
Abstract
Purpose: The urinary glucose tetrasaccharide, Glcα1-6Glcα1-4Glcα1-4Glc (Glc4), is a biomarker of glycogen accumulation and tissue damage and is elevated in patients with Pompe disease. We report baseline urinary Glc4 concentrations for patients with classic infantile-onset or late-onset Pompe disease, and those with a pseudodeficiency of acid alpha-glucosidase (GAA), identified through newborn screening (NBS) in Taiwan.
Methods: Infants identified through NBS with (1) classic infantile-onset Pompe disease (NBS-IOPD) (n = 7) defined as patients with evidence for hypertrophic cardiomyopathy by EKG, X-ray, and echocardiogram, (2) a late-onset phenotype (NBS-LOPD) (n = 13) defined as patients without evidence for cardiomyopathy, (3) a GAA pseudodeficiency (n = 58), and (4) one patient with LOPD diagnosed in infancy due to family history were consented to the study. Four infants diagnosed after the onset of clinical symptoms (CLIN-IOPD) were included for comparison. Glc4 concentrations in dried urine samples on filter paper were determined using tandem mass spectrometry.
Results: Baseline Glc4 concentrations were at or above the 90th centile of the age-matched reference range for the NBS-IOPD cohort. The median Glc4 level for this group was lower than that of the CLIN-IOPD group, although not at the level of significance (p = 0.07), but was significantly higher than that of the NBS-LOPD group (p < 0.05). Baseline Glc4 was not elevated for the NBS-LOPD and GAA pseudodeficiency cohorts and remained low for late-onset patients that did not require treatment before the age of three years.
Conclusion: Baseline urinary Glc4 is elevated in neonates with infantile-onset Pompe disease identified through NBS.
Competing interests: None declared
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
Introduction
Pompe disease (OMIM #232300) is caused by a deficiency of lysosomal acid α-glucosidase (GAA; EC 3.2.1.20), resulting in glycogen accumulation that has a destructive effect on muscle (Hirschhorn and Reuser 2001). Patients with a severe GAA deficiency present in infancy (infantile-onset Pompe disease; IOPD) with cardio- and skeletal myopathy and succumb to cardiorespiratory disease within 2 years (Kishnani et al. 2006). Patients with an attenuated phenotype (termed late-onset or later-onset Pompe disease; LOPD) usually have measurable residual enzyme activity and no cardiac involvement. They present anywhere from early childhood to adulthood with a myopathy that progresses to respiratory insufficiency if untreated (Hagemans et al. 2005).
Enzyme replacement therapy (ERT) with recombinant human alglucosidase alfa (rhGAA) (Myozyme®, Lumizyme®, Genzyme, Cambridge, MA) is available for IOPD and LOPD. Evidence suggests ERT has the most favorable clinical outcome when started early in the disease process (Kishnani et al. 2009; Chien et al. 2009). Recognition of the importance of early diagnosis and treatment of Pompe disease has led to the development of newborn screening (NBS) assays for this condition and a recommendation by the US Discretionary Advisory Committee for Heritable Disorders in Newborns and Children for its inclusion in the recommended uniform newborn screening panel. A NBS pilot program in Taiwan identified 7 infants with IOPD and 13 with LOPD out of 344,056 infants screened between 2005 and 2009 (Chien et al. 2008, 2009, 2011). Survival, particularly ventilator-free survival, was improved for patients with IOPD diagnosed by NBS compared with those diagnosed after onset of symptoms. A high false-positive rate was the result of a pseudodeficiency allele of the GAA gene, c.[1726A;2065A] which is common in the Chinese population, but has no known clinical effects (Kroos et al. 2008; Kumamoto et al. 2009; Labrousse et al. 2010).
The glucose tetrasaccharide, Glcα1-6Glcα1-4Glcα1-4Glc (Glc4), is a limit dextrin of glycogen (Kumlien et al. 1988) and was shown to correlate with glycogen content in quadriceps biopsies in patients with IOPD (Young et al. 2009). Glc4 can be useful in the diagnosis of Pompe disease and for monitoring the response to ERT (Young et al. 2009, 2012). We assessed the usefulness of urinary Glc4, measured as the total hexose tetrasaccharide (Hex4) fraction in urine, in the follow-up of infants with low GAA activity identified by the Taiwanese pilot NBS program.
Materials and Methods
Materials
Whatman grade 903 filter paper (VWR, Batavia, IL); Sep-Pak® Vac 100 mg C18 cartridges (Waters Corporation, Milford, MA); d3-creatinine (Cambridge Isotopes, Andover, MA); creatinine standards, sodium cyanoborohydride, butyl 4-aminobenzoate, and glacial acetic acid (Sigma-Aldrich, St. Louis, MO); Glc4 standard (Glycorex AB, Lund, Sweden); HPLC grade solvents (VWR, West Chester, PA); and [13C6]-labeled glucose tetrasaccharide internal standard which was synthesized as previously described (Young et al. 2003).
Patients
This study included infants identified by the Taiwan NBS program between 2005 and 2009 with IOPD (NBS-IOPD, n = 7) and LOPD (NBS-LOPD, n = 13). The diagnostic confirmation and clinical status have been reported for all patients except NBS9 (Chien et al. 2008, 2009, 2011), and subject designations (see Table Supplemental Digital Content 1) are consistent with these previous publications. Four of the thirteen patients in the NBS-LOPD cohort were started on treatment at or before the age of 3 years because of the severity of their clinical condition (NBS-LOPD early treated), whereas the remaining nine patients did not require treatment within the first 3 years of life (NBS-LOPD-Group 2) (Chien et al. 2011). An additional patient with LOPD (L14), identified at birth because of a positive family history, was also included.
Four patients with IOPD, who were not part of the newborn screening pilot study, were diagnosed in early infancy (<5 months age) after the onset of clinical symptoms (NBS-CLIN) and were included as a prospective comparison group for the NBS-IOPD patients (Chien et al. 2009).
Additionally, 58 infants identified by NBS who had a pseudodeficiency of GAA in DBS (Labrousse et al. 2010) were evaluated. These infants had the following combinations of the pseudodeficiency allele c.[1726A; 2065A] and known or putative disease-causing GAA mutations:
-
1.
One mutation plus one pseudodeficiency allele (pseudodeficiency group 1, n = 23)
-
2.
One mutation plus two pseudodeficiency alleles (pseudodeficiency group 2, n = 19)
-
3.
Two pseudodeficiency alleles only (pseudodeficiency group 3, n = 16)
Baseline urine samples were collected within the first 6 weeks of life, except for NBS-L4 and NBS-L11 in the NBS-LOPD cohort on whom samples were collected at 9 and 6 months, respectively (Table Supplemental Digital Content 1). Longitudinal urine samples were collected at regular clinic visits and stored at −20°C.
This study was approved by the Institutional Review Boards of National Taiwan University Hospital and Duke University Health System. Informed consent was obtained from parents of all patients.
Control Samples
Age-specific reference ranges were determined using anonymized clinical samples (n = 472, median age: 1.5 years, min–max: 0.0–68 years) from the Duke Biochemical Genetics laboratory and urine specimens collected from anonymous volunteers (n = 143, median age: 33 years (min–max: 3–78 years)) under a Duke University Health System IRB-approved protocol.
Glc4 Determination in Dried Urine Samples Using Stable Isotope Dilution-ESI-MS/MS
Urine specimens were soaked onto filter paper strips, dried, and mailed to the Duke Biochemical Genetics Laboratory at ambient temperature. None of the patients were on ERT at the time of the baseline sample collections. 2 × 2 cm diameter disks cut from the dried urine spot on filter paper were extracted with 1 mL DI-H2O by shaking at room temperature for 1 h. 500 μL of the filter paper extract was dried under nitrogen at 40°C, reconstituted with 50 μL DI-H2O and mixed with 25 μL 0.1 mmol/L [13C6]-labeled Glc4 internal standard. The mixture was derivatized with butyl-p-aminobenzoic acid, and Glc4 was determined as the total hexose tetrasaccharide fraction (Hex4) in urine by ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) on an Acquity UPLC-Quattro Micro tandem mass spectrometer system (Waters Corp, Milford, MA), as previously described by Young et al. (2003, 2009). Glc4 comprises ≥90 % of the total hexose tetrasaccharide fraction in most patient and control samples (unpublished observation). In keeping with previous publications on this biomarker, Hex4 measurements will be referred to as Glc4. Glc4 concentrations were normalized to creatinine determined in an aliquot of the same filter paper extract using stable isotope dilution-tandem mass spectrometry (Young et al. 2009). Comparison studies have shown the equivalency of this assay when applied to liquid urine specimens and extracts of urine specimens dried on filter paper (Young et al. 2003).
Statistical Analyses
Descriptive statistics and Mann–Whitney comparison of median Glc4 values for the study cohorts were calculated using GraphPad Prism 5.04 software (La Jolla, CA). The relationship of age to Glc4 in the control cohort was examined using univariable linear regression with STATA 11.0 (College Station, TX) software. p-values ≤0.05 were considered to be significant.
Results
Urinary Glc4 in Control Samples
A significant negative correlation with age was observed for urinary Glc4 values in controls under aged 3 years (−2.956 [95 % confidence interval; −3.680, −2.232], p < 0.001, n = 287). No correlation with age was observed for controls over 3 years of age (−0.006 [−0.012, 0.001], p = 0.09, n = 328). Controls younger than 3 years were divided into different age bins based on a visual inspection of the data and statistical analysis. Upper limits of the reference ranges were defined as the 95th centile and were stratified according to the following age groups: 0–6 months age (95th centile: 20 mmol/mol CN, n = 132), 6–12 months (95th centile: 14 mmol/mol CN, n = 78), 1–3 years (95th centile: 8.3 mmol/mol CN, n = 77), and > 3 years (95th centile: 3.0 mmol/mol CN, n = 328). No significant correlation with age was observed for controls younger than 6 months of age (−3.217 [−9.770, 3.335], p = 0.34, n = 132), and there were no significant differences between median Glc4 values for the following subgroups: 0–1 month, 1–3 months, and 3–6 months (Table 1).
Baseline Urinary Glc4 Concentrations
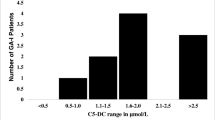
A comparison of the median values and ranges of urinary Glc4 concentrations and ages at baseline in the patient, pseudodeficiency, and control cohorts is shown in Table 1, and individual patient values are compared in Fig. 1 and Table Supplemental Digital Content 1. Urinary Glc4 concentrations were at or above the 90th centile (18 mmol/mol CN) of the age-matched reference range (Table Supplemental Digital Content 1) for the six full-term infants in the NBS-IOPD group. The remaining patient in this group, born at 29 weeks gestation and started on ERT at 40 days of age because of cardiomegaly, had pretreatment Glc4 concentrations of 32 and 16 mmol/mol CN at 0 and 1.2 months of age (not adjusted for prematurity), respectively. An appropriate age-matched control range has not been evaluated for pre-term infants. The NBS-IOPD group was significantly younger than the CLIN-IOPD group at the time of pretreatment assessment (p < 0.05) and had a lower median Glc4 concentration, although the difference was not statistically significant (p = 0.07) (Table 1).

Urinary Glc4 concentrations in infants with Pompe disease identified by newborn screening. Urinary Glc4 values at first evaluation (baseline) for individual patients with Pompe disease identified through newborn screening. Circles: Patients with infantile-onset Pompe disease (NBS-IOPD); Triangles: Patients with late-onset Pompe disease who were treated before the age of 3 years (NBS-LOPD early treated); Squares: Patients with late-onset Pompe disease who did not require treatment before 3 years of age (NBS-LOPD-Group 2). Dashed line represents the upper limit of the reference range for 0 to 6 months age (Glc4 < 20 mmol/mol CN)
Baseline Glc4 concentrations were within reference limits for all patients in the NBS-LOPD group, and median values were significantly lower than those of the NBS-IOPD cohort (p < 0.05). There was no significant difference in the median baseline Glc4 values for the NBS-LOPD early treated and the NBS-LOPD-Group 2 cohorts. Patients NBS-L3 and NBS-L9 in the early treated group had the highest values close to the upper limit of the reference range. Urinary Glc4 levels were within reference limits for infants with a GAA pseudodeficiency (Table 1).
Comparison of Urinary Glc4 and Serum Creatine Kinase at Baseline
Urinary Glc4 was significantly correlated with serum creatine kinase (CK) at the initial follow-up evaluation for patients with a confirmed diagnosis of Pompe disease (Pearson correlation coefficient = 0.624, p < 0.05; see Figure Supplemental Digital Content 2). As expected, the NBS-IOPD group had the highest CK values (excluding the premature infant, NBS9). Two of four patients in the NBS-LOPD early treated group (NBS-L3 and NBS-L9) had Glc4 and CK values that were comparable with those for three patients in the NBS-IOPD group. Infants in the pseudodeficiency groups had CK values within the reference intervals (data not shown).
Pretreatment Monitoring of Glc4 in Patients with a Late-Onset Phenotype
Longitudinal Glc4 measurements for 11 of the 13 NBS-LOPD patients on whom data were available are shown in Figure Supplemental Digital Content 3. Within the NBS-LOPD early treated cohort, Glc4 was elevated in two of the four patients prior to treatment. NBS-L3 had a persistent elevation of Glc4 prior to treatment at age 36 months and NBS-L9 had elevated Glc4 immediately prior to treatment at 1.5 months. In contrast, NBS-L1 and NBS-L6 did not have elevated Glc4 prior to initiation of ERT. These trends in Glc4 are consistent with the CK trends observed for these patients as previously reported (Chien et al. 2011), in that NBS-L3 and NBS-L9 had elevated CK prior to treatment and NBS-L1 and L6 did not. Glc4 elevations were not observed for the NBS-LOPD group 2 during the observation period of up to 4 years of age.
Discussion
The newborn screening program for Pompe disease in Taiwan has presented a unique opportunity to evaluate Glc4 in infants with infantile- and late-onset Pompe disease, prior to the appearance of clinical symptoms. Our results indicate that Glc4 concentrations correlated with phenotype early in the disease process; patients with IOPD had higher Glc4 concentrations than those with LOPD. Furthermore, median baseline Glc4 in the NBS-IOPD group was clearly lower than that of the slightly older (by approximately 4–8 weeks) clinical comparator CLIN-IOPD group. These observations are consistent with (1) baseline clinical manifestations in the NBS-IOPD group including cardiomyopathy and elevated CK, despite a normal physical exam and tone (Chien et al. 2009), (2) the notable increase in baseline Glc4 values with age in untreated patients with IOPD ascertained clinically before 12 months of age (Young et al. 2012), and (3) the rapidly progressive nature of the infantile form of the disease.
The variability of late-onset Pompe disease is demonstrated by differences in the age of onset of clinical signs and symptoms of the disease within the NBS-LOPD group, of which one third of patients required treatment with ERT before 3 years of age (Chien et al. 2011). Although these early treated patients with LOPD had baseline urinary Glc4 concentrations within the reference range, two had the highest values (86th and 90th centile of the reference range) observed within the entire NBS-LOPD cohort. This observation was concomitant with an elevation of the serum CK values at baseline in these two patients, interpreted as a sign of cell damage as previously reported (Chien et al. 2011).
In conclusion, our findings suggest that urinary Glc4 determination may be a useful component in the follow-up of a positive newborn screening result for Pompe disease, especially when the results of confirmatory enzyme and molecular testing are equivocal. An elevated Glc4 suggests an infantile-onset phenotype, whereas a value within the reference range is consistent with a late-onset phenotype or a pseudodeficiency of GAA. However, more studies are needed to determine whether inclusion of Glc4 in the follow-up algorithm, either as a first or later tier test, would provide additional benefit to other testing such as CK or echocardiogram. Glc4 is probably most useful for evaluating the disease status in newly diagnosed patients, especially when combined with clinical and other laboratory assessments and for periodic monitoring of asymptomatic patients. With the possible expansion of newborn screening for Pompe disease in the United States and elsewhere, more data from larger cohorts should be accessible and will allow further assessment of the sensitivity of Glc4 in the newborn period and its prognostic value for monitoring asymptomatic patients.
References
Chien YH, Chiang SC, Zhang XK et al (2008) Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics 122(1):e39–e45
Chien YH, Lee NC, Thurberg BL et al (2009) Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics 124(6):e1116–e1125
Chien YH, Lee NC, Huang HJ, Thurberg BL, Tsai FJ, Hwu WL (2011) Later-onset Pompe disease: early detection and early treatment initiation enabled by newborn screening. J Pediatr 158(6):1023–1027
Hagemans M, Winkel L, Van Doorn P et al (2005) Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain 128(Pt 3):671–677
Hirschhorn R, Reuser A (2001) Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly WS and Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 3389–3420
Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D (2006) A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 148(5):671–676
Kishnani PS, Corzo D, Leslie ND et al (2009) Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res 66(3):329–335
Kroos MA, Mullaart RA, Van Vliet L et al (2008) p.[G576S; E689K]: pathogenic combination or polymorphism in Pompe disease? Eur J Hum Genet 16(8):875–879
Kumamoto S, Katafuchi T, Nakamura K et al (2009) High frequency of acid alpha-glucosidase pseudodeficiency complicates newborn screening for glycogen storage disease type II in the Japanese population. Mol Genet Metab 97(3):190–195
Kumlien J, Chester MA, Lindberg BS, Pizzo P, Zopf D, Lundblad A (1988) Urinary excretion of a glucose-containing tetrasaccharide. A parameter for increased degradation of glycogen. Clin Chim Acta 176(1):39–48
Labrousse P, Chien YH, Pomponio RJ et al (2010) Genetic heterozygosity and pseudodeficiency in the Pompe disease newborn screening pilot program. Mol Genet Metab 99(4):379–383
Young SP, Stevens RD, An Y, Chen Y-T, Millington DS (2003) Analysis of a glucose tetrasaccharide elevated in Pompe disease by stable isotope dilution–electrospray ionization tandem mass spectrometry. Anal Biochem 316(2):175–180
Young SP, Zhang H, Corzo D et al (2009) Long-term monitoring of patients with infantile-onset Pompe disease on enzyme replacement therapy using a urinary glucose tetrasaccharide biomarker. Genet Med 11(7):536–541
Young SP, Piraud M, Goldstein JL et al (2012) Assessing disease severity in Pompe disease: the roles of a urinary glucose tetrasaccharide biomarker and imaging techniques. Am J Med Genet C Semin Med Genet 160C(1):50–58
Acknowledgments
The authors would like to thank the patients and their families for their participation in this study. The authors would also like to acknowledge Genzyme, A Sanofi Company for previous grant support for the development and evaluation of Glc4 as a biomarker in Pompe disease at DUMC and for support of the Taiwan pilot newborn screening program for Pompe disease.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Verena Peters
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
334361_1_En_366_MOESM2_ESM.docx
Figure Supplemental Digital Content 2. Comparison of urinary Glc4 and Ck values at initial evaluation (baseline) following an abnormal newborn screen (see Table, Supplemental Digital Content 1 for ages) for patients who were confirmed with infantile-onset Pompe disease (NBS-IOPD) shown as red squares or with a late-onset phenotype (NBS-LOPD). For the NBS-LOPD group, those who were treated before the age of 3 years are shown as blue triangles (NBS-LOPD-Early Treated) and those who were not treated before the age of 3 years are shown as open diamonds (NBS-LOPD-Group 2). Dashed lines represent the upper limit of the reference ranges. The arrow shows patient NBS9, born at 29 weeks gestation
334361_1_En_366_MOESM3_ESM.docx
Figure Supplemental Digital Content 3. (a) Urinary Glc4 concentrations with age for the four late onset patients identified through newborn screening who were treated at or before the age of 3 years (NBS-LOPD-Early Treated). The dotted lines represent the upper limit of the age-matched reference ranges. The solid black vertical lines represent the age at which treatment was initiated. The details on motor development and serum CK have been reported (Chien et al. 2011). Patients NBSL-1 and NBS-L6 had normal CK levels prior to treatment with ERT, whereas patients NBS-L3 and NBS-L9 had elevated CK. (b) Urinary Glc4 concentrations with age for 7 of 9 NBS-LOPD-Group 2 patients and patient L14 who were not treated at or before the age of 3 years. Monitoring data was not available for NBS-L5 and NBS-L8
Appendices
Synopsis
The glucose tetrasaccharide biomarker, Glc4, determined in dried urine spots, was elevated in neonates with infantile-onset Pompe disease identified through newborn screening, compared with infants with a late-onset phenotype or a pseudodeficiency of acid alpha-glucosidase.
Details of Funding
This study was funded in part by Genzyme, a Sanofi Company
Compliance with Ethics Guidelines
Conflict of Interest
Yin-Hsui Chien reports receiving research grant support, honoraria, and travel support from Genzyme, a Sanofi Company and BioMarin Pharmaceutical Inc.
Wuh-Liang Hwu reports receiving research grant support, honoraria, and travel support from Genzyme, a Sanofi Company and BioMarin Pharmaceutical Inc.
Ni-Chung Lee reports receiving travel support from Genzyme, a Sanofi Company.
Shu-Chuan Chiang reports receiving travel support from Genzyme, a Sanofi Company.
Sarah P. Young reports receiving research grant support from Genzyme, a Sanofi Company and Amicus Therapeutics.
David S. Millington reports receiving research grant support from Genzyme, a Sanofi Company and BioMarin Pharmaceutical Inc. David S. Millington was a member of the science advisory board of and received stock options from Advanced Liquid Logics, LLC.
Priya S. Kishnani reports receiving research grant support and honoraria from Genzyme, a Sanofi Company and Amicus Therapeutics, and consulting fees from Genzyme, a Sanofi Company. P.S. Kishnani is a member of the Pompe Disease and the Gaucher Disease Registry Advisory Boards for Genzyme, a Sanofi Company.
Haoyue Zhang reports receiving research grant support from Genzyme, a Sanofi Company and BioMarin Pharmaceutical Inc.
Jennifer L. Goldstein reports receiving research grant support from Genzyme, a Sanofi Company.
P. Brian Smith, Adviye A. Tolun, and Amie E. Vaisnins declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (Institutional Review Boards of National Taiwan University Hospital and Duke University Health System) and with the Helsinki Declaration of 1075, as revised in 2000 (5). Informed consent was obtained from parents of all patients.
Author Contributions
Yin-Hsiu Chien and Wuh-Liang Hwu planned the study, coordinated the collection of dried urine samples and the compliance with National Taiwan University Hospital Institutional Review Board, and participated in the data analysis and preparation and final approval of the manuscript.
Sarah P. Young supervised the analysis of Glc4, performed data analysis and interpretation, and participated in the preparation of the manuscript and final approval of the manuscript.
Jennifer Goldstein ensured compliance with Duke Health System Institutional Review Board and participated in the preparation and final approval of the manuscript.
P. Brian Smith performed statistical analysis of the data and participated in the preparation and final approval of the manuscript
Ni-Chung Lee contributed to patient care and data collection, revision, and final approval of the manuscript.
Shu-Chuan Chiang contributed to sample management, revision, and final approval of the manuscript.
Adviye A. Tolun, Haoyue Zhang, and Amie E. Vaisnins conducted Glc4 analysis and data processing and assisted with the data analysis and final approval of the manuscript.
David S. Millington and Priya S. Kishnani participated in data interpretation and the preparation and final approval of the manuscript.
Rights and permissions
Copyright information
© 2014 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Chien, YH. et al. (2014). Baseline Urinary Glucose Tetrasaccharide Concentrations in Patients with Infantile- and Late-Onset Pompe Disease Identified by Newborn Screening. In: Zschocke, J., Baumgartner, M., Morava, E., Patterson, M., Rahman, S., Peters, V. (eds) JIMD Reports, Volume 19. JIMD Reports, vol 19. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2014_366
Download citation
DOI: https://doi.org/10.1007/8904_2014_366
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-46189-1
Online ISBN: 978-3-662-46190-7
eBook Packages: MedicineMedicine (R0)