
Abstract
Hereditary tyrosinemia type 1 (HT1) (OMIM 276700) is a severe inherited metabolic disease affecting mainly hepatic and renal functions that leads to a fatal outcome if untreated. HT1 results from a deficiency of the last enzyme of tyrosine catabolism, fumarylacetoacetate hydrolase (FAH). Biochemical findings include elevated succinylacetone in blood and urine; elevated plasma concentrations of tyrosine, methionine and phenylalanine; and elevated tyrosine metabolites in urine. The HT1 frequency worldwide is about 1 in 100,000 individuals. In some areas, where the incidence of HT1 is noticeably higher, prevalence of characteristic mutations has been reported, and the estimated incidence of carriers of a specific mutation can be as high as 1 out of 14 adults. Because the global occurrence of HT1 is relatively low, a considerable number of cases may go unrecognized, underlining the importance to establish efficient prenatal and carrier testing to facilitate an early detection of the disease. Here we describe the 95 mutations reported so far in HT1 with special emphasis on their geographical and ethnic distributions. Such information should enable the establishment of a preferential screening process for mutations most predominant in a given region or ethnic group.
Competing interests: None declared
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Hereditary tyrosinemia type 1 (HT1) (OMIM 276700) is an inherited metabolic disease, mainly of childhood. This pathological condition was referred to as hereditary tyrosinemia type 1 in the mid-1960s (reviewed in Mitchell et al. 2001; Russo et al. 2001), and it was later shown to result from a deficiency in fumarylacetoacetate hydrolase (FAH), the last enzyme of the tyrosine catabolic pathway (Lindblad et al. 1977; Fällström et al. 1979; Berger et al. 1981; Kvittingen et al. 1981; Tanguay et al. 1990).
HT1 is an autosomal recessive disease characterized by severe liver dysfunction, impaired coagulation, neurological crises, renal tubular dysfunctions and a high risk of hepatocellular carcinoma (HCC). Three main clinical forms of HT1 have been described: the acute form, which presents itself in the first months of life and is associated with acute liver failure; the subacute form (second half of the first year) that manifests a similar but less severe clinical picture presenting usually with hepatomegaly or hypophosphatemic rickets (due to tubular dysfunction); and the chronic form which appears after the first year of age and shows a slower progression (Tanguay et al. 1990; van Spronsen et al. 1994; Bergman et al. 1998; Russo et al. 2001). Patients affected with HT1 generally show failure to thrive and hepatic damage including hepatomegaly, cirrhosis, hepatic failure and HCC. Complications associated with liver damage include jaundice, ascites and bleeding. HT1 also disrupts kidney function causing multiple tubular dysfunctions, Fanconi-like syndrome and glomerulosclerosis. In 1992, the introduction of NTBC (2-(2-nitro-trifluoromethylbenzoyl) 1,3-cyclohexanedione, also known as nitisinone) (Lindstedt et al. 1992) has proven to be highly effective in preventing the progression of liver damage, neurological crises and kidney damages (Larochelle et al. 2012; Bartlett et al. 2014). NTBC in combination with a low-tyrosine diet represents the only treatment available for this disease. However, one of the most severe complications occurring in HT1 patients remains the development of HCC (Mitchell et al. 2001). Indeed, although regular administration of NTBC in HT1 patients, combined with a protein-restricted diet, prevents liver and kidney dysfunction, recent reports have documented the presence of HCC even under therapy (de Laet et al. 2013). Effectiveness of this treatment depends on how early the disease is recognized and treated; thus, recent retrospective studies highly recommend the implementation of newborn screening in more areas (Zytkovicz et al. 2013; Dehghani et al. 2013; De Laet et al. 2013; Mayorandan et al. 2014). For example, Mayorandan and collaborators in their retrospective study point out the necessity of neonatal programmes borne by the government or health insurance companies to allow early diagnosis and access to adequate treatment. Indeed they report that patients, who were diagnosed after the neonatal period and consequently received NTBC treatment later, had a 2–12-fold higher risk (depending on age at start of therapy) of developing hepatocellular carcinoma compared to patients treated as neonates.
Detection of succinylacetone (SA) in urine, blood and amniotic fluid is the most reliable biochemical diagnostic for HT1. Assay of FAH enzyme activity in skin fibroblasts is possible but not readily available. Advent of molecular genetic testing has greatly improved the diagnostic power for this disease. Mutation analysis is not essential for clinical management but is useful for prenatal diagnosis and reproductive counselling. In fact targeted mutation analysis for diseased alleles and sequence analysis of the entire fah coding region can detect mutations in more than 95% of affected individuals (Sniderman King et al. 2011). The database of the GTR (Genetic Testing Registry: https://www.ncbi.nlm.nih.gov/gtr/conditions/C0268490) reports 56 clinical tests for diagnosis and monitoring of this condition. Carrier testing for at-risk relatives and prenatal diagnosis for pregnancies at increased risk are possible if both disease-causing alleles in a family are known.
Patients and Methods
The present review is based on a current compilation of all HT1 alleles reported worldwide including those from patients identified in the Laboratory of Cellular and Developmental Genetics (LGCD), Université Laval, Quebec, Canada (Dr RM Tanguay), and the Department of Clinical Chemistry at Birmingham Children’s Hospital (BCH), Birmingham, UK (Dr G Gray), mostly between 2001 and 2013 (unpublished data). Screening of genetic databases (e.g. HGMD, NCBI, ENSEMBL) and HT1 literature has been made to classify the reported mutations and to identify the ethnic group of patients. The mutations reported so far and the patients’ origins are listed in Table 1.
Since there are inconsistencies in the literature of names of the mutations in this gene, we have used the Human Genome Variation Society’s nomenclature for the description of sequence variations (http://www.hgvs.org/mutnomen/recs.html) as the basis of nomenclature (den Dunnen and Antonarakis 2000) and used the fah cDNA sequence given as GenBank accession number BT007160.1 as our reference sequence. For splice defects we have also added the historical mutation nomenclature, since this is the most common way in which they are named worldwide.
Results and Discussion
Fah Gene Characteristics and Mutations
The first mutation reported in the fah gene was the c.47A>T (p.Asn16Ile) in a French Canadian patient and was shown to be causative of FAH deficiency (Phaneuf et al. 1992).
The human fah gene is located on chromosome 15q23-q25, spans 30–35 kb and consists of 14 exons. The cDNA has an open reading frame of 1,257 bp encoding 419 amino acids (Phaneuf et al. 1991; Labelle et al. 1993). Identification of this gene (Phaneuf et al. 1991) led to mutation screening of patients and characterization of a number of disease-causing alleles, some of which were present at relatively high frequencies in specific populations (St-Louis and Tanguay 1997).
Eighty-three disease-causing mutations are presently reported on Human Gene Mutation Database (HGMD® Professional 2014.2, accessed in August 2014). Recently, two new mutations were uncovered at LGCD, Quebec, and in BCH, Birmingham (unpublished data). The first was the c.726G>A (p.Trp242X) nonsense mutation, obtained by screening one English adult patient at BCH. The second, the c.775G>C (p.Val259Leu) a potential missense mutation, was observed in an American patient at the Quebec laboratory. This patient was heterozygous for the new c.775G>C (p.Val259Leu) allele and the already reported c.554-1G>T (IVS6-1G>T) (Grompe et al. 1994). Western blot analysis of his liver obtained after transplantation revealed the absence of FAH protein and no activity was detected by enzymatic assay (data no shown). RNA analysis suggested a defect in splicing affecting exon 9, and this was confirmed using minigene constructs transfected in HeLa cells (Dreumont and Tanguay, unpublished).
Reclassification of HT1 Mutations
After cross-checking of genetic databases and the literature on HT1 from the oldest publications to the present day, we updated the number of allelic variants with the two found by our group and others recently reported (Fig. 1 and Table 1). Next we decided to reclassify them in a unique list containing number of known alleles from patients and geographical distribution of the mutations most predominant for each country (Fig. 2, and Table 1). Indeed, frequency of reported alleles and origin of patients could be useful in helping clinicians to focus on mutations specific of certain regions, facilitating the targeted detection of diseased alleles.

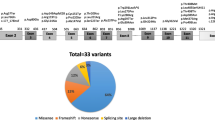
Location of the 95 mutations identified on the fah gene. Among the known HT1 alleles causing mutations, 45 are missense mutations, 23 are splicing mutations, 13 are nonsense mutations, 10 are deletions and 4 are frameshift. Intronic mutations are illustrated at the bottom of the figure

Geographical distribution of the most common HT1 alleles causing mutations worldwide. Pie chart representing distribution of ethnic groups in HT1 alleles. Where the patient provenance was not clear, the mutation is included in the continent of origin, i.e. undefined (un) on graphic. The top three mutations and the total number of alleles for each continent are reported. There are more than 894 HT1 alleles reported worldwide. The most frequent HT1 mutation encountered is the IVS12+5G>A splice mutation, which accounts for 33.7% of all HT1 alleles, followed by the IVS6-1G>T mutation (16.4%). The French Canadian population alone accounts for as much as a third of all HT1 alleles reported. Both mutations are the most reported globally
Overall 95 mutations are now reported within the fah gene in this review (Fig. 1 and Table 1). All 95 HT1 alleles are divided in 45 missense mutations, 23 splice defects, 13 nonsense mutations, 10 deletions and 4 frameshift (Table 1). In addition the missense c.1021C>T (p.Arg341Trp) sequence variant is described as a pseudodeficiency variant since individuals homozygous for this mutation are healthy (Rootwelt et al. 1994b; Bergeron et al. 2001).
Predominance of Ethnic Groups in HT1 Distribution
Despite the fact that the worldwide incidence of HT1 is relatively low with one affected individual in approximately 100,000 healthy individuals (Hutchesson et al. 1996), specific populations stand out as they represent small clusters of diseased alleles (Fig. 2). The population that possesses the highest incidence of HT1 is the French Canadian population of the SLSJ region, in the province of Quebec (Canada) (De Braekeleer and Larochelle 1990; Poudrier et al. 1996). The prevalence of HT1 in the SLSJ region was as high as 1/1,042 births in 1971 but dropped to 1/1,846 births in 1986, most likely due to the implementation of a screening programme for HT1 in 1970 conducted by the Quebec Network of Genetic Medicine. The most predominant mutation in this region is the c.1062 + 5G>A (IVS12 + 5G>A) accounting ~90% of all the disease-causing alleles. Furthermore, even though the Quebec population accounts for only approximately 0.12% of the world population (estimated today to number of 7 billion), it represents ~33% of all HT1 alleles worldwide. Although these data may be biased by the fact that all newborns in Quebec are screened for HT1, it is clear that this region represents the highest incidence of HT1 and that the c.1062 + 5G>A mutation is predominant in this region.
A second cluster of HT1 is found in Scandinavia (Kvittingen et al. 1981). In the Finnish population of Pohjanmaa, 1 individual out of 5,000 is affected with HT1 (St-Louis et al. 1994), whereas the overall incidence of HT1 in Finland is 1:60,000 (Mustonen et al. 1997). In this region, one single mutation (c.786G > A, p.Trp262X) represents ~88% of all reported HT1 alleles (St-Louis et al. 1994). Indeed 40 of the 46 European c.786G>A alleles have been reported in this country.
New findings show a peculiar pattern of HT1 mutations also in Norway. In a recent report, 19 Norwegian HT1 patients were investigated in the Hospital of Oslo University and three new small deletions were found: c.615delT, (p.Phe205LeufsX2), c.744delG (p.Pro249HisfsX55) and c.835delC (p.Gln279ArgfsX25). The novel mutations lead to frameshift and premature termination codons. FAH protein structure is affected, and normal folding, function and stability of the protein cannot be expected (Bliksrud et al. 2012). The c.615delT, c.744delG and the c.835delC are found in 13.5%, 3.8% and 1.9% of the alleles, respectively. Around 65% of the Norwegian HT1 patients are heterozygous for different mutations. The relatively high incidence of HT1 in Norway (1 in 74,800 live births) has not been connected with a single founder effects or high incidence of parental consanguinity as in the previous areas (Bliksrud et al. 2012).
Another cluster occurs in an immigrant population from Pakistan living in the UK, predominantly in Birmingham (Hutchesson et al. 1998). Birmingham is a city in the West Midlands Region, which has a total population of approximately 5.3 million of which nearly 3% are of Pakistani origin. We have diagnosed 44 patients from the West Midlands with this disorder of which 30 (68%) were of Pakistani origin. This is over 22-fold higher than the frequency of people of Pakistani origin in this region. Mutation analysis revealed that five out of 12 index patients (42%) in this ethno-geographic group had the c.192G>T (p.Gln64His) mutation. This mutation was not detected in patients from any other close-by region suggesting a founder effect from the region of origin of this population. Indeed the frequency of this mutation in Pakistani from the UK was comparable to that of the common pan-ethnic c.1062 + 5G>A mutation.
Most Frequent HT1 Alleles Around the World
Although Quebec, Finland, Norway and Pakistani in the UK stand out as populations with the higher frequency of HT1, reports highlight a specific tendency in mutational distribution among ethnic groups. The c.1062 + 5G>A (IVS12 + 5G>A) mutation is found frequently in patients from a wide range of ethnic groups over a large geographical distribution. Given the high frequency and wide spread of this mutation, it is likely to be a very old mutation and it was originally reported in a French Canadian patient and in two patients of Iranian origin (Grompe et al. 1994). Although this is the most frequent HT1 mutation encountered worldwide (302/894 HT1 alleles), the c.554-1G>T (IVS6-1G>T) splice mutation is also frequently observed (147/894 HT1 alleles), showing a high prevalence in the Mediterranean region and in southern Europe. In a recent cross-sectional retrospective study on 168 HT1 patients originating from Europe, Turkey and Israel, mutational analysis performed in 58/168 patients revealed the predominance of the IVS12 + 5G>A (11 patients) and IVS6-1G>T (13 patients) mutations in these ethnic groups (Mayorandan et al. 2014).
The mutation that ranks third in prevalence in Europe is the c.786G>A (p.Trp262X) nonsense mutation. This ranking is due to its predominance in the Finnish population. A number of others mutations have also been associated with specific ethnic or geographic groups, as described below (Table 1, Fig. 2). The c.1062 + 5G>A (IVS12 + 5G>A), the c.607-6T>G (IVS7-6T/G) and the c.554-1G>T (IVS6-1G>T) splicing mutations and the c.786G>A (p.Trp262X) nonsense mutation all together represent 60% of mutant alleles in the general US population (Sniderman King et al. 2011). Surprisingly, only one HT1 allele was reported until now in Mexico and this allele carried the c.1062 + 5G>A mutation most prevalent in Quebec (Table 1). 16 new cases have recently been described in Brazil (Neto et al. 2014), with only two alleles reported at this time, and these harboured the c.554-1G>T (IVS6-1G>T) mutation, most prevalent in the Mediterranean area (Table 1).
Arranz et al. in their work based on a panel of 29 patients mostly from southern Europe demonstrated a high homogeneity of the mutational spectrum in this region (Arranz et al. 2002). In a retrospective study on European HT1-affected individuals (Couce et al. 2011), mutational analysis on 34 Spanish patients reported nine different mutations in this population, documenting c.554-1G>T (IVS6-1G>T) as the most prevalent, in accordance with the previous literature (Arranz et al. 2002). Molecular genetics analysis of the fah gene in 11 Czech patients with HT1, diagnosed in the Medical Faculty of Charles University in Prague between 1982 and 2006, revealed three mutations not previously described: the c.579C>A nonsense mutation (p.Cys193X) and the c.680G>T (p.Gly227Val) and c.1210G>A (p.Gly404Ser) missense mutations (Vondrackova et al. 2010).
The Middle East is interesting in the sense that even though patients harbour the common c.1062 + 5G>A (IVS12 + 5G>A) and c.554-1G > T (IVS6-1G>T) mutations, many of the other mutations reported are typical to this region. One such example is the already described c.192G>T (p.Gln64His) mutation, which is thus far found only in people originating from Pakistan, the Middle East and North West India. This mutation accounts for over one third of all HT1 alleles in these populations (Rootwelt et al. 1994a; Rootwelt et al. 1996). Another mutation that is often detected in patients from the Middle East is the c.709C>T (p.Arg237X) mutation (Imtiaz et al. 2011). In Turkey, the c.698A>T (p.Asp233Val) mutation, which has not been reported elsewhere, accounts for 20% of the reported alleles (Rootwelt et al. 1994a; Rootwelt et al. 1996; Dursun et al. 2011). Moreover, other different mutations, although not at high frequency, are peculiar for this population (Table 1).
The c.782C>T (p.Pro261Leu) missense mutation was found in 100% of Ashkenazi-Jewish examined in Israel (Elpeleg et al. 2002). Direct sequencing in 43 HT1-affected patients originating from Saudi Arabia, Egypt and Iran identified a total of 17 different homozygous mutations. Eleven of these (8 missense, 1 nonsense, 1 splice site and 1 deletion) had not been reported previously (Imtiaz et al. 2011).
Little information about the epidemiology and molecular defects in HT1 patients from East Asia is available at this time. Sakai and Kitagawa (1957) reported the first case of HT1 in a two-month-old Japanese patient, but genetic analysis was not possible at that time. The c.185T>G (p.Phe62Cys) represents the first and the only allele reported in Japan to date (Awata et al. 1994). Recent findings start to describe HT1 mutations in China. The missense mutation c.1124T>C (p.Leu375Pro) represents the first case of HT1 analysed by molecular genetics in this area (Cao et al. 2012). This mutation, affecting the secondary protein structure, decreases the stability of FAH enzyme and compromises the protein’s functions. Another report represents the first case of HT1 in a two-month-old Hong Kong Chinese patient (Mak et al. 2013). Genetic analysis of this patient showed two novel mutations, the c.1063-1G>A splicing mutation and the c.1035_1037del. Recently, clinical data on 3 HT1 Chinese patients showed five mutations in the FAH gene: c.455G>A (p.Trp152X), c.520C>T (p.Arg174X), c.974_976delCGAinsGC, c.1027G>A (p.Gly343Arg) and c.1100G>A (p.Trp367X) (Yang et al. 2012; Dou et al. 2013). The c.455G>A, c.974_976delCGAinsGC and c.1100G>A mutations have not been described elsewhere. Currently, few cases of HT1 have been reported in Korea. Mutational analysis of two female neonates admitted to hospital for further work-up of an abnormal newborn screening test revealed three novel mutations (one deletion, one missense and one splice defect) that have not been reported elsewhere (Park et al. 2009; Choi et al. 2014).
To our knowledge no mutations in HT1 have yet been documented in Central America or in the Oceania continent.
Conclusions
The advent of neonatal screening, prenatal diagnosis and carrier tests for genetic disorders has shown the importance of establishing the population frequencies and ethno-geographic spread of mutations for the evaluation of future screening strategies. To highlight the prevalence of HT1 mutations in a geographical context, we compiled all reported HT1 alleles worldwide, including those not yet reported in the common databases, and another two, discovered in the screening of HT1 patients in our laboratories over a period between 2001 and 2013 (summarized in Table 1 and Fig. 2). Obvious conclusions can be drawn when we examine the incidence of HT1 worldwide (Fig. 2 and Table 1).
According to the data gathered so far, a preferential screening for those mutations in regions in which they show a higher prevalence could provide some improvement in carrier diagnostic efficiency and may enable the establishment of family pedigrees for adequate counselling in some cases. Currently, screening is carried out in Quebec, the USA and Europe (Morrissey et al. 2011; Barnby 2014). In this case it is obviously important to know the pattern of mutations in the respective populations.
However, it is necessary to bear in mind that this compilation may be partly biased by the fact that: (1) very few cases of HT1 are overlooked in some countries as in the province of Quebec due to a screening programme for HT1 established early in 1970 and (2) not all cases of HT1 are described in the literature. Many of these probably occur in countries with no or limited access to service for diagnosis of genetic disease and as a result remain undiagnosed (De Laet et al. 2013). This could lead to some geographical bias reflected in the fact that the majority of the patients whose mutations have been described are residents of Europe, the Middle East or North America. (3) Whilst we have carefully attempted to ensure that patients are not counted twice because they appear in more than one publication, this may occur in a few cases.
In summary, this report allows a detailed identification of the mutations causing HT1 worldwide, with diagnostic and methodological consequences implementing the groundwork for future carrier and prenatal testing, premarital screening and pre-implantation genetic diagnosis.
Abbreviations
- BCH:
-
Birmingham Children’s Hospital
- FAH:
-
Fumarylacetoacetate hydrolase
- GTR:
-
Genetic Testing Registry
- HCC:
-
Hepatocellular carcinoma
- HT1:
-
Hereditary tyrosinemia type 1
- LGCD:
-
Laboratory of Cell and Developmental Genetics
- NTBC:
-
2-(2-Nitro-trifluoromethylbenzoyl) 1,3-cyclohexanedione
- SLSJ:
-
Saguenay-Lac-St-Jean region
References
Al-Shamsi A, Hertecant JL, Al-Hamad S, Souid AK, Al-Jasmi F (2014) Mutation spectrum and birth prevalence of inborn errors of metabolism among Emiratis: a study from Tawam Hospital Metabolic Center, United Arab Emirates. Sultan Qaboos Univ Med J 14(1):e42–e49
Arranz JA, Pinol F, Kozak L et al (2002) Splicing mutations, mainly IVS6-1(G>T), account for 70% of fumarylacetoacetate hydrolase (FAH) gene alterations, including 7 novel mutations, in a survey of 29 tyrosinemia type I patients. Hum Mutat 20(3):180–188
Awata H, Endo F, Tanoue A, Kitano A, Nakano Y, Matsuda I (1994) Structural organization and analysis of the human fumarylacetoacetate hydrolase gene in tyrosinemia type I. Biochim Biophys Acta 1226(2):168–172
Barnby E (2014) Tyrosinemia type 1: an overview of nursing care. Pediatr Nursing 40(2):61–68
Bartlett DC, Lloyd C, McKiernan PJ, Newsome PN (2014) Early nitisinone treatment reduces the need for liver transplantation in children with tyrosinaemia type 1 and improves post-transplant renal function. J Inherit Metab Dis 37(5):745–752.
Berger R, Smit GP, Stoker-de Vries SA, Duran M, Ketting D, Wadman SK (1981) Deficiency of fumarylacetoacetase in a patient with hereditary tyrosinemia. Clin Chim Acta 114(1):37–44
Bergeron A, D’Astous M, Timm DE, Tanguay RM (2001) Structural and functional analysis of missense mutations in fumarylacetoacetate hydrolase, the gene deficient in hereditary tyrosinemia type 1. J Biol Chem 276(18):15225–15231
Bergman AJ, van den Berg IE, Brink W, Poll-The BT, Ploos van Amstel JK, Berger R (1998) Spectrum of mutations in the fumarylacetoacetate hydrolase gene of tyrosinemia type 1 patients in northwestern Europe and Mediterranean countries. Hum Mutat 12(1):19–26
Bliksrud YT, Brodtkorb E, Andresen PA, van den Berg IE, Kvittingen EA (2005) Tyrosinaemia type I–de novo mutation in liver tissue suppressing an inborn splicing defect. J Mol Med (Berl) 83(5):406–410
Bliksrud YT, Brodtkorb E, Backe PH, Woldseth B, Rootwelt H (2012) Hereditary tyrosinaemia type I in Norway: incidence and three novel small deletions in the fumarylacetoacetase gene. Scand J Clin Lab Invest 72(5):369–373
Cao YY, Zhang YL, Du J et al (2012) Compound mutations (R237X and L375P) in the fumarylacetoacetate hydrolase gene causing tyrosinemia type I in a Chinese patient. Chin Med J (Engl) 125(12):2132–2136
Cassiman D, Zeevaert R, Holme E, Kvittingen EA, Jaeken J (2009) A novel mutation causing mild, atypical fumarylacetoacetase deficiency (Tyrosinemia type I): a case report. Orphanet J Rare Dis 4:28
Choi HJ, Bang HI, Ki CS et al (2014) Two novel FAH gene mutations in a patient with hereditary tyrosinemia type I. Ann Clin Lab Sci 44(3):317–323
Couce ML, Dalmau J, del Toro M, Pintos-Morell G, Aldamiz-Echevarria L (2011) Tyrosinemia type 1 in Spain: mutational analysis, treatment and long-term outcome. Pediatr Int 53(6):985–989
De Braekeleer M, Larochelle J (1990) Genetic epidemiology of hereditary tyrosinemia in Quebec and in Saguenay-Lac-St-Jean. Am J Hum Genet 47(2):302–307
de Laet C, Dionisi-Vici C, Leonard JV et al (2013) Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis 8:8
Dehghani SM, Haghighat M, Imanieh MH, Karamnejad H, Malekpour A (2013) Clinical and para clinical findings in the children with tyrosinemia referring for liver transplantation. Int J Prev Med 4(12):1380–1385
den Dunnen JT, Antonarakis SE (2000) Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 15(1):7–12
Dou LM, Fang LJ, Wang XH et al (2013) Mutation analysis of FAH gene in patients with tyrosinemia type 1. Zhonghua Er Ke Za Zhi 51(4):302–307
Dreumont N, Poudrier JA, Bergeron A, Levy HL, Baklouti F, Tanguay RM (2001) A missense mutation (Q279R) in the fumarylacetoacetate hydrolase gene, responsible for hereditary tyrosinemia, acts as a splicing mutation. BMC Genet 2:9
Dursun A, Ozgul RK, Sivri S et al (2011) Mutation spectrum of fumarylacetoacetase gene and clinical aspects of tyrosinemia type I disease. JIMD Rep 1:17–21
Elpeleg ON, Shaag A, Holme E et al (2002) Mutation analysis of the FAH gene in Israeli patients with tyrosinemia type I. Hum Mutat 19(1):80–81
Fällström S-P, Lindblad B, Lindstedt S, Steen G (1979) Hereditary tyrosinemia-fumarylacetoacetase deficiency. Pediatr Res 13:78
Georgouli H, Schulpis KH, Michelakaki H, Kaltsa M, Sdogou T, Kossiva L (2010) Persistent coagulopathy during Escherichia coli sepsis in a previously healthy infant revealed undiagnosed tyrosinaemia type 1. BMJ Case Rep 2010. pii: bcr0720103150. doi:10.1136/bcr.07.2010.3150
Grompe M, al-Dhalimy M (1993) Mutations of the fumarylacetoacetate hydrolase gene in four patients with tyrosinemia, type I. Hum Mutat 2(2):85–93
Grompe M, St-Louis M, Demers SI, al-Dhalimy M, Leclerc B, Tanguay RM (1994) A single mutation of the fumarylacetoacetate hydrolase gene in French Canadians with hereditary tyrosinemia type I. N Engl J Med 331(6):353–357
Haghighi-Kakhki H, Rezazadeh J, Ahmadi-Shadmehri A (2014) Identification of a combined missense/splice-site mutation in FAH causing tyrosinemia type 1. J Pediatr Endocrinol Metab. doi:10.1515/jpem-2013-0489
Hahn SH, Krasnewich D, Brantly M, Kvittingen EA, Gahl WA (1995) Heterozygosity for an exon 12 splicing mutation and a W234G missense mutation in an American child with chronic tyrosinemia type 1. Hum Mutat 6(1):66–73
Heath SK, Gray RG, McKiernan P, Au KM, Walker E, Green A (2002) Mutation screening for tyrosinaemia type I. J Inherit Metab Dis 25(6):523–524
Hutchesson AC, Hall SK, Preece MA, Green A (1996) Screening for tyrosinaemia type I. Arch Dis Child Fetal Neonatal Ed 74(3):F191–F194
Hutchesson AC, Bundey S, Preece MA, Hall SK, Green A (1998) A comparison of disease and gene frequencies of inborn errors of metabolism among different ethnic groups in the West Midlands, UK. J Med Genet 35(5):366–370
Imtiaz F, Rashed MS, Al-Mubarak B et al (2011) Identification of mutations causing hereditary tyrosinemia type I in patients of Middle Eastern origin. Mol Genet Metab 104(4):688–690
Jitraruch S, Treepongkaruna S, Teeraratkul S et al (2011) Long-term outcome of living donor liver transplantation in a Thai boy with hereditary tyrosinemia type I: a case report. J Med Assoc Thai 94(10):1276–1280
Kim SZ, Kupke KG, Ierardi-Curto L et al (2000) Hepatocellular carcinoma despite long-term survival in chronic tyrosinaemia I. J Inherit Metab Dis 23(8):791–804
Kvittingen EA, Jellum E, Stokke O (1981) Assay of fumarylacetoacetate fumarylhydrolase in human liver-deficient activity in a case of hereditary tyrosinemia. Clin Chim Acta 115(3):311–319.
la Marca G, Malvagia S, Pasquini E et al (2011) Newborn screening for tyrosinemia type I: further evidence that succinylacetone determination on blood spot is essential. JIMD Rep 1:107–109
Labelle Y, Phaneuf D, Leclerc B, Tanguay RM (1993) Characterization of the human fumarylacetoacetate hydrolase gene and identification of a missense mutation abolishing enzymatic activity. Hum Mol Genet 2(7):941–946
Larochelle J, Alvarez F, Bussieres JF et al (2012) Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Quebec. Mol Genet Metab 107(1–2):49–54
Laszlo A, Rozsa M, Sallay E et al (2013) The fate of tyrosinaemic Hungarian patients before the NTBC aera. Ideggyogy Sz 66(11–12):415–419
Lindblad B, Lindstedt S, Steen G (1977) On the enzymic defects in hereditary tyrosinemia. Proc Natl Acad Sci U S A 74(10):4641–4645
Lindstedt S, Holme E, Lock EA, Hjalmarrson T, Strandvik B (1992) Treatment of hereditary tyrosinemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 340:813–817
Mak CM, Lam CW, Chim S, Siu TS, Ng KF, Tam S (2013) Biochemical and molecular diagnosis of tyrosinemia type I with two novel FAH mutations in a Hong Kong Chinese patient: recommendation for expanded newborn screening in Hong Kong. Clin Biochem 46(1–2):155–159
Mayorandan S, Meyer U, Gokcay G et al (2014) Cross-sectional study of 168 patients with hepatorenal tyrosinaemia and implications for clinical practice. Orphanet J Rare Dis 9(1):107
Mitchell GA, Grompe M, Lambert H, Tanguay RM (2001) Hypertyrosinemia.In: The metabolic and molecular bases of inherited diseases. McGrawHill, New York, pp 1777–1805
Mohamed S, Kambal MA, Al Jurayyan NA et al (2013) Tyrosinemia type 1: a rare and forgotten cause of reversible hypertrophic cardiomyopathy in infancy. BMC Res Notes 6(1):362
Morrissey MA, Sunny S, Fahim A, Lubowski C, Caggana M (2011) Newborn screening for Tyr-I: two years’ experience of the New York State program. Mol Genet Metab 103(2):191–192
Mustonen A, Ploos van Amstel HK, Berger R, Salo MK, Viinikka L, Simola KO (1997) Mutation analysis for prenatal diagnosis of hereditary tyrosinaemia type 1. Prenat Diagn 17(10):964–966
Park HD, Lee DH, Choi TY et al (2009) Clinical, biochemical, and genetic analysis of a Korean neonate with hereditary tyrosinemia type 1. Clin Chem Lab Med 47(8):930–933
Perez-Carro R, Sanchez-Alcudia R, Perez B et al (2013) Functional analysis and in vitro correction of splicing FAH mutations causing tyrosinemia type I. Clin Genet. doi:10.1111/cge.12243
Phaneuf D, Labelle Y, Berube D et al (1991) Cloning and expression of the cDNA encoding human fumarylacetoacetate hydrolase, the enzyme deficient in hereditary tyrosinemia: assignment of the gene to chromosome 15. Am J Hum Genet 48(3):525–535
Phaneuf D, Lambert M, Laframboise R, Mitchell G, Lettre F, Tanguay RM (1992) Type 1 hereditary tyrosinemia. Evidence for molecular heterogeneity and identification of a causal mutation in a French Canadian patient. J Clin Invest 90(4):1185–1192
Ploos van Amstel JK, Bergman AJ, van Beurden EA et al (1996) Hereditary tyrosinemia type 1: novel missense, nonsense and splice consensus mutations in the human fumarylacetoacetate hydrolase gene; variability of the genotype–phenotype relationship. Hum Genet 97(1):51–59
Poudrier J, St-Louis M, Lettre F, et al (1996) Frequency of the IVS12 + 5G>A splice mutation of the fumarylacetoacetate hydrolase gene in carriers of hereditary tyrosinaemia in the French Canadian population of Saguenay-Lac-St-Jean. Prenat Diagn 16(1): 59–64
Poudrier J, Lettre F, St-Louis M, Tanguay RM (1999) Genotyping of a case of tyrosinaemia type I with normal level of succinylacetone in amniotic fluid. Prenat Diagn 19(1):61–63
Prieto-Alamo MJ, Laval F (1998) Deficient DNA-ligase activity in the metabolic disease tyrosinemia type I. Proc Natl Acad Sci U S A 95(21):12614–12618
Rootwelt H, Berger R, Gray G, Kelly DA, Coskun T, Kvittingen EA (1994a) Novel splice, missense, and nonsense mutations in the fumarylacetoacetase gene causing tyrosinemia type 1. Am J Hum Genet 55(4):653–658
Rootwelt H, Brodtkorb E, Kvittingen EA (1994b) Identification of a frequent pseudodeficiency mutation in the fumarylacetoacetase gene, with implications for diagnosis of tyrosinemia type I. Am J Hum Genet 55(6):1122–1127
Rootwelt H, Chou J, Gahl WA et al (1994c) Two missense mutations causing tyrosinemia type 1 with presence and absence of immunoreactive fumarylacetoacetase. Hum Genet 93(6):615–619
Rootwelt H, Kristensen T, Berger R, Hoie K, Kvittingen EA (1994d) Tyrosinemia type 1–complex splicing defects and a missense mutation in the fumarylacetoacetase gene. Hum Genet 94(3):235–239
Rootwelt H, Hoie K, Berger R, Kvittingen EA (1996) Fumarylacetoacetase mutations in tyrosinaemia type I. Hum Mutat 7(3):239–243
Russo PA, Mitchell GA, Tanguay RM (2001) Tyrosinemia: a review. Pediatr Dev Pathol 4(3):212–221
Sakai K, Kitagawa T (1957) An atypical case of tyrosinosis (1-Parahydroxyphenyl-lactic aciduria) Part 1. Clinical and laboratory findings. Jikei Med J 2:1–5
Seda Neto J, Leite KM, Porta A et al (2014) HCC prevalence and histopathological findings in liver explants of patients with hereditary tyrosinemia type 1. Pediatr Blood Cancer. doi:10.1002/pbc.25094
Sheth JJ, Ankleshwaria CM, Pawar R, Sheth FJ (2012) Identification of novel mutations in FAH Gene and Prenatal Diagnosis of Tyrosinemia in Indian Family. Case Rep Genet 2012:428075
Sniderman King L, Trahms C, Scott CR (2011) Tyrosinemia type 1. Retrieved from GeneReviews® June 2014 http://www.ncbi.nlm.nih.gov/books/NBK1515/)
St-Louis M, Leclerc B, Laine J, Salo MK, Holmberg C, Tanguay RM (1994) Identification of a stop mutation in five Finnish patients suffering from hereditary tyrosinemia type I. Hum Mol Genet 3(1):69–72
St-Louis M, Poudrier J, Phaneuf D, Leclerc B, Laframboise R, Tanguay RM (1995) Two novel mutations involved in hereditary tyrosinemia type I. Hum Mol Genet 4(2):319–320
St-Louis M, Tanguay RM (1997) Mutations in the fumarylacetoacetate hydrolase gene causing hereditary tyrosinemia type I:overview. Hum Mutat 9(4):291–299.
Tanguay RM, Valet JP, Lescault A et al (1990) Different molecular basis for fumarylacetoacetate hydrolase deficiency in the two clinical forms of hereditary tyrosinemia (type I). Am J Hum Genet 47(2):308–316
Timmers C, Grompe M (1996) Six novel mutations in the fumarylacetoacetate hydrolase gene of patients with hereditary tyrosinemia type I. Hum Mutat 7(4):367–369
van Spronsen FJ, Thomasse Y, Smit GP et al (1994) Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology 20(5):1187–1191
Vondrackova A, Tesarova M, Magner M et al (2010) Clinical, biochemical and molecular characteristics in 11 Czech children with tyrosinemia type I. Cas Lek Cesk 149(9):411–416
Yang N, Han LS, Ye J et al (2012) Analysis of clinical data and genetic mutations in three Chinese patients with tyrosinemia type I. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 29(6):648–652
Zytkovicz TH, Sahai I, Rush A et al (2013) Newborn screening for hepatorenal tyrosinemia-I by tandem mass spectrometry using pooled samples: a four-year summary by the New England newborn screening program. Clin Biochem 46(7–8):681–684
Acknowledgements
Work on HT1 was supported by the Canadian Institutes for Health Research (grant to RMT, studentship to AB and postdoctoral fellowship to FA).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Bridget Wilcken
Appendices
Synopsis
Geographical and ethnic distribution of mutations in hereditary tyrosinemia type 1
Compliance with Ethics Guidelines
Conflict of Interest
Francesca Angileri, Anne Bergeron, Geneviève Morrow, Francine Lettre, George Gray, Tim Hutchin, Sarah Ball and Robert M. Tanguay declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration as revised in 2013.
Animal Rights
This article does not contain any studies with animal subjects performed by any of the authors.
Author’s Contributions
AB and FA contributed equally to this review. AB, FA and SB did the literature review and contributed to the draft of the manuscript. FL, TH and SB performed mutational analysis in some patients. RMT, GM and GG designed the review and worked on the draft of the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Copyright information
© 2014 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Angileri, F. et al. (2014). Geographical and Ethnic Distribution of Mutations of the Fumarylacetoacetate Hydrolase Gene in Hereditary Tyrosinemia Type 1. In: Zschocke, J., Baumgartner, M., Morava, E., Patterson, M., Rahman, S., Peters, V. (eds) JIMD Reports, Volume 19. JIMD Reports, vol 19. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2014_363
Download citation
DOI: https://doi.org/10.1007/8904_2014_363
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-46189-1
Online ISBN: 978-3-662-46190-7
eBook Packages: MedicineMedicine (R0)