
Abstract
This chapter presents a historical overview of the development and changes in scientific approaches to classifying members of the Agrobacterium genus. We also describe the changes in the inference of evolutionary relationships among Agrobacterium biovars and Agrobacterium strains from using the 16S rRNA marker to recA genes and to the use of multilocus sequence analysis (MLSA). Further, the impacts of the genomic era enabling low cost and rapid whole genome sequencing on Agrobacterium phylogeny are reviewed with a focus on the use of new and sophisticated bioinformatics approaches to refine phylogenetic inferences. An updated genome-based phylogeny of ninety-seven Agrobacterium tumefaciens complex isolates representing ten known genomic species is presented, providing additional support to the monophyly of the Agrobacterium clade. Additional taxon sampling within Agrobacterium genomovar G3 indicates potential exceptions to interpretation of the concept of bacterial genomics species as ecological species because the genomovar G3 genomic cluster, which initially includes clinical strains, now also includes plant-associated and cave isolates.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Agrobacterium Biovar
- Genome-based Phylogeny
- Rapid Whole-genome Sequencing
- Genome Species
- Agrobacterium Vitis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Since the first uses of DNA sequences to classify relationship among bacterial strains became routine (Janda and Abbott 2007; Stackebrandt and Goebel 1994), new and increasing amounts of single-copy protein-coding DNA markers have been employed to revaluate and revise the taxonomy of Agrobacterium. Phylogenetic analyses based on increased taxon and gene sampling have led to the reclassification of the traditional Agrobacterium biovars 1 and 3 to two new genera (Costechareyre et al. 2010; Mousavi et al. 2014). With the now common practice of sequencing whole bacterial genomes, large data sets are increasingly available, and these sequences have become linked to more sophisticated approaches to analyse data using multiple and linear bioinformatical approaches. These approaches have provided new and improved insight into the evolutionary relationships among Agrobacterium species. In this review chapter, we first provide a historical overview of the molecular systematics of the genus Agrobacterium which led to an intense debate among the scientific community during the 16S rRNA era. We next review changes to the Agrobacterium taxonomy which is gradually embraced by the scientific community in the light of more recent and refined phylogenetic analyses using improved gene and taxon sampling. The unprecedented genetic information about Agrobacterium derived from the advent of next-generation sequencing and its impacts on the inference and delineation of Agrobacterium at the strain level is summarized. We also provide a genome-based phylogeny of ninety-seven Agrobacterium tumefaciens complex isolates, representing a significant increase in taxon sampling compared to a previous phylogenomic study (Ormeno-Orrillo et al. 2015). The validity of bacterial genome species being ecological species (Lassalle et al. 2011) is briefly assessed and discussed in the light of new phylogenomic inferences and observed ecological niche diversity among recently sequenced strains belonging to Agrobacterium genomovar 3.
2 Pre-2006 Agrobacterium Taxonomy
The use of 16S rRNA sequence as a genetic marker for microbial taxonomy brought about both chaos and order within the taxonomy of Agrobacterium. The availability of universal 16S rRNA primers and the inherent high copy number of 16S rRNA in most bacterial genomes are two of the main attributes promoting the inclusion of the 16S rRNA sequence as part of the developed polyphasic taxonomy for bacteria (Janda and Abbott 2007; Woo et al. 2008). Furthermore, the high sequence conservation of the 16S rRNA gene makes it a very powerful genetic marker when inferring deep relationships. However, at the species or genus level, the use of the 16 s rRNA gene to discriminate among species tends to be modest if not inferior to other universal genetic markers (Kisand and Wikner 2003; Stackebrandt and Goebel 1994).
It is important to note that 16S rRNA gene substitution rates appear to vary among different groups of bacteria (Ochman et al. 1999; Smit et al. 2007). In other words, if the 16S rRNA gene substitution rates are lower in the family Rhizobiaceae, this will translate into low 16S rRNA gene nucleotide divergence and/or phylogenetic signals among members of the Rhizobiaceae. This may negatively affect phylogenetic interpretation, raising doubts about the veracity of their inferred evolutionary relationships. An initial proposal by Young et al. (2001) to incorporate all species of Agrobacterium and Allorhizobium into the genus Rhizobium due to the lack of concordance between DNA hybridization, biochemical traits, and fatty acid profiles among members of the described genera sparked an intense response from the scientific community (Farrand et al. 2003; Young et al. 2001). Farrand et al. (2003) claimed that members of the genus Agrobacterium and Rhizobium can be distinguished based on chromosomal structure and phenotype (as an individual species but not genera). Young et al. (2001) replied to Farrand et al. (2003) defending the initial proposal in addition to highlighting that the proposal is in accordance with the rules/codes set out by the International Code of Nomenclature of Bacteria. Young et al. (2001) further cautioned that bending the codes to retain the genus Agrobacterium may trigger a potential return to unregulated and chaotic bacterial nomenclature. The initial classification of Agrobacterium species based on their pathogenicity has been problematic, as it is now well established that the virulence factors are usually encoded on plasmids and some of these can even be lost relatively easily through growth at elevated temperature (Genetello et al. 1977). For further reading on the change and development in Agrobacterium taxonomy until 2006, we direct reader to a comprehensive review by Young (2008).
3 Alternative Views of the Agrobacterium Phylogeny
3.1 The recA Gene as an Alternative Genetic Marker to 16S rRNA for Inferring Agrobacterium Phylogeny
The recA gene encodes a multifunctional and important enzyme involved in homologous recombination and DNA repair (Kowalczykowski et al. 1994). A recA mutant is therefore characterized by its high sensitivity to UV light in addition to being recombination-deficient, a desirable trait for genetic studies involving trans-complementation of mutations located on a chromosome or plasmid (Kanie et al. 2007; Kuzminov and Stahl 1997). The importance of a recA mutant is well recognized among Agrobacterium geneticists, leading to the construction of strains LBA4301 and UIA143, recA mutants of Agrobacterium tumefaciens Ach5, and Agrobacterium tumefaciens C58, respectively (Farrand et al. 1989). Beyond molecular genetics, the recA gene is also well known in molecular systematics (Lloyd and Sharp 1993) and has been incorporated as one of the main genes for multilocus typing (MLSA) (Bennasar et al. 2010; Delamuta et al. 2012; Huo et al. 2017; Martens et al. 2008; Menna et al. 2009; Sakamoto and Ohkuma 2011). Phylogenetic analysis based on the recA gene of 138 strains from 13 genomic species of Agrobacterium lends support to the use of this marker gene for speciation of the genus Agrobacterium (Costechareyre et al. 2010). Genomic species is a concept of bacterial species based on similarities among bacterial chromosomal DNAs as determined by DNA–DNA hybridization or alternatively by in silico calculation of pair-wise average nucleotide identity (ANI) using whole genome sequences (Konstantinidis et al. 2006; Stackebrandt and Goebel 1994). A genomic species is defined as a group of bacterial strains with DNA–DNA reassociation values of more than 70%, which corresponds closely to ~95% ANI (Konstantinidis et al. 2006). A recA-based phylogenetic analysis indicates that Agrobacterium biovar 2, typically represented by Agrobacterium rhizogenes, and biovar 3 represented by Agrobacterium vitis are distantly related to Agrobacterium biovar 1. In addition, inclusion of recA sequences from several Rhizobium type strains in the analysis showed a stronger affiliation of Agrobacterium rhizogenes and Agrobacterium vitis to the Rhizobium clade.
3.2 Four (or Six) Is Better Than One: Refining and Revising the Agrobacterium Genus Through Multilocus Sequence Analysis (MLSA)
Phylogenetic tree construction based on six protein-coding housekeeping genes consisting of ATP synthase F1, beta subunit (atpD), glutamine synthetase type I (glnA), glutamine synthetase type II (glnII), recombinase A (recA), RNA polymerase beta subunit (rpoB), and threonine synthase (thrC) from 114 rhizo- and agrobacteria reinforced the monophyly of the genus Agrobacterium which was previously reestablished based on the recA gene. In addition to resolving other pending taxonomic issues related to the family Rhizobiaceae, the substantial increase in gene and taxon sampling also lent support to the reclassification of Agrobacterium vitis to an existing genus Allorhizobium (Mousavi et al. 2014). Once belonging to three different biovars of the same genus, the phythopathogenic Agrobacterium tumefaciens, Agrobacterium vitis (now Allorhizobium vitis), and Agrobacterium rhizogenes (now Rhizobium rhizogenes) now belong to three different genera. Furthermore, with the creation of the genus Neorhizobium which is a sister group to Agrobacterium, Agrobacterium can now remain a suitable genus name for a monophyletic clade within the Rhizobiaceae family. A follow-up study based on three housekeeping genes and the 16S rRNA gene again supported the monophyly of the revised Agrobacterium clade in addition to expanding the membership of the genus Allorhizobium to include R. taibanshenase, R. paknamense, R. oryzae, R.psuedoryzae, R. qilianshanense, and R. borbori. However, in contrast to a previous study based on six housekeeping genes, a sister grouping of Agrobacterium–Neorhizobium was not observed. The Agrobacterium clade instead shared a sister grouping with the R. aggregatum complex (Mousavi et al. 2015). Mousavi et al., however, did not suggest the reclassification of members from the R. aggregatum complex to the genus Agrobacterium as members of this sister clade, citing the lack of Agrobacterium-specific genome architecture (linear chromosome and the presence of the protelomerase-coding gene, telA (Ramirez-Bahena et al. 2014).
4 Agrobacterium and the Genomic Era
4.1 Pre-next-Generation Sequencing Period
Whole genome sequencing provides an unprecedented view into the evolutionary relationships of microorganisms. With a repertoire of single-copy and near-universal genes, usually in the range of hundreds, that can be used for phylogenetic inference, there is no longer a limitation to gene sampling, one of the main requirements for accurate phylogenetic analysis (Hedges 2002; Rosenberg and Kumar 2003). Agrobacterium tumefaciens C58 (now Agrobacterium fabrum C58) is the first Agrobacterium strain to have its complete genome sequenced by two separate research groups using conventional Sanger sequencing (Goodner et al. 2001; Wood et al. 2001) and subsequently revised with improved annotation (Slater et al. 2013). Approximately nine years later, the complete genome for members from the remaining two biovars, e.g. Agrobacterium vitis (biovar 3, now Allorhizobium vitis) and Agrobacterium radiobacter (biovar 2, now Rhizobium sp.; Slater et al. 2009), was reported. In addition, for the first time a high-resolution phylogeny of Agrobacterium was constructed based on the concatenated protein alignment of 507 single-copy orthologous gene families encoded on the primary chromosomes. Phylogenetic clustering patterns indicated that biovar 2 should be grouped to the genus Rhizobium, whereas biovar 3 and biovar 1 are still members of the Agrobacterium genus. The limited taxon sampling resulting from the high cost of whole genome sequencing at the time unfortunately prevented Slater et al. (2009) from inferring the delineation of biovar 3 and biovar 1 into two separate genera.
4.2 Next-Generation Sequencing and Agrobacterium
The advent of next-generation sequencing brought about a revolution in microbial genomics by enabling the whole genome sequence of a pure culture to be obtained at a small fraction of the cost and time initially required by Sanger sequencing (MacLean et al. 2009; Metzker 2010). Coupled with advances in algorithms for quick and accurate microbial genome assembly and annotation (Bankevich et al. 2012; Seemann 2014), the scientific community is now blessed with an explosion of publicly available microbial genomic resources which naturally invite a new investigation of the phylogeny of Agrobacterium. Ormeno-Orrillo and workers used a sophisticated and reproducible bioinformatics pipeline (Segata et al. 2013) to reconstruct the Agrobacterium phylogeny based on the concatenated alignment of 384 universal proteins identified from 113 sequenced strains from the family Rhizobiaceae (Ormeno-Orrillo et al. 2015). In contrast to the previously inferred whole genome phylogeny, Agrobacterium vitis S4 no longer formed a tight cluster with Agrobacterium tumefaciens C58. Instead, the increased taxon sampling supported previous recA and MLSA-based analyses indicating the monophyletic clustering of Agrobacterium vitis S4 with members of the genus Allorhizobium such as Allorhizobium undicola (de Lajudie et al. 1998), lending further support to the revival of Allorhizobium as a genus within the Rhizobiaceae (Mousavi et al. 2014). By reclassifying Agrobacterium biovars 2 and 3 into separate genera (Mousavi et al. 2014, 2015; Velázquez et al. 2010), a monophyletic cluster consisting solely of members from the genus Agrobacterium can be obtained with maximal support, indicating that at the genomic level, Agrobacterium is a definable genus of the family Rhizobiaceae (Ormeno-Orrillo et al. 2015). The author noted, however, the exclusion of an important Agrobacterium genome, e.g. Agrobacterium radiobacter NCPPB 3001 = DSM30147T (accession number ASXY01, Bioproject PRJNA212112; Zhang et al. 2014) from their analysis, citing unusual genomic anomalies such as low sequence homology (<97%) to some of its published gene sequences. Leveraging the recent availability of key Agrobacterium species genomes, Kim and Gan (2017) performed a smaller scale phylogenomic analysis of the genus Agrobacterium showing the monophyletic clustering of A. tumefaciens B6 and A. radiobacter NCPPB 3001T = DSM30147T with high pair-wise ANI value (>95%), providing conclusive genomic evidence that both strains are identical species (Kim and Gan 2017).
4.3 Updating the Agrobacterium Phylogeny in the Light of More Publicly Available Genomic Resources
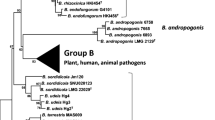
In this chapter, we present an updated phylogeny of Agrobacterium and more generally the Rhizobiaceae using a similar PhyloPhlAn approach implemented by Ormeno-Orrillo et al. (2015). PhyloPhlAn is a bioinformatics pipeline which takes the predicted proteomes from multiple microbial strains in fasta format as input and uses an ultra-fast protein similarity search (Edgar 2010) to identify more than 400 single-copy and conserved proteins within each predicted proteome. The identified proteins are aligned individually using MUSCLE (Edgar 2004), concatenated, and used for maximum likelihood tree reconstruction with FastTree2 (Price et al. 2010). Consistent with previous reports, a cluster consisting of mainly Agrobacterium strains could be recovered with maximal support, with Agrobacterium rubi and Agrobacterium larrymoorei being basal to the rest of Agrobacterium (Figs. 1 and 2). The presence of a substantial number of Rhizobium strains in the Agrobacterium clade (Fig. 2) is an aftermath of Young et al.’s initial proposal (2001) for merging Agrobacterium with the genus Rhizobium. In addition, the phylogenetic placement of A. radiobacter DSM 30147T basal to the rest of agrobacteria genomovar 4, which now includes a more recent and improved genome of A. radiobacter DSM 30147T (=NCPPB3001T; WGS Accession: LMVJ01; Lee et al., unpublished), is unusual, suggesting a genome assembly anomaly as previously noted (Ormeno-Orrillo et al. 2015). Another notable anomaly revealed by increased taxon sampling is the unexpected clustering of strain LBA4404, a disarmed derivative of the wild-type Ach5 Tn904 mutant (strain LBA4213), with members from the genovar 8 containing Agrobacterium fabrum C58 (Ooms et al. 1982). Recently, both strains Ach5 and LBA4213 have been sequenced by two independent groups (Henkel et al. 2014; Huang et al. 2015) and in contrast to strain LBA4404, both strains resided in the genomovar 1 clade, forming a monophyletic group. Given the known divergence between strain Ach5 and strain C58, this strongly indicates that the currently deposited whole genome sequence of strain LBA4404 is incorrect and warrants future investigation. The abnormal phylogenetic placement of strain LBA4404 was similarly observed but not explicitly mentioned in a study by Ormeno-Orrillo et al. (2015). Clustering based on genospecies is apparent; albeit the relationships among some of the genospecies are not strongly supported, suggesting the limitation of amino acid-based phylogenomic analysis for fully resolving strain, subspecies, and/or species-level relationships similarly observed in a recent genome-based phylogeny of Pseudomonas (Tran et al. 2017). To infer accurately the phylogeny of the currently well-supported Agrobacterium clade, future work utilizing the newly published phylogenetic-aware pan-genome analysis tool (Ding et al. 2017) to improve the recovery of core Agrobacterium single-copy genes, coupled with complementary analysis based on pair-wise average nucleotide identity (ANI) (Richter et al. 2016), will be instructive.

Reconstruction of the Rhizobiaceae phylogeny using maximum likelihood inference based on the concatenated amino acid alignment of universal single-copy genes as implemented in the PhyloPhlAn pipeline (Segata et al. 2013). Members of the family Sphingomondaceae were rooted as the outgroup. Values along branch indicate SH-like aLRT support values (Shimodaira and Hasegawa 1999) calculated using FastTree2 (Price et al. 2010)

Expanded Agrobacterium clade from Fig. 1 depicting the evolutionary relationships among Agrobacterium strains. First text strings are the WGS accession numbers, and the first letters after the strings represent the submitted genus name (R = Rhizobium; A = Agrobacterium). Taxon name is as per species name deposited into the NCBI whole genome shotgun database. Taxa coloured green: Agrobacterium rubi; taxa coloured red: Agrobacterium tumefaciens; taxa coloured blue: Agrobacterium fabrum. Nodes were coloured according to their SH-like local support values, and genomic species clusters were indicated by the vertical lines or arrows next to the tree. Asterisk signs indicate taxa that were included in a previous large-scale phylogenomic analysis by Ormeno-Orrillo et al. (2015). The tree was constructed using a whole genome-based (400 universal single-copy genes) approach
5 Genomic Species Within Agrobacterium
Traditionally, genome–genome hybridization has been used to establish genomic relatedness among strains, and a hybridization ratio of approximately 70% between two strains usually indicates a species-level relationship (Wayne et al. 1987; Stackebrandt et al. 2002). Average nucleotide calculation (ANI) is becoming increasingly popular for in silico species delineation in the light of genomic data availability. An initial genomic comparison indicated 95% pair-wise ANI as correlated with 70% DNA–DNA hybridization (DDH), and this correlation was consistently observed in various subsequent studies (Auch et al. 2010; Colston et al. 2014). Using the established 70% DDH criterion in addition to a follow-up validation based on mathematical models and amplified fragment length polymorphism (AFLP) data, members within the Agrobacterium tumefaciens complex were classified into ten distinct genomic species with a non-continuous genomovar numbering, e.g. G1–G9 followed by G13, as a consequence of the reclassification of some initially established genomovars to a different genus, e.g. Agrobacterium rhizogenes (genomovar 10) to Rhizobium rhizogenes or to a greater extent Agrobacterium clade, e.g. Agrobacterium rubi (genomovar 11; clade 2 in Fig. 2). To date, most of the genomovars have not received official Latin binomials due to the lack of differentiating biochemical features that are traditionally used to describe new bacterial species. Lassalle et al. (2011) took one of the first initiatives to differentiate the Agrobacterium tumefaciens species complex by identifying the gene repertoire specific to Agrobacterium genospecies 8 which includes strain C58, a widely used strain among Agrobacterium geneticists that has had its genome sequenced and annotated. By comparing the C58 genome against 25 strains from different Agrobacterium genospecies based on hybridization to DNA microarrays spanning the whole genome of strain C58, genes relevant to the speciation and ecological isolation of genomovar G8 were identified. Phenotypic traits specific to genomovar G8 initially inferred from microarray data, such as ferulic acid degradation and curdlan production, were subsequently validated using HPLC and Congo red assays, respectively. As a result, the species name Agrobacterium fabrum was suggested for strains of Agrobacterium genomovar G8, from the Latin plural genitive of smith, in reference to the pioneer isolator of an Agrobacterium strain (Smith and Townsend 1907).
Based on identification of a gene repertoire unique to genomovar G8 that is associated with commensal interactions with plants, and by citing several similar studies linking ecological niche and genomic species beyond the genus Agrobacterium (Cai et al. 2009; Johnson et al. 2006; Lefébure et al. 2010; Porwollik et al. 2002), Lassalle et al. (2011) suggested the generalization of the concept of bacterial genomic species as ecological species. A potential exception to this generalization is currently emerging within Agrobacterium genomovar G3. The Agrobacterium genomovar G3 initially consisted of strains isolated from clinical environments, e.g. human host and antiseptic flask (Popoff et al. 1984). However, based on the newly constructed phylogenomic tree, in addition to the classical Agrobacterium sp. CFBP 6623, the agrobacteria G3 clade now consists of strains LC34, SUL3, and Root651 which were isolated from a diverse and non-clinical environment. Notably, Agrobacterium sp. LC34 originated from the rock surface of the Lechuguilla Cave which has been isolated from humans for over four million years (Bhullar et al. 2012), an environment that substantially differs from that of strain CFBP 6623. On the contrary, Agrobacterium sp. Root651 may share a similar ecological niche with that of G8 agrobacteria given that it is a member of the Arabidopsis plant root microbiota (Bai et al. 2015). Agrobacterium sp. SUL3 was isolated from a laboratory culture of the hydrocarbon-producing Botryococcus braunii, a non-plant photosynthetic organism (green microalga; (Jones et al. 2016). Taken together, it will be hard to convince microbial ecologists that members of the Agrobacterium genomovar G3 are a single ecological species despite their high genomic relatedness.
6 Concluding Remarks
The progress of using whole genome sequence data for establishing relatedness among members of the Rhizobiaceae family is presented. As additional whole genome sequences of these members are elucidated, further insight into the complex phylogeny of Agrobacterium will become available. Further and rigorous analysis of large data sets will validate or further contest the concept of bacterial genomic species as ecological species.
References
Auch AF, von Jan M, Klenk H-P, Göker M (2010) Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2:117–134
Bai Y et al (2015) Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528:364–369
Bankevich A et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comp Biol 19:455–477
Bennasar A, Mulet M, Lalucat J, García-Valdés E (2010) PseudoMLSA: a database for multigenic sequence analysis of Pseudomonas species. BMC Microbiol 10:118
Bhullar K et al (2012) Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS ONE 7:e34953
Cai H, Thompson R, Budinich MF, Broadbent JR, Steele JL (2009) Genome sequence and comparative genome analysis of Lactobacillus casei: insights into their niche-associated evolution. Genome Bio Evol 1:239–257
Colston SM, Fullmer MS, Beka L, Lamy B, Gogarten JP, Graf J (2014) Bioinformatic genome comparisons for taxonomic and phylogenetic assignments using Aeromonas as a test Case. mBio 5
Costechareyre D et al (2010) Rapid and efficient identification of Agrobacterium species by recA allele analysis. Microb Ecol 60:862–872
de Lajudie P et al (1998) Allorhizobium undicola gen. nov., sp. nov., nitrogen-fixing bacteria that efficiently nodulate Neptunia natans in Senegal. Int J Syst Bacteriol 4:1277–1290
Delamuta JRM, Ribeiro RA, Menna P, Bangel EV, Hungria M (2012) Multilocus sequence analysis (MLSA) of Bradyrhizobium strains: revealing high diversity of tropical diazotrophic symbiotic bacteria. Braz J Microbiol 43:698–710
Ding W, Baumdicker F, Neher RA (2017) panX: pan-genome analysis and exploration. Nucleic Acids Res 25
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Farrand SK, O’Morchoe SP, McCutchan J (1989) Construction of an Agrobacterium tumefaciens C58 recA mutant. J Bacteriol 171:5314–5321
Farrand SK, van Berkum PB, Oger P (2003) Agrobacterium is a definable genus of the family Rhizobiaceae. Int J Syst Evol Microbiol 53:1681–1687
Genetello C, Van Larebeke N, Holsters M, De Picker A, Van Montagu M, Schell J (1977) Ti plasmids of Agrobacterium as conjugative plasmids. Nature 265:561–563
Goodner B et al (2001) Genome sequence of the plant pathogen and biotechnology agent Agrobacterium tumefaciens C58. Science 294:2323–2328
Hedges SB (2002) The origin and evolution of model organisms. Nat Rev Gen 3:838–849
Henkel CV, den Dulk-Ras A, Zhang X, Hooykaas PJJ (2014) Genome sequence of the octopine-type Agrobacterium tumefaciens strain Ach5. Genome Announcements 2:e00225–00214
Huang Y-Y, Cho S-T, Lo W-S, Wang Y-C, Lai E-M, Kuo C-H (2015) Complete genome sequence of Agrobacterium tumefaciens Ach5. Genome Announc 3:e00570–00515
Huo Y-B, Chan Y, Lacap-Bugler DC, Mo S, Woo PC, Leung WK, Watt RM (2017) Multilocus sequence analysis of phylogroup 1 and 2 oral treponeme strains. Appl Environ Microbiol 83:e02499–02416
Janda JM, Abbott SL (2007) 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol 45:2761–2764
Johnson ZI, Zinser ER, Coe A, McNulty NP, Woodward EMS, Chisholm SW (2006) Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science 311:1737–1740
Jones KJ, Moore K, Sambles C, Love J, Studholme DJ, Aves SJ (2016) Draft genome sequences of Achromobacter piechaudii GCS2, Agrobacterium sp. Strain SUL3, Microbacterium sp. Strain GCS4, Shinella sp. Strain GWS1, and Shinella sp. strain SUS2 isolated from consortium with the hydrocarbon-producing alga Botryococcus braunii. Genome Announc 4
Kanie S et al (2007) Roles of RecA protein in spontaneous mutagenesis in Escherichia coli. Genes Genet Syst 82:99–108
Kim K, Gan HM (2017) A glimpse into the genetic basis of symbiosis between Hydrogenophaga and their helper strains in the biodegradation of 4-aminobenzenesulfonate. J Genomics 5:77–82
Kisand V, Wikner J (2003) Limited resolution of 16S rDNA DGGE caused by melting properties and closely related DNA sequences. J Microbiol Meth 54:183–191
Konstantinidis KT, Ramette A, Tiedje JM (2006) The bacterial species definition in the genomic era. Philos Trans R Soc Lond B Biol Sci 361:1929–1940
Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM (1994) Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev 58:401–465
Kuzminov A, Stahl FW (1997) Stability of linear DNA in recA mutant Escherichia coli cells reflects ongoing chromosomal DNA degradation. J Bacteriol 179:880–888
Lassalle F et al (2011) Genomic species are ecological species as revealed by comparative genomics in Agrobacterium tumefaciens. Genome Bio Evol 3:762–781
Lefébure T, Pavinski Bitar PD, Suzuki H, Stanhope MJ (2010) Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Bio Evol 2:646–655
Lloyd AT, Sharp PM (1993) Evolution of the recA gene and the molecular phylogeny of bacteria. J Mol Evol 37:399–407
MacLean D, Jones JD, Studholme DJ (2009) Application of next-generation sequencing technologies to microbial genetics. Nat Rev Gen 7:287–296
Martens M, Dawyndt P, Coopman R, Gillis M, De Vos P, Willems A (2008) Advantages of multilocus sequence analysis for taxonomic studies: a case study using 10 housekeeping genes in the genus Ensifer (including former Sinorhizobium). Int J Syst Evol Microbiol 58:200–214
Menna P, Barcellos FG, Hungria M (2009) Phylogeny and taxonomy of a diverse collection of Bradyrhizobium strains based on multilocus sequence analysis of the 16S rRNA gene, ITS region and glnII, recA, atpD and dnaK genes. Int J Syst Evol Microbiol 59:2934–2950
Metzker ML (2010) Sequencing technologies—the next generation. Nat Rev Gen 11:31–46
Mousavi SA et al (2014) Phylogeny of the Rhizobium-Allorhizobium-Agrobacterium clade supports the delineation of Neorhizobium gen. nov. Syst Appl Microbiol 37:208–215
Mousavi SA, Willems A, Nesme X, de Lajudie P, Lindstrom K (2015) Revised phylogeny of Rhizobiaceae: proposal of the delineation of Pararhizobium gen. nov., and 13 new species combinations. Syst Appl Microbiol 38:84–90
Ochman H, Elwyn S, Moran NA (1999) Calibrating bacterial evolution. PNAS 96:12638–12643
Ooms G, Hooykaas PJ, Van Veen RJ, Van Beelen P, Regensburg-Tuïnk TJ, Schilperoort RA (1982) Octopine Ti-plasmid deletion mutants of Agrobacterium tumefaciens with emphasis on the right side of the T-region. Plasmid 7:15–29
Ormeno-Orrillo E et al (2015) Taxonomy of rhizobia and agrobacteria from the Rhizobiaceae family in light of genomics. Syst Appl Microbiol 38:287–291
Popoff MY, Kersters K, Kiredjian M, Miras I, Coynault C (1984) Taxonomic position of Agrobacterium strains of hospital origin. Ann Microbiol 3:427–442
Porwollik S, Wong RM-Y, McClelland M (2002) Evolutionary genomics of Salmonella: gene acquisitions revealed by microarray analysis. PNAS 99:8956–8961
Price MN, Dehal PS, Arkin AP (2010) FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490
Ramirez-Bahena MH et al (2014) Single acquisition of protelomerase gave rise to speciation of a large and diverse clade within the Agrobacterium/Rhizobium supercluster characterized by the presence of a linear chromid. Mol Phylogenet Evol 73:202–207
Richter M, Rossello-Mora R, Oliver Glockner F, Peplies J (2016) JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32:929–931
Rosenberg MS, Kumar S (2003) Taxon sampling, bioinformatics, and phylogenomics. Syst Biol 52:119
Sakamoto M, Ohkuma M (2011) Identification and classification of the genus Bacteroides by multilocus sequence analysis. Microbiology 157:3388–3397
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069
Segata N, Bornigen D, Morgan XC, Huttenhower C (2013) PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat Commun 4
Shimodaira H, Hasegawa M (1999) Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol 16:1114–1114
Slater SC et al (2009) Genome sequences of three Agrobacterium biovars help elucidate the evolution of multichromosome genomes in bacteria. J Bacteriol 191:2501–2511
Slater S et al (2013) Reconciliation of sequence data and updated annotation of the genome of Agrobacterium tumefaciens C58, and distribution of a linear chromosome in the genus Agrobacterium. Appl Environ Microbiol 79:1414–1417
Smit S, Widmann J, Knight R (2007) Evolutionary rates vary among rRNA structural elements. Nucleic Acids Res 35:3339–3354
Smith EF, Townsend CO (1907) A plant-tumor of bacterial origin. Science 25:671–673
Stackebrandt E, Goebel B (1994) Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Evol Microbiol 44:846–849
Stackebrandt E, Frederiksen W, Garrity GM, Grimont PAD, Kamper P, Maiden MCJ, Nesme X, Rossello-Mora R, Swings J et al (2002) Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int J Syst Evol Microbiol 52:1043–1047
Tran PN, Savka MA, Gan HM (2017) In-silico taxonomic classification of 373 genomes reveals species misidentification and new genospecies within the genus Pseudomonas. Front Microbiol 8:1296
Velázquez E et al (2010) Analysis of core genes supports the reclassification of strains Agrobacterium radiobacter K84 and Agrobacterium tumefaciens AKE10 into the species Rhizobium rhizogenes. Syst Appl Microbiol 33:247–251
Wayne LG, Brenner DJ, Colwell RR et al (1987) International committee on systematic bacteriology. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol 37:463–464
Woo P, Lau S, Teng J, Tse H, Yuen KY (2008) Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect 14:908–934
Wood DW et al (2001) The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science 294:2317–2323
Young J, Kuykendall L, Martinez-Romero E, Kerr A, Sawada H (2001) A revision of Rhizobium Frank 1889, with an emended description of the genus, and the inclusion of all species of Agrobacterium Conn 1942 and Allorhizobium undicola de Lajudie et al. 1998 as new combinations: Rhizobium radiobacter, R. rhizogenes, R. rubi, R. undicola and R. vitis. Int J Syst Evol Microbiol 51:89–103
Young JM (2008) Agrobacterium—Taxonomy of plant-pathogenic Rhizobium species. In: Tzfira T, Citovsky V (eds) Agrobacterium: from biology to biotechnology. Springer, New York, pp 183–220. https://doi.org/10.1007/978-0-387-72290-0_5
Zhang L, Li X, Zhang F, Wang G (2014) Genomic analysis of Agrobacterium radiobacter DSM 30147(T) and emended description of A. radiobacter (Beijerinck and van Delden 1902) Conn 1942 (Approved Lists 1980) emend. Sawada et al. 1993. Stand Genomic Sci 9:574–584
Acknowledgements
M.A.S. and H.M.G acknowledge funding for research provided by the Gosnell School of Life Science at Rochester Institute of Technology. H.M.G was partially supported by a research start-up grant from the Deakin University Centre of Integrative Ecology.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Gan, H.M., Savka, M.A. (2018). One More Decade of Agrobacterium Taxonomy. In: Gelvin, S. (eds) Agrobacterium Biology. Current Topics in Microbiology and Immunology, vol 418. Springer, Cham. https://doi.org/10.1007/82_2018_81
Download citation
DOI: https://doi.org/10.1007/82_2018_81
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-03256-2
Online ISBN: 978-3-030-03257-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)