
Abstract
Tyrosinemia type I is a genetic disorder characterized by accumulation in the blood and urine of the toxic metabolite succinylacetone (SUAC), not detectable in healthy samples. In many countries, newborns are screened for tyrosinemia type I using tyrosine as a primary marker. Unfortunately, tyrosine accumulation may take longer to occur and it may be not obvious when specimens are collected, in the first few days of life, as for newborn screening. In 2008, we reported changes to simultaneously measure acylcarnitines, amino acids, and SUAC during expanded newborn screening. We established the usefulness of this method after identifying a first asymptomatic newborn affected by tyrosinemia type I. Now we report a second infant with positive SUAC screening result (14.1 μmol/L, n.v. < 2) and normal tyrosine concentration (74 μmol/L; n.v. < 250). We also performed molecular analysis of FAH gene in both patients after diagnosis at newborn screening. They had consanguineous parents and were both homozygous for two known disease-causing mutations of the FAH gene. The outcome of patients detected in the MS/MS screening is significantly favorable. We also report our results of newborn screening for tyrosinemia type I before and after inclusion of SUAC as a primary marker for this disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Tyrosinemia type I (MIM 276700) affects 1 in every 100,000 to 120,000 babies worldwide, although the real incidence could be higher. It is an autosomal recessive disorder caused by mutations in the FAH gene that leads to deficiency of the fumarylacetoacetic hydrolase (FAH; EC 3.7.1.2), the last enzyme in the tyrosine degradation pathway. The consequence of the metabolic block is the conversion of the catabolic intermediates maleylacetoacetate and fumarylacetoacetate to the toxic metabolites SUAC and succinylacetoacetate. The presence of SUAC in urine or blood is pathognomonic for tyrosinemia type I (Mitchell et al. 2001), which, if untreated, is usually fatal within age 10 years. Affected children may exhibit diarrhea, vomiting, jaundice, liver or kidney failure, neurological crisis, rickets, failure to thrive, and hepatocellular carcinoma (Mitchell et al. 2001). With treatment, many patients can lead normal lives with few restrictions. Treatment usually involves 2-(2-nitro-4-3 trifluoro-methylbenzoyl)-1,3-cyclohexanedione (NTBC) administration and a diet low in tyrosine and phenylalanine (Lindstedt et al. 1992). Early diagnosis and initiation of therapy may thus be crucial in improving outcome (Joshi and Venugopalan 2004).
In many countries, newborns are screened for tyrosinemia type I using tyrosine as a primary marker (US Department of Health and Human Services, Maternal and Child Health Bureau 2005). An elevated concentration of tyrosine, however, is not sensitive enough to detect all cases (Wilcken et al. 2003). Some newborn screening programs have recently introduced the determination of SUAC as a reliable and valid marker for the identification of tyrosinemia type I (Allard et al. 2004; Sander et al. 2006; la Marca et al. 2008, Turgeon et al. 2008; Al-Dirbashi et al. 2008, Adam et al. 2009).
In 2008, we reported an efficient method to simultaneously measure acylcarnitines, amino acids, and SUAC during expanded newborn screening (la Marca et al. 2008). We established the usefulness of this method after identifying a first asymptomatic newborn with by tyrosinemia type I (la Marca et al. 2009). We now present a second infant with positive SUAC screening and tyrosine within the normal range. We also report our results of newborn screening for tyrosinemia type I before and after inclusion of SUAC as a primary marker for this disease.
Case Report
The patient was a Moroccan boy, born at full term after an uneventful pregnancy and delivery with a weight of 3,620 g. He was the first child of first degree cousins. Both parents were healthy, but the family reported a positive history for deaths (three babies) in early childhood of unknown causes. Blood for newborn screening, collected on the third day of life, revealed an elevated SUAC level of 14.1 μmol/L (n.v. < 2). Tyrosine value was normal (74 μmol/L; n.v. < 250). On the sixth day of life, the boy was hospitalized for further testing. The diagnosis of tyrosinemia type I was confirmed by detection of SUAC in the urine (70 mmol/mol of creatinine) and plasma (11 μmol/L). Plasma tyrosine level was increased (791 μmol/L; n.v. < 123). The child was in good condition and no clinical manifestations were apparent. Liver transaminases, alkaline phosphatase, bilirubin, coagulation factors, and ammonia were within normal levels. NTBC treatment was started at a dose of 1 mg/kg/die body weight combined with dietary restriction of tyrosine and phenylalanine.
Results and Discussion
In many countries (Canada, USA, Latin America), to reduce hospital costs, newborn screening specimen collection is done within 24–48 h after birth in response to the early discharge of mothers and their infants. This tendency causes a serious problem for metabolic newborn screening since accumulation of some metabolites, used as markers, may occur only in some days after delivery. The success of newborn screening depends on the type of marker used. Hence, the challenge is to find and to include in newborn screening programs appropriate diagnostic markers for the early detection of metabolic disorders.
Up to now, many newborn screening programs worldwide use tyrosine levels as a marker for tyrosinemia type I. Unfortunately this metabolic condition cannot be detected when specimens are collected in the first few days of life, as tyrosine accumulation may take longer to occur (Mitchell et al. 2001). The increased SUAC levels, the metabolite immediately upstream of the enzymatic block, is a reliable, appropriate, and early marker of this disorder.
The case presented here, as also the previously reported case (la Marca et al. 2009), provides further evidence on the importance of using SUAC as the primary metabolic marker for detecting tyrosinemia type I in newborn screening.
Experience in various laboratories, including our own, suggests that tyrosine was not a sensitive diagnostic marker for the timely identification of patients and decreased the specificity of the test while increasing the unnecessary medical cost for false-positive recall rate. Indeed, the most common cause of increased blood tyrosine levels is benign transient tyrosinemia of the newborn.
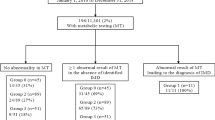
The inclusion of SUAC in our newborn screening program dates back to January 2007, subsequently to a false-negative result. Among about 136,000 newborns screened, two patients with tyrosinemia type I were identified. No false positive was on record. Both patients had consanguineous parents (from Turkey and Morocco) and were homozygous for the known c.709C>T (p.Arg237X) and IVS6-1G>T disease-causing mutations of the FAH gene (Rootwelt et al. 1996; Ploos van Amstel et al. 1996). The IVS6-1G>T leads to a splicing defect and has been reported as common in the European and Mediterranean area (Arranz et al. 2002). The results for tyrosinemia type I in the Tuscan newborn screening program are reported in the Tables 1 and 2.
It is very difficult to estimate the incidence of this disease in the population but, based on our own experience, we are inclined to believe that it is generally underestimated. Furthermore, in Turkey as well as Morocco and other North African and Arabic countries, the elevated rates of consanguinity may have an impact on the incidence of rare autosomal recessive disorders (Al-Gazali et al. 2006; Jaouad et al. 2009).
There are various methods for SUAC determination on DBS, with significant modifications in the newborn screening procedures. The method we had previously proposed (la Marca et al. 2008) consists of a simple and fast protocol for newborn screening sample preparation and allows identifying this metabolic defect in the neonatal period with 100% sensitivity. No cost for additional equipment or staff members is required for applying such testing.
References
Adam BW, Lim TH, Hall EM, Hannon WH (2009) Preliminary proficiency testing results for succinylacetone in dried blood spots for newborn screening for tyrosinemia type I. Clin Chem 55:2207–2213
Al-Dirbashi OY, Rashed MS, Jacob M, Al-Ahaideb LY, Al-Amoudi M, Rahbeeni Z, Al-Sayed MM et al (2008) Improved method to determine succinylacetone in dried blood spots for diagnosis of tyrosinemia type I using UPLC-MS/MS. Biomed Chromatogr 22:1181–1185
Al-Gazali L, Hamamy H, Al-Arrayad S (2006) Genetic disorders in the Arab world. BMJ 333:831–834
Allard P, Grenier A, Korson MS, Zytkovicz TH (2004) Newborn screening for hepatorenal tyrosinemia by tandem mass spectrometry: analysis of succinylacetone extracted from dried blood spots. Clin Biochem 37:1010–1015
Arranz JA, Piñol F, Kozak L, Pérez-Cerdà C, Cormand B, Ugarte M, Riudor E (2002) Splicing mutations, mainly IVS6–1(G>T), account for 70% of fumarylacetoacetate hydrolase (FAH) gene alterations, including 7 novel mutations, in a survey of 29 tyrosinemia type I patients. Hum Mutat 20:180–188
Jaouad IC, Elalaoui SC, Sbiti A, Elkerh F, Belmahi L, Sefiani A (2009) Consanguineous marriages in Morocco and the consequence for the incidence of autosomal recessive disorders. J Biosoc Sci 41:575–581
Joshi SN, Venugopalan P (2004) Experience with NTBC therapy in hereditary tyrosinemia type I: an alternative to liver transplantation. Ann Trop Paediatr 24:259–265
la Marca G, Malvagia S, Pasquini E, Innocenti M, Fernandez MR, Donati MA, Zammarchi E (2008) The inclusion of succinylacetone as marker for tyrosinemia type I in expanded newborn screening programs. Rapid Commun Mass Spectrom 22:812–818
la Marca G, Malvagia S, Funghini S, Pasquini E, Moneti G, Guerrini R, Zammarchi E (2009) The successful inclusion of succinylacetone as a marker of tyrosinemia type I in Tuscany newborn screening program. Rapid Commun Mass Spectrom 23:3891–3893
Lindstedt S, Holme E, Lock EA, Hjalmarson O, Strandvik B (1992) Treatment of hereditary tyrosinemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 340:813–817
Mitchell GA, Grope M, Lambert M, Tanguay RM (2001) Hypertyrosinemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 1777–1805
Ploos van Amstel JK, Bergman AJ, van Beurden EA, Roijers JF, Peelen T, van den Berg IE, Poll-The BT, Kvittingen EA, Berger R (1996) Hereditary tyrosinemia type 1: novel missense, nonsense and splice consensus mutations in the human fumarylacetoacetate hydrolase gene; variability of the genotype-phenotype relationship.Hum Genet 97:51–59
Rootwelt H, Høie K, Berger R, Kvittingen EA (1996) Fumarylacetoacetase mutations in tyrosinaemia type I. Hum Mutat 7:239–243
Sander J, Janzen N, Peter M, Sander S, Steuerwald U, Holtkamp U, Schwahn B et al (2006) Newborn screening for hepatorenal tyrosinemia: tandem mass spectrometric quantification of succinylacetone. Clin Chem 52:482–487
Turgeon C, Magera MJ, Allard P, Tortorelli S, Gavrilov D, Oglesbee D, Raymond K et al (2008) Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots. Clin Chem 54:657–664
US Department of Health and Human Services, Maternal and Child Health Bureau (2005) Newborn screening: toward a uniform screening panel and system report for public comment. Fed Regist 70:308–329
Wilcken B, Wiley V, Hammond J, Carpenter K (2003) Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med 348:2304–2312
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Synopsis
Synopsis
Succinylacetone determination on dried blood spot.
Rights and permissions
Copyright information
© 2011 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
la Marca, G. et al. (2011). Newborn Screening for Tyrosinemia Type I: Further Evidence that Succinylacetone Determination on Blood Spot Is Essential. In: JIMD Reports - Case and Research Reports, 2011/1. JIMD Reports, vol 1. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2011_24
Download citation
DOI: https://doi.org/10.1007/8904_2011_24
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-17707-1
Online ISBN: 978-3-642-17708-8
eBook Packages: MedicineMedicine (R0)