Helsinki Policemen Study. Circulation. 1998;98:398–404.
Pyörälä M, Miettinen H, Laakso M, Pyörälä K. Hyperinsulinemia and the risk of stroke in healthy middle-aged men: the 22-year follow-up results of the Helsinki Policemen Study. Stroke. 1998;29:1860–1866.
Yarnell JW, Patterson CC, Bainton D, Sweetnam PM. Is metabolic syndrome a discrete entity in the general population? Evidence from the Caerphilly and Speedwell population studies. Heart. 1998;79:248–252.
Welin L, Bresäter LE, Eriksson H, Hansson PO, Welin C, Rosengren A. Insulin resistance and other risk factors for coronary heart disease in elderly men. The Study of Men Born in 1913 and 1923. Eur J Cardiovasc Prev Rehabil. 2003;10:283–288.
Sarwar N, Sattar N, Gudnason V, Danesh J. Circulating concentrations of insulin markers and coronary heart disease: a quantitative review of 19 Western prospective studies. Eur Heart J. 2007;28:2491–2497. Review.
Chen W, Srinivasan SR, Li S, Xu J, Berenson GS. Metabolic syndrome variables at low levels in childhood are beneficially associated with adulthood cardiovascular risk: the Bogalusa Heart Study. Diabetes Care. 2005;28:126–131.
Modan M, Halkin H, Almog S, et al. Hyperinsulinemia. A link between hypertension obesity and glucose intolerance. J Clin Invest. 1985;75:809–817.
Reaven GM, Lerner RL, Stern M, et al. Role of insulin in hypertriglyceridemia. J Clin Invest. 1974;57:551–560.
Jeppesen J, Hollenbeck CB, Zhou M.-Y, et al. Relation between insulin resistance, hyperinsulinemia, postheparin plasma lipoprotein lipase activity, and postprandial lipemia. Arterioscler Thromb Vasc Biol. 1995;15:320–324.
Tobey TA, Greenfield M, Kraemer F, Reaven GM. Relationship between insulin resistance, insulin secretion, very low density lipoprotein kinetics and plasma triglyceride levels in normotriglyceridemic man. Metabolism. 1981;30:165–171.
Zavaroni I, Bonora E, Pagliara M, Dall’Aglio E, Luchetti L, Buonanno G, et al. Risk factors for coronary artery disease in healthy persons with hyperinsulinemia and normal glucose tolerance. N Engl J Med. 1989;320:702–706.
Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428.
Reaven GM. The individual components of the metabolic syndrome: is there a raison d‘etre? J Am Coll Nutr. 2007;26:191–195.
WHO Consultation Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications. Part 1: Diagnosis and Classification of Diabetes Mellitus. Geneva: World Health Organization; 1999.
Balkau B, Charles MA. Comment on the provisional report from the WHO consultation. European Group for the Study of Insulin Resistance (EGIR). Diab Med. 1999;16:442–443.
Executive Summary of the Third Report of The National Cholesterol Education Program (NCEP) Expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). J Am Med Assoc. 2001;285:2486–2497.
Einhorn D, Reaven GM, Cobin RH, et al. American College of Endocrinology position statement on the insulin resistance syndrome. Endocr Pract. 2003;9:237–252.
Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–2752.
Alberti KG, Zimmet P, Shaw J. Metabolic syndrome—a new world-wide definition: a consensus statement from the International Diabetes Federation. Diab Med. 2006;23:469–480. Also International Diabetes Federation. The IDF consensus worldwide definition of the metabolic syndrome. Available at: http://www.idf.org/wedata/docs/Meta_def-final.pdf. Accessed December 6, 2008.
Kahn R, Buse J, Ferrannini E, Stern M. The metabolic syndrome: a time for critical reappraisal-joint statement from the Americal Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2005;28:2289–2304.
Ford ES. Prevalence of the metabolic syndrome defined by the International Diabetes Federation among adults in the U.S. Diabetes Care. 2005;28:2745.
Ford ES, Giles WH, Mokdad AH. Increasing prevalence of the metabolic syndrome among U.S. adults. Diabetes Care. 2004;27:2444–2449.
Lorenzo C, Williams K, Hunt KJ, Haffner SM. Trend in the prevalence of the metabolic syndrome and its impact on cardiovascular disease incidence: the San Antonio Heart Study. Diabetes Care. 2006;29:625–630.
Lorenzo C, Williams K, Gonzalez-Villalpando C, Haffner SM. The prevalence of the metabolic syndrome did not increase in Mexico City between 1990–1992 and 1997–1999 despite more central obesity. Diabetes Care. 2005;28:2480–2485.
Hu G, Lindström J, Jousilahti P, et al. The increasing prevalence of metabolic syndrome among Finnish men and women over a decade. J Clin Endocrinol Metab. 2008;93:832–836.
Goodman E, Daniels SR, Meigs JB, Dolan LM. Instability in the diagnosis of metabolic syndrome in adolescents. Circulation. 2007;115:2316–2322.
Wilson PW, Meigs JB, Sullivan L, Fox CS, Nathan DM, D‘Agostino RB Sr. Prediction of incident diabetes mellitus in middle-aged adults: the Framingham Offspring Study. Arch Intern Med. 2007;28(167):1068–1074.
Lorenzo C, Williams K, Hunt KJ, Haffner SM. The National Cholesterol Education Program-Adult Treatment Panel III, International Diabetes Federation, and World Health Organization definitions of the metabolic syndrome as predictors of incident cardiovascular disease and diabetes. Diabetes Care. 2007;30:8–13.
Wilson PW, D‘Agostino RB, Parise H, Sullivan L, Meigs JB. Metabolic syndrome as a precursor of CV disease and type 2 diabetes mellitus. Circulation. 2005;112:3066–3072.
Ford ES, Li C, Sattar N. Metabolic syndrome and incident diabetes: current state of the evidence. Diabetes Care. 2008;31:1898.
Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care. 2005;28:1769–1778.
Wang J, Ruotsalainen S, Moilanen L, Lepistö P, Laakso M, Kuusisto J. The metabolic syndrome predicts cardiovascular mortality: a 13-year follow-up study in elderly non-diabetic Finns. Eur Heart J. 2007;28:857–864.
Wang J, Ruotsalainen S, Moilanen L, Lepistö P, Laakso M, Kuusisto J. The metabolic syndrome predicts incident stroke: a 14-year follow-up study in elderly people in Finland. Stroke. 2008;39:1078–1083.
Nilsson PM, Engström G, Hedblad B. The metabolic syndrome and incidence of cardiovascular disease in non-diabetic subjects – a population-based study comparing three different definitions. Diab Med. 2007;24:464–472.
Katzmarzyk PT, Janssen I, Ross R, Church TS, Blair SN. The importance of waist circumference in the definition of metabolic syndrome: prospective analyses of mortality in men. Diabetes Care. 2006;29:404–409.
Qiao Q; DECODE Study Group. Comparison of different definitions of the metabolic syndrome in relation to cardiovascular mortality in European men and women. Diabetologia. 2006;49:2837–2846.
Sattar N, McConnachie A, Shaper AG, et al. Can metabolic syndrome usefully predict cardiovascular disease and diabetes? Outcome data from two prospective studies. Lancet. 2008;371:1927–1935.
Gami AS, Witt BJ, Howard DE, et al. Metabolic syndrome and risk of incident cardiovascular events and death: a systematic review and meta-analysis of longitudinal studies. J Am Coll Cardiol. 2007;49:403–414.
Bonora E, Targher G, Formentini G, et al. The metabolic syndrome is an independent predictor of cardiovascular disease in type 2 diabetic subjects. Prospective data from the Verona Diabetes Complications Study. Diab Med. 2004;21:52–58.
Guzder RN, Gatling W, Mullee MA, Byrne CD. Impact of metabolic syndrome criteria on cardiovascular disease risk in people with newly diagnosed type 2 diabetes. Diabetologia. 2006;49:49–55.
Tong PC, Kong AP, So WY, et al. The usefulness of the International Diabetes Federation and the National Cholesterol Education Program’s Adult Treatment Panel III definitions of the metabolic syndrome in predicting coronary heart disease in subjects with type 2 diabetes. Diabetes Care. 2007;30:1206–1211.
Sone H, Mizuno S, Fujii H, et al.; Japan Diabetes Complications Study. Is the diagnosis of metabolic syndrome useful for predicting cardiovascular disease in asian diabetic patients? Analysis from the Japan Diabetes Complications Study. Diabetes Care. 2005;28:1463–1471.
Bruno G, Merletti F, Biggeri A, et al.; Casale Monferrato Study. Metabolic syndrome as a predictor of all-cause and cardiovascular mortality in type 2 diabetes: the Casale Monferrato Study. Diabetes Care. 2004;27:2689–2694.
Cull CA, Jensen CC, Retnakaran R, Holman RR. Impact of the metabolic syndrome on macrovascular and microvascular outcomes in type 2 diabetes mellitus: United Kingdom Prospective Diabetes Study 78. Circulation. 2007;116:2119–2126.
Hunt KJ, Resendez RG, Williams K, Haffner SM, Stern MP. NCEP versus WHO metabolic syndrome in relation to all cause and CV mortality in the San Antonio Heart Study. Circulation. 2004;110:1251–1257.
Wilson PW, D’Agostino RB, Parise H, Sullivan L, Meigs JB. Metabolic syndrome as a precursor of CV disease and type 2 diabetes mellitus. Circulation. 2005;112:3066–3072.
Malik S, Wong ND, Franklin SS, et al. Impact of the metabolic syndrome on mortality from CHD, CVD and all causes in United States adults. Circulation. 2004;110:1245–1250.
Stern MP, William K, Hunt KJ. Impact of diabetes/metabolic syndrome in patients with established CVD. Atherosclerosis Suppl. 2005;6:3–6.
Stern MP, Williams K, Gonzalez-Villalpando C, Hunt KJ, Haffner SM. Does the metabolic syndrome improve identification of individuals at risk of type 2 diabetes and/or cardiovascular disease? Diabetes Care. 2004;27:2676–2681.
Cheal KL, Abbasi F, Lamendola C, McLaughlin T, Reaven GM, Ford ES. Relationship to insulin resistance of the adult treatment panel III diagnostic criteria for identification of the metabolic syndrome. Diabetes. 2004;53:1195.
Liao Y, Kwon S, Shaughnessy S, et al. Critical evaluation of adult treatment panel III criteria in identifying insulin resistance with dyslipidemia. Diabetes Care. 2004;27:978–983.
Meigs JB, Rutter MK, Sullivan LM, Fox CS, D‘Agostino RB Sr, Wilson PW. Impact of insulin resistance on risk of type 2 diabetes and cardiovascular disease in people with metabolic syndrome. Diabetes Care. 2007;30:1219–1225.
Rosenzweig JL, Ferrannini E, Grundy SM, et al. Primary prevention of cardiovascular disease and type 2 diabetes in patients at metabolic risk: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:3671–3689.
Ridker PM, Danielson E, Fonseca FA, et al.; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207.
Hayashi T, Boyko EJ, McNeely MJ, Leonetti DL, Kahn SE, Fujimoto WY.Visceral adiposity, not abdominal subcutaneous fat area, is associated with an increase in future insulin resistance in Japanese Americans. Diabetes. 2008;57:1269–1275. Epub 2008 Feb 25.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Banerji, M.A., Chaiken, R.L. (2010). Insulin Resistance and the Metabolic Syndrome. In: Poretsky, L. (eds) Principles of Diabetes Mellitus. Springer, Boston, MA. https://doi.org/10.1007/978-0-387-09841-8_34
Download citation
DOI: https://doi.org/10.1007/978-0-387-09841-8_34
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-0-387-09840-1
Online ISBN: 978-0-387-09841-8
eBook Packages: MedicineMedicine (R0)