
Abstract
Diabetes is a very common cause of hospitalization and is listed as a diagnosis in 12% of hospital discharges. This diagnosis has a substantial impact on medical costs since hospital stay for diabetic patients is 1–3 days longer than non-diabetics. Cardiovascular complications are a common cause of hospitalizations for diabetic patients, and diabetes is a listed diagnosis in 29% of cardiac surgery patients.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Oxidative Stress
- Diabetic Patient
- Diabetic Complication
- Reactive Oxygen Species
- Advanced Glycation Endproducts
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Diabetes is a very common cause of hospitalization and is listed as a diagnosis in 12% of hospital discharges. This diagnosis has a substantial impact on medical costs since hospital stay for diabetic patients is 1–3 days longer than non-diabetics. Cardiovascular complications are a common cause of hospitalizations for diabetic patients, and diabetes is a listed diagnosis in 29% of cardiac surgery patients.1 – 4
The underlying pathogenesis of cardiovascular disease in diabetes includes increased oxidant stress (OS) and inflammation. The manifestations of the diabetic state include metabolic abnormalities, increased OS, and a chronic inflammatory state, which serve to accentuate each other and result in a cycle of increasing organ damage. Since data obtained over the past few years support the new and novel concept that the inflammatory response seen in diabetes results from the cumulative and sustained pressure from oxidant stress, it is important to decrease OS from all potential sources.4 – 9 The origin of oxidants in diabetes was initially considered to be entirely endogenous, but there is now an agreement that the environment, especially the diet, is a substantial source of oxidants.10 This being the case it is important to understand the significant contribution of the diet to OS in diabetics and seek to reduce this input.
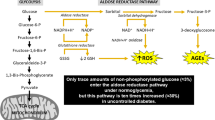
Advanced glycation endproducts (AGEs) are among the most commonly encountered oxidants in food.10 They belong to a class of toxic oxidant molecules, also called glycoxidants. High levels of toxic oxidant AGEs are thought to underlie many of the complications of diabetes.11 , 12 One of the ways by which AGEs induce these changes is by generating reactive oxidant species (ROS), which promote the formation of more AGEs, in a vicious action/reaction cycle, which progressively increases oxidative stress (OS) and the risk for both micro- and macrovascular disease (Fig. 20.1). Activation of receptors which recognize AGEs results in activation of downstream signaling pathways (including NFκB) which induce the production of pro-inflammatory cytokines and pro-angiogenic factors, leading to increased OS.9

Cellular/tissue injury in chronic disease is due to increased oxidant stress from exogenous and endogenous sources
AGE deposition in aging blood vessels and other tissues is well documented, and its role in the pathogenesis of cardiovascular disease and diabetic nephropathy is supported by the fact that structurally unrelated inhibitors of AGE formation provide protection against these diseases.13 – 33 The number and breadth of these studies are an indication of the importance of blocking the formation and actions of AGEs. Understanding how AGEs interact with cells and tissues (Fig. 20.2), and how they can be prevented or treated, is an important challenge in the management of diabetes and its complications, across all medical disciplines. This chapter focuses on a non-pharmacological approach to the management of OS in diabetic patients.

AGEs interact with most cell types in the body, eliciting a variety of responses, depending on cell type and underlying state of OS
2 Toxic Oxidants (AGEs) and Cellular Responses
The glycoxidation pathway makes a fundamental contribution to OS, which underlies aging-mediated vascular disease and diabetes-related vascular and renal complications.3 , 7 , 8 , 11 , 12 , 21 , 34 – 39 There are two sources of AGEs, endogenous and exogenous (diet and smoking 34). It was previously thought that most AGEs are generated endogenously in diabetics by spontaneous reactions between the carbonyl groups of reducing sugars, ascorbate, and other carbohydrates and amino acids (lysine, arginine) or cystine-containing amino-peptides, nucleic acids, and lipids.39 , 40 – 46 However, regular foods are now known to be a major source of AGEs in diabetics, as well as in non-diabetics.47 , 48 The term AGEs, while often referring to non-reactive terminal products, such as\(^\epsilon\) N-carboxymethyllysine (CML) or pentosidine, also includes a broad range of reactive precursors, including 1- or 3-deoxyglucosone as well as methylglyoxal (MG) and their derivatives, a common one being hydroimidazolone, MG-H1. The latter are formed largely by non-oxidative mechanisms from triose phosphate intermediates during anaerobic glycolysis and are elevated in diabetes and aging.49 , 50 Amine-containing lipids also form advanced lipoxidation endproducts (ALE), such as 4-hydroxy-nonenal or CML, and other lipid analogues.11 , 30 , 40 , 41 , 49 – 53 Glycoxidation products of proteins and lipids (AGE/ALE) can accelerate the generation of reactive oxygen species (ROS), leading to oxidative and “carbonyl” stress.38 Autoxidation of glucose is also accompanied by the generation of reactive oxygen species (ROS), such as superoxide radicals.4 , 54 ROS enhance glycation and both mechanisms can promote atherogenesis and other complications related to diabetes or aging.4 In fact, the direct cellular and tissue toxicity of certain AGEs, such as 1- or 3-deoxyglucosone or methylglyoxal derivatives, is now well established.54
Other non-glucose dependent AGE pathways involve activated white blood cells, i.e., neutrophils, monocytes, and macrophages. Activated white blood cells produce enzymes, including myeloperoxidase and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, causing AGE formation by oxidation of amino acids.4 , 26 , 44 , 55 – 59 Cell activation by AGEs, i.e. via binding to the AGE receptor RAGE can also promote ROS and AGE formation via the NADPH oxidase pathway, the myeloperoxidase pathway or possibly through the nuclear protein amphoterin (also termed High Mobility Group Box 1) which can activate RAGE and toll-like receptor 4, and thus amplify AGE formation.11 , 25 , 60 – 63
Another mechanism of AGE formation is the aldose reductase-mediated polyol pathway. Glucose entering the polyol pathway may directly form AGEs via reactive intermediates, i.e., glyoxal, methyl-glyoxal, or 3-deoxyglucosone, as well as via depletion of NADPH or glutathione, which result in raised intracellular ROS.56 These changes indirectly result in the further formation of AGEs.61 Since these two mutually enhancing processes are tightly linked, interventions targeting one will inevitably have an impact on the other.
3 Sources of Toxic Oxidants (AGEs) in Diabetic Patients
Endogenous Sources: Hyperglycemia promotes metabolic activity and is the best known endogenous pathway by which the levels of AGEs and OS are increased in cells.9 , 24 , 27 , 44 , 48 , 52 , 60 , 64 – 68 However, increased OS leads to the oxidation of other sugars and/or lipids, which create dicarbonyl compounds that use highly reactive carbonyl groups to bind amino acids and form AGEs.24 , 52 While hyperglycemia may be one cause of increased OS in diabetic patients, as noted above, other pathways may increase the levels and activity of enzymes, such as NADPH oxidase, which induce AGE formation by oxidizing amino acids in both inflammatory and parenchymal cells.27 , 59 , 64 MG may be formed as an intracellular toxic product of glycolysis. It is metabolized to lactate by enzymes (glyoxylase I and II) that require the non-enzymatic conjugation of glutathione with MG.49 , 69 Thus, MG is neutralized when the levels of glutathione are normal, a state that may be compromised in the presence of high OS, such as diabetes.
AGE-modified moieties, especially long-lived proteins and lipids, as well as nucleic acids may be removed by proteolytic digestion, degraded to inactive molecules, and then excreted by the kidneys. However, AGE-derived cross-links are particularly resistant to degradation.70 – 72 AGE crosslinks may contribute to the increased levels and delayed clearance of oxidized lipoproteins, the inactivation of immune components, and increased sensitivity of diabetic patients to drug toxicity, infection, and ischemia.1 – 4 , 8 , 22 , 58 , 73 – 77 In particular, the toxic effects of AGEs on tissue structure and function may include the formation of chemical cross-links within and between connective tissue components or between these elements and plasma constituents, which can impair vasodilation or LDL removal (due to the retention of molecules trapped in the sub-endothelium and/or by impairing recognition and uptake of AGE-modified LDL by the LDL receptor).20 , 26 , 29 , 52 , 53 , 67 , 78 , 79 Since AGE-LDL is a particular form of glycoxidized lipoprotein which is cross linked and is thus retained in the aortic wall, it recruits macrophages and promotes their conversion to foam cells and/or the accumulation of smooth muscle cells. As such it is thought to be an efficient pro-atherogenic substance.5 , 12 , 13 , 25 , 26 , 28 , 40 , 45 , 46 , 49 , 58 , 63 , 80 – 87
By creating cross-links between components of the extracellular matrix, and thereby changing their physical properties, AGEs affect both their distensibility and elasticity of arteries. Both type 1 diabetes and type 2 diabetes subjects have increased arterial disease, which is associated with increased cardiovascular mortality based on diastolic dysfunction,73 , 88 increased pulse-wave velocity, decreased arterial compliance, and formation of aneurysms. Decreased vascular elasticity due to cross-links, which are resistant to degradation and the normal turnover of collagen and elastin, may play a larger role than previously appreciated.5 , 28 , 38 , 72 , 89 This interpretation is reinforced by the observation that AGE inhibition restores these properties in rodents.13 , 26 , 28
Exogenous Sources: Many studies suggest that the modern diet is also a significant source of toxic oxidants (AGEs) in both man and experimental animals and contribute to the development of diabetes and diabetic complications.65 , 90 While it has long been appreciated that AGEs are present in food,67 , 68 they were not considered to be an important contaminant, since bioavailability studies showed that “only a small amount” of AGEs was taken into the body.32 , 66 , 86 , 87 , 91 This point of view now has to be revised for two reasons. First, the cumulative oxidant properties of AGEs are well known. Second, the amount of oxidants in the modern diet has dramatically increased, particularly the amount of AGEs, as documented throughout this review. In part, this can be traced to the widespread consumption of red meat, processed foods, and soft drinks, as well as the near universal use of pasteurization and other forms of food preservation using heat.86 , 87 , 9 Thus, the amount of toxic oxidants, including AGEs, that is present in the diet is now cause for concern, even if “only” a fraction is absorbed. This fact led to several recent studies of the bioavailability, kinetics, and renal elimination of dietary-derived AGE in healthy adults90 and diabetic patients, with or without impaired renal function66, as well as in animals.32 We found that ∼10% of the AGEs in the diet are absorbed, of which 2/3 is incorporated into tissues and turn over very slowly; the other ∼1/3 is excreted via the kidneys.32 , 66 Since the amount of AGEs in food in the last 50 years has increased, and the lifespan of the population has increased, this amount of absorption assumes greater importance clinically.
While increasedOS and the propensity to develop cardiovascular diseases have been in part attributed to excess fat in the diet, it is now recognized that even diets with a modest amount of lipids may contain high levels of oxidants, generated when proteins or lipids are mixed with reducing sugars and processed under elevated temperatures, as in standard cooking (Table 20.1).65 , 92 , 93 Prime examples of the resultant dietary oxidants include reactive carbonyl compounds, advanced glycation (AGE), and lipoxidation endproducts (ALE), containing CML and MG derivatives in large concentrations.44 , 51 , 94 , 95 The compounds formed in food are identical to those formed endogenously. Together with the exogenous AGEs the endogenous AGEs contribute to the chronic inflammatory response associated with the complications of aging and diabetes.8 , 41 , 49 , 52 , 54 , 86 , 91 , 96
The data on the effect of cooking temperature on the formation of AGEs, and perhaps other toxic oxidants, are based on direct measurements of CML, MG, and MDA (a lipid peroxide product) in food prepared by different methods.65 , 96 Grilling or frying meat increases the amounts of toxic oxidants significantly more than meat prepared by boiling. Frying results in a vast increase in AGEs, compared to boiling. For instance, so-called French fries contain 100-fold more AGEs, compared to boiled potatoes. This results from the addition of lipids to carbohydrates and proteins under conditions of dry heat. The amount of AGEs in the diet largely depends on the temperature during cooking and the degree of hydration (i.e., the amount of water present). For instance, cooking fish or chicken in water results in 4–6 times less AGEs, compared to cooking these foods in the absence of water. In addition, the amount of AGEs in cooked food is directly proportional to the fat content.
The question is often raised as to the levels of specific AGEs in food. Therefore, we extracted red meat, prepared by different cooking methods and found that the amount of CML correlated with MG (Fig. 20.3a), that the amount of CML and MG correlated with the levels of MDA (an oxidized lipid) (Fig. 20.3b). In addition, both grilled and fried meat contained increased amount of both CML and MG, compared to either raw or boiled meat (Fig. 20.3c). While a relationship between the AGE content of foods, OS, and diabetic complications had previously been found, we directly tested whether food AGEs have biological activity, by preparing soluble extracts of these meat preparations, and then adding them to endothelial cells in vitro. The amount of intracellular anti-oxidants (i.e., glutathione) negatively correlated with the amount of AGEs in the preparations (Fig. 20.3d). The extracts also caused a decrease in anti-oxidant levels (GSH/GSSG ratio) (Fig. 20.3e), and promoted the release of TNFα (Fig. 20.3f). These data are of particular importance because they clearly show that AGEs are present in the food, and that their levels depend on the method of preparation. In addition, the data show that the AGEs present in the food are biologically active, since they directly cause increased OS and depletion of anti-oxidant defenses. Thus, both the type of food and the means by which it is prepared is critical in determining the total amount of oxidant AGEs present in food. We recently obtained direct proof that AGEs in the diet cause increased OS and raise the levels of circulating AGEs in mice. This was accomplished by adding a well-characterized AGE (MG) to a diet prepared with low heat, so that the initial content of AGEs in the food was reduced and that any changes observed would be directly related to the added AGEs. Examination of the mice from birth to 6 months of age revealed a linear rise in serum AGEs and increased OS levels in the mice fed the diet supplemented with MG (Fig. 20.4).11 This critical finding provided direct proof of the ability of AGEs ingested via the diet to raise AGEs in the blood and lead to increased OS.

Pro-oxidant and pro-inflammatory activity of food AGEs. (a) Correlation between food-derived protein AGEs (CML and MG-derivatives); (b) between CML and malondialdehyde (MDA, a lipid oxidation product); (c) addition of different amounts of food extracts to human endothelial cell (EC) cultures. CML levels inversely correlate with intracellular levels of glutathione, indicating that food AGEs induce cellular oxidant stress; (d) the amount of AGEs (CML and MG) in food (red meat) varies with method of cooking; (e) endothelial cell levels of anti-oxidant GSH vary, following the addition of food extracts prepared by different methods; (f) levels of the inflammatory cytokine, TNFα in EC, also vary with method of cooking following the addition of identical amounts of food extracts

Methyl-glyoxal (MG) derivatives in diet increase serum AGEs and OS. Pups from dams fed a low-AGE diet during gestation and nursing were pair-fed a low diet, a Low+MG, or a regular (Reg) diet at weaning. (a) Serum MG levels from weaning to 6 months of age. Low vs. Reg, * p<0.05, * * p<0.001; Low vs. Low+MG, §p<0.01, §§p<0.001; Low+MG vs. Reg, + p<0.05. (b) 8-Isoprostane levels. Low vs. Low+MG, * * p<0.01; Low vs. Reg, #p<0.01
4 Reduction of Toxic Oxidants (AGEs) in Mice
Recent evidence indicates that the cycle of increased levels of AGEs and elevated OS can be interrupted by reducing glycation, either in the body (endogenous) or in the food (exogenous). While anti-oxidant or anti-AGE agents may prevent the formation of AGEs, a non-pharmaceutical intervention such as reducing the amount of AGEs in the food by simply reducing the heat applied may be the most cost-efficient, effective, and widely applicable way of decreasing exposure to oxidants. The reduction of dietary oxidants (AGEs) prevents many of the diabetic complications in several animal models and reinforces the need to continue the search for optimal interventions in the clinical setting.
We have previously shown that sustained parenteral administration of AGEs, or feeding a high-AGE diet, reproduces many of the complications associated with diabetes.48 , 66 , 68 , 78 , 97 – 100 The accumulation of advanced glycation endproducts (AGEs) begins in early life and progressively increases with time.101 In this context, avoidance of early exposure to AGEs may determine susceptibility to certain diseases, including type 1 and type 2 diabetes. This concept was examined in non-obese diabetic (NOD) mice which have a high propensity to develop type 1 DM (∼80% of young NOD females develop diabetes), which is attributed to an autoimmune process (Fig. 20.5, left upper panel).98 We found that when these mice were exposed to a low-AGE diet the structure of their islets remained normal (Fig. 20.5, right upper panel), and the propensity to develop diabetes was sharply reduced. In fact, this “loss of susceptibility” could be traced to the maternal diet, since offspring of dams maintained on a low-AGE diet during gestation had a greatly reduced incidence of diabetes in the F1 generation, and diabetes was nearly absent in the F2 progeny.98 The absence of diabetes resulted from preservation of intact pancreatic islets, the loss of several abnormalities in T lymphocytes, and the maintenance of low levels of OS (Fig. 20.6). This phenomenon was epigenetic in nature, since the progeny became diabetic, if they were returned to regular mouse chow, which has a high AGE content, similar to that of the modern human diet.10 , 102

Effect of dietary AGEs on islet morphology. (a) Non-diabetic F1 NOD mice, and (b) db/db +/+ mice were exposed to either regular (left) or low-AGE diet (right panels). NOD mouse pancreatic tissues stained by H&E (a) showed absence of the severe mononuclear cell infiltration after a low-AGE diet (for >12 mos). Islets of age-matched db/db+/+ mice (b), after L-AGE (×5 mos), stained for insulin showed intact insulin production compared to those on regular diet. Mag. ×400

A low-AGE diet suppresses age-associated diabetes. a, b: Serum and tissue AGE levels are reduced in normal mice pair-fed a low-AGE diet for life. Reg-AGE (open symbols), low-AGE diet (closed symbols). (b) Kidney, spleen, and liver AGE at 24 mos of age (n=8/group). c–e: Changes in glucose, insulin response to IGTT and GIR at 4 and 24 mos (n=6/group). Note: none of the low-AGE fed mice became diabetic by 3 years of age. All Reg-fed mice were diabetic at various age points before death (n=20/group)
We also tested other models of Type 2 DM, including db/db+/+ and high fat diet-induced type 2 DM in normal C57B6 mice, and found that both diabetes and complications were reduced.22 , 28 , 48 , 97 Thus, the development of diabetes was blocked in both genetic and non-genetic models by reducing the intake of AGEs in the diet. A conclusion reached from these genetically and pathogenetically diverse models of diabetes was that the maintenance of lower levels of OS, by reducing the intake of AGEs (oxidants) in the diet may be a critical factor in the preservation of normal beta-cell function. Furthermore, the prevention of diabetes was effective in both those with a genetic background prone to the development of diabetes (type 1 and type 2), and the prevention of diabetes by diabetogenic interventions in otherwise normal strains. Furthermore, the data show that the levels of OS may be partly driven by the levels of toxic oxidants in the diet, and showed that the maternal diet was also an important source of oxidant AGEs in the fetus. Finally, the data show that these changes may be entirely preventable by modifying the preparation of the diet, without altering either the calorie content or particular nutrients. The latter point may be critical to normal fetal and post-natal development of diabetic mothers, and possibly even those with gestational diabetes. It also raises the question as to whether this may be important to the prevention of diabetes itself. Nonetheless, the conclusions from these initial studies are that consumption of diets that contain high amounts of AGEs can promote OS in apparently normal adults and that this can begin in the unborn fetus or in childhood. For instance, we and others found that infant formulae contain 28–389 fold (median=70) higher levels of toxic oxidant AGEs than maternal milk.65 , 103 The CML in the infant formulae was absorbed and appeared in the urine.103 Finally, the effect of oxidants in the diet may have other untoward effects as they may neutralize anti-oxidants in normal food, or anti-oxidants added for nutritional or pharmacologic purposes, and many vitamins are altered by oxidants or heat.96
Reduction of these toxic dietary AGE oxidants in diabetic mice attenuated diabetic vascular and kidney disease, acute vascular injury, and promoted wound healing.48 , 67 , 68 , 75 , 76 , 104 Diabetic mice fed with standard laboratory diets (which have a high content of AGEs) developed vascular and renal lesions.105 However, their age-matched cohorts fed with low-AGE isocaloric diets remain largely free of these changes, despite persistence of hyperlipidemia and diabetes.47 , 54 , 67 , 68 , 75 , 76 , 97 , 106 A low-AGE diet also led to significant suppression of atherosclerotic plaque formation at the aortic root of diabetic/hyperlipidemic (ApoE–/–) mice (Fig. 20.7).67 In addition, a low-AGE diet provided protection of post-injury arterial inflammation and re-stenosis in ApoE–/– mice despite sustained hyperlipidemia.78 As noted above, feeding diabetic mice a diet with lower levels of toxic oxidants (AGEs) prevented the structural and functional changes of diabetic nephropathy in genetic models of type 1 and type 2 diabetes.77 , 107 These studies support the proposed synergism between the levels of toxic oxidants in exogenous (diet) and the endogenously derived OS burden in diabetes. In addition, AGEs are antigenic, a property which may contribute to inflammation and atherosclerosis.77 , 101

Atheromatous lesion and inflammatory infiltrate are both inhibited in hypercholesterolemic mice (ApoE–/–) after feeding on a low-AGE diet for 5 mos. Aortic root sections from ApoE–/– mice fed a regular diet (a) or an AGE-restricted diet (b) are stained by a macrophage surface antigen-specific antibody (MOMA). Note the markedly decreased inflammatory cell infiltrate (Mag. ×200)
In normally aging mice, long-term reduction in the intake of exogenous oxidants, without altering caloric intake, ameliorated OS, insulin resistance, and the incidence of diabetes, as well as significantly extending lifespan.108 These diverse positive effects were attributed to the functional preservation of AGER1, anti-oxidant reserves, and the amelioration of aging-induced OS-response genes, including p66Shc and FOXO-1.57 , 108 , 109
Interventions which reduce the formation of ROS, such as calorie restriction, have long been known to promote lifespan extension.43 , 110 – 112 Since calorie restriction is accomplished by reducing food intake, this effectively reduces the intake of AGEs as an obligate concomitant. We recently found that the well-known beneficial effects of a calorie-restricted diet could be due to the 40% reduction in food intake, primarily because it also restricts the intake of AGEs. In fact, when the AGE content of a calorie-restricted diet was increased, the benefits of calorie restriction on reducing OS, CVD, and CKD were lost.12 We have shown that a long-term reduction of the intake of toxic AGEs decreases ROS, the expression of OS-response genes, cardiovascular and renal disease, and leads to an increased life span.76 , 80 , 83 , 102 , 108 , 113 Furthermore, these effects were independent of calorie intake.
These experimental studies suggest that while factors such as heredity or nutrient intake play a large role in diabetes, the level of oxidants in the diet may be a key link to both the induction of diabetes and diabetic complications. AGEs may be a major contributor to these lesions, since they cause a wide spectrum of vascular abnormalities, including basement membrane thickening and endothelial injury, resulting in increased vascular permeability, a pro-thrombotic state, and decreased blood flow; all of which are traits of microvascular disease affecting the retina, kidneys, and peripheral nerves.9 , 114 – 119 The specific role that AGEs play in causing microvascular disease is well documented in the kidney glomeruli.100 However, the role of exogenous AGEs in retinopathy or neuropathy is not yet defined.
Elevated AGEs are also associated with macrovascular abnormalities in diabetic subjects, including coronary atherosclerosis.14 , 27 , 73 , 120 AGEs decrease both endothelial cell nitric oxide levels and activity, by inhibiting endothelial nitric oxide synthase and prostacyclin or by quenching NO–.38 , 80 , 120 These changes, in conjunction with the effects attributed to protein kinase C activation, can further increase vasoconstriction.117 , 118 , 121 , 122 AGEs increase expression of angiotensin II and endothelin in vascular smooth muscle cells, which also contribute to vasoconstriction, enhancing pro-inflammatory processes, and mitogenesis.121 , 122 Activation of NFκB and activator protein-1 (AP-1) and other transcription factors by AGEs may lead to increased expression of adhesion molecules (e.g., ICAM-1, VCAM-1) and plasminogen activator inhibitor 1 (PAI-1), and contribute to chronic vascular dysfunction.117
In summary, these studies on diabetes and its complications introduce the view that the accumulation of advanced glycation endproducts (AGEs) may begin in utero (during the fetal period), continue in childhood, and progressively increases with normal aging. If the intake of AGEs is high, i.e., similar to that in the average modern diet, the baseline level of OS may be higher than “normal” and the rate of rise would be accelerated. This would reduce anti-oxidant defenses and result in an inflammatory state that may significantly alter innate immune responses to inflammatory processes. The end result could be the earlier onset of obesity and type 1 or type 2 diabetes, accompanied by macrovascular (atherosclerosis) and microvascular (kidney, retinal) diseases.4 , 117 , 118
5 Cellular Receptors that Recognize Toxic AGE Oxidants
Among the receptors that bind AGEs, AGER1 is the most extensively studied receptor involved in AGE endocytosis and processing (Fig. 20.8).52 , 55 , 123 , 124 AGER1 is the principal receptor mediating the removal of excess extracellular AGEs, from both endogenous and exogenous origin. AGER1 is a ã50 kDa integral surface membrane protein that inhibits AGE-induced OS (as depicted in Fig. 20.9), via inhibition of RAGE, MAPK and Ras activation and phosphorylation of EGFR and ERK;26 inhibition of phosphorylation of ser-36 of p66Shc, a key pro-apoptotic adaptor protein involved in the phosphorylation and inactivation of members of the FOXO pathway,109 which normally enhances anti-oxidants such as MnSOD and increases resistance to OS; and also via reduction of AGE-induced mitochondrial OS and mitochondrial injury in endothelial cells. Increased expression of the AGER1 transgene blocks ROS in vitro55 , 57 and prevents vascular and kidney injury in mice.137 These observations point to AGER1 as a potentially important multifunctional component of the anti-OS defense system. The levels of AGER1 are directly correlated with the levels of circulating AGEs in healthy adults, but they are reduced in those with chronically high levels of AGEs, such as found in patients with diabetes.123 , 125 Our studies in both human subjects and experimental models show that AGEs and AGE receptors are a part of normal homeostasis.10 , 62 , 102 , 125 When the levels of AGEs exceed the capacity of the body to detoxify them, they accumulate and raise the levels of OS. If the levels of AGER1 are sufficient, AGER1 actively counteracts this increase and maintains the normal oxidant balance. Elevated levels of AGEs, combined with low AGER1 expression, are a signal that the capacity for removal of AGEs has been exceeded, i.e., in diabetes and chronic kidney disease.123 The sustained nature of the responses induced by AGEs, coupled with the continued intake of excess dietary oxidants in the Western diet, may constitute the basis for the progressive depletion of innate defenses in diabetes. These changes may include, or be due to, reduced levels of AGER1 (Fig. 20.9). Decreased AGER1 function, allowing AGEs to accumulate, can result in depletion of anti-oxidants and promote high OS. Thus, the maintenance of normal AGER1 levels may be critical to redox homeostasis.

Major AGE receptors and functions: AGER1 is localized on cell surface, mitochondria, and ER. RAGE is principally a cell surface receptor

Schematic representation of putative AGE-receptor interactions and their contribution to oxidant balance. (a) In the normal state AGER1 can suppress AGE- and ROS-dependent increased RAGE/Ras/MAPK and NFκB activity, as well as p66shc-dependent inactivation of FOXO. These oppose the formation of excess ROS from AGEs. (b) Chronic, poorly controlled diabetes, and/or sustained influxes of exogenous oxidants can reduce levels of AGER1 and its anti-OS actions, which will further increase ROS/AGEs
Among the AGE receptors that trigger an inflammatory response, the best known is a multi-ligand protein, RAGE and its variants.66 , 81 HMGB1 (amphoterin) has recently been identified as a major RAGE ligand.60 Activated RAGE leads to the induction of ROS and an inflammatory response.126 – 128 RAGE does not participate in AGE removal, but its extracellular domain (sRAGE) is present in circulation. Since, blood levels of sRAGE inversely correlate with the OS state, it has been suggested that sRAGE may bind and assist in clearing AGEs and other oxidants.129 – 132 While sRAGE levels have generally been found to be low in diabetic individuals, some have found that they are increased in diabetic patients with coronary vascular disease.23 , 82
Other entities that bind AGEs include AGER2, which is an 80–90 kDa protein possibly involved in early AGE signaling, and AGER3, a 30–35 kDa protein, which may make a contribution to both AGE removal and cell activation.52 , 107 AGEs are also bound by ScR-II, CD36, lysozyme and other defensins.133 While several AGE-receptor gene polymorphisms have been identified, none have been strongly linked to diabetes.134 Toll-like receptor 4 also binds RAGE ligands (HMGB-1), although it is not known if it also binds AGEs.60
6 Reduction of Exposure to Toxic Oxidants (AGEs) in Humans
Several epidemiologic studies show that a rise in OS among clinically normal subjects may be important in the pathogenesis of insulin resistance and other metabolic diseases.4 , 6 , 7 , 39 , 135 We found that the AGE content of regular meals or meals with a low AGE content given to normal subjects is readily reflected in the serum levels of AGEs and in the amount of AGEs excreted in the urine.66 , 86 , 87 In addition, we recently found that serum AGEs correlated with oxidant stress in a cross-sectional study in normal non-diabetic adults of different ages (Fig. 20.10a).88 , 125 Those who ate a diet with a low AGE content, had lower levels of markers of inflammation and oxidant stress, i.e., hsCRP, TNFα, and fibrinogen, whereas these levels were much higher in those consuming a diet with a higher AGE content. Of interest, a significant association was noted between serum AGE and HOMA, an indicator of insulin resistance.88 , 125 , 136 The associations between serum AGEs, 8-isoprostane, and HOMA suggest that sAGE may be an important contributor to OS, prior to the onset of diabetes. Hyperglycemia has been assumed to be the source of the increased AGE levels found in diabetes, a condition associated with systemic OS.3 , 64 None of the normal subjects in this study were hyperglycemic in the fasting state. Therefore, their higher fasting levels of serum AGE could not be explained on the basis of glycemia. This data provided strong additional evidence that the intake of AGEs in the diet very likely contributes to the increased levels of serum AGEs, and the subsequent increased systemic OS.

a. Serum AGEs (CML) correlate with plasma lipid peroxidation products (8-isoprostanes) in non-diabetic subjects. b. Levels of inflammatory factors, hsCRP and TNFα, are elevated in healthy subjects consuming AGE-rich diets (>23 Eq/d), but not in those consuming a low-AGE diets (<15.4 Eq/d), * p=0.025. Note: high-AGE consumers had a BMI >33
These data are consistent with previous observations in patients with diabetes and chronic kidney disease.47 , 106 The use of CML-like AGEs as surrogates for other oxidants or carbonyl-rich products in the diet seems justified by the correspondence between the levels of CML-like AGEs in the diet and CML or MG in the circulation, as well as the highly significant correlation (r=0.7, p=0.0001) between sAGEs and the endogenous lipid peroxidation derivatives, 8-isoprostanes.125
Thus, the intake of AGEs contained in the usual adult diet may deliver excessive amounts of AGE oxidants to the body, which may promote cumulative changes in oxidant homeostasis with time. These changes may presage the emergence of metabolic and cardiovascular disturbances, heretofore believed to be part of the normal aging process.4 , 6 – 8 , 35 , 37 , 88 , 137 , 138 The above data suggest that reduction of the intake of AGEs via the diet may inhibit the accumulation of toxic AGEs, increased oxidant stress, and slow or reduce the emergence of the diabetes and diabetes-related complications. Indeed, a reduced intake of AGEs via the diet in diabetic persons was associated with lowered serum AGEs, OS, and inflammatory markers, e.g., TNFα, hsCRP, and VCAM-1.see references in 90 , 102 These findings led to the hypothesis that dietary AGEs, together with those made endogenously, could promote an excessive systemic glycoxidant burden, oxidant stress, and cell activation, all of which would enhance the ”vulnerability” of tissues to injury. This is especially true of diabetic patients, since they have elevated levels of AGEs and OS, accompanied by decreased anti-oxidant reserves, at baseline. It is imperative that studies be undertaken in diabetic patients to determine if this approach would also improve diabetic complications.
7 The Link Between Toxic AGEs and Oxidant Stress (OS)
For the last few decades, the focus has been on the oxidation and glycoxidation products generated by hyperglycemia. Extended studies, including those in type 1 and type 2 diabetes, have lent support to this view, at least in part.89 However, very recently two other long-term trials raised serious doubts about the utility of strict normalization of glycemic levels in reducing diabetic complications.1 , 2 These studies may redirect our thinking to embrace a more diverse realm of controls of risk factors in diabetic patients, perhaps with a focus on OS. It may include prospective on oxidants generated by other mechanisms, including the contribution of dietary AGEs as toxic oxidants. The evidence is increasing that the generation of OS is the basis of many diabetes complications and that it can be lowered by reducing the levels of toxic oxidants from either exogenous or endogenous sources.64 , 66 , 106 The fact that reduction of AGE intake prevents type 1 and type 2 diabetes, as well as diabetic complications in different animal models, noted above corroborates the importance of external sources of AGEs in their pathogenesis and provides a strong impetus for novel clinical interventions.
Diabetes increases the incidence of large vessel diseases such as strokes, myocardial infarction, aortic atherosclerosis, aneurysms, and limb ischemia requiring amputation.1 – 4 , 73 These complications often have catastrophic effects in the short term. Based on the studies in animals discussed above, it is reasonable to postulate that a high dietary intake of toxic AGEs in the usual Western diet may be a major contributor to atherosclerotic lesion severity in the presence of high circulating glucose levels in diabetic patients, an effect that may be preventable by reducing the amount of toxic oxidant AGEs in the diet. Clinical studies to examine this point are now in progress.
8 Acute Effects of Dietary AGEs in Normal and Diabetic Subjects
After normal subjects are given an AGE-rich meal, the levels of serum AGEs peak within 4–6 h, and return to baseline within 12 h (Fig. 20.11a, solid lines, right panel) and the urinary levels follow.66 In these individuals, ∼70% of the absorbed oxidants in the diet remain in cells and tissues, and 30% is excreted in the urine.66 These data point to the unrecognized potential for toxic oxidants from the diet to progressively accumulate over time, particularly in persons consuming a high-AGE diet. As discussed above, even persons who are “normal” in all other aspects can develop high levels of serum AGEs. Importantly, this suggests that the tissue levels of toxic oxidants may progress with time, if the intake remains high.

(a) Serum and urine AGE kinetics in normal adults after a regular meal (high AGE) or a meal with 50% lower AGEs (low-AGE). (b) Diabetic patients with different degrees of diabetic nephropathy were fed the same amount of diet AGEs (regular meal): higher baseline and prolonged elevation of serum AGE levels corresponded to significantly reduced urine AGE levels in those with a lower GFR (solid lines). Note, neither group had severe disease but AGE excretion was markedly reduced in both groups
Since diabetic patients often have long-term elevation of OS and a concomitant decrease in anti-oxidant reserves, it is logical that they would have a decreased ability to deal with a high dietary load of oxidant AGEs. In fact, this is the case as shown by a study of diabetics with either microalbuminuria or overt proteinuria. Long before any clinical or laboratory signs of renal disease are apparent, diabetic patients with microalbuminuria have an impaired ability to excrete the increased load of toxic AGEs presented within the normal diet. Namely, after a high-AGE meal, diabetic patients with microalbuminuria and an estimated GFR in the normal range (Fig. 20.11b, broken line) had sharply increased levels of serum AGEs, and that these levels remained elevated for more than 30 hours. The far smaller peak of AGEs in the urine fell only when the serum AGE levels returned to baseline levels. On the other hand, patients with overt proteinuria, but with only a modestly decreased GFR (20%) (Fig. 20.11b, solid line), had a marked prolongation of the high levels of serum AGEs, while the amount excreted in the urine was significantly decreased. Note that since serum creatinine levels were in the normal range at this level of GFR, these patients would have been considered to have normal renal function by most physicians. Thus, diabetic patients with kidney lesions, as evidenced by albuminuria of any level, appear to have a marked impairment in their ability to handle oxidants presented in the diet. Importantly, this occurs long before physicians generally consider them to have “kidney damage.” In addition, these data suggest that the kidney is a major site for the handling of toxic oxidants, and current clinical measures of kidney damage do not accurately measure critical abnormalities in this aspect of kidney function. Since toxic AGEs within the diet directly contribute to elevated OS, and to diabetic complications, the data provided by the studies presented above suggest that it is critical to lower the exposure of patients with diabetes to AGEs within the diet prior to the time they develop clinical evidence of renal disease. This intervention should be in addition to inhibiting the further increase of endogenous AGEs in the body due to elevated OS.
9 Conclusions
Oxidant stress (OS) is now recognized to be one of the major factors predisposing to diabetic complications. The sources of OS include both the intake of toxic oxidants (AGEs) and their formation intracellularly as part of the hypermetabolic state and other sources of OS. This chapter emphasizes the fact that the intake of toxic oxidants contained in food is a major factor in the pathogenesis of diabetic complications. Furthermore, we show that the amount of toxic oxidants present within the food can be very simply modified by changing the methods by which meals are prepared. The results of decreasing the intake of toxic oxidants by diabetic patients include a decrease in markers of inflammation, vascular injury, and OS. In animals, this is associated with a sharp reduction in the severity of established diabetic complications, the presence of an inflammatory state, vascular injury, insulin resistance, and diabetes. Since reducing the level of toxic oxidants within the diet does not add to health care costs, does not change nutrient intake, and promises to substantially reduce diabetic complications, this intervention should be strongly considered by both physicians and patients in the day-to-day management of diabetic complications.
References
Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–2572.
Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559.
Abbatecola AM, Ferrucci L, Grella R, et al. Diverse effect of inflammatory markers on insulin resistance and insulin-resistance syndrome in the elderly. J Am Geriatr Soc. 2004;52:399–404.
de Rekeneire N, Peila R, Ding J, et al. Diabetes, hyperglycemia, and inflammation in older individuals: the health, aging and body composition study. Diabetes Care. 2006;29:1902–1908.
Zhang L, Zalewski A, Liu Y, et al. Diabetes-induced oxidative stress and low-grade inflammation in porcine coronary arteries. Circulation. 2003;108:472–478.
Reuben DB, Cheh AI, Harris TB, et al. Peripheral blood markers of inflammation predict mortality and functional decline in high-functioning community-dwelling older persons. J Am Geriatr Soc. 2002;50:638–644.
Harris TB, Ferrucci L, Tracy RP, et al. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med. 1999;106:506–512.
Ershler WB, Ferrucci L, Finch C, et al. Inflammation, inflammatory mediators and aging. In: Sherman RFS, Carrington J, Miller M, Monjan A, eds. NIA Inflammation and Aging Workshop. Bethesda, MD: NIH; 2004.
Vlassara H, Cai W, Crandall J, et al. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc Natl Acad Sci USA. 2002;99:15596–15601.
Vlassara H, Uribarri J, Cai W, Striker G. Advanced glycation end product homeostasis: exogenous oxidants and innate defenses. Ann N Y Acad Sci. 2008;1126:46–52.
Cai W, He JC, Zhu L, et al. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am J Pathol. 2008;173:327–336.
Cai W, He JC, Zhu L, et al. High levels of dietary advanced glycation end products transform low-density lipoprotein into a potent redox-sensitive mitogen-activated protein kinase stimulant in diabetic patients. Circulation. 2004;110:285–291.
Wolffenbuttel BH, Boulanger CM, Crijns FR, et al. Breakers of advanced glycation end products restore large artery properties in experimental diabetes. Proc Natl Acad Sci USA. 1998;95:4630–4634.
Williams SB, Cusco JA, Roddy MA, Johnstone MT, Creager MA. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1996;27:567–574.
Williams ME, Bolton WK, Khalifah RG, Degenhardt TP, Schotzinger RJ, McGill JB. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol. 2007;27:605–614.
Waanders F, van den Berg E, Nagai R, van Veen I, Navis G, van Goor H. Renoprotective effects of the AGE-inhibitor pyridoxamine in experimental chronic allograft nephropathy in rats. Nephrol Dial Transplant. 2008;23:518–524.
Voziyan PA, Hudson BG. Pyridoxamine as a multifunctional pharmaceutical: targeting pathogenic glycation and oxidative damage. Cell Mol Life Sci. 2005;62:1671–1681.
Thornalley PJ. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch Biochem Biophys. 2003;419:31–40.
Stracke H, Hammes HP, Werkmann D, et al. Efficacy of benfotiamine versus thiamine on function and glycation products of peripheral nerves in diabetic rats. Exp Clin Endocrinol Diabetes. 2001;109:330–336.
Onorato JM, Jenkins AJ, Thorpe SR, Baynes JW. Pyridoxamine, an inhibitor of advanced glycation reactions, also inhibits advanced lipoxidation reactions. Mechanism of action of pyridoxamine. J Biol Chem. 2000;275:21177–21184.
Zheng F, Zeng YJ, Plati AR, et al. Combined AGE inhibition and ACEi decreases the progression of established diabetic nephropathy in B6 db/db mice. Kidney Int. 2006;70:507–514.
Jain SK, Lim G. Pyridoxine and pyridoxamine inhibits superoxide radicals and prevents lipid peroxidation, protein glycosylation, and (Na+ + K+)-ATPase activity reduction in high glucose-treated human erythrocytes. Free Radic Biol Med. 2001;30:232–237.
Nakamura S, Li H, Adijiang A, Pischetsrieder M, Niwa T. Pyridoxal phosphate prevents progression of diabetic nephropathy. Nephrol Dial Transplant. 2007;22:2165–2174.
Nagaraj RH, Sarkar P, Mally A, Biemel KM, Lederer MO, Padayatti PS. Effect of pyridoxamine on chemical modification of proteins by carbonyls in diabetic rats: characterization of a major product from the reaction of pyridoxamine and methylglyoxal. Arch Biochem Biophys. 2002;402:110–119.
Liu H, Zheng F, Uribarri J, et al. Reduced acute vascular injury and atherosclerosis in hyperlipidemic mice transgenic for lysozyme. Am J Pathol. 2006;169:303–313.
Li YM, Steffes M, Donnelly T, et al. Prevention of cardiovascular and renal pathology of aging by the advanced glycation inhibitor aminoguanidine. Proc Natl Acad Sci USA. 1996;93:3902–3907.
Kang Z, Li H, Li G, Yin D. Reaction of pyridoxamine with malondialdehyde: mechanism of inhibition of formation of advanced lipoxidation end-products. Amino Acids. 2006;30:55–61.
Huijberts MS, Wolffenbuttel BH, Boudier HA, et al. Aminoguanidine treatment increases elasticity and decreases fluid filtration of large arteries from diabetic rats. J Clin Invest. 1993;92:1407–1411.
Degenhardt TP, Alderson NL, Arrington DD, et al. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002;61:939–950.
Davies SS, Brantley EJ, Voziyan PA, et al. Pyridoxamine analogues scavenge lipid-derived gamma-ketoaldehydes and protect against H2O2-mediated cytotoxicity. Biochemistry. 2006;45:15756–15767.
Chetyrkin SV, Zhang W, Hudson BG, Serianni AS, Voziyan PA. Pyridoxamine protects proteins from functional damage by 3-deoxyglucosone: mechanism of action of pyridoxamine. Biochemistry. 2008;47:997–1006.
He C, Sabol J, Mitsuhashi T, Vlassara H. Dietary glycotoxins: inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes. 1999;48:1308–1315.
Booth AA, Khalifah RG, Hudson BG. Thiamine pyrophosphate and pyridoxamine inhibit the formation of antigenic advanced glycation end-products: comparison with aminoguanidine. Biochem Biophys Res Commun. 1996;220:113–119.
Cerami C, Founds H, Nicholl I, et al. Tobacco smoke is a source of toxic reactive glycation products. Proc Natl Acad Sci USA. 1997;94:13915–13920.
Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–1224.
Smith CD, Carney JM, Tatsumo T, Stadtman ER, Floyd RA, Markesbery WR. Protein oxidation in aging brain. Ann N Y Acad Sci. 1992;663:110–119.
Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15.
Finkel TH, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247.
Cappola AR, Xue QL, Ferrucci L, Guralnik JM, Volpato S, Fried LP. Insulin-like growth factor I and interleukin-6 contribute synergistically to disability and mortality in older women. J Clin Endocrinol Metab. 2003;88:2019–2025.
Sobenin IA, Tertov VV, Koschinsky T, et al. Modified low density lipoprotein from diabetic patients causes cholesterol accumulation in human intimal aortic cells. Atherosclerosis. 1993;100:41–54.
Requena JR, Ahmed MU, Fountain CW, et al. Carboxymethylethanolamine, a biomarker of phospholipid modification during the maillard reaction in vivo. J Biol Chem. 1997;272:17473–17479.
Menini S, Amadio L, Oddi G, et al. Deletion of p66Shc longevity gene protects against experimental diabetic glomerulopathy by preventing diabetes-induced oxidative stress. Diabetes. 2006;55:1642–1650.
Masoro EJ. Overview of caloric restriction and ageing. Mech Ageing Dev. 2005;126:913–922.
Cai W, Gao QD, Zhu L, Peppa M, He C, Vlassara H. Oxidative stress-inducing carbonyl compounds from common foods: novel mediators of cellular dysfunction. Mol Med. 2002;8:337–346.
Bucala R, Makita Z, Vega G, et al. Modification of low density lipoprotein by advanced glycation end products contributes to the dyslipidemia of diabetes and renal insufficiency. Proc Natl Acad Sci USA. 1994;91:9441–9445.
Bucala R, Makita Z, Koschinsky T, Cerami A, Vlassara H. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc Natl Acad Sci USA. 1993;90:6434–6438.
Uribarri J, Peppa M, Cai W, et al. Dietary glycotoxins correlate with circulating advanced glycation end product levels in renal failure patients. Am J Kidney Dis. 2003;42:532–538.
Sandu O, Song K, Cai W, Zheng F, Uribarri J, Vlassara H. Insulin resistance and type 2 diabetes in high-fat-fed mice are linked to high glycotoxin intake. Diabetes. 2005;54:2314–2319.
Bechmann J. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe Seyler. 1994;375:81–88.
Hamada Y, Araki N, Koh N, Nakamura J, Horiuchi S, Hotta N. Rapid formation of advanced glycation end products by intermediate metabolites of glycolytic pathway and polyol pathway. Biochem Biophys Res Commun. 1996;228:539–543.
Henle T. A food chemists view of advanced glycation end-products. Perit Dial Int. 2001;21:S125–S130.
Li YM, Mitsuhashi T, Wojciechowicz D, et al. Molecular identity and cellular distribution of advanced glycation endproduct receptors: relationship of p60 to OST-48 and p90 to 80 K-H membrane proteins. Proc Natl Acad Sci USA. 1996;93:11047–11052.
Thornalley PJ. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification--a role in pathogenesis and antiproliferative chemotherapy. Gen Pharmacol. 1996;27:565–573.
Requena JR, Baynes JW. Studies in animal models on the role of glycation and advanced glycation end-products (AGEs) in the pathogenesis of diabetic complications: pitfalls and limitations. In: Sima AAF, ed. Lessons From Animal Models of Diabetes. Vol VII. Boston, MA: Birkhauser; 2001.
Lu C, He JC, Cai W, Liu H, Zhu L, Vlassara H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc Natl Acad Sci USA. 2004;101:11767–11772.
Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C – dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945.
Cai W, He JC, Zhu L, Lu C, Vlassara H. Advanced glycation end product (AGE) receptor 1 suppresses cell oxidant stress and activation signaling via EGF receptor. Proc Natl Acad Sci USA. 2006;103:13801–13806.
Black PH. The inflammatory response is an integral part of the stress response: Implications for atherosclerosis, insulin resistance, type II diabetes and metabolic syndrome X. Brain Behav Immun. 2003;17:350–364.
Anderson MM, Heinecke JW. Production of N(epsilon)-(carboxymethyl)lysine is impaired in mice deficient in NADPH oxidase: a role for phagocyte-derived oxidants in the formation of advanced glycation end products during inflammation. Diabetes. 2003;52:2137–2143.
van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis. 2008;11:91–99.
Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–368.
Dougan M, Dranoff G. Inciting inflammation: the RAGE about tumor promotion. J Exp Med. 2008;205:267–270.
Negrean M, Stirban A, Stratmann B, et al. Effects of low- and high-advanced glycation endproduct meals on macro- and microvascular endothelial function and oxidative stress in patients with type 2 diabetes mellitus. Am J Clin Nutr. 2007;85:1236–1243.
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820.
Goldberg T, Cai W, Peppa M, et al. Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc. 2004;104:1287–1291.
Koschinsky T, He CJ, Mitsuhashi T, et al. Orally absorbed reactive glycation products (glycotoxins): an environmental risk factor in diabetic nephropathy. Proc Natl Acad Sci USA. 1997;94:6474–6479.
Lin RY, Choudhury RP, Cai W, et al. Dietary glycotoxins promote diabetic atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 2003;168:213–220.
Zheng F, He C, Cai W, Hattori M, Steffes M, Vlassara H. Prevention of diabetic nephropathy in mice by a diet low in glycoxidation products. Diabetes Metab Res Rev. 2002;18:224–237.
Shinohara M, Thornalley PJ, Giardino I, et al. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J Clin Invest. 1998;101:1142–1147.
Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 2004;63:582–592.
Monnier VM, Sell DR, Nagaraj RH, et al. Maillard reaction-mediated molecular damage to extracellular matrix and other tissue proteins in diabetes, aging, and uremia. Diabetes. 1992;41(Suppl 2):36–41.
Sell DR, Monnier VM. End-stage renal disease and diabetes catalyze the formation of a pentose-derived crosslink from aging human collagen. J Clin Invest. 1990;85:380–384.
Berg TJ, Snorgaard O, Faber J, et al. Serum levels of advanced glycation end products are associated with left ventricular diastolic function in patients with type 1 diabetes. Diabetes Care. 1999;22:1186–1190.
McCance DR, Dyer DG, Dunn JA, et al. Maillard reaction products and their relation to complications in insulin-dependent diabetes mellitus. J Clin Invest. 1993;91:2470–2478.
Peppa M, Brem H, Ehrlich P, et al. Adverse effects of dietary glycotoxins on wound healing in genetically diabetic mice. Diabetes. 2003;52:2805–2813.
Peppa M, Uribarri J, Vlassara H:. Advanced glycoxidation. A new risk factor for cardiovascular disease?. Cardiovasc Toxicol. 2002;2:275–287.
Shamshi FA, Partal A, Sady C, Glomb MA, Nagaraj RH. Immunological evidence for methylglyoxal-derived modifications in vivo: determination of antigenic epitopes. J Biol Chem. 1998;273:6928–6936.
Lin RY, Reis ED, Dore AT, et al. Lowering of dietary advanced glycation endproducts (AGE) reduces neointimal formation after arterial injury in genetically hypercholesterolemic mice. Atherosclerosis. 2002;163:303–311.
Klein RL, Laimins M, Lopes-Virella MF. Isolation, characterization, and metabolism of the glycated and nonglycated subfractions of low-density lipoproteins isolated from type I diabetic patients and nondiabetic subjects. Diabetes. 1995;44:1093–1098.
Linden E, Cai W, He JC, et al. Endothelial dysfunction in patients with chronic kidney disease results from advanced glycation end products (AGE)-mediated inhibition of endothelial nitric oxide synthase through RAGE activation. Clin J Am Soc Nephrol. 2008;3:691–698.
Gao X, Zhang H, Schmidt AM, Zhang C. AGE/RAGE Produces Endothelial Dysfunction in Coronary Arterioles in Type II Diabetic Mice. Am J Physiol Heart Circ Physiol. 2008;295:H491–H498.
Nakamura K, Yamagishi S, Adachi H, et al. Elevation of soluble form of receptor for advanced glycation end products (sRAGE) in diabetic subjects with coronary artery disease. Diabetes Metab Res Rev. 2007;23:368–371.
Peppa M, Uribarri J, Vlassara H. The role of advanced glycation end products in the development of atherosclerosis. Curr Diab Rep. 2004;4:31–36.
Stirban A, Negrean M, Stratmann B, et al. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care. 2006;29:2064–2071.
Stirban A, Negrean M, Gotting C, et al. Dietary advanced glycation endproducts and oxidative stress: in vivo effects on endothelial function and adipokines. Ann N Y Acad Sci. 2008;1126:276–279.
Thorpe SR, Baynes JW. Role of the Maillard reaction in diabetes mellitus and diseases of aging. Drugs Aging. 1996;9:69–77.
Uribarri J, Stirban A, Sander D, et al. Single oral challenge by advanced glycation end products acutely impairs endothelial function in diabetic and nondiabetic subjects. Diabetes Care. 2007;30:2579–2582.
Vlassara H, Cai W, Goodman S, et al. Protection against loss of innate defenses in adulthood by low AGE intake; role of a new anti-inflammatory AGE-Receptor-1. J Clin Endocrinol Metab. 2009;94:4483–4491.
Sell DR, Lapolla A, Odetti P, Fogarty J, Monnier VM. Pentosidine formation in skin correlates with severity of complications in individuals with long-standing IDDM. Diabetes. 1992;41:1286–1292.
Huebschmann AG, Regensteiner JG, Vlassara H, Reusch JE. Diabetes and advanced glycoxidation end products. Diabetes Care. 2006;29:1420–1432.
Tan D, Wang Y, Lo CY, Sang S, Ho CT. Methylglyoxal: its presence in beverages and potential scavengers. Ann N Y Acad Sci. 2008;1126:72–75.
Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40:405–412.
Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999;48:1–9.
O‘Brien J. Nutritional and toxicological aspects of the Maillard browning reaction in foods. Crit Rev Food Sci Nutr. 1989;28:211–248.
Lee TC, Kimiagar M, Pintauro SJ, Chichester CO. Physiological and safety aspects of Maillard browning of foods. Prog Food Nutr Sci. 1981;5:243–256.
Pouillart P, Mauprivez H, Ait-Ameur L, et al. Strategy for the study of the health impact of dietary Maillard products in clinical studies: the example of the ICARE clinical study on healthy adults. Ann N Y Acad Sci. 2008;1126:173–176.
Yang CW, Vlassara H, Peten EP, He CJ, Striker GE, Striker LJ. Advanced glycation end products up-regulate gene expression found in diabetic glomerular disease. Proc Natl Acad Sci USA. 1994;91:9436–9440.
Peppa M, He C, Hattori M, McEvoy R, Zheng F, Vlassara H. Fetal or neonatal low-glycotoxin environment prevents autoimmune diabetes in NOD mice. Diabetes. 2003;52:1441–1448.
Vlassara H, Striker LJ, Teichberg S, Fuh H, Li YM, Steffes M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc Natl Acad Sci USA. 1994;91:11704–11708.
Vlassara H, Bucala R, Striker L. Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Invest. 1994;70:138–151.
Booth AA, Khalifah RG, Todd P, Hudson BG. In vitro kinetic studies of formation of antigenic advanced glycation end products (AGEs). Novel inhibition of post-Amadori glycation pathways. J Biol Chem. 1997;272:5430–5437.
Vlassara H, Striker G. Glycotoxins in the diet promote diabetes and diabetic complications. Curr Diab Rep. 2007;7:235–241.
Birlouez-Aragon I, Pischetsrieder M, Leclere J, et al. Assessment of protein glycation markers in infant formulas. Food Chem. 2004;87:253–259.
Sugiyama S, Miyata T, Horie K, et al. Advanced glycation end-products in diabetic nephropathy. Nephrol Dial Transplant. 1996;11(Suppl 5):91–94.
Sebekova K, Faist V, Hofmann T, Schinzel R, Heidland A. Effects of a diet rich in advanced glycation end products in the rat remnant kidney model. Am J Kidney Dis. 2003;41:S48–S51.
Uribarri J, Peppa M, Cai W, et al. Restriction of dietary glycotoxins reduces excessive advanced glycation end products in renal failure patients. J Am Soc Nephrol. 2003;14:728–731.
He C, Zheng F, Sabol J, et al. Differential expression of renal AGE-receptor genes in NOD mouse kidneys: possible role in non-obese diabetic renal disease. Kidney Int. 2000;58:1931–1940.
Cai W, He JC, Zhu L, et al. Reduced oxidant stress and extended lifespan in mice exposed to a low glycotoxin diet: association with increased AGER1 expression. Am J Pathol. 2007;170:1893–1902.
Cai W, He JC, Zhu L, Chen X, Striker GE, Vlassara H. AGE-receptor-1 counteracts cellular oxidant stress induced by AGEs via negative regulation of p66shc-dependent FKHRL1 phosphorylation. Am J Physiol Cell Physiol. 2008;294:C145–C152.
Mattson MP, Wan R. Beneficial effects of intermittent fasting and caloric restriction on the cardiovascular and cerebrovascular systems. Nutr Biochem. 2005;16:129–137.
Spindler SR, Dhahbi JM. Conserved and tissue-specific genic and physiologic responses to caloric restriction and altered IGFI signaling in mitotic and postmitotic tissues. Annu Rev Nutr. 2007;27:193–217.
Zimmerman JA, Malloy V, Krajcik R, Orentreich N. Nutritional control of aging. Exp Gerontol. 2003;38:47–52.
Peppa M, Uribarri J, Vlassara H. Aging and glycoxidant stress. Hormones (Athens). 2008;7:123–132.
Chou SM, Han CY, Wang HS, Vlassara H, Bucala R. A receptor for advanced glycosylation endproducts (AGEs) is colocalized with neurofilament-bound AGEs and SOD1 in motoneurons of ALS: immunohistochemical study. J Neurosci. 1999;169:87–92.
Fosmark DS, Torjesen PA, Kilhovd BK, et al. Increased serum levels of the specific advanced glycation end product methylglyoxal-derived hydroimidazolone are associated with retinopathy in patients with type 2 diabetes mellitus. Metabolism. 2006;55:232–236.
Makita Z, Radoff S, Rayfield EJ, et al. Advanced glycosylation end products in patients with diabetic nephropathy. N Engl J Med. 1991;325:836–842.
Murata T, Nagai R, Ishibashi T, Inomuta H, Ikeda K, Horiuchi S. The relationship between accumulation of advanced glycation end products and expression of vascular endothelial growth factor in human diabetic retinas. Diabetologia. 1997;40:764–769.
Stitt AW, Moore JE, Sharkey JA, et al. Advanced glycation end products in vitreous: Structural and functional implications for diabetic vitreopathy. Invest Ophthalmol Vis Sci. 1998;39:2517–2523.
Boehm BO, Schilling S, Rosinger S, et al. Elevated serum levels of N(epsilon)-carboxymethyl-lysine, an advanced glycation end product, are associated with proliferative diabetic retinopathy and macular oedema. Diabetologia. 2004;47:1376–1379.
Zeiher AM, Fisslthaler B, Schray-Utz B, Busse R. Nitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cells. Circ Res. 1995;76:980–986.
Yao D, Taguchi T, Matsumura T, et al. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J Biol Chem. 2007;282:31038–31045.
Quehenberger P, Bierhaus A, Fasching P, et al. Endothelin 1 transcription is controlled by nuclear factor-kappaB in AGE-stimulated cultured endothelial cells. Diabetes. 2000;49:1561–1570.
He CJ, Koschinsky T, Buenting C, Vlassara H. Presence of diabetic complications in type 1 diabetic patients correlates with low expression of mononuclear cell AGE-receptor-1 and elevated serum AGE. Mol Med. 2001;7:159–168.
Skolnik EY, Yang Z, Makita Z, Radoff S, Kirstein M, Vlassara H. Human and rat mesangial cell receptors for glucose-modified proteins: potential role in kidney tissue remodelling and diabetic nephropathy. J Exp Med. 1991;174:931–939.
Uribarri J, Cai W, Peppa M, et al. Circulating glycotoxins and dietary advanced glycation endproducts: two links to inflammatory response, oxidative stress, and aging. J Gerontol A Biol Sci Med Sci. 2007;62:427–433.
Galichet A, Weibel M, Heizmann CW. Calcium-regulated intramembrane proteolysis of the RAGE receptor. Biochem Biophys Res Commun. 2008;370:1–5.
Schmidt AM, Hori O, Brett J, Yan SD, Wautier JL, Stern D. Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arterioscler Thromb. 1994;14:1521–1528.
Schmidt AM, Yan SD, Yan SF, Stern DM. The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta. 2000;1498:99–111.
Nakamura K, Yamagishi S, Adachi H, et al. Serum levels of soluble form of receptor for advanced glycation end products (sRAGE) are positively associated with circulating AGEs and soluble form of VCAM-1 in patients with type 2 diabetes. Microvasc Res. 2008;76:52–56.
Basta G, Sironi AM, Lazzerini G, et al. Circulating soluble receptor for advanced glycation end products is inversely associated with glycemic control and S100A12 protein. J Clin Endocrinol Metab. 2006;91:4628–4634.
Emanuele E, D‘Angelo A, Tomaino C, et al. Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch Neurol. 2005;62:1734–1736.
Gohda T, Tanimoto M, Moon JY, et al. Increased serum endogenous secretory receptor for advanced glycation end-product (esRAGE) levels in type 2 diabetic patients with decreased renal function. Diabetes Res Clin Pract. 2008;81:196–201.
Li YM, Tan AX, Vlassara H. Antibacterial activity of lysozyme and lactoferrin is inhibited by binding of advanced glycation-modified proteins to a conserved motif. Nat Med. 1995;1:1057–1061.
Poirier O, Nicaud V, Vionnet N, et al. Polymorphism screening of four genes encoding advanced glycation end-product putative receptors. Association study with nephropathy in type 1 diabetic patients. Diabetes. 2001;50:1214–1218.
Ershler WB. Biological interactions of aging and anemia: a focus on cytokines. J Am Geriatr Soc. 2003;51:S18–S21.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419.
Bokov A, Chaudhuri A, Richardson A. The role of oxidative damage and stress in aging. Mech Ageing Dev. 2004;125:811–826.
Peppa M, Uribarri J, Cai W, Lu M, Vlassara H. Glycoxidation and inflammation in renal failure patients. Am J Kidney Dis. 2004;43:690–695.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Vlassara, H., Striker, G.E. (2010). Intake of Advanced Glycation Endproducts: Role in the Development of Diabetic Complications. In: Poretsky, L. (eds) Principles of Diabetes Mellitus. Springer, Boston, MA. https://doi.org/10.1007/978-0-387-09841-8_20
Download citation
DOI: https://doi.org/10.1007/978-0-387-09841-8_20
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-0-387-09840-1
Online ISBN: 978-0-387-09841-8
eBook Packages: MedicineMedicine (R0)