
Abstract
Objective: Barth syndrome is an X-linked recessive disorder characterized by dilated cardiomyopathy, neutropenia, 3-methylglutaconic aciduria, abnormal mitochondria, variably expressed skeletal myopathy, and growth delay. The disorder is caused by mutations in the tafazzin (TAZ/G4.5) gene located on Xq28. We report a novel exonic splicing mutation in the TAZ gene in a patient with atypical Barth syndrome.
Patient & Methods: The 4-month-old proband presented with respiratory distress, neutropenia, and dilated cardiomyopathy with reduced ejection fraction of 10%. No 3-methylglutaconic aciduria was detected on repeated urine organic acid analyses. Family history indicated that his maternal uncle died of endocardial fibroelastosis and dilated cardiomyopathy at 26 months. TAZ DNA sequencing, mRNA analysis, and cardiolipin analysis were performed.
Results: A novel nucleotide substitution c.553A>G in exon 7 of the TAZ gene was identified in the proband, predicting an amino acid substitution p.Met185Val. However, this mutation created a new splice donor signal within exon 7 causing mis-splicing of the message, producing two messages that only differ in the presence/absence of exon 5; these retain intron 6 and have only 11 bases of exon 7. Cardiolipin analysis confirmed the loss of tafazzin activity. The proband’s mother, maternal aunt, and grandmother carry the same mutation.
Conclusions: The identification of a TAZ gene mutation, mRNA analysis, and monolysocardiolipin/cardiolipin ratio determination were important for the diagnosis and genetic counseling in this family with atypical Barth syndrome that was not found to be associated with 3-methylglutaconic aciduria.
Competing interests: None declared
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Barth syndrome (OMIM 302060, BTHS), first described in 1983 by Barth and colleagues, is manifested clinically by an array of characteristics varying in severity and presentation including dilated cardiomyopathy, skeletal muscle weakness, growth delay with delayed puberty, and neutropenia (Barth et al. 1983; Kelley et al. 1991). Other features include abnormal mitochondria associated with the reduced concentration and altered composition of cardiolipin, hypocholesterolemia, and elevated urinary levels of 3-methylglutaconate (3-MGCA), 3-methylglutarate, or 2-ethylhydracrylate (Kelley et al. 1991; Schlame and Ren 2006). Considerable variability in the age of onset and progression of the disease is observed, and while mortality is highest during the first 4 years, the survival curve for BTHS patients has improved thanks to discovery of the gene (Bione et al. 1996) and to earlier detection and treatments. While previous reports (Barth et al. 2004) cited lifespan peaking at puberty, a current update cites much reduced mortality leading to extended lifespan (Clarke et al. 2013).
Barth syndrome is associated with mutations in the TAZ (Tafazzin) gene located on chromosome Xq28; this gene produces four main transcripts: full length, delta5 (lacking exon 5), delta7 (lacking exon 7), and delta5/7 (lacking both exons 5 and 7) (Gonzalez 2005). We report a case of BTHS caused by an exonic splice mutation in exon 7 of the TAZ (or G4.5) gene; this case lacks the characteristic 3-methylglutaconic aciduria.
Patients and Methods
The proband of German/Irish descent was first brought to the emergency room at the age of 7 weeks with shortness of breath, tachypnea, tachycardia, and cyanosis. Upon physical examination, he had microcephaly (HC = 37 cm, < 3rd percentile) and short stature (Height = 57 cm, 3rd percentile; Weight = 3.89 kg, 10th–25th percentile) with no craniofacial dysmorphic features or other notable organomegaly. A chest x-ray revealed significant cardiomegaly, while an echocardiogram demonstrated a dilated left ventricle with an ejection fraction of only 10%. A second echocardiogram at 11 weeks confirmed the presence of severe systolic dysfunction of the dilated left ventricle. Additionally, there was moderate dilation of the left atrium, moderate insufficiency of the mitral valve, and mild tricuspid valve insufficiency at high velocity, which indicated a moderate degree of pulmonary hypertension. Further work-up disclosed normal liver function tests with normal levels of lactate, glucose, and electrolytes. Acetylcarnitine levels were found to be elevated (35.01 umol/L, normal: 1.62–16.06) and plasma amino acids, namely, Ala, Val, Leu, Ile, Lys, and Trp, were slightly below the lower limit of normal; however, these were due to levocarnitine supplementation and reduced protein intake. Multiple urine organic acid analyses did not demonstrate any abnormalities, even with special attention paid to assess for 3-methylglutaconic, 3-methylglutaric, and 2-ethylhydracrylic aciduria. A complete blood cell count displayed a low white blood cell count (3.3 K/uL, normal 4.9–17.8) and mild neutropenia (15.4%, normal: 17–65% of WBC; absolute neutrophil count: 508), while normal levels of CPK and MB CPK were obtained. Additionally, B-type natriuretic peptide was greater than 5,000 (normal: <100 pg/mL), and Troponin I and high sensitivity CRP were found to be very high.
Over the next 5 months, our proband’s condition worsened until he received a heart transplant from a 17-month-old donor. An echo performed a week prior to his surgery showed severe dilation of the left atrium and ventricle with global severe depression of left ventricular function and wall motion, severe mitral valve regurgitation, and an ejection fraction of less than 20% (Fig. 1), while an echo the day of his surgery additionally revealed a normal aorta, a dilated mitral valve annulus, regurgitation of both the mitral and tricuspid valves, and an ejection fraction of <10%. His final pre-transplant chest x-ray reported persistent cardiomegaly with a known dilated left atrium and mild pulmonary edema (data not shown).

An echocardiogram displaying severe left ventricular and atrial dilation. LA left atrium, LV left ventricle, RA right atrium, RV right ventricle
He successfully underwent cardiac transplantation, and at 2 months post-transplant, his echocardiogram showed normal left ventricular size with increased wall thickness, normal wall motion and systolic function, normal right ventricular size and systolic function, an ejection fraction of 64%, and abnormal septal motion consistent with a post-cardiac transplant heart. His chest x-ray showed stable cardiomediastinal structures, right parahilar subsegmental atelectasis, and normal lung aeration. His laboratory investigations (also at 2 months post-transplant) showed a brain natriuretic peptide (BNP) of 43 (normal: <100 pg/mL); a high sensitivity C-reactive protein (hs-CRP) of 0.3 (low risk: <1.0 mg/L); a completely normal electrolyte panel, an HA1c of 4.7 (normal: 4.4–5.9); a normal kidney panel with slightly elevated alkaline phosphatase of 158 (normal: 31–103 U/L); and a normal complete blood count with exceptions, including a neutrophil percent of 11.6% (normal: 40.1–75.9%) and an absolute neutrophil count of 0.5 (normal: 1.3–7.0 × 103/uL). For his neutropenia, following sampling of his bone marrow, he was started on GCSF treatments of 20 mcg SC 3x/week; 5 mcg/kg/dose. Post-transplant urine organic acid analysis at a second trustworthy lab again failed to demonstrate any 3-methylglutaconic, 3-methylglutaric, or 2-ethylhydracrylic aciduria.
The explanted heart weighed 64 g. There was noncompaction of the left ventricle with secondary endocardial fibroelastosis confirmed by trichrome stain and normal glycogen content confirmed with PAS stain with and without diastase (Figs. 2, 3, and 4). Electron micrographs revealed marked myofibrillar disarray; widening of Z-bands; and fragmented, disorganized, intercalated disks. Loss of myofibrils with perinuclear pools of glycogen particles were noted, and his mitochondria showed marked variation in size, shape, and cristae. Additionally, many greatly enlarged mitochondria (giant forms) displayed stacks of closely packed cristae or had tubular or concentric cristae, and intramitochondrial glycogen was frequently noted (Fig. 5a–d).

Cross sections from the 64.0 g explanted heart exhibit prominent trabeculations involving greater than 50% of the left ventricular wall thickness. Also note thickened fibroelastotic endocardium

Hematoxylin- and eosin-stained section of left ventricle shows hypertrophic cardiac myocytes (x10)

This photomicrograph shows significantly thickened endocardium with duplication of elastic fibers (red) and collagen fibers (blue) highlighted with Trichrome-EVG stain (Elastic Van Gieson Stain). (x10). In general, the normal endocardium has fewer than five layers of elastic fibers

(a) Noticeable onion skinning of the mitochondrial cristae, fusion of intracristae inner mitochondrial membrane preventing transport, and marked variation in size of mitochondria. (b) Note many inclusions of glycogen in intramitochondrial vacuoles replacing sarcomeres. (c, d) Notice disorganized stacks of cristae, intramitochondrial glycogen, and glycogen surrounding mitochondria
Upon review of the family history, the proband has no siblings, and other notable phenotypes include a maternal uncle who displayed similar symptoms to the proband before his death at 2 years old. Examination of the uncle’s past medical records revealed an initial admission for respiratory distress, fever, and diaphoresis at 5 months of age. A chest x-ray and echocardiogram displayed cardiomegaly with increased pulmonary vascular markings and poor function of the left ventricle resulting in a diagnosis of endocardial fibroelastosis. All growth measurements were noted below the 5th percentile at that time. Subsequent admissions to the hospital over the next year and a half revealed an EKG displaying tall R waves and inverted T waves, gastroenteritis, mild generalized hypotonia, jerking movements of the head, severe failure to thrive, pneumonia and eventual cardiac arrest, coma and severe encephalopathy before his death. Genetic testing was not available at the time of his death, and it is uncertain if urine organic acid analysis was performed. Our proband also had a second maternal aunt who was found to have a very unusual right hypoplastic heart complex and received a prostaglandin E1 infusion. Cardiac findings included tricuspid stenosis, pulmonary valve atresia, a small hypertrophied right ventricle, marked left ventricular hypertrophy, and an atrial septal defect. A Gore-Tex shunt from the right subclavian to the right pulmonary artery was created and she was discharged on post-operation day 7. At 2½ months of age, she was readmitted with cyanotic heart disease and expired despite massive resuscitative efforts. Unfortunately, no karyotype analysis appears to have been done and we were not able to conduct any further studies on her due to unavailability of saved frozen tissue.
Results
Mutation analysis for the tafazzin gene (OMIM 300394, TAZ) in our proband revealed a novel adenine to guanine transition mutation at position c.553 in exon 7, resulting in a predicted p.Met185Val amino acid substitution. Family studies showed that the proband’s mother, maternal aunt, and grandmother carry the same mutation (Figs. 6, 7). Subsequent bioinformatic analyses by both PolyPhen-2 and PANTHER algorithms (http://genetics.bwh.harvard.edu/pph/; http://www.pantherdb.org/tools/csnpScoreForm.jsp) predicted that this sequence alteration would be deleterious. The substitution c.553A>G found in the proband was not observed in 115 X chromosomes from 81 ethnically matched control subjects (47 males and 34 females); it is also known that methionine is evolutionarily conserved at position 185 in all vertebrate classes and many invertebrate classes as well. However, SplicePort analysis (http://spliceport.cs.umd.edu; Dogan et al. 2007) predicted that c.553A>G generates a strong splice donor signal within exon 7 which would result in a truncated protein; this was supported by another splice site prediction tool (http://www.fruitfly.org/seq_tools/splice.html). To determine whether this mutation leads to amino acid substitution or to mis-splicing, we obtained RNA from the patient’s lymphoblast cell line. RT-PCR with labeled primers showed only two abnormally sized TAZ mRNA fragments instead of the four known splice variants. The RT-PCR fragments were subjected to capillary electrophoresis separation to obtain precise fragment sizing and were also sequenced. These analyses showed two main mRNAs differing in the presence/absence of exon 5, both of which retain intron 6 and use a new splice donor site within exon 7 after only 11 bases of the exon. Thus, c.553A>G results in r.[541+1_542-1ins;r.553_583del]; p.Lys182Glnfs*4 (Fig. 8).

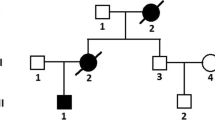
A pedigree of our family with atypical Barth syndrome. Filled symbols indicate clinically affected individuals; open symbols indicate unaffected individuals; a slash through a circle or square indicates a deceased individual; symbol with “?” indicates an individual who did not have clinical evaluation and genetic testing; squares indicate males; circles indicate females. The proband patient is marked with an arrow

TAZ sequencing of the family members revealed that the proband’s mother, maternal aunt, and grandmother all carry the c.553A>G substitution

(Top) TAZ gene diagram showing mutation location and its effect on splicing. (Bottom) Processed mRNA including complete intron 6 retention and only 11 bases of exon 7
Biochemical testing for monolysocardiolipin/cardiolipin (Kulik et al. 2008; Houtkooper et al. 2009) was performed in order to confirm the loss of tafazzin activity. The MLCL/CL results are shown in Fig. 9, yielding an m/z 582.4 to m/z 723.5 ratio of 91, consistent with BTHS.

MLCL and CL analysis performed on patient lymphoblast cell line shows the monolysocardiolipin and cardiolipin profiles with their characteristic doubly charged ions. The ratio of monolysocardiolipin/cardiolipin species is strongly increased as compared to normal controls (see Houtkooper et al. 2009). m/z mass/charge
Discussion
The c.553A>G exonic splice mutation is being reported for the first time and is considered to be a pathogenic mutation for the following reasons: (a) abnormal splicing of the TAZ message, (b) abnormal MLCL/CL ratio, (c) absence of this change in 115 control chromosomes, and (d) family history of an early male infant death.
If this single base pair change resulted in p.Met185Val, it would only affect splice variants that contain exon 7 (full length and delta 5). Instead, this mutation generated a strong donor splice signal within exon 7 causing retention of intron 6 and partial deletion of exon 7 (r.[541+1_542-1ins; r.553_583del]), resulting in p.Lys182Glnfs*4. BTHS-causing mutations have been found in all TAZ exons, and known mutations include small deletions and insertions, termination mutations, and splice signal mutations. The Human Tafazzin (TAZ) Gene Mutation and Variation Database lists only one other exonic splice mutation (http://www.barthsyndrome.org/).
To date, no clear genotype-phenotype correlation has emerged (Bione et al. 1996; D’Adamo et al. 1997; Johnston et al. 1997; Sakamoto et al. 2001; Chen et al. 2002; Roberts et al. 2012). In fact, to the authors’ best knowledge, there is also no clear genotype-phenotype correlation between the six reported cases of Barth syndrome not displaying 3-methylglutaconic aciduria (Table 1). There is significant variation in the severity of presentation between the patients as noted in the table, with varying degrees of neutropenia – heart defects and outcome with three patients passing before the age of 5 months and three patients still alive at publication. However, it is noted that BTHS not displaying elevated 3-MGCA may be more common than reported, in that there is considerable variability in laboratory methods used to detect 3-MGCA. There is also noteworthy variability seen in two very similar cases reported by Brady et al. in 2006 and Sakamoto et al. in 2002. Brady presented a proband with an Arg94His missense mutation in exon 3 with dilated cardiomyopathy, neutropenia, and no organic aciduria leading to death within 12 days, while Sakamoto reported an Arg94Ser missense mutation in exon 3 in a functioning 17-year-old boy with cardiac manifestations, organic aciduria, and a normal neutrophil count. However, it is possible that Brady’s proband may have deceased before 3-methylglutaconic acids could be detected, or that the difference is due to the different amino acid substitutions in their cases.
While Bione had predicted that tafazzins may function as membrane anchors or soluble cytoplasmic proteins (Bione et al. 1996), more recent studies have demonstrated that they are phospholipid acyltransferases involved in the acyl-specific remodeling of cardiolipin (Vreken et al. 2000; Valianpour et al. 2002; Schlame et al. 2002). Tetralinoleoyl cardiolipin is a phospholipid found in the inner membrane of mitochondria that is essential for mitochondrial membrane structure and correct insertion and function of electron transport components; it has been shown that incorrect remodeling due to mutated tafazzin may compromise the assembly and stability of the electron transport chain (Schlame et al. 2000). Thus, the resultant mitochondrial dysfunction is an underlying mechanism of the skeletal and cardiac myopathy seen in Barth syndrome. This is supported by Drosophila studies where Taz mutants showed an 80% reduction in cardiolipin, abnormal mitochondria, and weakness in their indirect flight muscles (Xu et al. 2006).
However, the complex array of symptoms seen in patients with BTHS is much more sophisticated than a breakdown in energy metabolism, and continued research is imperative in determining the function of multiple tafazzin proteins and in elucidating the pathogenesis of BTHS. Further, the variability in presentation and severity of the syndrome not only suggest that multiple factors during multiple time periods may be influencing the expression of the phenotype. For example, TAZ expression has been found to decline with age, suggesting that tafazzins may be more essential during fetal and early postnatal periods (Marziliano et al. 2007; Malhotra et al. 2009). Intrafamilial phenotypic variability and the variability between families that carry the same mutation further suggest that there are modifier genes that modulate severity (Malhotra et al. 2009; Steward et al. 2010).
Conclusion
We have presented a novel substitution mutation in the TAZ gene that is associated with an atypical presentation of BTHS lacking one of the cardinal signs: 3-methylglutaconic aciduria. There have only been five other patients reported in the literature possibly because (a) it truly is a rare variant of Barth syndrome or (b) its actual prevalence is presented falsely low either because of variability in lab methods, or because molecular testing for mutations in the TAZ gene or biochemical testing of urine organic acids are not considered by health providers. These six cases highlight the current lack of genotype-phenotype correlation. Tafazzin gene mutations are now recognized (Barth et al. 2004; Steward et al. 2010; Roberts et al. 2012) as being implicated in a spectrum of cardiac presentations that include endocardial fibroelastosis, isolated noncompaction of left ventricular myocardium, hypertrophic cardiomyopathy, ventricular arrhythmia, which are also part of syndromes caused by various other genes. The variability in clinical presentation and severity complicate early recognition and may lead to underdiagnosis (Cantlay et al. 1999; Barth et al. 1999; Spencer et al. 2006; Steward et al. 2010) delaying appropriate treatment for BTHS patients. We propose that every male child with dilated cardiomyopathy should be tested for Barth syndrome.
Abbreviations
- 3-MGCA:
-
3-methylglutaconic acid
- BNP:
-
Brain natriuretic peptide
- BTHS:
-
Barth Syndrome
- hs-CRP:
-
High sensitivity C-reactive protein
- MLCL/CL:
-
Monolysocardiolipin/cardiolipin
- TAZ :
-
Tafazzin
References
Barth PG, Scholte HR, Berden JA et al (1983) An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci 62(1–3):327–355
Barth PG, Wanders RJ, Vreken P (1999) X-linked cardioskeletal myopathy and neutropenia (Barth syndrome)-MIM 302060. J Pediatr 135(3):273–276
Barth PG, Valianpour F, Bowen VM et al (2004) X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am J Med Genet A 126A(4):349–354
Bione S, D'Adamo P, Maestrini E et al (1996) A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet 12(4):385–389
Bleyl SB, Mumford BR, Thompson V et al (1997) Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am J Hum Genet 61(4):868–872
Brady AN, Shehata BM, Fernhoff PM (2006) X-linked fetal cardiomyopathy caused by a novel mutation in the TAZ gene. Prenat Diagn 26(5):462–465
Cantlay AM, Shokrollahi K, Allen JT et al (1999) Genetic analysis of the G4.5 gene in families with suspected Barth syndrome. J Pediatr 135(3):311–315
Chen R, Tsuji T, Ichida F et al (2002) Mutation analysis of the G4.5 gene in patients with isolated left ventricular noncompaction. Mol Genet Metab 77(4):319–325
Clarke SL, Bowron A, Gonzalez IL et al (2013) Barth syndrome. Orphanet J Rare Dis 8:23
D'Adamo P, Fassone L, Gedeon A et al (1997) The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet 61(4):862–867
Dogan RI, Getoor L, Wilbur WJ et al (2007) SplicePort–an interactive splice-site analysis tool. Nucleic Acids Res 35(Web Server issue):W285–W291, Epub 2007 Jun 18
Gedeon AK, Wilson MJ, Colley AC et al (1995) X linked fatal infantile cardiomyopathy maps to Xq28 and is possibly allelic to Barth syndrome. J Med Genet 32:383–388
Gonzalez IL (2005) Barth syndrome: TAZ gene mutations, mRNAs, and evolution. Am J Med Genet A 134(4):409–414
Houtkooper RH, Rodenburg RJ, Thiels C et al (2009) Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography-mass spectrometry as a diagnostic test for Barth syndrome. Anal Biochem 387(2):230–237
Johnston J, Kelley RI, Feigenbaum A et al (1997) Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am J Hum Genet 61(5):1053–1058
Kelley RI, Cheatham JP, Clark BJ et al (1991) X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria. J Pediatr 119(5):738–747
Kulik W, van Lenthe H, Stet FS et al (2008) Bloodspot assay using HPLC-tandem mass spectrometry for detection of Barth syndrome. Clin Chem 54(2):371–378
Malhotra A, Edelman-Novemsky I, Xu Y et al (2009) Role of calcium-independent phospholipase A2 in the pathogenesis of Barth Syndrome. Proc Natl Acad Sci U S A 106(7):2337–2341
Marziliano N, Mannarino S, Nespoli L et al (2007) Barth syndrome associated with compound hemizygosity and heterozygosity of the TAZ and LDB3 genes. Am J Med Genet A 143A(9):907–915
Roberts AE, Nixon C, Steward CG et al (2012) The Barth Syndrome Registry: distinguishing disease characteristics and growth data from a longitudinal study. Am J Med Genet Part A 158A:2726–2732
Sakamoto O, Ohura T, Katsushima Y et al (2001) A novel intronic mutation of the TAZ ( G4.5) gene in a patient with Barth syndrome: creation of a 5' splice donor site with variant GC consensus and elongation of the upstream exon. Hum Genet 109(5):559–563
Sakamoto O, Kitoh T, Ohura T et al (2002) Novel missense mutation (R94S) in the TAZ (G4.5) gene in a Japanese patient with Barth syndrome. J Hum Genet 47(5):229–231
Schlame M, Rua D, Greenberg ML (2000) The biosynthesis and functional role of cardiolipin. Prog Lipid Res 39(3):257–288
Schlame M, Towbin JA, Heerdt PM et al (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol 51(5):634–637
Schlame M, Ren M (2006) Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett 580(23):5450–5455
Schmidt MR, Birkebaek N, Gonzalez I et al (2004) Barth syndrome without 3-methylglutaconic aciduria. Acta Paediatr 93(3):419–421
Spencer CT, Bryant RM, Day J et al (2006) Cardiac and clinical phenotype in Barth syndrome. Pediatrics 118(2):e337–e346
Steward CG, Newbury-Ecob RA, Hastings R et al (2010) Barth Syndrome: an X-linked cause of fetal cardiomyopathy and stillbirth. Prenal Diagn 30(10):970–976
Valianpour F, Wanders RJ, Overmars H et al (2002) Cardiolipin deficiency in X-linked cardioskeletal myopathy and neutropenia (Barth syndrome, MIM 302060): a study in cultured skin fibroblasts. J Pediatr 141(5):729–733
Vreken P, Valianpour F, Nijtmans LG et al (2000) Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun 279(2):378–382
Xu Y, Condell M, Plesken H et al (2006) A Drosophila model of Barth syndrome. Proc Natl Acad Sci U S A 103(31):11584–11588
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Additional information
Communicated by: Eva Morava, MD PhD
Appendices
Synopsis
We present a novel exonic splicing mutation in the TAZ gene in a patient with an atypical presentation of Barth syndrome lacking one of the cardinal signs: 3-methylglutaconic aciduria.
Conflicts of Interest
There are no conflicts of interest.
Rights and permissions
Copyright information
© 2013 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Fan, Y. et al. (2013). A Novel Exonic Splicing Mutation in the TAZ (G4.5) Gene in a Case with Atypical Barth Syndrome. In: Zschocke, J., Gibson, K., Brown, G., Morava, E., Peters, V. (eds) JIMD Reports - Volume 11. JIMD Reports, vol 11. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2013_228
Download citation
DOI: https://doi.org/10.1007/8904_2013_228
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-37327-5
Online ISBN: 978-3-642-37328-2
eBook Packages: MedicineMedicine (R0)