
Abstract
During the last decade, small noncoding RNAs (ncRNAs) have emerged as essential post-transcriptional regulators in bacteria. Nearly all important physiological and stress responses are modulated by ncRNA regulators, such as riboswitches, trans-acting small RNAs (sRNAs), and cis-antisense RNAs. Recently, three RNA-seq studies identified a total of 1534 candidate ncRNAs from Agrobacterium tumefaciens, a pathogen and biotechnology tool for plants. Only a few ncRNAs have been functionally characterized in A. tumefaciens, and some of them appear to be involved in virulence. AbcR1 regulates multiple ABC transporters and modulates uptake of a quorum-sensing inhibitor produced by plants. RNA1111, a Ti plasmid-encoded sRNA, might regulate the dispersal of the Ti plasmid and virulence. In addition, a chromosomally encoded sRNA Atr35C is induced by the vir gene regulator VirG and its expression is affected by iron, manganese, and hydrogen peroxide, suggesting a possible role in oxidative stress responses and Agrobacterium–plant interactions. Progress in ncRNA functional analysis is slow, likely resulting from innate challenges, such as poor sequence conservation and imperfect base-pairing between sRNAs and mRNAs, which make computational target predictions inefficient. Advances in single-cell-based RNA-seq and proteomics approaches would provide valuable tools to reveal regulatory networks involving ncRNA regulators.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
1.1 Discovery of ncRNAs
Regulatory noncoding RNAs (ncRNAs) have emerged as important regulators of physiological responses in bacteria to survive in ever-changing environments. RNA-mediated response regulation is more advantageous for bacteria than is regulation by proteins (e.g., transcription factors) because it requires less time and energy for synthesis (transcription only vs. transcription and translation) and the responses can be rapidly reversed when needed thanks to a short ncRNA turnover time. Numerous RNA molecules have been discovered that modulate most biological processes and stress responses via various mechanisms. The first studied bacterial small ncRNAs were exosome-encoded antisense RNAs that block plasmid replication (Stougaard et al. 1981; Tomizawa et al. 1981) and inhibit transposon movement (Simons and Kleckner 1983). Although these findings precede the discovery of microRNAs (miRNAs) and small interfering RNAs (siRNAs), the importance of bacterial ncRNAs as regulators had not been much appreciated until the early 2000s when genome-wide identification of chromosomally encoded ncRNAs from E. coli and other bacteria were reported (reviewed in Livny and Waldor 2007). Since then, tens to hundreds of candidate ncRNAs have been identified from diverse bacterial species including plant-associated bacteria (reviewed in Becker et al. 2014; Harfouche et al. 2015).
1.2 Classification and Mode of Action
Ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs) are well-characterized ncRNAs regulating protein translation but they are not discussed in this review. We will focus on riboswitches, trans-acting small RNAs (sRNAs), and cis-antisense RNAs (asRNAs) in this chapter (Waters and Storz 2009). Each group of RNAs uses a variety of mechanisms to modulate physiological and stress responses. Below, we review how these ncRNAs exert regulatory effects in the model bacterial species.
1.2.1 Riboswitches
Riboswitches are part of untranslated regions (UTRs) of mRNAs and affect cognate gene expression at the transcriptional or post-transcriptional levels. Riboswitches are evolutionarily conserved among distantly related bacteria and their functional counterparts are also found in archaea, plants, and fungi. Bacterial riboswitches were first discovered in 2002 as sensors of intracellular small molecules (Mironov et al. 2002; Nahvi et al. 2002; Winkler et al. 2002). Three independent studies reported that part of the mRNA binds to vitamin B derivatives and affects downstream gene expression via transcription attenuation or translation inhibition. Since then, many different riboswitches have been identified and functionally characterized in various bacteria (Winkler and Breaker 2005; Serganov and Nudler 2013). Most riboswitches have two distinct parts: the ligand-binding aptamer domain and the expression platform domain. The aptamer is the sensor region which binds to a specific ligand or metabolite, and the expression platform domain is the response region which adopts alternative structures to affect gene expression. Most well-characterized riboswitches are metabolite sensors and are located in the 5′ UTR of mRNAs encoding enzymes responsible for the biosynthesis the metabolites. Under normal conditions, the ribosome binding site (RBS) is open and accessible to the translation machinery, resulting in the production of functional proteins. Under high metabolite conditions, however, a metabolite binds to the aptamer domain, leading to a conformational change. This change can result in transcriptional attenuation by forming a terminator or translational inhibition by masking the RBS. This type of negative feedback loop prevents overproduction of a specific metabolite, ensuring balanced resource utilization.
Riboswitches are highly selective and many different types of riboswitches have been discovered (reviewed in Serganov and Nudler 2013). A wide range of ligands can be sensed by riboswitches: fluoride anions, metals, purines and their derivatives, cofactors, and amino acids. Recent studies showed that these cis-acting regulatory elements can also affect ncRNA expression and modulate RNA–protein interactions (reviewed in Mellin and Cossart 2015). In addition, some riboswitches can act as catalytic enzymes (Tinsley et al. 2007) or as trans-acting sRNAs (Loh et al. 2009), suggesting that bacteria have unexpectedly complex regulatory networks involving different classes of ncRNAs.
1.2.2 Trans-encoded sRNAs
sRNAs are 50–500 nt in size and are often encoded in intergenic regions. This group of ncRNAs represents the most well-known RNA regulators and are involved in many physiological and stress responses (Waters and Storz 2009; Gottesman and Storz 2011). sRNAs appear to evolve rapidly, because primary sequence conservation is very limited among closely related bacterial species (Gottesman and Storz 2011). Many sRNAs modulate gene expression via imperfect base-pairing with their target mRNAs, which are transcribed from distinct genomic locations (Waters and Storz 2009). Another feature of sRNAs is their requirement for the global RNA chaperone Hfq for optimal sRNA–target interactions (De Lay et al. 2013). Hfq-binding affects RNA secondary structure and might facilitate sRNA–mRNA interactions, presumably by binding to both molecules (Gottesman and Storz 2011). Interestingly, however, Hfq is not required for sRNA functions in some bacteria, such as Staphylococcus aureus and Bacillus subtilis. A recent study by Smirnov et al. (2016) discovered another major sRNA-binding protein, ProQ, which forms stable complexes with small RNAs. It is possible that evolutionarily distant bacteria might have adopted different RNA-binding proteins to facilitate sRNA–mRNA interactions. The exact mechanisms of how sRNAs select and interact with their target mRNAs are still largely unknown.
Another group of sRNAs modulate RNA-binding proteins by sequestering them or directly affecting enzymatic activity (reviewed in Storz et al. 2011). For example, 6S RNA sequesters the house-keeping RNA polymerase (Wassarman and Storz 2000) and the CsrB family sRNAs negatively regulate the activity of CsrA (carbon storage regulator), the regulator of secondary metabolism, by sequestering multiple subunits (Romeo 1998). Many protein-binding sRNAs contain multiple protein-binding sequences, and direct competition by mimicry is the underlying mechanism of these sRNAs (Storz et al. 2011). The presence of multiple protein-binding sRNAs and RNA-binding proteins suggests that bacteria utilize complex sRNA-protein pairs to fine-tune the regulatory networks.
1.2.3 Cis-antisense RNAs
asRNAs are transcribed from the complementary strand of a target gene, and thus exert effects via base-pairing with perfect complementarity (Waters and Storz 2009). The most well-studied asRNAs are encoded on mobile elements, such as plasmids, transposons, and bacteriophages, and maintain proper copy numbers via various mechanisms (Waters and Storz 2009). Common mechanisms are to inhibit plasmid replication by blocking replication primer formation and to inhibit the translation of transposases and protein toxins encoded by these mobile elements (Brantl 2007; Wagner and Simons 1994).
There are an increasing number of asRNAs discovered from diverse bacteria (Georg and Hess 2011; Thomason et al. 2015). However, it is not clear how many of these are actually regulatory asRNAs. Because a low level of pervasive transcription occurs throughout the entire genome (reviewed in Wade and Grainger 2014; Lloréns-Rico et al. 2016), systematic approaches need to be developed to distinguish regulatory asRNAs from these pervasive antisense transcripts. Adding another level of complexity, recent studies discovered that genes and operons encoding proteins performing opposing functions can modulate the expression of genes encoded on the opposite strand (reviewed in Sesto et al. 2013). The total number of asRNAs inventories in bacterial genomes will likely increase in the near future. However, it remains challenging to study how these asRNAs exert regulatory effects, if any, to counter ever-changing environmental stresses.
1.3 Genome-Wide Identification of ncRNAs
Early genome-wide ncRNA identification studies utilized whole genome sequencing data and searched for conserved bacterial promoter and rho-independent terminator sequences in the conserved intergenic regions of E. coli (reviewed in Livny and Waldor 2007; Livny et al. 2008). The predicted ncRNAs were experimentally validated to prove the effectiveness of the computational predictions. However, the bias toward intergenic regions and low throughput validation procedures limited the thorough discovery of inventories of regulatory RNAs in a genome. High-resolution tiling arrays were successfully used to identify 20–50 sRNAs (Landt et al. 2008; Toledo-Arana et al. 2009) before whole transcriptome sequencing (RNA-seq) became a powerful tool to discover many hundreds of candidate ncRNAs from various bacterial species (Sharma and Vogel 2009). In this chapter, we review recent advances in regulatory ncRNA research in the “natural genetic engineer” A. tumefaciens C58 and discuss current challenges and remaining questions.
2 Identification of Small ncRNAs in Agrobacterium
Three RNA-seq studies have identified numerous small ncRNAs from A. tumefaciens strain C58 thus far (Table 1; Wilms et al. 2012a; Lee et al. 2013; Dequivre et al. 2015). Each study employed some unique approaches, and therefore provided nonredundant identification of novel candidate ncRNAs.
The first genome-wide identification study was done by Wilms et al. (2012a), who used a Roche FLX platform and identified 228 novel ncRNAs. Briefly, total RNA was extracted from A. tumefaciens C58 cultures grown under two different conditions: AB minimal medium in the presence (+Vir) or absence (−Vir) of the vir gene inducer acetosyringone (AS). To identify transcription start sites (TSS), each total RNA sample was treated or not treated with the Terminator™ 5′-Phosphate-Dependent Exonuclease (TEX), which selectively degrades transcripts containing a 5′-mono-phosphate, to enrich primary transcripts which contain a 5′-tri-phosphate. Four cDNA libraries were constructed and sequenced using a Roche FLX sequencer. The resulting 422,204 cDNA sequences were compared to the C58 reference genome and 348,998 sequences longer than 18 nt were mapped. Sequences mapping to intergenic regions or complementary to protein-coding genes were manually analyzed for ncRNA discovery. Putative ncRNAs were identified if there were a minimum of five cDNA reads in at least one of the four cDNA libraries. A total of 228 candidate ncRNAs were identified from all four replicons: 129 on the circular chromosome, 59 on the linear chromosome, 20 on the pAt plasmid, and 20 on the Ti plasmid. The list also included widely conserved ncRNAs, such as 6S RNA, SRP RNA 4.5S, RNase P, and tmRNA, as well as the previously published Agrobacterium ncRNAs RepE, AbcR1, and AbcR2. Twenty-two ncRNAs were validated by RNA-blot hybridization: 10 from the circular chromosome, six from the linear chromosome, two from the pAt plasmid, and four from the pTi plasmid. Among these, 152 were intergenic sRNAs, whereas 76 were antisense to known protein-coding genes (asRNAs). Several independently validated ncRNAs were differentially expressed under varying growth conditions such as medium, temperature, and pH. One ncRNA encoded by the Ti plasmid, Ti2, was highly induced under Vir gene induction conditions, and its expression was diminished in virA and virG deletion mutants.
Lee et al. (2013) identified 475 highly expressed candidate ncRNAs under four different growth conditions: YEP logarithmic and stationary phases and AB minimal medium in the presence or absence of AS. Because ribosomal RNAs represent the vast majority of the total cellular RNA (He et al. 2010), two commercial kits were used to deplete rRNAs and tRNAs, the TEX and MICROBExpressTM kits. All four total RNA samples were treated with reagents in the MICROBExpressTM kit, which removed ~55% of the 16S and 23S rRNAs, followed by TEX treatment (±TEX). RNA samples were fragmented to ~200–300 nt before cDNA library construction. A total of eight cDNA libraries (four growth conditions; ±TEX) were sequenced using the Illumina GAII platform. A total of 842 million 50-bp reads were obtained and 48.3 million reads were mapped exactly once to the reference genome (Uniquely Mapped Reads, UMRs). These UMRs were used for data analysis. The use of TEX treatment substantially improved the UMR ratio from 7.5 (−TEX) to 12.5% (+TEX), indicating RNA-seq sensitivity was considerably enhanced. The highly expressed ncRNA identification procedure began with calculating the depth of coverage data for each individual nucleotide position on both forward/reverse strands of all four replicons. Candidate ncRNAs were identified in the intergenic regions or complementary sequences of protein-coding genes, where the average depth of coverage of a candidate ncRNA region was at least 10 times greater than those of immediate upstream and downstream regions. A total of 101 sRNAs and 310 asRNAs were identified, as well as 20 5′ UTR leader sequences. Thirty-six ncRNAs were experimentally validated by RNA-blot hybridization and RACE (Rapid Amplification of cDNA Ends)-PCR (Gerhart et al. 2014). Twenty-two ncRNAs were differentially expressed by Vir gene induction: 15 were up-regulated and seven were down-regulated. Fourteen of the 15 AS-induced ncRNAs contain a putative vir box, a conserved motif for VirG binding, in the promoter regions. In addition to the identified ncRNAs, a stranded RNA-seq approach also revealed interesting features of Agrobacterium transcripts: (i) highly expressed antisense transcripts from the complementary strands of important vir genes and operons and (ii) novel transcripts within the known protein-coding genes (e.g., virD4* internal transcript; Lee et al. 2013). It is likely that many putative asRNAs might have been ignored due to the high stringency of the informatics cutoff, i.e., a minimum of 10 times higher expression level compared to adjacent regions, whereas internal transcripts had not been considered for ncRNA identification.
Most recently, Dequivre et al. (2015) conducted another RNA-seq study using size-fractionated RNA samples (25–500 nt). A. tumefaciens strain C58 was grown under four different growth conditions: logarithmic/stationary phases in YPG rich medium and in AB minimal medium. tRNAs were depleted using a First Strand cDNA synthesis kit. tRNA-specific primers were used to synthesize the first strand cDNAs, and RNaseH was used to degrade tRNAs in the RNA–DNA duplex, followed by DNase I treatment. A total of 193.1 million reads were obtained and 28.4 million reads were mapped once to the reference genome (UMRs). Genomic regions whose average depth was at least 10 times greater than the adjacent regions were considered transcribed, and candidate ncRNAs were identified only when a transcript was presented in all four libraries. A total of 1108 candidate ncRNAs were evenly distributed among all four replicons: 602 on the circular chromosome, 291 on the linear chromosome, 140 on the pAt plasmid, and 75 on the Ti plasmid. Four hundred and seven were intergenic sRNAs and 262 were asRNAs. Additionally, 402 and 37 were derived from 5′ and 3′ UTRs, respectively. Seventeen candidate ncRNAs were independently validated by RACE-PCR. An intergenic sRNA encoded by the Ti plasmid, RNA1111, was conserved among other Ti plasmids, and the deletion mutant exhibited reduced tumorigenicity in tomato, suggesting the involvement of this sRNA in bacterial virulence.
These three genome-wide RNA-seq studies identified a total of 1534 candidate ncRNAs from A. tumefaciens C58. As summarized in Table 2, 54 (3.5%) candidate ncRNAs were identified by all three studies, whereas 170 (11.1%) and 1310 (85.4%) ncRNAs were identified by two and one studies, respectively. The numbers presented in Table 2 are slightly different from those presented in the Venn Diagram of Dequivre et al. (2015) due to two small differences. First, one sRNA identified by Lee et al. (2013) corresponds to two tandemly encoded sRNAs identified by Wilms et al. (2012a); thus the total number of ncRNAs identified by Wilms et al. (2012a) was 228, not 227. The second was a simple calculation error, as the total number of ncRNAs is 1534 (=98 + 300 + 912 + 28 + 48 + 94 + 54; Table 2) not 1560 (Dequivre et al. 2015). As each RNA-seq investigation employed unique approaches, the collective efforts led to a thorough inventory of Agrobacterium ncRNAs. Functional analyses of these ncRNAs, however, have not been comprehensive; only several conserved ncRNAs have been characterized in Agrobacterium and other closely related species.
3 Functions of Agrobacterium ncRNAs
Although over a thousand candidate ncRNAs have been discovered from A. tumefaciens, the regulatory functions of all but a few remain unknown. Only a handful of ncRNAs have been functionally characterized thus far: repE (Chai and Winans 2005), AbcR1 (Wilms et al. 2011), a TPP riboswitch (Lee et al. 2013), and RNA1111 (Dequivre et al. 2015). Here we describe how these ncRNAs have been discovered and how they exert regulatory functions via various mechanisms. We also report how a chromosomally encoded and AS-induced sRNA, Atr35C (Lee et al. 2013), is expressed under iron deficiency and oxidative stress conditions.
3.1 Thi-Box Riboswitch
Thiamine, also known as vitamin B1, is an essential coenzyme for carbohydrate and branched-chain amino acid metabolism in all living cells. Maintaining a proper level of thiamine is critical and a highly conserved RNA structure called the Thi-box riboswitch or TPP (thiamine pyrophosphate) riboswitch regulates the biosynthesis and transport of thiamine in bacteria, archaea, and eukaryotes (Serganov and Nudler 2013; RF00059 in Rfam database). The Thi-box riboswitch binds to TPP to cause RNA structural changes which can lead to transcriptional attenuation or translational inhibition (Serganov and Nudler 2013). Three TPP riboswitches have been identified in A. tumefaciens C58, two on the circular and one on the linear chromosome (Rfam database: http://rfam.xfam.org/search?q=Agrobacterium%20fabrum%20AND%20rna_type:%22riboswitch%22%20and%20TPP%20AND%20alignment_type:%22full%22). All three Thi-box riboswitches are located in the 5′ UTR of operons encoding putative thiamine biosynthesis enzymes or transporters. Lee et al. (2013) demonstrated, using Northern Blot analysis, that a Thi-box riboswitch (C1_2541934R) located in the 5′ UTR of the thiamine biosynthesis operon thiCOGG indeed regulates gene expression via transcriptional attenuation. The thiCOGG mRNA was detected when A. tumefaciens was grown in minimal medium lacking thiamine, but only the ~110 nt riboswitch accumulated when grown in nutrient-rich medium-containing thiamine, suggesting that the thiCOGG promoter has constitutive activity and a transcriptional attenuator is formed to block transcription of the full-length mRNA of the thiamine biosynthesis genes (Lee et al. 2013). Thi-box riboswitch-mediated transcriptional attenuation was also observed in the nitrogen-fixing bacterium R. etli (Miranda-Ríos et al. 2001).
In addition to the Thi-box riboswitches, the A. tumefaciens C58 genome was predicted to encode six Cobalamin (vitamin B12), two SAM (S-Adenosyl Methionine), one Flavin mononucleotide (FMN; vitamin B2), and one glycine riboswitches (http://rfam.xfam.org/search?q=Agrobacterium%20fabrum%20AND%20rna_type:%22riboswitch%22), but their functional roles have not yet been confirmed.
3.2 RepE
The first characterized sRNA in Agrobacterium was RepE, a sRNA that regulates the replication of an octopine-type tumor-inducing Ti plasmid (Chai and Winans 2005). RepE is encoded in the intergenic region of the repABC operon, whose products are responsible for the replication of the Ti plasmid (Chai and Winans 2005). RepABC-type replication is widespread among plasmids found in alpha-proteobacteria, especially in Rhizobiales (Palmer et al. 2000). All known repABC operons include at least three genes: repA, repB, and repC (reviewed in Cevallos et al. 2008). RepA and RepB are involved in plasmid partitioning and segregation, whereas RepC is responsible for initiation of the DNA synthesis. Chai and Winans (2005) demonstrated that RepE is ~54 nt in size and suppresses the replication of a mini-Ti plasmid when expressed in trans (Chai and Winans 2005). In addition, mutations introduced at the promoter region resulted in downregulation of RepE, which subsequently increased plasmid copy number, further suggesting that RepE is a negative regulator of Ti plasmid replication. RepE likely form duplexes with repC mRNAs resulting in transcriptional attenuation (Brantl et al. 2002). Because the repE-encoded intergenic region is highly conserved in other repABC-type plasmids, it is likely that repE-mediated transcriptional attenuation is an important mechanism to maintain plasmid copy numbers in Rhizobiales.
3.3 AbcR1
The AbcR1 (ABC regulator) was the first studied chromosomally encoded sRNA in α-proteobacteria (Wilms et al. 2011). AbcR1 was discovered by a computational search (Wilms et al. 2011) in the conserved intergenic region between atu2186 and atu2187, in tandem with a homologous sRNA AbcR2. Both AbcR1 and AbcR2 are well conserved in α-proteobacteria and belong to the αr15 sRNA family (del Val et al. 2012). AbcR1/AbcR2 orthologues have been identified in other α-proteobacteria: Sinorhizobium meliloti (SmrC15/SmrC16; del Val et al. 2007), Rhizobium etli (ReC58/ReC59; Vercruysse et al. 2010), and Brucella abortus 2308 (AbcR1/AbcR2; Caswell et al. 2012). Hfq is likely required for AbcR1-meditated negative regulation of atleast for some target genes because their expression levels were elevated in both hfq and abcR1 knockout mutants (Wilms et al. 2012b).
AbcR1 regulons have been identified by one- and two-dimensional PAGE analysis (Wilms et al. 2011; Overlöper et al. 2014) or computational predictions using the CopraRNA algorithm (Wright et al. 2013). AbcR1 regulates at least 16 mRNAs including several periplasmic substrate-binding proteins required for sugar and amino acid ABC transporters (Wilms et al. 2011, 2012b; Overlöper et al. 2014): AtpH, AttC, Atu0857, Atu1879, Atu2422, Atu3114, Atu4046, Atu4259, Atu4431, Atu4577, Atu4678, ChvE, DppA, FrcB, and NocT. Among these, several target genes are involved in A. tumefaciens virulence. Atu2422 encodes a periplasmic protein which is responsible for uptake of the plant defense molecule γ-amino butyric acid (GABA) (Chevrot et al. 2006). GABA can suppress the quorum-sensing signal within A. tumefaciens, thus attenuating bacterial virulence (Chevrot et al. 2006). ChvE is a sugar-binding protein that senses host-released sugars and directly interacts with the VirA/VirG two-component system to induce vir gene expression (He et al. 2009; Hu et al. 2013). AttC and NocT are responsible for the uptake of spermidine/putrescine and nopaline, respectively (Matthysse et al. 1996).
AbcR1 possesses two separate target-binding regions (Overlöper et al. 2014) and each region binds to a set of target mRNAs either near the ribosomal binding site (RBS) to block translation and accelerate target mRNA turnover, or the coding DNA sequence (CDS) to cause transcriptional attenuation (Wilms et al. 2011, 2012b; Overlöper et al. 2014). Further studies may greatly expand the AbcR1 regulon because a large number of proteins differentially expressed by an avcR1 deletion have yet to be validated (Overlöper et al. 2014).
Recent studies showed that regulation by AbcR1/AbcR2 orthologues has become diversified in α-proteobacteria. In A. tumefaciens, AbcR1/AbcR2 have near identical promoter sequences and are highly expressed in late stationary phase, but only AbcR1 has regulatory functions (Wilms et al. 2011). Conversely, AbcR1/AbcR2 orthologues in the human pathogen B. abortus 2308 have some redundant functions, because only the abcR1/abcR2 double knockout mutant exhibited reduced survival in cultured murine macrophages (Caswell et al. 2012). B. abortus AbcR1/AbcR2 have multiple unique and shared target genes (Caswell et al. 2012). In the nitrogen-fixing bacterium S. meliloti Rm1021, AbcR1/AbcR2 orthologues (SmrC15/SmrC16) are divergently expressed: AbcR1 was expressed in actively growing cells but was not detected in stationary phase, whereas AbcR2 was highly expressed in the stationary phase and under various stress conditions (Torres-Quesada et al. 2014). Together, these data suggest that AbcR1/AbcR2 orthologues may have evolved rapidly in α-proteobacteria, but it is not yet known whether AbcR1 regulons in the plant pathogenic A. tumefaciens are also evolutionarily conserved in the human pathogen B. abortus or the nitrogen-fixing symbionts S. meliloti and R. etli.
3.4 RNA1111
RNA1111 is a recently identified sRNA from the intergenic region between atu6186 (virE3) and Atu6188 (virE0) on the complementary strand (Dequivre et al. 2015). RNA1111 is ~173 nt in length and highly conserved among the nopaline-type Ti plasmids. Although RNA1111 was located within the vir gene region, its expression level was not affected by vir gene induction conditions (Dequivre et al. 2015). Interestingly, however, an rna1111 deletion mutant exhibited reduced virulence on tomato plants: an rna1111 mutant strain harboring an empty expression vector produced an average of two tumors per plant, whereas the wild-type and rna1111 mutant harboring the complementation construct produced 20 and 9.5 tumors per plant, respectively. The complementation construct alone does not restore a full level of virulence, presumably because the deleted rna1111 gene region contains the vir box of virE0.
Because RNA1111 may be involved in A. tumefaciens virulence, the next step was to identify the regulatory targets of this sRNA. Three sRNA target search programs (RNApredator, sTarPicker, and IntaRNA) were utilized to identify a total of eight putative target genes, which were predicted by all three programs. Six candidate target genes were encoded on the pTiC58 plasmid, including three virulence-related genes (6b, virC2, and virD3), two conjugal transfer genes (traA and trbD), and a gene encoding a hypothetical protein (atu6072). Interestingly, Möller et al. (2014) found that virC2, virD3, and traA mRNAs were enriched by Hfq tagged by 3xFlag. Further studies are needed to determine if RNA1111 interacts with Hfq to regulate its putative target genes.
Quantitative reverse transcription PCR (qRT-PCR) analyses showed that trbD RNA was not detectable, and virC2/virD3 did not show altered expression in the rna1111 mutant compared to the wild-type strain. Three genes, 6b, traA, and atu6072, however, exhibited significantly lower expression levels in the rna1111 mutant. Importantly, 6b, traA, and atu6072 expression levels were not different in the rna1111 mutant strain harboring the complementation construct from those in the wild-type strain, further suggesting that RNA1111 might stabilize these target mRNAs or protect them from degradation. Together, these results suggest that RNA1111 might regulate genes involved in A. tumefaciens–plant interactions as well as in the dispersal of the Ti plasmid.
3.5 Atr35C
In our previous RNA-seq study, we identified 475 candidate ncRNAs from A. tumefaciens C58 (Lee et al. 2013). Fifteen of these were up-regulated by the vir gene inducer acetosyringone, and among these was a chromosomally encoded sRNA, C2_132595F (=Atr35C), which belongs to the αr35 sRNA family (http://rfam.xfam.org/family/ar35). Atr35C is encoded in the intergenic region between atu3124 (hypothetical protein) and atu3126 (hypothetical protein) on the linear chromosome. The first αr35 RNA family member, Smr35B, was identified from the symbiotic bacterium S. meliloti 1021 by computational prediction and experimental validation (del Val et al. 2007). A comparative genomics approach suggested that this sRNA family is conserved among certain members of the order Rhizobiales, which include both symbiotic (e.g., R. etli and R. leguminosarum) and pathogenic species (e.g., A. tumefaciens and Ochrobactrum anthropi; del Val et al. 2012).
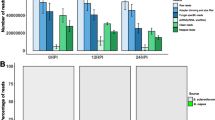
The expression of αr35 RNA was first reported in S. meliloti 1021 (del Val et al. 2007) and interestingly, it was induced by luteolin, the plant flavone that induces nodulation genes, suggesting a possible role during host–bacterial interactions. Similarly, our previous RNA-seq study found that Atr35C is induced by the vir gene inducer AS (Lee et al. 2013). qRT-PCR analysis confirmed that Atr35C is indeed induced by AS (Fig. 1a). To verify further that Atr35C is regulated by VirG, a virG mutant was generated as previously described (Lee et al. 2013), and A. tumefaciens strains were grown in induction medium (IM) containing (+Vir) or lacking AS (−Vir). qRT-PCR analysis showed that Atr35C expression was 21-fold lower in the virG mutant than in the wild-type strain in the presence of AS, and Atr35C expression was 15-fold higher in the presence of AS in the wild-type strain (Fig. 1a). By comparison, in our previous RNA-seq study, the Atr35C level was 6.1-fold higher in the presence of AS in the wild-type strain C58 (Lee et al. 2013, Table S4A. C2_132595F). Together, these results strongly suggest that the chromosomally encoded sRNA Atr35C is regulated by VirG.

Atr35C transcript levels were estimated by RT-qPCR using the 2−ΔΔCT method (Livak and Schmittgen 2001) as described previously (Lee et al. 2013). a Atr35C transcript level was not induced by AS in the virG mutant, suggesting VirG-dependent expression. b Atr35C expression was negatively correlated with the concentration of iron and manganese ions. c Atr35C transcript level was further enhanced by 9 mM hydrogen peroxide (H2O2). Error bars represent standard errors
Because Atr35C appears to be part of the VirG regulon, we examined if an atr35C mutant has altered virulence. However, neither transient GUS expression using an Arabidopsis seedling assay (Wu et al. 2014) nor tumorigenicity assay using tobacco leaf disks (Clemente 2006) showed significant differences between the atr35C mutant and wild-type C58 strains.
In search of environmental/stress stimuli that trigger Atr35C expression, several transition metals were added to the IM, and qRT-PCR assays were used to monitor Atr35C transcript levels. When added to a concentration of 100 µM, FeCl3 and MnCl2 greatly reduced Atr35C expression levels in the presence of AS (Fig. 1b), whereas CuSO4 and ZnSO4 did not have a significant impact. Because typical IM contains 10 μM FeSO4 (Gelvin 2006), we tested if there were a dosage effect of iron and manganese. Addition of 10 µM ferric (FeCl3) and ferrous (FeSO4) irons to IM resulted in a mild reduction of Atr35C expression levels by about 3.8- (27.5 vs. 7.3) and 1.5-fold (27.5 vs. 18.9), respectively. A higher iron concentration further reduced Atr35C transcript levels: addition of 50 µM FeCl3 and FeSO4 reduced Atr35C transcript levels by 35.3- (27.5 vs. 0.8) and 12.9-fold (27.5 vs. 2.1), respectively. Thus, A. tumefaciens is more responsive to ferric than to ferrous iron (3.8- vs. 1.5-fold changes at 10 µM; 35.3- vs. 12.9-fold change at 50 µM). Addition of 50 µM MnCl2 reduced Atr35C transcript levels by 55-fold (27.5 vs. 0.5). These results strongly suggest that Atr35C might be involved in iron and manganese homeostasis.
Interestingly, iron and manganese play important roles in oxidative stress responses and virulence in A. tumefaciens (Saenkham et al. 2008; Kitphati et al. 2007). We therefore tested if hydrogen peroxide (H2O2), a primary defense molecule of plants (Wojtaszek 1997; Dan et al. 2015), affects Atr35C expression. Wild-type A. tumefaciens C58 was grown in the presence of AS for 24 h and 9 mM H2O2 was added to the culture and further incubated for 30 min. Atr35C transcript levels increased by ~twofold in the presence of 9 mM H2O2 compared to the control with 0, 10, or 50 µM FeCl3 or FeSO4 (Fig. 1c). However, Atr35C transcript levels were not affected by 9 mM H2O2 in the presence of 50 µM MnCl2. Taken together, our results suggest that there is cross-talk between the Ti plasmids and the chromosomally encoded sRNA Atr35C, which might be involved in oxidative stress responses or iron/manganese homeostasis. Further studies are needed to identify the target genes regulated by Atr35C and to elucidate how this sRNA exerts regulatory functions.
4 Challenges
Regulatory ncRNAs are versatile and provide bacteria many adaptive advantages in rapidly changing environments. As mentioned above, however, the biological functions of most ncRNAs remain largely unknown; only a small number of ncRNAs have been functionally characterized in A. tumefaciens. This can be attributed to the characteristics of ncRNAs and their interactions with targets: (1) poor sequence conservation in homologous ncRNAs, (2) imperfect complementarity in sRNA–mRNA base-pairing, and (3) quantitative changes in target gene expression.
Bacterial ncRNA homologs have a low level of primary sequence conservation among evolutionarily distantly related species. Consequently, most ncRNA homologs are only found among closely related bacteria. This observation significantly limits data mining, which can provide useful information such as conserved domains, putative functions, and interactions with putative targets and transcription factors. For instance, although 1534 candidate ncRNAs have been discovered in A. tumefaciens C58 thus far, only 45 families are found in the Rfam database (http://rfam.xfam.org/search?q=UP000000813%20AND%20entry_type:%22Family%22). Among these are 5S rRNA (RF00001), RNase P RNA (RF00010), SRP RNA (RF00169), and 6S RNA (RF00013). Other than these broadly conserved ncRNAs, α-proteobacterial ncRNA families, such as αr7 (RF02342), αr9 (RF02343), αr14 (RF02344), αr15 (RF02345), αr35 (RF02346), and αr45 (RF02347) still lack known functions. Extended searches for conserved secondary structures and adjacent protein-coding genes have proven useful to facilitate homologous ncRNA discovery (Barrick et al. 2005), but it still remains challenging to identify functional analogs among distantly related bacteria.
In contrast to eukaryotic miRNAs and siRNAs that base-pair with target mRNAs with near-perfect complementarity (Brodersen et al. 2008), bacterial ncRNAs, especially sRNAs, interact with target mRNAs via base-pairing with a less perfect complementarity and with gaps (Storz et al. 2011). This finding poses a difficult challenge to identify sRNA targets using existing computational algorithms (Pain et al. 2015). Even validated sRNA-mRNA target pairs are not predicted as top candidates (Pain et al. 2015), which strongly suggests that there are unknown crucial factors determining sRNA-mRNA specificities, or that current computational algorithms need further optimization. Many RNA-seq-based approaches have recently been developed to identify sRNA targets (reviewed in Saliba et al. 2017), but these approaches are costly and time consuming for extensive optimization and data analyses. Undoubtedly, additional experimentally validated ncRNA–target interactions will improve ncRNA target prediction algorithms in the future, but it is important to expand the search algorithms to include protein databases because some ncRNAs directly interact with protein targets.
As post-transcriptional regulators, some ncRNAs do not dramatically alter target gene transcript levels, whereas others only affect target mRNA translation without altering mRNA stability (Storz et al. 2011). In addition, as demonstrated by Levine et al. (2007), ncRNA-mediated gene regulation is largely affected by the rate of transcription of the target genes. Therefore, it is crucial to define the conditions under which a specific ncRNA exerts regulatory effects on target gene expression. Moreover, a high level of heterogeneity exists among the individual cells in bacterial colonies (Martins and Locke 2015), but standard procedures measure only the average levels in a population. In this regard, single-cell-based analyses may provide useful platforms to measure precisely the regulatory effects of ncRNAs on target gene expression. Recent advances in single-cell-based RNA-seq and proteomics look promising to provide more accurate genome-wide pictures of complex regulatory networks, including ncRNA regulators (Shapiro et al. 2013; Martins and Locke 2015).
5 Conclusions
RNA-seq approaches allowed identification of 1534 candidate ncRNAs from A. tumefaciens C58 (Table 2). This is, however, only the beginning of the regulatory ncRNA era, and a number of questions remain unanswered. For example, how many ncRNAs are true regulators? Do asRNAs represent important regulators or mere by-products of transcriptional noise? Which ncRNAs, if any, modulate Agrobacterium–plant interactions? Although there are many challenges for ncRNA research, accumulating evidence has solidified the importance of ncRNA regulators for many aspects of biological reactions and stress responses. Technical advances, such as single-cell-based RNA-seq and proteomics, will provide new tools to reveal how ncRNAs specify targets, both RNAs and proteins, and how multiple layers of regulatory networks interact harmoniously with one another to maximize bacterial fitness. This in turn offers an excellent opportunity to improve the efficiency and host-range of A. tumefaciens-mediated plant genetic transformation.
References
Aiba H, Adhya S, de Crombrugghe B (1981) Evidence for two functional gal promoters in intact Escherichia coli cells. J Biol Chem 256:11905–11910
Barrick JE, Sudarsan N, Weinberg Z et al (2005) 6S RNA is a widespread regulator of eubacterial RNA polymerase that resembles an open promoter. RNA 11:774–784
Becker A, Overlöper A, Schlüter JP et al (2014) Riboregulation in plant-associated-proteobacteria. RNA Biol 11:550–562
Brantl S (2007) Regulatory mechanisms employed by cis-encoded antisense RNAs. Curr Opin Microbiol 10:102–109
Brantl S, Wagner E, Gerhard H (2002) An antisense RNA-mediated transcriptional attenuation mechanism functions in Escherichia coli. J Bacteriol 184:2740–2747
Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M et al (2008) Widespread translational inhibition by plant miRNAs and siRNAs. Science 30:1185–1190
Caswell CC, Gaines JM, Ciborowski P et al (2012) Identification of two small regulatory RNAs linked to virulence in Brucella abortus 2308. Mol Microbiol 85:345–360
Cevallos MA, Cervantes-Rivera R, Gutiérrez-Ríos RM (2008) The repABC plasmid family. Plasmid 60:19–37
Chai Y, Winans SC (2005) A small antisense RNA downregulates expression of an essential replicase protein of an Agrobacterium tumefaciens Ti plasmid. Mol Microbiol 56:1574–1585
Chevrot R, Rosen R, Haudecoeur E et al (2006) GABA controls the level of quorum-sensing signal in Agrobacterium tumefaciens. Proc Natl Acad Sci USA 103:7460–7464
Clemente T (2006) Nicotiana (Nicotiana tobaccum, Nicotiana benthamiana) In: Wang K (ed) Agrobacterium protocols, 2nd edn. Humana Press Inc., New Jersey, pp 143–154
Dan Y, Zhang S, Zhong H et al (2015) Novel compounds that enhance Agrobacterium-mediated plant transformation by mitigating oxidative stress. Plant Cell Rep 34:291–309
De Lay N, Schu D, Gottesman S (2013) Bacterial small RNA-based negative regulation: Hfq and its accomplices. J Bio Chem 288:7996–8003
del Val C, Rivas E, Torres-Quesada O et al (2007) Identification of differentially expressed small non-coding RNAs in the legume endosymbiont Sinorhizobium meliloti by comparative genomics. Mol Microbiol 66:1080–1091
del Val C, Romero-Zaliz R, Torres-Quesada O et al (2012) A survey of sRNA families in α-proteobacteria. RNA Biol 9:119–129
Dequivre M, Diel B, Villard C et al (2015) Small RNA deep-sequencing analyses reveal a new regulator of virulence in Agrobacterium fabrum C58. Mol Plant-Microbe Interact 28:580–589
Gelvin SB (2006) Agrobacterium virulence gene induction. In: Wang K (ed) Agrobacterium protocols, 2nd edn. Humana Press Inc, New Jersey, pp 77–84
Georg J, Hess WR (2011) cis-antisense RNA, another level of gene regulation in bacteria. Microbiol Mol Biol Rev 75:286–300
Gerhart E, Wagner H, Vogel J (2014) Approaches to identify novel non-messenger RNAs in bacteria to investigate their biological functions: functional analysis of identified non-mRNAs. In: Hartmann RK, Bindereif A, Schön A, Westhof E (eds) Handbook of RNA biochemistry, 2nd edn. Wiley-VCH Verlag GmbH & Co., Weinheim, pp 719–786
Gottesman S, Storz G (2011) Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Bio 3:1–16
Harfouche L, Haichar F, Achouak W (2015) Small regulatory RNAs and fine-tuning of plant-bacteria interactions. New Phytol 206:98–106
He F, Nair GR, Soto CS et al (2009) Molecular basis of ChvE function in sugar binding, sugar utilization, and virulence in Agrobacterium tumefaciens. J Bacteriol 191:5802–5813
He S, Wurtzel O, Singh K et al (2010) Validation of two ribosomal RNA removal methods for microbial metatranscriptomics. Nat Methods 7:807–812
Hu X, Zhao J, DeGrado WF et al (2013) Agrobacterium tumefaciens recognizes its host environment using ChvE to bind diverse plant sugars as virulence signals. Proc Natl Acad Sci USA 110:678–683
Kitphati W, Ngok-Ngam P, Suwanmaneerat S et al (2007) Agrobacterium tumefaciens fur has important physiological roles in iron and manganese homeostasis, the oxidative stress response, and full virulence. Appl Environ Microbiol 73:4760–4768
Landt SG, Abeliuk E, McGrath PT et al (2008) Small non-coding RNAs in Caulobacter crescentus. Mol Microbiol 68:600–614
Lee K, Huang X, Yang C et al (2013) A genome-wide survey of highly expressed non-coding RNAs and biological validation of selected candidates in Agrobacterium tumefaciens. PloS One 8:e70720
Levine E, Zhang Z, Kuhlman T et al (2007) Quantitative characteristics of gene regulation by small RNA. PLoS Biol 5(9):e229
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408
Livny J, Waldor MK (2007) Identification of small RNAs in diverse bacterial species. Curr Opin Microbiol 10:96–101
Livny J, Teonadi H, Livny M et al (2008) High-throughput, kingdom-wide prediction and annotation of bacterial non-coding RNAs. PLoS ONE 3:e3197
Lloréns-Rico V, Cano J, Kamminga T et al (2016) Bacterial antisense RNAs are mainly the product of transcriptional noise. Sci Adv 2(3):e1501363
Loh E, Dussurget O, Gripenland J et al (2009) A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 139:770–779
Maes M, Messens E (1992) Phenol as grinding material in RNA preparations. Nucleic Acids Res 20:4374
Martins BMC, Locke JCW (2015) Microbial individuality: how single-cell heterogeneity enables population level strategies. Curr Opin Microbiol 24:104–112
Matthysse AG, Yarnall HA, Young N (1996) Requirement for genes with homology to ABC transport systems for attachment and virulence of Agrobacterium tumefaciens. J Bacteriol 178:5302–5308
Mellin JR, Cossart P (2015) Unexpected versatility in bacterial riboswitches. Trends Genet 31:150–156
Miranda-Ríos J, Navarro M, Soberón M (2001) A conserved RNA structure (thi box) is involved in regulation of thiamin biosynthetic gene expression in bacteria. Proc Natl Acad Sci USA 98:9736–9741
Mironov AS, Gusarov I, Rafikov R et al (2002) Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell 111:747–756
Mӧller P, Overlöper A, Förstner KU et al (2014) Profound impact of Hfq on nutrient acquisition, metabolism and motility in the plant pathogen Agrobacterium tumefaciens. PLoS ONE 9:e110427
Nahvi A, Sudarsan N, Ebert M et al (2002) Genetic control by a metabolite binding mRNA. Chem Biol 9:1043–1049
Overlöper A, Kraus A, Gurski R et al (2014) Two separate modules of the conserved regulatory RNA AbcR1 and address multiple target mRNAs in and outside of the translation initiation region. RNA Biol 11:624–640
Pain A, Ott A, Amine H et al (2015) An assessment of bacterial small RNA target prediction programs. RNA Biol 12:509–513
Palmer KM, Turner SL, Young JPW (2000) Sequence diversity of the plasmid replication gene repC in the Rhizobiaceae. Plasmid 44:209–219
Romeo T (1998) Global regulation by the small RNA-binding proteins CsrA and the non-coding RNA molecule CsrB. Mol Microbiol 29:1321–1330
Saenkham P, Utamapongchai S, Vattanaviboon P et al (2008) Agrobacterium tumefaciens iron superoxide dismutases have protective roles against singlet oxygen toxicity generated from illuminated Rose Bengal. FEMS Microbiol Lett 289:97–103
Saliba AE, Santos SC, Vogel J (2017) New RNA-seq approaches for the study of bacterial pathogens. Curr Opin in Microbiol 35:78–87
Serganov A, Nudler E (2013) A decade of riboswitches. Cell 152:17–24
Sesto N, Wurtzel O, Archambaud C et al (2013) The excludon: A new concept in bacterial antisense RNA-mediated gene regulation. Nat Rev Microbiol 11:75–82
Shapiro E, Biezuner T, Linnarsson S (2013) Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet 14:618–630
Sharma CM, Vogel J (2009) Experimental approaches for the discovery and characterization of regulatory small RNAs. Curr Opin Microbiol 12:536–546
Simons RW, Kleckner N (1983) Translational control of IS10 transposition. Cell 34:683–691
Smirnov A, Förstner KU, Holmqvist E et al (2016) Grad-seq guides the discovery of ProQ as a major small RNA-binding protein. Proc Natl Acad Sci USA 113:11591–11596
Storz G, Vogel J, Wassarman KM (2011) Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43:880–891
Stougaard P, Molin S, Nordström K (1981) RNAs involved in copy number control and incompatibility of plasmid R1. Proc Natl Acad Sci USA 78:6008–6012
Thomason MK, Bischler T, Eisenbart SK et al (2015) Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol 197:18–28
Tinsley RA, Furchak JRW, Walter NG (2007) Trans-acting glmS catalytic riboswitch: locked and loaded. RNA 13:468–477
Toledo-Arana A, Dussurget O, Nikitas G et al (2009) The Listeria transcriptional landscape from saprophytism to virulence. Nature 459:950–956
Tomizawa J, Itoh T, Selzer G et al (1981) Inhibition of ColE1 RNA primer formation by a plasmid-specified small RNA. Proc Natl Acad Sci USA 78:1421–1425
Torres-Quesada O, Reinkensmeier J, Schlűter JP et al (2014) Genome-wide profiling of HFq-binding RNAs uncovers extensive post-transcriptional rewiring of major stress response and symbiotic regulons in Sinorhizobium meliloti. RNA Biol 11:563–579
Vercruysse M, Fauvart M, Cloots L et al (2010) Genome-wide detection of predicted non-coding RNAs in Rhizobium etli expressed during free-living and host-associated growth using a high resolution tiling array. BMC Genom 11:53
Wade JT, Grainger DC (2014) Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat Rev Microbiol 12:647–653
Wagner EG, Simons RW (1994) Antisense RNA control in bacteria, phages, and plasmids. Annu Rev Microbiol 48:713–742
Wassarman KM, Storz G (2000) 6S RNA regulates E. coli RNA polymerase activity. Cell 101:613–623
Waters LS, Storz G (2009) Regulatory RNAs in bacteria. Cell 13:615–628
Wilms I, Voss B, Hess WR et al (2011) Small RNA-mediated control of the Agrobacterium tumefaciens GABA binding protein. Mol Microbiol 80:492–506
Wilms I, Overlöper A, Nowrousian M et al (2012a) Deep sequencing uncovers numerous small RNAs on all four replicons of the plant pathogen Agrobacterium tumefaciens. RNA Biol 9(4):446–457
Wilms I, Möller P, Stock AM et al (2012b) Hfq influences multiple transport systems and virulence in the plant pathogen Agrobacterium tumefaciens. J Bacteriol 194:5209–5217
Winkler WC, Breaker RR (2005) Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol 59:487–517
Winkler W, Nahvi A, Breaker RR (2002) Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 419:952–956
Wojtaszek P (1997) Oxidative burst: an early plant response to pathogen infection. Biochem J 322:681–692
Wright PR, Richter AS, Papenfort K et al (2013) Comparative genomics boosts target prediction for bacterial small RNAs. Proc Natl Acad Sci USA 110:E3487–E3496
Wu HY, Liu KH, Wang YC et al (2014) AGROBEST: an efficient Agrobacterium-mediated transient expression method for versatile gene function analyses in Arabidopsis seedlings. Plant Methods 10:19
Acknowledgements
The authors thank Abbagail Johnson, Juan Carlos Martinez-Nicolas, Alan Eggenberger, and Jonah Miller for their assistances. This work was partially supported by the USDA National Institute of Food and Agriculture, Hatch project number #IOW05162, by State of Iowa funds, and by Charoen Pokphand Indonesia.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Lee, K., Wang, K. (2018). Small Noncoding RNAs in Agrobacterium tumefaciens. In: Gelvin, S. (eds) Agrobacterium Biology. Current Topics in Microbiology and Immunology, vol 418. Springer, Cham. https://doi.org/10.1007/82_2018_84
Download citation
DOI: https://doi.org/10.1007/82_2018_84
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-03256-2
Online ISBN: 978-3-030-03257-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)