
Abstract
Metabolomic profiling is an emerging technology in the clinical setting with immediate diagnostic potential for the population of patients with Inborn Errors of Metabolism. We present the metabolomics profile of two ABAT deficiency patients both pre- and posttreatment with flumazenil. ABAT deficiency, also known as GABA-transaminase deficiency, is caused by recessive mutations in the gene ABAT and leads to encephalopathy of variable severity with hypersomnolence, hypotonia, hypomyelination, and seizures. Through metabolomics screening of multiple patient tissues, we identify 2-pyrrolidinone as a biomarker for GABA that is informative in plasma, urine, and CSF. These data will enable noninvasive diagnostic testing for the population of patients with disorders of GABA metabolism.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
Introduction
Metabolomic profiling is a recent advancement in diagnostic technologies introduced into the clinical diagnostic setting. Metabolomic profiling consists of measurement of hundreds of small molecules and can be conducted through noninvasive sampling of blood and urine. These data can provide insights into biological processes and pathways and the effects drugs, environmental exposures, and genetic variants may have on them.
ABAT deficiency (OMIM 613163), also termed GABA-T deficiency, is a recessively inherited, rare single gene disorder caused by loss of function mutations in the gene ABAT (Jaeken et al. 1984; Medina-Kauwe et al. 1999). ABAT encodes for the enzyme GABA Transaminase (GABA-T, EC 2.6.1.19), which is a dual function enzyme essential for both the metabolism of GABA in the GABA shunt and for converting dNDPs to dNTPs in the mitochondrial nucleoside salvage pathway. Elevation of GABA in brain and CSF as well as reduction of mtDNA in patient tissues has been observed (Besse et al. 2015, 2016). GABA is a neurotransmitter which is excitatory in utero and becomes inhibitory directly after birth. As a result subjects with ABAT deficiency have hypersomnolence due to excess GABA in the brain. Most reported patients also experience intractable seizures with onset in the first months of life; however, a milder case of ABAT deficiency was reported who did not experience clinically overt seizures (Besse et al. 2016). It is unclear which aspects of clinical presentation are due to each of the two major functions of ABAT.
Diagnosis of ABAT deficiency typically relies on a combination of clinical and diagnostic testing including molecular genetic and biochemical testing as well as neuroimaging. Relatively few patients and consequently few pathogenic alleles have been reported for this rare disorder, as a result a definitive diagnosis often relies on other modalities. As with other IEMs, gold standard for diagnosis is assessment of relevant metabolite levels, in this case GABA, which in current practice is most often measured through biochemical testing in the cerebral spinal fluid (CSF) or proton magnetic resonance spectroscopy (MRS) of the brain. However, GABA is typically not included on diagnostic neurotransmitter panels measured in CSF and may not be screened for in MRS unless specified by ordering physician.
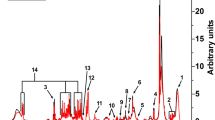
GABA can be detected in multiple forms in various human tissues. For example, the measurement of total CSF GABA detects several forms of GABA including “free GABA,” homocarnosine, 2-pyrrolidinone, and small amounts of other GABA-containing peptides (Fig. 1). In fact, homocarnosine and 2-pyrrolidinone are the major components of CSF, exceeding the amount of free GABA by more than 1,000-fold (Grove et al. 1982; Haegele et al. 1987). Homocarnosine is a dipeptide of GABA and histidine that is synthesized from GABA. 2-pyrrolidinone is the lactam of GABA, and while it is more abundant in CSF than GABA, it readily converts to GABA in the brain. Brain concentrations of homocarnosine and 2-pyrrolidinone are low with the human occipital lobe showing 2-pyrrolidinone concentrations half that of free GABA (Hyder et al. 1999; Kish et al. 1979). Moreover, intravenous infusion (Callery et al. 1979) or chronic oral administration (Fasolato et al. 1988) results in elevations of GABA in the brain from the conversion of 2-pyrrolidinone into free GABA.

2-Pyrrolidinone is elevated in ABAT deficiency. (a) Citric acid cycle, GABA shunt, and GABA metabolites: 2-pyrrolidinone and homocarnosine. (b) 2-Pyrrolidinone measured in Subject 1 plasma and Subject 2 plasma, urine, and CSF. (c) 2-Pyrrolidinone measured in plasma in Subject 2 before (“S2 pre”) and during treatment (“S2 post”) with flumazenil
We present the metabolomics profile of ABAT deficiency patients in multiple tissues and demonstrate the utility of metabolomics profiling for this patient population. We show that 2-pyrrolidinone is a biomarker for GABA that can be leveraged to identify elevations of GABA whether due to genetic or pharmacologic cause. Through study of multiple patient tissues we confirm that 2-pyrrolidinone is accessible and informative by noninvasive sampling of plasma and urine. Additionally, we show GABA metabolite levels pre- and posttreatment with flumazenil, the only available treatment for this disorder (Koenig et al. 2017).
Methods
Human Subjects
All subjects were consented to an Institutional Review Board approved protocol for participation in research study. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Metabolomic Profiling
Metabolomic screening was conducted at Baylor Genetics Laboratories (Houston, Texas) and Metabolon, Inc. (Durham, North Carolina). Metabolomic profiling was performed as described previously (Dehaven et al. 2010; Evans et al. 2009). Small molecules were extracted and subjected to four analyses: two LC-MS/MS analyses in positive mode and two LC-MS/MS analyses in negative mode. Metabolites were identified with known chemical structure by matching the ion chromatographic retention index, nominal mass, and mass spectral fragmentation signatures with reference library entries created from authentic standard metabolites under the identical analytical procedure as the experimental samples (Dehaven et al. 2010). Semi-quantitative analysis was achieved by comparing patient samples to a set of invariant anchor specimens included in each batch. Raw spectral intensity values were normalized to the anchor samples, log transformed, and compared to a normal reference population to generate z-score values.
Results
Clinical Description
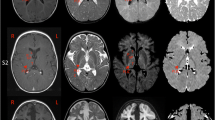
Two patients previously published representing the spectrum of mild and severe ABAT deficiency patients were studied for metabolomics profile. Subject 1 presented in infancy with a severe encephalopathy including intractable seizures, hypotonia, and lethargy (Besse et al. 2015). Brain MRI showed extensive symmetric signal abnormalities in several regions including posterior limbs of the internal capsule, cerebral peduncles, dorsal tegmentum and pons, medulla, and dentate nuclei indicating significant hypomyelination and brain atrophy. This child never achieved any developmental milestones and remains alive in a static state at age 9. This child is one of three similarly affected siblings all of whom were demonstrated to be homozygous for ABAT NM_000663.4 c.631C>T; NP_000654.2 p.Leu211Phe (Besse et al. 2015, 2016).
Subject 2 presented to clinic at 6 months with global developmental delay, hypersomnolence, hypotonia, bilateral opthalmoplegia, and mild choreiform movements without apparent epilepsy (Besse et al. 2016). Oculomotor apraxia was appreciated by age 18 months and cranial MRI conducted showed extensive signal abnormalities in the deep white matter indicative of diffuse, mild hypomyelination. In contrast to Subject 1, no brain atrophy was apparent on MRI. Genetics studies showed this child was compound heterozygous for ABAT NM_000663.4 c.454C>T;p.Pro152Ser and c.1393G>C;p.Gly465Arg.
Metabolic Testing
For both subjects prior to molecular genetic studies, CSF was tested for a panel of neurotransmitters that was reported normal; however, GABA was not included in this panel testing. After molecular studies pointed to ABAT as the pathogenic gene, Subject 1 was tested by proton NMR spectroscopy which showed elevated GABA in the brain (Besse et al. 2015). Subject 2 exome studies identified compound heterozygous mutations in ABAT that were reported as variants of uncertain significance (VUS). Subsequent studies of cerebral spinal fluid (CSF) in this patient demonstrated free GABA level of 247 nmol/L (17–67) and total GABA level of 33.4 μmol/L (4.2–13.4).
Additional metabolic testing on these patients showed no other abnormalities. Subject 1 had normal metabolic testing results for: Krabbe disease, hexosaminidase A and B, CSF protein, glycine, CSF lactate and plasma lactate, urine organic acids, plasma amino acids, very long-chain fatty acids, acylcarnitine profile, urine purines, and urine oligosaccharides. Subject 2 exhibited normal plasma and CSF amino acids.
Metabolomic Profiling
Metabolomics screening showed abnormalities in both patients. Subject 1 had metabolomics screening conducted on plasma when he was 5 years and was not receiving flumazenil treatment. This testing showed 2-pyrrolidinone elevated more than 2 standard deviations (SD) above normal (z-score = 3.7) (Fig. 1b). Similarly, at age 1 year, Subject 2 had metabolomics screening in plasma, urine, and CSF all of which showed 2-pyrrolidinone elevated above normal (z-score = 6.1, 4.24, and 7.1) (Fig. 1b). Glutamate, which is just prior to GABA in the GABA shunt, was normal in all samples, as was alpha-ketoglutarate, which is just prior to glutamate and the metabolite that flows from the citric acid cycle to the GABA shunt (Fig. 1). Succinic semialdehyde, which is the product of GABA-T catabolism of GABA in the GABA shunt, was not measured. However, metabolites of the Kreb’s cycle that are just post GABA shunt were in the normal range: succinate, fumarate, and malate.
Subject 2 currently receives treatment for ABAT deficiency of flumazenil, 1.7 mg/kg/day. He was 2 years old at the time of initiation of treatment and was able to make significant gains in alertness, developmental milestones, and some amelioration of oculomotor apraxia. Metabolomics was conducted on patient plasma while the child was receiving treatment and the same elevation in 2-pyrrolidinone was observed (Fig. 1c). This reconciles with the mechanism of action of flumazenil as a GABA receptor antagonist that would not be expected to decrease the levels of GABA or its metabolites.
Discussion
We observed elevated GABA in the brain of Subject 1 while also observing elevated 2-pyrrolidinone in this subject’s plasma. Similarly, Subject 2 showed elevated GABA in CSF (brain was not measured) and plasma alongside elevated 2-pyrrolidinone in both of these tissues. Notably, treatment with flumazenil did not appreciably reduce levels of 2-pyrrolidinone in Subject 2, which is in accordance with its mechanism of action as a negative allosteric modulator of GABA receptor rather than an agent of GABA degradation.
Lundgren and Hankins first suggested 2-pyrrolidinone as a “transport form” of GABA in 1978 (Lundgren and Hankins 1978) and subsequent work supports this theory. 2-pyrrolidinone crosses the blood–brain barrier more readily than GABA. 2-pyrrolidinone is more stable in plasma (Fasolato et al. 1988; Nakamura et al. 1991) and CSF (Grove et al. 1982; Haegele et al. 1987) than GABA, while readily converting to free GABA in the brain (Callery et al. 1979; Fasolato et al. 1988) where GABA exerts its neuromodulatory effects. 2-pyrrolidinone has been shown to have anticonvulsant properties (Sasaki et al. 1991) which may be a result of its conversion to GABA. Additionally, Vigabatrin, an irreversible inhibitor of ABAT, has been demonstrated to elevate CSF and brain GABA, homocarnosine, and pyrrolidinone (Hyder et al. 1999; Petroff et al. 1996; Petroff and Rothman 1998; Riekkinen et al. 1989; Rothman et al. 1997).
These studies support what we observed in our ABAT deficiency patients and we conclude that 2-pyrrolidinone is an excellent biomarker for GABA in plasma, urine, and CSF. As a biomarker for GABA, 2-pyrrolidinone would be a relevant metabolite to assess in individuals suspected of having a genetic disorder resulting in GABA elevations, such as ABAT deficiency or SSADHD, or individuals taking therapeutic interventions that would be expected to elevate GABA, such as Vigabatrin. The ability to distinguish between genetic and other causes of GABA/2-pyrrolidinone elevations would require the integration of genetic and metabolic testing.
References
Besse A, Wu P, Bruni F et al (2015) The GABA transaminase, ABAT, is essential for mitochondrial nucleoside metabolism. Cell Metab 21:417–427
Besse A, Petersen AK, Hunter JV et al (2016) Personalized medicine approach confirms a milder case of ABAT deficiency. Mol Brain 9:93
Callery PS, Stogniew M, Geelhaar LA (1979) Detection of the in vivo conversion of 2-pyrrolidinone to gamma-aminobutyric acid in mouse brain. Biomed Mass Spectrom 6:23–26
Dehaven CD, Evans AM, Dai H et al (2010) Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J Cheminform 2:9
Evans AM, DeHaven CD, Barrett T et al (2009) Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem 81:6656–6667
Fasolato C, Bertazzon A, Previero A et al (1988) Effect of 2-pyrrolidone on the concentration of GABA in rat tissues. Pharmacology 36:258–264
Grove J, Schechter PJ, Tell G et al (1982) Artifactual increases in the concentration of free GABA in samples of human cerebrospinal fluid are due to degradation of homocarnosine. J Neurochem 39:1061–1065
Haegele KD, Schwartz JJ, Schoun J et al (1987) 2-Pyrrolidinone in human cerebrospinal fluid: a major constituent of total gamma-aminobutyric acid. J Neurochem 49:1402–1406
Hyder F, Petroff OA, Mattson RH et al (1999) Localized 1H NMR measurements of 2-pyrrolidinone in human brain in vivo. Magn Reson Med 41:889–896
Jaeken J, Casaer P, de Cock P et al (1984) Gamma-aminobutyric acid-transaminase deficiency: a newly recognized inborn error of neurotransmitter metabolism. Neuropediatrics 15:165–169
Kish SJ, Perry TL, Hansen S (1979) Regional distribution of homocarnosine, homocarnosine-carnosine synthetase and homocarnosinase in human brain. J Neurochem 32:1629–1636
Koenig MK, Hodgeman R, Riviello JJ et al (2017) Phenotype of GABA-transaminase deficiency. Neurology 88:1919–1924
Lundgren DW, Hankins J (1978) Metabolism of putrescine to 2-pyrrolidone by rat liver slices. J Biol Chem 253:7130–7133
Medina-Kauwe LK, Tobin AJ, De Meirleir L et al (1999) 4-Aminobutyrate aminotransferase (GABA-transaminase) deficiency. J Inherit Metab Dis 22:414–427
Nakamura J, Miwa T, Mori Y et al (1991) Comparative studies on the anticonvulsant activity of lipophilic derivatives of gamma-aminobutyric acid and 2-pyrrolidinone in mice. J Pharmacobiodyn 14:1–8
Petroff OA, Rothman DL (1998) Measuring human brain GABA in vivo: effects of GABA-transaminase inhibition with vigabatrin. Mol Neurobiol 16:97–121
Petroff OA, Behar KL, Mattson RH et al (1996) Human brain gamma-aminobutyric acid levels and seizure control following initiation of vigabatrin therapy. J Neurochem 67:2399–2404
Riekkinen PJ, Ylinen A, Halonen T et al (1989) Cerebrospinal fluid GABA and seizure control with vigabatrin. Br J Clin Pharmacol 27(Suppl 1):87S–94S
Rothman DL, Behar KL, Prichard JW et al (1997) Homocarnosine and the measurement of neuronal pH in patients with epilepsy. Magn Reson Med 38:924–929
Sasaki H, Mori Y, Nakamura J et al (1991) Synthesis and anticonvulsant activity of 1-acyl-2-pyrrolidinone derivatives. J Med Chem 34:628–633
Acknowledgments
Research reported in this publication was supported by National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number R01NS083726 to PEB. We thank the families for participating in this study.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Saskia Brigitte Wortmann, M.D., Ph.D.
Appendices
Author Contributions
MKK conceived the study and treated the patients. PEB conceived and led the study and wrote the manuscript.
Conflict of Interest
MKK declares no conflict of interest. PEB declares no conflict of interest.
Rights and permissions
Copyright information
© 2018 Society for the Study of Inborn Errors of Metabolism (SSIEM)
About this chapter

Cite this chapter
Koenig, M.K., Bonnen, P.E. (2018). Metabolomics Profile in ABAT Deficiency Pre- and Post-treatment. In: Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V. (eds) JIMD Reports, Volume 43. JIMD Reports, vol 43. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2018_94
Download citation
DOI: https://doi.org/10.1007/8904_2018_94
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-58613-6
Online ISBN: 978-3-662-58614-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)