
Abstract
Neurotransmitter expression has long been thought of as a defining phenotypic property of adult neurons. However, it has now been shown that most neurons co-release multiple signaling molecules. Many examples of neurons that co-release a classical transmitter (e.g., acetylcholine, norepinephrine or glutamate) and neuromodulators have been demonstrated, but neurons can also co-release more than one classical transmitter. Defining the mechanisms that determine released transmitter(s) is important for understanding neural function since this largely determines the influence of neural activity. This chapter details evidence showing that mammalian sympathetic neurons co-release acetylcholine (ACh) and norepinephrine (NE). Sympathetic neurons project to body tissues including blood vessels and heart to control functions such as regulation of blood pressure and cardiac output. Transmitter choice in sympathetic neurons is controlled by target-derived, soluble growth factors. Current data suggests that these factors may operate to regulate the relative amounts of ACh and NE released by sympathetic neurons, which may play an important role in homeostatic regulation of essential physiological processes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
4.1 Introduction
It has historically been held that the identity of a released neurotransmitter is a defining and unchanging characteristic of differentiated adult neurons (Dale 1935); Eccles 1976). However, more recent evidence suggests that neurons can release different classical transmitters under different physiological situations and even, in some cases, co-release multiple transmitters. The existence of co-transmission of classical transmitters (e.g., acetylcholine, norepinephrine, glutamate and GABA) with neuropeptides or neuromodulators is well established in many types of neurons (Kupfermann 1991; Elfvin et al. 1993). Co-transmission of two classical transmitters has now also been demonstrated in both invertebrate and vertebrate preparations (Kupfermann 1991; Marder et al. 1995; Seal and Edwards 2006). In this chapter we will summarize the evidence that mammalian sympathetic neurons coexpress the classical transmitters acetylcholine (ACh) and norepinephrine (NE) and that, under certain circumstances, co-release both of these transmitters in a regulated manner.
Co-transmission is biologically more costly than utilizing a single transmitter since multiple molecular regulatory systems are necessary to generate different transmitter molecules, package them in vesicles and recycle or break them down in the synaptic cleft. Since it would seem more parsimonious to utilize a single transmitter, the question arises as to why neurons would go through the trouble of using multiple neurotransmitters. It seems that there must be an evolutionary advantage to co-transmission. One possibility is that it limits crosstalk between nearby neurons. Transmission between closely apposed pairs of connected neurons might interact if all cells used the same transmitter-receptor system. Similarly unwanted interactions may occur if closely apposed neurons received common input from a single afferent source. However, co-release of multiple transmitters coupled with distinct transmitter receptor expression patterns in target cells would ensure exclusivity of signaling and remove a potential restriction on the structural complexity of neural circuits.
Another advantage of co-transmission is that multiple transmitter systems allow more complex signaling. For example, if one transmitter were contained in a more readily releasable vesicle pool then a weaker stimulus would release only one transmitter while a stronger stimulus might cause release of both transmitters. Therefore different patterns or levels of activity could have different physiological effects. An example of this is seen in sympathetic neurons controlling vasoconstriction in the rat tail where the release of norepinephrine, adenosine triphosphate, and neuropeptide Y is differentially affected by stimulus burst duration (Bradley et al. 2003).
Co-transmission may also allow for local modulation of signaling. Thus, the release of retrograde signals from innervated tissue could be an important determinant of relative release of coexpressed neurotransmitters. In this way target cells could directly regulate afferent input through the regulation of co-transmission. As will be discussed later, this type of signaling appears to play an important role in co-transmission of ACh and NE in the sympathetic nervous system.
4.2 Developmental Regulation of Neurotransmitter Expression in Sympathetic Neurons by Target-Derived Signals
Sympathetic neurons project from ganglia located in the neck and on either side of the spinal column to innervate somatic organs and tissues. The sympathetic nervous system regulates metabolic, contractile and secretory functions that are essential for physiological homeostasis. Developmentally, sympathetic neurons derive from neural crest precursors and express noradrenergic markers (i.e., enzymes necessary to produce the neurotransmitter norepinephrine) early in development (Ernsberger and Rohrer 1996; Francis and Landis 1999; Ernsberger 2000). Cholinergic markers are coexpressed somewhat later in development in a subpopulation of neurons (Schafer et al. 1997; Ernsberger and Rohrer 1999). These expression patterns occur relatively early in development before neurons contact target tissue, suggesting that developing sympathetic neurons have an autonomous capacity to express both cholinergic and noradrenergic markers.
The expression of neurotransmitter phenotype in postnatal sympathetic neurons is developmentally influenced by interactions with target tissues. Most adult sympathetic neurons utilize norepinephrine as their principal neurotransmitter at synaptic contacts with body tissues (Elfvin et al. 1993). However, some tissues, such as sweat glands, periosteum and some vascular beds are cholinergically innervated by sympathetic nerves (Schotzinger and Landis 1988; Elfvin et al. 1993; Francis and Landis 1999; Asmus et al. 2000). Sympathetic innervation of these tissues is initially noradrenergic and gradually switches to cholinergic over the first one to two weeks after birth (Francis and Landis 1999).
The final transmitter profile of noradrenergic and cholinergic sympathetic neurons is dependent upon the target of innervation. In rodents, the glabrous skin of the footpad contains sweat glands that receive cholinergic sympathetic innervation, whereas hairy skin is noradrenergically innervated. Several studies have demonstrated that interactions between the neurons and target tissue are necessary to cause the cholinergic neurotransmitter switch (Schotzinger and Landis 1988; Schotzinger and Landis 1990; Asmus et al. 2000). Schotzinger and Landis 1988 removed a patch of hairy skin from newborn rats and transplanted a section of glabrous skin from a donor animal. They were able to observe the initial innervation of the transplanted skin since rat sympathetic fibers do not enter the dermis until several days after birth. Sympathetic fibers that would have innervated hairy skin and remained noradrenergic switched to a cholinergic phenotype when they terminated on transplanted glabrous skin. Catecholaminergic histofluorescence in the sympathetic fibers innervating sweat glands, but not surrounding hairy skin or blood vessels, decreased sharply. Conversely, immunostaining for acetylcholinesterase and the activity of choline acetyltransferase (markers of cholinergic transmission) gradually increased in fibers innervating the transplanted glabrous skin. Together, these experiments demonstrated that target interactions play an instructive role in setting the neurotransmitter phenotype of innervating sympathetic neurons.
In an analogous set of experiments, the parotid salivary gland, which is innervated by noradrenergic fibers, was transplanted under glabrous skin in the footpad of newborn rats. Sympathetic fibers innervating the transplanted salivary gland retained noradrenergic markers, while fibers innervating surrounding sweat glands switched to a cholinergic phenotype (Schotzinger and Landis 1990). Finally, transplantation of periosteum, the connective tissue surrounding bone that is cholinergically innervated in the adult, to hairy skin in newborn rats promoted a switch to a cholinergic phenotype in sympathetic fibers innervating the transplanted tissue (Asmus et al. 2000). These studies strongly suggest that target-derived signaling plays a crucial developmental role in the determination of sympathetic neurotransmitter phenotype.
The switch from noradrenergic to cholinergic neurotransmission that occurs in sympathetic neurons innervating the sweat glands is dependent on a soluble factor released by the target tissue (Rao and Landis 1990; Rao et al. 1992; Rohrer 1992). Whole protein extracts purified from rat glabrous skin induced a dose-dependent cholinergic shift in cultured sympathetic neurons, while cultures treated with liver, salivary gland or hairy skin extracts, or with extracts from mutant mice that lack sweat glands remained noradrenergic (Rao and Landis 1990; Habecker et al. 1995).
Several growth factors have been identified that induce cholinergic and reduce noradrenergic marker expression in cultured sympathetic neurons. These include ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), cardiotrophin -1 (CT-1), and neurotrophin-3 (NT-3) (Saadat et al. 1989; Yamamori et al. 1989; Habecker et al. 1995; Brodski et al. 2000). It was therefore initially proposed that one or more of these secreted proteins might be the cholinergic differentiating factor released by sweat gland tissue (Rao et al. 1992; Rohrer 1992). However, several lines of evidence suggest that the in vivo cholinergic differentiating factor is not one of these proteins and that the relevant activity has not yet been identified. Pre-treating glabrous skin extracts with antisera to CNTF resulted in a 55 to 80 % decrease in cholinergic differentiation in cultured sympathetic neurons as measured by acetylcholinesterase activity (Rao et al. 1992; Rohrer 1992), However, Rao et al. (1992) found no evidence for expression of CNTF protein or mRNA in sweat glands. This led to the conclusion that CNTF in glabrous skin extracts was likely derived from sensory neurons innervating the dermis and that it probably played no physiological role in the determination of sympathetic neurotransmitter phenotype at sweat glands. Further, cholinergic sympathetic innervation of sweat glands developed normally in mice lacking either or both the CNTF and LIF genes (Francis et al. 1997), supporting the hypothesis that CNTF is not the cholinergic differentiation factor. Similarly, antisera directed against LIF or CT-1 did not prevent cholinergic differentiation of cultured sympathetic neurons induced by glabrous skin extract (Rao et al. 1992; Habecker et al. 1995). Thus, none of the factors that have been identified as a cholinergic differentiation factor has been shown to play that role during the cholinergic switch of sympathetic neurons in vivo.
Whatever the identity of the released cholinergic differentiation factor turns out to be it does seem to act through the same receptor and signaling systems as CNTF, LIF and CT-1. These three growth factors show structural similarities and signal through LIFRβ and gp130 receptors. The cholinergic differentiation activity of glabrous skin extract was blocked by pharmacological blockade of LIFRβ signaling and the extract induced gp130 specific signaling cascades in cell culture (Bazan 1991; Habecker et al. 1997). Furthermore, deletion of the gp130 gene in mice led to a failure of sympathetic neurons innervating sweat glands to switch to a cholinergic transmitter phenotype in vivo (Stanke et al. 2006). Thus, the physiologically relevant cholinergic differentiation factor is likely to be related to CNTF, LIF, and CT-1 and may be a new member of this cytokine family.
The release of the cholinergic differentiation factor by sweat glands appears to be dependent upon release of NE from sympathetic neurons upon initial synaptic contact during postnatal development. Extracts of glabrous skin selectively deprived of noradrenergic sympathetic input by treatment with the neurotoxin 6-hydroxydopamine were unable to promote a cholinergic transmitter switch in cultured sympathetic neurons (Habecker and Landis 1994). Sweat glands cultured alone or with sensory neurons (which do not produce NE) do not produce cholinergic differentiation factor as assayed by the ability of conditioned media to induce the expression of cholinergic markers in sympathetic neuronal cultures (Habecker et al. 1995). However, sweat gland tissue produced a cholinergic differentiation factor when cultured with sympathetic neurons. Production of this factor was blocked by pharmacological inhibition of NE transmission (Habecker and Landis 1994; Habecker et al. 1995). These results demonstrate that initial noradrenergic synaptic contact is necessary to induce release of the cholinergic differentiation factor and affect a transmitter phenotype switch in the sympathetic neurons.
The neurotrophins are another family of target-derived growth factors that play a key role in regulating physiology in both developing and mature sympathetic neurons (Bibel and Barde 2000). Neurotrophin signaling may also play an important role in determining mature sympathetic neurotransmitter phenotypes. The expression pattern of the high affinity receptor for neurotrophin-3 (NT-3), tropomyosin-related kinase receptor C (TrkC) overlaps with the expression pattern of cholinergic markers in sympathetic ganglia (Brodski et al. 2000), suggesting a possible role for this pathway in setting the cholinergic phenotype. Consistent with this idea, culturing sympathetic chain explants in the presence of NT-3 increased the expression of cholinergic markers while decreasing the expression of noradrenergic markers (Brodski et al. 2000).
Interestingly, the effects of NT-3 could be reversed and were antagonized by another neurotrophin, nerve growth factor (NGF) (Brodski et al. 2000). NGF is a sympathetic neuron survival factor that has been demonstrated to promote noradrenergic properties in the neurons (Chun and Patterson 1977; Bibel and Barde 2000). Expression of noradrenergic markers in the sympathetic chain overlap with the expression of the NGF high affinity tropomyosin-related kinase receptor A (TrkA) (Brodski et al. 2002), consistent with earlier studies showing that NGF increased the expression of noradrenergic markers in cultured sympathetic neurons (Chun and Patterson 1977). Additionally, NGF rapidly increases the activity of tyrosine hydroxylase, an enzyme necessary for the generation of NE, in rat PC12 cell line cultures (Greene et al. 1984). These results suggest that target-derived signaling plays a crucial, yet complex role in determining transmitter expression in sympathetic neurons and raises the possibility of acute changes in neurotransmitter properties as a result of interaction with target-derived factors.
This section has summarized studies that demonstrate that sympathetic neurons in vivo have the ability to express and release two classical transmitters: ACh and NE. The determination of transmitter phenotype appears to be dependent on bi-directional signaling between the sympathetic neurons and their targets via soluble molecules. One question that remains from these studies is whether individual sympathetic neurons are capable of co-releasing these two different classical transmitters. The next section will present evidence from cell culture studies that show that individual sympathetic neurons indeed co-release both ACh and NE and that this co-release is regulated by target-derived soluble signaling molecules.
4.3 Plasticity of Sympathetic Neurotransmitter Phenotype: Cell Culture Studies
A switch in neurotransmitter status from noradrenergic to cholinergic was first observed in sympathetic neurons grown in culture (Bunge et al. 1974; O’Lague et al. 1974). A series of papers published in 1978 described a careful electrophysiological and pharmacological analysis of this phenomenon in cultured sympathetic neurons (O’Lague et al. 1978; O’Lague et al. 1978; O’Lague et al. 1978). These studies used neurons taken from the superior cervical sympathetic ganglion of neonatal rats, which are almost exclusively noradrenergic in vivo (Elfvin et al. 1993). Surprisingly, culturing neurons either with cardiac myocytes or under conditions that allowed proliferation of non-neuronal ganglionic cells (including Schwann cells and fibroblasts) resulted in the formation of functional cholinergic synapses between the sympathetic neurons. When neurons were cultured in media that inhibited non-neuronal cell growth they remained noradrenergic (O’Lague et al. 1978; O’Lague et al. 1978; O’Lague et al. 1978).
Several lines of evidence supported the conclusion that cholinergic transmission occurred in these cultures. Electrophysiological recordings in pairs of neurons demonstrated that stimulation of action potentials in one neuron resulted in synaptic responses in a second neuron (O’Lague et al. 1978; O’Lague et al. 1978; O’Lague et al. 1978). These responses were blocked by decreasing extracellular calcium concentration, which blocks synaptic transmission, and by cholinergic antagonists. Iontophoresis of ACh onto small regions of individual neurons caused depolarizations similar to the synaptic events elicited by electrical stimulation, further supporting the idea that endogenous ACh release could account for the observed synaptic events. In contrast, application of noradrenergic antagonists had no effect on these cholinergic synaptic events, although iontophoretic application of catecholaminergic agonists caused membrane hyperpolarization. This showed that the neurons expressed functional noradrenergic receptors that did not contribute to the cholinergic transmission.
The cholinergic effects of non-neuronal cells appeared to be mediated by a released soluble factor since media conditioned with these cells could also elicit a cholinergic shift in the cultured neurons (Patterson and Chun 1977). Thus, these studies established the ability of sympathetic neurons to form functional cholinergic connections and demonstrated that these cholinergic properties were regulated by extrinsic factors in vitro.
While non-neuronal cells promoted the formation of cholinergic synaptic connections, NE continued to be produced in these cultures. This meant that culture with non-neuronal cells altered the balance of ACh and NE expression in sympathetic neuron co-cultures. Noradrenergic sympathetic neurons grown in the absence of non-neuronal cells produced only small amounts of ACh as assayed by incorporation of radioactive precursors (Patterson and Chun 1974). Addition of increasing numbers of non-neuronal cells increased the amount of ACh produced while decreasing NE levels (Patterson and Chun 1974). While these results suggested that the cultures produced both ACh and NE, it was not clear whether different subpopulations of neurons were responsible for production of each transmitter or whether individual neurons could produce variable amounts of both transmitters.
ACh and NE expression needed to be unambiguously determined in individual neurons to establish if sympathetic neurons were capable of simultaneously expressing both transmitters. The use of microisland cultures containing single sympathetic neurons together with target cells made it possible to address this issue (Furshpan et al. 1976; Furshpan et al. 1986; Potter et al. 1986). The target cells used in these cultures were neonatal rat cardiac myocytes. Sympathetic neurons innervate the heart in vivo to provide excitatory regulation of heart beat rate and cardiac function via noradrenergic transmission (Elghozi and Julien 2007). Interestingly, although sympathetic innervation of the heart is noradrenergic, cardiac myocytes produce cholinergic differentiation factors, and promote the development of cholinergic properties in co-cultured sympathetic neurons (Fukada 1980; Marvin et al. 1984; Furshpan et al. 1986). Thus, co-culture with cardiac myocytes provided a system in which both noradrenergic transmission and the development of cholinergic properties could be examined.
Cardiac myocytes were cultured together with sympathetic neurons in small (300 to 500 μm) microislands on the surface of a culture dish (Furshpan et al. 1976; Furshpan et al. 1986; Potter et al. 1986). In these cultures it was possible to identify individual microislands that contained only a single neuron that grew over the cardiac myocytes, but did not grow past the edge of the microisland (Furshpan et al. 1976; Furshpan et al. 1986). Neurons grown in this way formed functional synapses on themselves and onto the myocytes (Furshpan et al. 1976; Furshpan et al. 1986). This preparation allowed the determination of the identity of released transmitter(s) from individual sympathetic neurons.
One way to determine the identity of the released transmitters in neuron-myocyte microislands was to examine the postsynaptic response of the myocytes to neuronal activity. Myocytes cultured in microislands form electrical junctions with one another and contract, or beat, in a coordinated fashion across the extent of the microisland. These contractions are modulated by neuronal activity via synaptic transmission in a manner similar to the autonomic regulation of heart rate in vivo (Furshpan et al. 1976; Furshpan et al. 1986; Elghozi and Julien 2007). Myocytes have an excitatory response to NE released by sympathetic neurons that is mediated via activation of β–adrenergic receptors. Conversely, myocyte beat rate is inhibited by ACh via activation of muscarinic ACh receptors (Furshpan et al. 1976; Furshpan et al. 1986). Using electrophysiological recording techniques it is possible to monitor the myocyte response to stimulation of the neuron by recording the myocyte action potentials, which correspond to beat rate. Additionally, NE depolarizes and ACh hyperpolarizes myocytes, which can also be monitored by electrophysiological techniques. Therefore by stimulating neurons to release transmitter and recording the myocyte response it is possible to determine whether ACh or NE is being released by a neuron.
In addition to measuring the postsynaptic responses of the myocytes, information about the neurotransmitter identity of the neurons could be gained by examining neuronal postsynaptic responses to synaptic transmission taking place at autapses. While sympathetic neurons grown in microislands released NE, no evidence was found for a noradrenergic neuronal synaptic response. The neurons were excited, however, by synaptically released ACh through activation of nicotinic receptors (Furshpan et al. 1976; Furshpan et al. 1986). Thus, in these microislands, the neurons showed responses to cholinergic transmission, while the myocytes responded differentially to ACh and NE release by the neurons. Thus, recording from individual neurons and myocytes grown in microisland cultures provided an approach to investigate whether individual sympathetic neurons utilized exclusively ACh or NE, or could co-release both transmitters (Furshpan et al. 1976; Furshpan et al. 1986; Potter et al. 1986).
Sympathetic neurons in microislands were found to vary greatly in release of transmitter with some cells expressing almost exclusively ACh or NE while other cells expressed both transmitters in varying proportions. In some neurons, stimulation resulted in excitatory synaptic events recorded in the neuron itself and an inhibition of the myocyte, seen as a membrane hyperpolarization and decrease in spiking and contraction rate. Nicotinic antagonists such as curare and hexamethonium blocked the neuronal synaptic events, while the muscarinic antagonist, atropine blocked the myocyte inhibition. These data demonstrated that the synaptic events in these neurons were due to cholinergic transmission (Furshpan et al. 1976; Furshpan et al. 1986; Potter et al. 1986).
In some neurons stimulation resulted in no synaptic events in the neuron, but caused an increase in spiking and contraction rate of the myocyte. This type of response was found to be sensitive to β–adrenergic antagonists such as propranolol, confirming a noradrenergic phenotype for those neurons. In the majority of cells however, the myocyte response was mixed, showing slower, noradrenergic-mediated excitation preceded by a more rapid cholinergic-mediated inhibition of the myocytes. These effects could be pharmacologically separated using cholinergic or adrenergic antagonists. These data provided evidence for co-release of NE and ACh in individual neurons.
Further evidence for dual neurotransmitter coexpression in sympathetic neurons came from ultrastructural examination. Electron microscopic analysis of microisland neurons revealed the existence of small granular and small clear vesicles, presumably representing noradrenergic and cholinergic vesicles, respectively. In elegant experiments in which physiological responses were linked to ultrastructure in individual neurons, the authors observed that neurons with mixed physiological responses showed both types of vesicles at synaptic endings, while single-function neurons showed mostly one or the other type of vesicle. These studies unambiguously demonstrated that cultured sympathetic neurons have the capacity to co-release both ACh and NE (Furshpan et al. 1976; Furshpan et al. 1986; Potter et al. 1986).
As described in the previous section, initially noradrenergic sympathetic neurons that innervate sweat glands demonstrate a developmental switch to cholinergic transmission over a period of time following the formation of synaptic contacts with target. It is thus possible that co-transmission in sympathetic neurons grown in microislands represents a transitory state that occurs while neurons are switching from a noradrenergic to a cholinergic phenotype in response to soluble signals from non-neuronal cells. Indeed, by recording multiple times on successive days from individual neurons it was found that transmitter phenotype tended to shift from noradrenergic to cholinergic over time. However, many neurons retained dual transmitter status for up to several months in culture, suggesting that dual transmitter expression could be a stable condition (Potter et al. 1986).
Sympathetic neurons contacting sweat glands switch transmitter phenotype in the first one to two postnatal weeks in vivo, raising the question of whether neurotransmitter plasticity is a transient developmental phenomenon. The microisland recordings described above were performed using neonatal sympathetic neurons, which are still in the process of forming final target contacts and hence may be more plastic in respect to transmitter phenotype expression (Francis and Landis 1999). However, adult superior cervical sympathetic neurons grown on microislands also demonstrated neurotransmitter plasticity, albeit in a smaller proportion of cells and with more cells tending to remain noradrenergic (Potter et al. 1986). These results demonstrate that transmitter plasticity, while more pronounced in younger neurons, is not lost in the adult.
4.4 Neurotrophins Induce a Rapid Switch in Neurotransmitter Status of Sympathetic Neurons
Thus far, the neurotransmitter plasticity displayed by sympathetic neurons has been described as a developmental process that takes place over a period of days or weeks. Recent studies however, show that the regulation of neurotransmitter properties can also take place over a rapid time scale. In addition to their role in the developmental regulation of neurotransmitter phenotype (described above), neurotrophins are able to rapidly regulate the co-release properties of sympathetic neurons.
As will be described in detail below, neurotrophins have been shown to cause rapid differential regulation of neurotransmitter release in sympathetic neurons by acting through different receptors. Therefore it will be helpful to detail the types of neurotrophin receptors expressed by sympathetic neurons. Neurotrophins act through two types of receptors; the pan-neurotrophin receptor (p75), which binds all family members with similar affinity, and the tropomyosin-related kinases (Trks), which show specific binding affinities to various neurotrophin family members (Reichardt 2006). Sympathetic neurons express p75 and two of the three tropomyosin-related kinases; TrkA, the NGF receptor and TrkC, which specifically binds NT-3. These neurons do not express the TrkB receptor, the specific receptor for brain-derived neurotrophic factor (BDNF) and neurotrophin 4/5 (Dixon and McKinnon 1994; Wyatt and Davies 1995; Bamji et al. 1998; Reichardt 2006). Trk receptors are thought to underlie many described neuronal responses to neurotrophins including neuronal differentiation, growth, survival and synaptic modulation (Bibel and Barde 2000; Reichardt 2006). However, in recent years a number of distinct and sometimes opposing functions have been described for the p75 receptor. These include promoting apoptotic neuronal death, modulation of growth dynamics, and the regulation of long-term depression (Chao and Hempstead 1995; Lu et al. 2005; Woo et al. 2005). For neurons expressing both Trk and p75 receptors, including sympathetic neurons, responses to neurotrophins may reflect the output of complex interactions between the p75 and Trk signaling pathways (Chao and Hempstead 1995; Huber and Chao 1995; Lu et al. 2005; Woo et al. 2005).
Evidence for differential regulation of co-transmission by p75 and Trk receptors has been provided by analysis of synaptic transmission in neuron-myocyte co-cultures. Superior cervical sympathetic neurons form functional synapses onto the co-cultured cardiac myocytes and these connections are initially almost entirely noradrenergic. Electrophysiological stimulation of neurons in young, noradrenergic neuron-myocyte co-cultures results in an increase in beat rate of myocytes that are connected to the stimulated neuron. This increase can be observed visually and quantified by counting contractions (Conforti et al. 1991; Lockhart et al. 1997). Treatment of the cultures with NGF for ten minutes caused a rapid and reversible potentiation of synaptic transmission that was observed as an increase in myocyte beat rate in response to neuronal stimulation (Lockhart et al. 1997).
The effect of NGF on excitatory transmission was likely to be due to a presynaptic action of NGF on neuronally expressed TrkA receptors. Cardiac myocytes do not express appreciable levels of TrkA and the myocyte response to pressure ejection application of NE was not altered in the presence of NGF (Lockhart et al. 1997). The potentiation of neuronal regulation of myocyte beat rate was blocked by application of the Trk antagonist K252a suggesting that the effect was mediated via TrkA and not p75 activation (Lockhart et al. 1997). This study provided evidence that NGF, acting through TrkA, promotes noradrenergic neurotransmission in sympathetic neurons.
Further investigation into the actions of neurotrophins on sympathetic presynaptic properties revealed the surprising result that, in addition to potentiating the release of NE, neurotrophin signaling regulated co-release of ACh. Treatment of young, noradrenergic neuron-myocyte co-cultures with BDNF rapidly and reversibly resulted in the inhibition of myocyte beat rate during neuronal stimulation (Yang et al. 2002). This result was in contrast to the excitation of myocyte beat rate seen during neuronal stimulation in the presence of NGF, a related member of the neurotrophin family. In these neuron-myocyte co-cultures a fifteen minute application of BDNF was sufficient to switch the myocyte beat rate response during neuron stimulation from excitation to inhibition. The effect of BDNF was reversible and was blocked by the muscarinic cholinergic antagonist, atropine. Similar to the finding with NGF, the effect of BDNF was likely to be presynaptic since myocyte responses to pressure ejection application of muscarine, a muscarinic ACh receptor agonist, or NE were not altered in BDNF. BDNF did not induce expression of cholinergic markers over the time frame of the experiment and, unlike classic cholinergic differentiation factors such as CNTF, did not promote the expression of cholinergic markers even over a three-day culture period (Slonimsky et al. 2003). These experiments suggest that BDNF acts to rapidly promote ACh release through a mechanism independent of the cholinergic differentiation factors described in the previous section.
Evidence suggests that the switch from excitatory to inhibitory neurotransmission in neuron-myocyte co-cultures is mediated through p75 signaling. BDNF is likely to be a selective ligand for p75 in sympathetic neurons as TrkB is not expressed and BDNF does not activate the TrkA or TrkC receptors (Dixon and McKinnon 1994; Wyatt and Davies 1995; Bamji et al. 1998; Yang et al. 2002; Reichardt 2006). In contrast to the finding for potentiation of noradrenergic transmission by NGF, the BDNF-dependent induction of inhibitory synaptic function was not blocked by application of K252a, which blocks the function of Trk family receptors. This finding suggested that BDNF was not acting through a Trk receptor (Yang et al. 2002). Application of C2-ceramide, a second messenger produced in response to p75 activation mimicked the BDNF response however, suggesting that BDNF acted to modulate cholinergic transmission via p75 receptor signaling (Yang et al. 2002). Further evidence implicating p75 came from overexpression of human p75 in neuron-myocyte co-cultures which led to inhibitory transmission even in the absence of exogenous BDNF (Yang et al. 2002). Finally, the effect of BDNF was not seen in neuron-myocyte co-cultures derived from mice carrying a targeted deletion of the p75 receptor gene (Lee et al. 1992; Yang et al. 2002). Together, these data suggest that even in apparently noradrenergic sympathetic neurons there exists a releasable cholinergic vesicle pool that is mobilized via cell signaling through the p75 receptor.
Pharmacological evidence suggests that the relative level Trk to p75 signaling determines the ratio of ACh and NE released. Sympathetic neurons in myocyte co-cultures co-released ACh and NE to varying degrees after being cultured for three days in different growth factors (Yang et al. 2002). Again, this was assessed by stimulating neurons and observing changes in beat rate of connected myocytes. Growth in control media, containing 5 ng/ml NGF, or media containing 50 ng/ml NGF, resulted in only excitatory responses. Application of the β–adrenergic antagonist propranolol blocked the excitatory responses and revealed a small inhibitory response suggesting that under these conditions neurons were predominately noradrenergic, but did release some ACh. Conversely, growth in BDNF resulted in only inhibitory myocyte responses when neurons were stimulated. This effect was blocked by the muscarinic ACh antagonist, atropine that revealed a small excitatory response presumably due to co-release of NE. These experiments demonstrated the co-release of ACh and NE and the modulation of that release by neurotrophic signaling.
The rapid effects of BDNF are specific to neurotrophin signaling pathways. Growth in CNTF required three weeks before neurons showed a significant functional cholinergic shift in transmission, suggesting that CNTF and BDNF work through different pathways to regulate cholinergic transmission (Yang et al. 2002; Slonimsky et al. 2003).
In addition to a requirement for p75 signaling, probably via the ceramide pathway, the effects of BDNF on promoting cholinergic transmission appear to be dependent on activation of calcium/calmodulin-dependent protein kinase II (CamKII) (Slonimsky et al. 2006). Inhibition of CamKII in the presynaptic neuron blocked the induction of cholinergic neurotransmission by BDNF, whereas transfection of constitutively active CamKII into presynaptic neurons induced a cholinergic shift in the absence of exogenous BDNF (Slonimsky et al. 2006). However an interaction between the p75 receptor and CamKII appears to be necessary since transfection of constitutively active CamKII into p75 knockout animals did not cause a cholinergic shift in neurotransmission (Slonimsky et al. 2006).
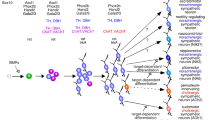
This section has described studies that show that neurotrophins can rapidly modulate the amount of ACh and NE being co-released at sympathetic terminals by signaling through two receptors (Fig. 4.1). Increased activation of the TrkA receptor enhances noradrenergic transmission and increased activation of the p75 receptor enhances cholinergic transmission. This provides a potential mechanism for the regulation of the function of sympathetic innervation via target-derived signaling. Sympathetically innervated targets express distinct profiles of neurotrophin species in vivo (Bierl et al. 2005; Randolph et al. 2007) and sympathetic neurons express both p75 and Trk receptors. Increasing the ratio of Trk to p75 activation would be predicted to increase relative release of NE, and conversely increasing the p75 activation compared to Trk activation would increase ACh release.

Target-derived neurotrophins rapidly regulate functional neuronal properties differentially through p75 and TrkA signaling pathways. Sympathetic neurons express both TrkA and p75. When TrkA activation is higher than p75 activation the neurons tend to release norepinephrine and fire in a tonic pattern (left). When p75 activity predominates cholinergic transmission is potentiated and cells fire in a phasic pattern. Although the signaling pathways involved are incompletely understood, evidence shows that the second messenger molecule, ceramide, is involved in promotion of both phasic firing and potentiation of acetylcholine release. Calcium/calmodulin-dependent protein kinase II (CamKII) also has been implicated in promoting cholinergic transmission. (See Color Plate 2)
While the mechanisms underlying the changes in neurotransmitter release properties are not known, firing pattern has been found to influence co-release of classical transmitters and neuromodulators in different cell types (Bradley et al. 2003; Fulop et al. 2005). Recent evidence shows that in addition to their effects on transmitter choice, neurotrophins also influence sympathetic neuronal firing patterns. In the next section we will explore the possibility that changes in neuronal firing pattern are important determinants of transmitter release.
4.5 Neurotrophins Regulate the Firing Properties of Sympathetic Neurons via Differential Activation of Trk and p75 Receptors
Neurotrophin signaling also plays a role in regulating the membrane electrical properties of sympathetic neurons. Sympathetic neurons fire in one of two characteristic patterns in response to electrophysiological stimulation and the proportion of cells firing in each of these patterns varies among sympathetic ganglia projecting to different targets (Cassell et al. 1986; Wang and McKinnon 1995; Jobling and Gibbins 1999; Anderson et al. 2001). Neurons are found to fire either phasically, that is, they fire once and then go silent, or fire tonically throughout the duration of the stimulus (Cassell et al. 1986). When firing patterns were surveyed over a population of cultured neonatal superior cervical neurons it was found that most cells fired tonically with firing rates being distributed evenly around a mean (Luther and Birren 2006). Application of NGF to these cultures resulted in a shift of firing pattern to a bimodal distribution: increasing the number of phasic cells and increasing the number of high-frequency tonic cells (Luther and Birren 2006).
As mentioned previously, NGF acts through both TrkA and p75 receptors, and therefore, the bimodal shift in firing patterns could be due to differential signaling through those two receptor types in different populations of cells (Fig. 4.1). Indeed, it was found that application of C2-ceramide, a second messenger generated subsequent to p75 activation, induced phasic firing in most neurons (reported in abstract form, Society for Neuroscience Abstracts, 32, 2006). Application of NGF in cultures prepared from p75 knockout mice resulted in most cells firing tonically with higher firing rates compared to control, suggesting that TrkA signaling increased tonic firing (unpublished observations). These results demonstrate that, in addition to their role in determining neurotransmitter phenotype, soluble, target-derived signaling molecules can also influence repetitive firing in sympathetic neurons.
Firing pattern has been shown to play a role in co-release of transmitters and neuromodulators; however, this has not previously been shown for multiple classical transmitters. For example, adrenal chromaffin cells release catecholamines, but when stimulated more strongly they also release neuropeptides (Fulop et al. 2005). Co-release of NE, adenosine triphosphate and neuropeptide Y from sympathetic neurons are differentially modulated by firing frequency and spike number per burst (Bradley et al. 2003). It is tempting to speculate that regulation of the neurotransmitter phenotype and firing pattern by neurotrophins are related. Perhaps p75 mediated changes in firing pattern lead to conditions that favor release of ACh containing vesicles, and conversely TrkA mediated changes in firing pattern favor release of NE containing vesicles. However, it is unknown whether transmitter status and firing pattern correspond in cultured sympathetic neurons. Future studies will be needed to determine if firing pattern is an important determinant of differential release of ACh and NE in sympathetic neurons.
4.6 Future Directions
Sympathetic neurons have the capacity to switch between cholinergic and noradrenergic phenotypes in vivo and to co-release both transmitters in cell culture. Neurotransmitter status in sympathetic neurons appears to depend on soluble, target-derived signaling molecules. This raises the possibility that sympathetic neuronal properties (i.e., neurotransmitter phenotype and firing patterns) are fine-tuned to the physiological needs of the specific target tissue through local signaling interactions. Target-derived signaling varies with physiological state, age and in response to tissue damage. For example, evidence suggests that a heart disease condition such as congestive heart failure can result in abnormalities of neurotrophin release from cardiac tissue including release of BDNF, which is normally not produced in heart tissue (Cai et al. 2006; Kreusser et al. 2007). Neurotrophin production also changes with age in sympathetically innervated tissues (Cai et al. 2006; Bierl and Isaacson 2007). These physiological changes could bring about adaptive (or maladaptive) shifts in neurotransmitter co-release and firing patterns in sympathetic neurons. Indeed, changes in sympathetic nerve activity occur in both hypertension and heart disease (Esler et al. 2001; Watson et al. 2006). A deeper understanding of how target-derived signaling regulates sympathetic neuronal properties could lead to clinical treatments for these diseases and to a better general understanding of how functional neuronal properties are regulated.
Sympathetic neurons co-release ACh and NE in culture conditions, and even apparently noradrenergically committed cells can rapidly be induced to release ACh via p75 signaling. It is less clear however, whether or not this actually occurs in vivo in mature animals. Interestingly, in adult humans, but not rodents, parasympathetic neurons innervating the heart and sympathetic neurons innervating sweat glands and vascular specializations in the hands and feet, called Hoyer-Grosser organs, were found to express all necessary proteins to co-release ACh and NE (Weihe et al. 2005). Data suggestive of potential co-transmission properties of sympathetic neurons in vivo is sparse however, and experiments that directly address co-transmission in physiologically realistic contexts are technically difficult. However, determining the role of co-transmission in the sympathetic system is an important step for understanding both diseases related to the sympathetic nervous system and neurotransmission in general.
References
Anderson RL, Jobling P and Gibbins IL (2001) Development of electrophysiological and morphological diversity in autonomic neurons. J Neurophysiol 86(3):1237--1251
Asmus SE, Parsons S and Landis SC (2000) Developmental changes in the transmitter properties of sympathetic neurons that innervate the periosteum. J Neurosci 20(4):1495--1504
Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG and Miller FD (1998) The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol 140(4):911--923
Bazan JF (1991) Neuropoietic cytokines in the hematopoietic fold. Neuron 7(2):197--208
Bibel M and Barde YA (2000) Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev 14(23):2919--2937
Bierl MA, Jones EE, Crutcher KA and Isaacson LG (2005) “Mature” nerve growth factor is a minor species in most peripheral tissues. Neurosci Lett 380(1-2):133--137
Bierl MA and Isaacson LG (2007) Increased NGF proforms in aged sympathetic neurons and their targets. Neurobiol Aging 28(1):122--134
Bradley E, Law A, Bell D and Johnson CD (2003) Effects of varying impulse number on cotransmitter contributions to sympathetic vasoconstriction in rat tail artery. Am J Physiol Heart Circ Physiol 284(6):H2007--14
Brodski C, Schnurch H and Dechant G (2000) Neurotrophin-3 promotes the cholinergic differentiation of sympathetic neurons. Proc Natl Acad Sci U S A 97(17):9683--9688
Brodski C, Schaubmar A and Dechant G (2002) Opposing functions of GDNF and NGF in the development of cholinergic and noradrenergic sympathetic neurons. Mol Cell Neurosci 19(4):528--538
Bunge RP, Rees R, Wood P, Burton H and Ko C (1974) Anatomical and physiological observations on synapses formed on isolated autonomic neurons in tissue culture. Brain Research 66:401--412
Cai D, Holm JM, Duignan IJ, Zheng J, Xaymardan M, Chin A, Ballard VL, Bella JN and Edelberg JM (2006) BDNF-mediated enhancement of inflammation and injury in the aging heart. Physiol Genomics 24(3):191--197
Cassell JF, Clark AL and McLachlan EM (1986) Characteristics of phasic and tonic sympathetic ganglion cells of the guinea-pig. J Physiol 372:457--483
Chao MV and Hempstead BL (1995) p75 and Trk: a two-receptor system. Trends Neurosci 18(7):321--326
Chun LL and Patterson PH (1977) Role of nerve growth factor in the development of rat sympathetic neurons in vitro. I. Survival, growth, and differentiation of catecholamine production. J Cell Biol 75(3):694--704
Conforti L, Tohse N and Sperelakis N (1991) Influence of sympathetic innervation on the membrane electrical properties of neonatal rat cardiomyocytes in culture. J Dev Physiol 15(4):237--246
Dale H (1935) Pharmacology and nerve-endings. Proc. R. Soc. Med. 28:319--332
Dixon JE and McKinnon D (1994) Expression of the trk gene family of neurotrophin receptors in prevertebral sympathetic ganglia. Brain Res Dev Brain Res 77(2):177--182
Eccles J (1976) From electrical to chemical transmission in the central nervous system. Notes Rec R Soc Lond 30(2):219--230
Elfvin LG, Lindh B and Hokfelt T (1993) The chemical neuroanatomy of sympathetic ganglia. Annu Rev Neurosci 16:471--507
Elghozi JL and Julien C (2007) Sympathetic control of short-term heart rate variability and its pharmacological modulation. Fundam Clin Pharmacol 21(4):337--347
Ernsberger U and Rohrer H (1996) The development of the noradrenergic transmitter phenotype in postganglionic sympathetic neurons. Neurochem Res 21(7):823--829
Ernsberger U and Rohrer H (1999) Development of the cholinergic neurotransmitter phenotype in postganglionic sympathetic neurons. Cell Tissue Res 297(3):339--361
Ernsberger U (2000) Evidence for an evolutionary conserved role of bone morphogenetic protein growth factors and phox2 transcription factors during noradrenergic differentiation of sympathetic neurons. Induction of a putative synexpression group of neurotransmitter-synthesizing enzymes. Eur J Biochem 267(24):6976--6981
Esler M, Rumantir M, Kaye D, Jennings G, Hastings J, Socratous F and Lambert G (2001) Sympathetic nerve biology in essential hypertension. Clin Exp Pharmacol Physiol 28(12):986--989
Francis NJ, Asmus SE and Landis SC (1997) CNTF and LIF are not required for the target-directed acquisition of cholinergic and peptidergic properties by sympathetic neurons in vivo. Dev Biol 182(1):76--87
Francis NJ and Landis SC (1999) Cellular and molecular determinants of sympathetic neuron development. Annu Rev Neurosci 22:541--566
Fukada K (1980) Hormonal control of neurotransmitter choice in sympathetic neurone cultures. Nature 287(5782):553--555
Fulop T, Radabaugh S and Smith C (2005) Activity-dependent differential transmitter release in mouse adrenal chromaffin cells. J Neurosci 25(32): 7324--7332
Furshpan EJ, MacLeish PR, O’Lague PH and Potter DD (1976) Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: evidence for cholinergic, adrenergic, and dual-function neurons. Proc Natl Acad Sci U S A 73(11):4225--4259
Furshpan EJ, Landis SC, Matsumoto SG and Potter DD (1986) Synaptic functions in rat sympathetic neurons in microcultures. I. Secretion of norepinephrine and acetylcholine. J Neurosci 6(4):1061--1079
Greene LA, Seeley PJ, Rukenstein A, DiPiazza M and A Howard (1984) Rapid activation of tyrosine hydroxylase in response to nerve growth factor. J Neurochem 42(6):1728--1734
Habecker BA and Landis SC (1994) Noradrenergic regulation of cholinergic differentiation. Science 264(5165):1602--1604
Habecker BA, Pennica D and Landis SC (1995) Cardiotrophin-1 is not the sweat gland-derived differentiation factor. Neuroreport 7(1):41--44
Habecker BA, Tresser SJ, Rao MS and Landis SC (1995) Production of sweat gland cholinergic differentiation factor depends on innervation. Dev Biol 167(1):307--316
Habecker BA, Symes AJ, Stahl N, Francis NJ, Economides A, Fink JS, Yancopoulos GD and Landis SC (1997) A sweat gland-derived differentiation activity acts through known cytokine signaling pathways. J Biol Chem 272(48):30421--30428
Huber LJ and Chao MV (1995) A potential interaction of p75 and trkA NGF receptors revealed by affinity crosslinking and immunoprecipitation. J Neurosci Res 40(4):557--563
Jobling P and Gibbins IL (1999) Electrophysiological and morphological diversity of mouse sympathetic neurons. J Neurophysiol 82(5):2747--2764
Kreusser MM, Buss SJ, Krebs J, Kinscherf R, Metz J, Katus HA, Haass M and Backs J (2007) Differential expression of cardiac neurotrophic factors and sympathetic nerve ending abnormalities within the failing heart. J Mol Cell Cardiol
Kupfermann I (1991) Functional studies of cotransmission. Physiol Rev 71(3):683--732
Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV and Jaenisch R (1992) Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell 69(5):737--749
Lockhart ST, Turrigiano GG and Birren SJ (1997) Nerve growth factor modulates synaptic transmission between sympathetic neurons and cardiac myocytes. J Neurosci 17(24):9573--9582
Lu B, Pang PT and Woo NH (2005) The yin and yang of neurotrophin action. Nat Rev Neurosci 6(8):603--614
Luther JA and Birren SJ (2006) Nerve growth factor decreases potassium currents and alters repetitive firing in rat sympathetic neurons. J Neurophysiol 96(2):946--958
Marder E, Christie AE and Kilman VL (1995) Functional organization of cotransmission systems: lessons from small nervous systems. Invert Neurosci 1(2):105--112
Marvin WJ, Jr., Atkins DL, Chittick VL, Lund DD and Hermsmeyer K (1984) In vitro adrenergic and cholinergic innervation of the developing rat myocyte. Circ Res 55(1):49--58
O’Lague PH, Obata K, Claude P, Furshpan EJ and Potter DD (1974) Evidence for cholinergic synapses between dissociated rat sympathetic neurons in cell culture. Proc Natl Acad Sci U S A 71(9):3602--3606
O’Lague PH, Furshpan EJ and Potter DD (1978) Studies on rat sympathetic neurons developing in cell culture. II. Synaptic mechanisms. Dev Biol 67(2):404--423
O’Lague PH, Potter DD and Furshpan EJ (1978) Studies on rat sympathetic neurons developing in cell culture. I. Growth characteristics and electrophysiological properties. Dev Biol 67(2):384--403
O’Lague PH, Potter DD and Furshpan EJ (1978) Studies on rat sympathetic neurons developing in cell culture. III. Cholinergic transmission. Dev Biol 67 (2):424--443
Patterson PH and Chun LL (1974) The influence of non-neuronal cells on catecholamine and acetylcholine synthesis and accumulation in cultures of dissociated sympathetic neurons. Proc Natl Acad Sci U S A 71 (9):3607--3610
Patterson PH and Chun LL (1977) The induction of acetylcholine synthesis in primary cultures of dissociated rat sympathetic neurons. I. Effects of conditioned medium. Dev Biol 56 (2):263--280
Potter DD, Landis SC, Matsumoto SG and Furshpan EJ (1986) Synaptic functions in rat sympathetic neurons in microcultures. II. Adrenergic/cholinergic dual status and plasticity. J Neurosci 6 (4):1080--1098
Potter DD, Matsumoto SG, Landis SC, Sah DW and Furshpan EJ (1986) Transmitter status in cultured sympathetic principal neurons: plasticity, graded expression and diversity. Prog Brain Res 68:103--120
Randolph CL, Bierl MA and Isaacson LG (2007) Regulation of NGF and NT-3 protein expression in peripheral targets by sympathetic input. Brain Res 1144:59--69
Ro, MS and Landis SC (1990) Characterization of a target-derived neuronal cholinergic differentiation factor. Neuron 5 (6):899--910
Rao MS, Patterson PH and Landis SC (1992) Multiple cholinergic differentiation factors are present in footpad extracts: comparison with known cholinergic factors. Development 116 (3):731--744
Reichardt LF (2006) Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361 (1473):1545--1564
Rohrer H (1992) Cholinergic neuronal differentiation factors: evidence for the presence of both CNTF-like and non-CNTF-like factors in developing rat footpad. Development 114 (3):689--698
Saadat S, Sendtner M and Rohrer H (1989) Ciliary neurotrophic factor induces cholinergic differentiation of rat sympathetic neurons in culture. J Cell Biol 108 (5):1807--1816
Schafer MK, Schutz B, Weihe E and Eiden LE (1997) Target-independent cholinergic differentiation in the rat sympathetic nervous system. Proc Natl Acad Sci U S A 94 (8):4149--4154
Schotzinger RJ and Landis SC (1988) Cholinergic phenotype developed by noradrenergic sympathetic neurons after innervation of a novel cholinergic target in vivo. Nature 335 (6191):637--639
Schotzinger RJ and Landis SC (1990) Acquisition of cholinergic and peptidergic properties by sympathetic innervation of rat sweat glands requires interaction with normal target. Neuron 5 (1):91--100
Seal RP and Edwards RH (2006) Functional implications of neurotransmitter co-release: glutamate and GABA share the load. Curr Opin Pharmacol 6 (1):114--119
Slonimsky JD, Yang B, Hinterneder JM, Nokes EB and Birren SJ (2003) BDNF and CNTF regulate cholinergic properties of sympathetic neurons through independent mechanisms. Mol Cell Neurosci 23 (4):648--660
Slonimsky JD, Mattaliano MD, Moon JI, Griffith LC and Birren SJ (2006) Role for calcium/calmodulin-dependent protein kinase II in the p75-mediated regulation of sympathetic cholinergic transmission. Proc Natl Acad Sci U S A 103 (8):2915--2919
Stanke M, Duong CV, Pape M, Geissen M, Burbach G, Deller T, Gascan H, Otto C, Parlato R, Schutz G and Rohrer H (2006) Target-dependent specification of the neurotransmitter phenotype: cholinergic differentiation of sympathetic neurons is mediated in vivo by gp 130 signaling. Development 133 (1):141--150
Wang HS and McKinnon D (1995) Potassium currents in rat prevertebral and paravertebral sympathetic neurones: control of firing properties. J Physiol 485 ( Pt 2):319--335
Watson AM, Hood SG and May CN (2006) Mechanisms of sympathetic activation in heart failure. Clin Exp Pharmacol Physiol 33 (12):1269--1274
Weihe E, Schutz B, Hartschuh W, Anlauf M, Schafer MK and Eiden LE (2005) Coexpression of cholinergic and noradrenergic phenotypes in human and nonhuman autonomic nervous system. J Comp Neurol 492 (3):370--379
Woo NH, Teng HK, Siao CJ, Chiaruttini C, Pang PT, Milner TA, Hempstead BL and Lu B (2005) Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci 8 (8):1069--1077
Wyatt S and Davies AM (1995) Regulation of nerve growth factor receptor gene expression in sympathetic neurons during development. J Cell Biol 130 (6):1435--1446
Yamamori T, Fukada K, Aebersold R, Korsching S, Fann MJ and Patterson PH (1989) The cholinergic neuronal differentiation factor from heart cells is identical to leukemia inhibitory factor. Science 246 (4936):1412--1416
Yang B, Slonimsky JD and Birren SJ (2002) A rapid switch in sympathetic neurotransmitter release properties mediated by the p75 receptor. Nat Neurosci 5 (6):539--545
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Luther, J.A., Birren, S.J. (2009). Co-Release of Norepinephrine and Acetylcholine by Mammalian Sympathetic Neurons: Regulation by Target-Derived Signaling. In: Gutierrez, R. (eds) Co-Existence and Co-Release of Classical Neurotransmitters. Springer, Boston, MA. https://doi.org/10.1007/978-0-387-09622-3_4
Download citation
DOI: https://doi.org/10.1007/978-0-387-09622-3_4
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-0-387-09621-6
Online ISBN: 978-0-387-09622-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)