
Abstract
Infections caused by opportunistic human fungal pathogens are a source of increasing medical concern, due to their growing incidence, the emergence of novel pathogenic species, and the lack of effective diagnostics tools. Fungal pathogens are phylogenetically diverse, and their virulence mechanisms can differ widely across species. Despite extensive efforts, the molecular bases of virulence in pathogenic fungi and their interactions with the human host remain poorly understood for most species. In this context, next-generation sequencing approaches hold the promise of helping to close this knowledge gap. In particular, high-throughput transcriptome sequencing (RNA-Seq) enables monitoring the transcriptional profile of both host and microbes to elucidate their interactions and discover molecular mechanisms of virulence and host defense. Here, we provide an overview of transcriptome sequencing techniques and approaches, and survey their application in studying the interplay between humans and fungal pathogens. Finally, we discuss novel RNA-Seq approaches in studying host–pathogen interactions and their potential role in advancing the clinical diagnostics of fungal infections.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Introduction
Over the last two decades, the incidence of fungal infections (also known as mycoses) has increased dramatically (Bitar et al. 2014; Pfaller and Diekema 2007; Oren and Paul 2014), particularly in hospital-associated (nosocomial) conditions (Turner and Butler 2014; Chapman et al. 2017). Fungal infections range from superficial skin lesions to life-threatening invasive infections, including fungemia. Superficial skin or mucosal infections affect around 25% of the global population (Havlickova et al. 2008), and although they are relatively easy to manage, they collectively constitute a high burden. Invasive mycoses are life-threatening and can be associated to high mortality rates of up to 38–63%, depending on several factors such as the health status of the patient and the infecting strain (Klingspor et al. 2015; Flevari et al. 2013). It has been estimated that invasive fungal infections kill around 1.5 million people worldwide every year (Brown et al. 2012).
The phylogenetic diversity of pathogenic fungal species is high—from estimated 2.2–3.8 million fungal species, nearly 300 can cause human infections, which is likely to be an underestimation (Nature Microbiology Editorial 2017; O’Brien et al. 2005; Hawksworth and Lücking 2017; Blackwell 2011). Although most common fungal pathogens belong to several major clades, such as Candida (Pfaller and Diekema 2007, 2010), Aspergillus (Dagenais and Keller 2009), and Cryptococcus (May et al. 2016), each of these groups can comprise numerous distinct pathogenic lineages. For example, Candida species, which are considered to be the most frequent and invasive opportunistic fungal pathogens (Guinea 2014), are spread across the Saccharomycotina phylogenetic tree (Gabaldón et al. 2016). C. glabrata, which is usually the second most abundant Candida pathogen, after C. abicans (Guinea 2014), is phylogenetically much closer to the biotechnology workhorse Saccharomyces cerevisiae than to other pathogenic Candidas (Gabaldón and Carreté 2016). Similarly, pathogenic members of the so-called CTG clade, such as C. albicans, (Kim and Sudbery 2011), C. parapsislosis, and others, have numerous non-pathogenic sister species and clades. This phylogenetic dispersion points out that the ability to infect humans has emerged multiple times independently in genetically distinct backgrounds (Gabaldón et al. 2016). Due to the high diversity and the difficulties to classify these pathogens from their physiological or morphological traits, the phylogenetic relationships between these fungi have been poorly resolved. Only the recent advent of molecular and genomic sequencing technologies has enabled accurate resolution of phylogenetic relationships and, as consequence, the taxonomic nomenclature of these species is still undergoing major revisions (Brandt and Lockhart 2012).
Novel pathogenic species are identified regularly, and the increase of incidence of previously rare species has been documented multiple times (Papon et al. 2013; Rhodes et al. 2017; Short et al. 2014). It is as yet unclear what factors drive the emergence of novel pathogens. Changes in the use of chemical products in industry or clinical care, or movement of products related to international commerce, can favor the global spread of certain species. In addition, biological factors resulting from evolutionary adaptation of microbes to novel niches can trigger the emergence of novel pathogens. One of the proposed mechanisms of emergence of novel pathogenic species in fungi is hybridization (Mixão and Gabaldón 2017), which has been related to the formation of pathogenic lineages such as Cryptococcus neoformans x Cryptococcus gatii (D’Souza et al. 2011), Malassezia furfur (Wu et al. 2015), C. metapsilosis (Pryszcz et al. 2015) and C. orthopsilosis (Pryszcz et al. 2014; Schröder et al. 2016).
Another major challenge posed by fungal pathogens is the increasing rate at which drug and multidrug resistance (MDR) is reported (Pfaller et al. 2009), which is often caused by the ability of fungi to evolve resistance phenotypes (Sanglard 2016). The problem of resistance to one or several drugs is worsened by the limited number of available antimycotic agents, which is currently restricted to few chemical families (Kathiravan et al. 2012). For example, the incidence of C. glabrata invasive infections has increased from 18% (in 1992–2001) to 25% (in 2001–2007), with concomitant fluconazole resistance rates increasing from 9% to 14%, respectively, in the USA (Pfaller et al. 2009). Candida auris is another striking example of MDR pathogenic yeast, which exhibits resistance to the main classes of antifungals (Sarma and Upadhyay 2017). Being first described in 2009 in Japan (Satoh et al. 2009), C. auris rapidly became a notorious pathogen causing outbreaks in hospitals throughout the world (Chowdhary et al. 2017). While the concern of research and medical community toward this pathogen is high, we still lack sufficient knowledge and effective approaches for controlling C. auris infections, highlighting the importance for public health of the emergence of antimycotic resistance.
As a consequence of high diversity, emergence of new pathogens, and increasing drug resistance, the diagnostic arsenal for the detection of the causative agent and the determination of the best treatment is limited (reviewed in Kozel and Wickes 2014; Griffin and Hanson 2014). Classical diagnostics methods have serious limitations. Culture-based methods using blood samples can take several days and do not provide high specificity and sensitivity, missing, for example, over 50% of the cases of documented candidiasis (Berenguer et al. 1993). Moreover, some fungal species are non-culturable in conventionally used media. To overcome these issues, recently novel molecular-based diagnostic tools have been developed mainly including those based on polymerase chain reaction (Khot and Fredricks 2009) or mass spectrometry (Chalupová et al. 2014). More recently, with the rapid development of nucleic acid sequencing technologies, next-generation sequencing (NGS) might become a promising tool for microbial diagnostics (Smeekens et al. 2016; Zoll et al. 2016). Nevertheless, all of the aforementioned methods have their limitations from both clinical and economical perspectives (described in Kozel and Wickes 2014; Griffin and Hanson 2014).
Considering all these factors, it is evident that investigation of host–fungus interactions is crucial for overcoming the threats that pathogenic fungi currently impose. Firstly, knowing the specific host-evasion and virulence mechanisms used by diverse fungal pathogens may pave the way for the discovery of novel drug targets or the design of new treatment approaches. Secondly, many fungal pathogens are also commensal species that are part of the normal human microbiota. Hence, there is a need to understand what triggers may turn a commensal behavior into an invasive and virulent one. In addition, response from host cells and tissues toward different fungal pathogens may also provide important clues toward more efficient ways to avoid and control infection. Finally, understanding host–pathogen interactions may open new avenues for diagnostic approaches that are able to differentiate between commensal or infective behavior by detecting specific biomarkers. Although several studies have advanced our understanding of host–pathogen interactions for some of the more common species, the interplay between humans and fungal pathogens is, overall, still poorly understood.
NGS techniques, which allows obtaining sequence data on unprecedented scales and low costs, have represented a revolution in biological research (Goodwin et al. 2016), and the investigation of host–pathogen interaction is no exception (Hu et al. 2011; Westermann et al. 2012, 2017). In particular, whole transcriptome analysis by means of RNA-Seq has opened a new window to understanding gene regulation and how it changes as a result of interactions between the host and the pathogen, which potentially can shed light on the mechanisms of pathogenicity, host defense, and their interplay in various conditions (Wolf et al. 2018). In this review, we focus on the application of whole transcriptome sequencing in addressing host–fungus interactions during infection. We will first discuss the methodological concepts and peculiarities of RNA-Seq in the context of host–microbe interaction studies, then survey past studies of human–fungus interactions based on transcriptome sequencing. Finally, future perspectives in the field including the potential of emerging technologies for the study or diagnosis of fungal infections will be discussed.
2 Whole Transcriptome Analysis Methods
RNA plays a key role in the majority of cellular processes. Hence, investigation of the identity, function, and abundance of transcribed RNA molecules (i.e., transcripts) is crucial for understanding cellular behavior. Advances in the field of RNA biology were mainly driven by the development of novel technologies and methods allowing researchers to study different aspects of transcripts in an increasingly efficient way. A brief chronological overview of those techniques is discussed below, and a more in-depth comparison is provided in Table 1.
Initial studies of RNA molecules were performed using methods such as northern blotting (Alwine et al. 1977), reverse transcriptase qPCR (Rappolee et al. 1988), and expressed sequence tags (ESTs) (Adams et al. 1991), which enabled to investigate individual molecules or small sets of transcripts. The first studies of transcriptomes, i.e., the whole set of RNA transcripts in a cell (or bulk of cells) at a given time point, begun in the mid 1990s, with the development of the serial analysis of gene expression (SAGE) method (Velculescu et al. 1995), based on Sanger sequencing technology (Sanger et al. 1977). SAGE and its derivatives (e.g., LongSAGE, RL-SAGE, SuperSAGE) in the beginning of 2000s were largely replaced by fluorescent hybridization-based RNA microarray technologies (Schena et al. 1995), which proved to be more cost-effective, as compared to previous methods. Finally, in the late 2000s, the advent of NGS superseded microarrays by RNA-Seq which provided unprecedented levels of resolution in a high throughput, unbiased, and relatively cheap manner (Bainbridge et al. 2006; Wang et al. 2009). In addition to considerations of throughput and cost, RNA-Seq presented the advantage over microarrays in that it did not require the design of probes and could explore the entire transcriptome in an unbiased manner, enabling the discovery of novel transcripts, even in the absence of a reference genome. In the last decade, RNA-Seq has been further developed, incorporating longer sequencing reads as well as increasing its versatility by being coupled to other approaches such as, for instance, target-enrichment (Amorim-vaz et al. 2015) or structure-specific digestion (Wan et al. 2013; Saus et al. 2018). Today, RNA-Seq is the major method used in transcriptomics studies.
As many other NGS-based techniques, RNA-Seq comprises two major steps: The first includes the study design and sequencing of the samples, whereas the second includes all downstream bioinformatics analysis. With current technologies, the particularities of each of the two stages can vary significantly depending on the main goal of the study. Hence, there is no universal procedure for addressing all possible biological questions that can be addressed with a transcriptomics approach (Conesa et al. 2016). Nevertheless, some generalities can be drawn. In the following sections, we will focus on the main principles of each of the two steps, underscoring, when applicable, the peculiarities that are most relevant for host–fungus interaction studies. We will first discuss steps usually performed for the dominating sequencing-by-synthesis NGS technology, implemented by Illumina, while other emerging approaches, such as nanopore and PacBio sequencing, will be discussed later in our review.
2.1 Study Design
Study design refers to the initial setup of the project, which is a crucial prerequisite for any RNA-Seq study. The project has to be planned carefully according to its main goals and taking into account the peculiarities of the addressed biological problem. Formally, the study design can be divided into experimental design and sequencing design.
2.1.1 Experimental Design
Experimental design refers to the overall type of the study (“control-vs-treatment,” timecourse, observational study, and different combinations of these types) and how it is planned and performed from both logical and technical perspectives. A poorly planned experimental design can possibly result in spurious and/or misleading results. For instance, in a “control-vs-treatment” in vivo study of the influence of antimycotic drug on the gene expression of the fungus, “control” and “treatment” cases need to be selected. If “control” samples were obtained only from young people whereas the “treatment” samples came from significantly older people, then the age of the donors could be a potential confounding factor, preventing from distinguishing whether the observed gene expression changes in fungus were due to the antimycotic agent or the age of the host. To avoid such kind of confounding effects, study donors for both control and treatment group have to be as similar as possible from various perspectives, controlling for factors such as age, sex, diet, or the presence of concomitant diseases. Another example illustrating poorly planned experimental design could be a time series study of a fungal pathogen interacting with host cells in vitro. Without performing time-matched controls for the fungus, it might not be possible to differentiate between the effect of the host–microbe interactions from the potential effect of time or growth of the fungus in the given medium. For instance, some nutrients in the medium may be exhausted, triggering physiological changes in the fungal cells, which may be wrongly interpreted as a consequence of interaction with the host. To overcome this limitation, ideally host cell-free controls have to be made at the corresponding time points of the experiment (Fig. 1).

A graphical representation of a time series experimental design of interaction between human cell line and a pathogenic fungus. Left-most sample represents a zero time-point control, from left to right samples—5-, 10-, and 25-h time points are the host–fungus interacting samples (above) and time-matched host cell-free fungal controls (below). Bar plots represent the expression levels of four fungal genes A, B, C, and D. The overall scheme illustrates the importance of time-matched fungal controls in the experimental design: In case of their absence, gene A at time point 5, 10, 15 h and gene B at time points 10 and 15 h could be spuriously interpreted as up- and down-regulated, respectively, as a consequence of host–fungus interaction. Controls samples allow to distinguish the effect of media and time from the effect of host–pathogen interaction, showing that only gene C is specifically up-regulated at 15 h as a consequence of interaction
Thus, as illustrated by the examples above, a well-planned experimental design is a crucial first step for a meaningful RNA-Seq-based study. Critically assessing previous studies investigating similar questions (perhaps on other pathogens or hosts) and extensive discussions between all project partners (e.g., clinical doctors treating the patients, personnel collecting the samples, personnel responsible for the statistical comparisons) can help to achieve a good planning and avoid potential design flaws.
2.1.2 Sequencing Design
The design of the sequencing approach itself refers to the main factors to consider for sample collection, storage, preparation, and sequencing per se. While sample collection and storage methods heavily depend on a particular project, a general recommendation is to perform these two steps in the same way for all samples within a project to avoid possible confounding effects. For further sequencing, the important aspects to consider are RNA extraction protocol, library preparation protocol, read type and length, number of replicates, sequencing depth, randomization of samples and sequencing runs. The combination of the aforementioned parameters entirely depends on the specific goal of the project, and some general recommendations are given in Table 2 and are discussed in more details in the text.
For fungi, several commercial kits are available for high-quality and high-yield RNA extraction. An important factor to consider on this step is whether rRNA depletion or poly-(A) selection is required, since usually transcriptome studies are focused on mRNAs, while ribosomal RNA can constitute the vast majority of RNA in a cell (i.e., up to 60% in an exponentially growing Saccharomyces cerevisiae cell (Warner 1999), and its removal will provide higher resolution for the mRNAs (Zhao et al. 2014; O’Neil et al. 2013). Moreover, different strategies to enrich specific mRNA molecules have been recently developed, which will be discussed in more detail in the following section of our review.
The specific sequencing library preparation protocol is yet another factor to consider, especially with regards to its ability to generate strand-specific data. Early library preparation protocols were not capable of preserving information about the strand of DNA from which a transcript originated, thus biasing, for example, gene expression analysis by anti-sense transcription (Zhao et al. 2015). Today several so-called strand-specific (or stranded) protocols are available, such as dUTP (Borodina et al. 2011; Parkhomchuk et al. 2009), RNA-ligation (Lister et al. 2008), SMART (Zhu et al. 2001) for retaining strand information of transcripts. A comprehensive benchmark of these protocols is given in Levin et al. (2010).
The length of the reads (short stretches of cDNA that are actually sequenced) is one of the major parameters of the sequencing design. The read length of Illumina-based sequencing varies from 25 to 300 bases depending on the model of sequencing machine (Kwon-Chung et al. 2011). As an additional option to increase the capabilities of obtained data, one can perform paired-end (PE) sequencing instead of single-end (SE). In the former case, the cDNA fragment is sequenced from both ends, hence doubling the amount of the information obtained from it. As a general rule, longer reads coupled with paired-end long-insert size sequencing provide higher mapping rates to reference genomes, more accurate transcript discovery, the ability to detect larger indels, among other advantages. However, PE sequencing and longer reads come with higher price, which may compromise the number of replicates if budget is limited. Moreover, some particular goals could be sensitively achieved even with short SE reads (Chhangawala et al. 2015). For example, considering that in Candida albicans introns are not abundant in the genome and their length is usually short (Mitrovich et al. 2007), long PE reads are not critical for most of the typical downstream analysis.
In the context of typical RNA-Seq applications, such as differential gene expression (DGE), replicates and sequencing depth are two crucial and interconnected parameters. When choosing the number of biological replicates one has to consider the intrinsic biological variability of the studied system, the technical variability of the experimental procedures, and the desired statistical power of the experiment. As a general rule, the number of biological replicates included in the study should be at least 3, while the recommended number ranges from 6 to 12 biological replicates (based on S. cerevisiae data) depending on the specific goals of the study (Schurch et al. 2016). Several approaches and calculators have been implemented to perform RNA-Seq power analysis to help deciding the number of replicates in an RNA-Seq design (Hart et al. 2013; Yu et al. 2017; Guo et al. 2014).
Sequencing depth (or library size) denotes the total number of reads for each sample to be sequenced. Higher sequencing depth allows more precise transcript detection and expression quantification, but also might suffer from transcriptional noise and false-positive calls of DGE (Tarazona et al. 2011). Thus, once again the optimal sequencing depth depends on the addressed question and the system under study.
As mentioned above, replication and library size are interconnected parameters and for a particular sequencing design one can wonder whether for a fixed budget it is more advisable to add more replicates to the study or to perform deeper sequencing. Liu et al. (2014b) have evaluated the impact of both factors on DGE analysis. The study revealed that, in the case of human transcriptomic data, increasing the sequencing depth over 10 million reads has diminishing incremental effects for power of detection of differentially expressed (DE) genes, whereas an increase in the number of biological replicates significantly enhances the power of detection. Authors of the study also suggest a metric of cost-effectiveness of RNA-Seq design as a trade-off between number of replicates and sequencing depth. The last but not least step in experimental design in the case of projects considering a high number of samples is to randomize the distribution of samples on different sequencing lanes or runs. This step is meant to avoid possible confounding factors such as different instrumental biases (lane effects, PCR duplicates, etc.) as well as difficult to control human factors.
2.2 Bioinformatics Data Analysis
As any high-throughput sequencing technology, RNA-Seq generates massive amounts of data (i.e., a typical DGE RNA-Seq analysis of yeast with two conditions and three replicates yields at least 100–150 million reads) which have to be thoroughly analyzed using bioinformatics approaches. Considering that RNA-Seq has numerous applications, the complete pipeline for bioinformatics analysis varies depending on the specific goals of the project. With regards to host–pathogen interaction studies, one of the most frequent RNA-Seq applications is the analysis of differential gene/transcript expression (Westermann et al. 2017). Here, we will briefly describe the main steps of this bioinformatics approach (Fig. 2).

General representation of RNA-Seq differential expression analysis pipeline. The numbers correspond to different steps of the analysis: 1—quality control of raw data; 2—read trimming (if necessary), 3a—read mapping to a reference genome, 3b—de novo assembly of transcripts 3c—pseudomapping strategy (requires reference transcriptome), 4a—read summarization, 4b—reference-guided transcriptome assembly (used for transcript identification), 4c—transcript quantification, 5—transcript-level quantifications can be converted to gene-level count, which improves gene-level inferences, 6a—differential gene/transcript/exon/feature analysis based on read counts, 6b—differential gene/transcript/exon/feature analysis based on relative expression values. Shades indicate different major strategies of the analysis: blue—general step of raw data quality control and read trimming; pink—downstream analysis when the reference genome is available, mainly includes gene-level inferences, green—when the reference genome is not available and includes transcript-level inferences
The general initial step for any NGS-based data analysis is quality control (QC) of the raw data produced by the sequencing machine. As a general rule, raw reads are stored in the standard fastq format (Cock et al. 2009), which provides associated per base quality scores. For basic QC, several software solutions can be used, such as FastQC (Andrews 2010), HTQC (Yang et al. 2013), or NGS QC (Patel and Jain 2012). The main parameters to assess include, among others, per base sequence quality, GC content, or the presence of overrepresented sequences and/or those corresponding to the library adapters. When some quality parameters are not satisfactory, one can perform several actions including read trimming to cut out low-quality reads or bases, or removing adapter sequences coming from the library preparation step. Popular software to perform read trimming are Trimmomatic (Bolger et al. 2014), Skewer (Jiang et al. 2014), among others. However, in the case of DGE analysis trimming has to be performed gently since the harsh trimming can affect the results of read mapping and differential expression calls (Williams et al. 2016). For this reason, if the quality of the data is still unsatisfactory after trimming, it is advisable to resequence the sample or the library.
After ensuring the high quality of the sequencing data, downstream analysis depends on the presence or absence of a reference genome or transcriptome of the studied organism. In the case of human fungal pathogens, most common species like those belonging to Candida, Aspergillus, or Cryptococcus groups have available reference genomes in both specialized and generic public databases such as Candida Genome Database (Binkley et al. 2014), Aspergillus Genome Database (Cerqueira et al. 2014), FungiDB (Stajich et al. 2012), or RefSeq (Pruitt et al. 2007). Alternatively, in the case of absence of reference, the transcriptome can be reconstructed de novo, using, for example, the Trinity package (Haas et al. 2013) or SOAPdenovo-Trans (Xie et al. 2014).
When a reference genome or transcriptome is available, downstream bioinformatics analysis implies mapping of reads to this reference. This computationally demanding task can be achieved by splice-aware read mappers (despite the fact that splicing is not as common in yeasts as in more complex eukaryotes). Numerous RNA-Seq mappers exist today, and choosing one might not be a trivial task. In fact, many researchers have addressed this question by performing benchmarks comparing different mappers, sometimes reaching contradicting conclusions (Otto et al. 2014; Kim et al. 2013; Dobin and Gingeras 2013; Baruzzo et al. 2017). Nevertheless, the most popular mappers include STAR (Dobin et al. 2013), TopHat2 (Kim et al. 2013), HISAT2 (Kim et al. 2015). After reads are mapped to the reference, quality control of the overall mapping is required. Most software tools provide basic mapping statistics, which include, among others, overall mapping rate, unique mapping rate, rate of multimappers (reads that map equally well to different locations in the reference), number of identified splices. Unique mapping rate is one of the crucial parameters and usually with high-quality raw data and a good enough reference genome the optimal values range from 85% to 95% of uniquely mapped reads. If the value is significantly lower, it might indicate low quality of reads, poor quality of reference genome assembly or the nature of reference genome itself—for instance, genomes with large amount of repeats might result in increased numbers of multimapped reads. The next step after read mapping is read summarization, which involves the calculation of the number of reads that overlap particular genes, which is proportional to the expression levels. This procedure relies on gene annotations (gff or gtf files) and popular software to accomplish this task are htseq-count (Anders et al. 2015) and featureCounts (Liao et al. 2014), with the latter being more flexible in dealing with multimapped reads.
The two previous steps described gene-level analysis to obtain the information about expression values based on genome alignments. However, today several so-called pseudo-alignment algorithms are available that allow the assessment of expression levels of individual transcripts, rather than of genes. Examples of such algorithmic implementations include Salmon (Patro et al. 2017) and kallisto (Bray et al. 2016). For organisms with high rates of alternatively spliced transcripts, it has been recently demonstrated that transcript-level estimates could improve gene-level inferences (Soneson et al. 2015). The final step of DGE bioinformatics analysis is the assessment of DGE between groups of samples. Many studies have been performed to evaluate the most effective models and corresponding software for assessing differential expression (Schurch et al. 2016; Soneson and Delorenzi 2013; Bullard et al. 2010; Seyednasrollah et al. 2015; Rapaport et al. 2013). Readers are referred to (Conesa et al. 2016) for more details on this matter. When sufficient number of biological replicates (three or more) are available, software tools such as DESeq2 (Love et al. 2014), edgeR (Robinson et al. 2010), and limma (Ritchie et al. 2015) generally perform well in most of circumstances. To choose the genes that are differentially expressed, one has to set a cutoff for both fold change of gene expression between conditions and p value of statistical significance of this change, and usually, these thresholds are arbitrary and depend on the desired level of stringency.
Since overall RNA-Seq analysis consists of many wet-lab and data analysis steps, it is advisable to confirm a subset of the obtained results with an alternative approach. For instance, one can perform quantitative PCR on a subset of DE genes to ensure the reliability of the obtained results. Once the DE genes have been identified, researchers can proceed with further in-depth analysis, which, among others, usually includes gene ontology (GO, The Gene Ontology Consortium 2017) or gene set enrichment analysis (GSEA, Subramanian et al. 2005), pathway analysis (Emmert-Streib and Glazko 2011) and gene co-expression and network analysis (Schulze et al. 2016). GO and GSEA are two different approaches addressing a similar question—whether DE set or subset of genes is enriched in a specific biological function, process or cellular location. For instance, when differential expression analysis reveals hundreds of DE genes, these methods help to get an overall insight which biological functions are altered in a given condition. Similarly, pathway analysis allows to identify specific molecular pathways which are dysregulated in the studied system. As in GO and GSEA, pathway analysis is performed based on statistical test of enrichment between sets of gene lists.
Gene co-expression and network analysis on the other hand allows to quantitatively assess genes which are changing the expression levels systematically in a similar manner, revealing gene-gene interactions. Especially this kind of analysis is relevant when a time series dual RNA-Seq data is available, which enables to detect the interacting co-expressed genes of the host and the pathogen (Schulze et al. 2015).
For a more detailed discussion about the best practices of RNA-Seq-related analysis, including both study design and bioinformatics data analysis, readers are referred to the recent review by Conesa et al. (2016).
2.3 Dual RNA Sequencing
Dual RNA sequencing (dual RNA-Seq) is a relatively new methodology (Westermann et al. 2012) of simultaneous sequencing of RNA that originates from two (or more) organisms. Originally dual RNA-Seq was developed in the context of host–pathogen interaction studies, allowing to profile the gene expression of both counterparts at the same time, but in principle can be used to study the interactions between any cohabiting organisms. The main idea behind this method is to sequence the mixture of RNA that contains transcripts from two or more organisms. The mixture of RNA could be obtained by direct extraction of RNA from both species (e.g., when studying interaction between co-cultured bacterial species) or it can be extracted separately for each species and then mixed into one sample. The latter approach is more suitable for host–fungus interaction studies, since RNA extraction protocols for fungi include a cell wall disruption step, which can degrade the RNA content of the host cells. After sequencing a mixed sample, the reads from both species are separated bioinformatically by mapping the mixture of reads to both reference genomes simultaneously. When the reads are successfully separated, the data analysis is largely the same as in the case of standard RNA-Seq. Hence, the above-mentioned recommendations of experimental and sequencing design for common RNA-Seq are also applicable in dual RNA-Seq.
Despite the fact dual RNA-Seq methods are in their infancy, they have already been proven to be an efficient tool for dissecting the interplay between hosts and pathogens (Bruno et al. 2015; Aprianto et al. 2016; Dutton et al. 2016; Nuss et al. 2017; Thänert et al. 2017). Nevertheless, this approach has some technical limitations that need to be overcome. Firstly, for in vivo studies, particularly those involving fungi, the amount of microbial cells and its corresponding RNA is extremely low, as compared to the host side. The RNA-Seq of the sample, heavily shifted toward the host, yields a negligible amount of fungal reads, precluding detailed analysis of fungal transcriptome. To date, the most efficient way to overcome this problem is by using targeted enrichment of fungal transcripts (Amorim-vaz et al. 2015), which we discuss later. Second, since the dual RNA-Seq method is new, specific software and data analysis pipelines still do not exist. The major bioinformatics problem that can arise in dual RNA-Seq experiments is cross-mapping of reads to the wrong reference genome, since the mixture of reads is mapped to both references simultaneously, biasing downstream analysis. Thus, specific data analysis pipelines should be implemented in order to remove that kind of reads. Despite these difficulties, dual RNA-Seq holds a great potential in resolving interactions between species on a transcriptome-wide manner.
3 RNA-Seq-Based Studies to Understand Human–Fungus Interactions in Candida, Aspergillus and Cryptococcus Clades
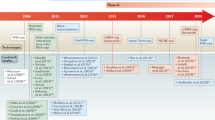
RNA-Seq has emerged as a versatile tool for studying host–pathogen interactions at the transcriptomic level. The majority of transcriptomic studies for elucidating pathogenic mechanisms in fungi so far has been performed in vitro by exposing the pathogen to different experimental conditions that try to mimic stress factors encountered in the host. These include, among many others, low pH, oxidative stress, or different temperatures (Cottier et al. 2015; Yang et al. 2016; Brown et al. 2016; Lin et al. 2013; Cheon et al. 2017). However, a limited number of transcriptomic studies have been performed in vivo, readily characterizing transcriptome responses of the pathogen, host, or both during their direct contact as it takes place during a real infection. Although this approach faces numerous challenges, it is still crucial for disentangling genuine human–fungal interactions. Here, we provide an overview of significant insights gained from transcriptomic studies. For simplicity, we will focus on the three major clades of fungal pathogens, namely Candida, Cryptococcus, and Aspergillus, as research on other fungal pathogens generally lag behind. A schematic summary of surveyed studies is given in Fig. 3.

Host–pathogen interactions transcriptomic studies of Candida, Aspergillus, and Cryptococuss species in different experimental models
3.1 Candida
The most well-studied opportunistic pathogen from Candida species is Candida albicans, and its virulence mechanisms and host–fungus interactions have been extensively reviewed in Wilson and Hube (2014). Briefly, primary pathogenic mechanisms of C. albicans explored to date include hyphae formation (Sudbery et al. 2004) alongside with the expression of virulence factors, such as candidalysin (Moyes et al. 2016), adhesins (e.g., HWP1, HGT2) (Nobile et al. 2006; Martin et al. 2013), invasins (e.g., ALS3) (Liu and Filler 2011), and secreted proteases (e.g., SAP4-6) (Naglik et al. 2003). Hyphae formation and the expression of virulence factors promote initial adherence to the host tissue followed by invasion (either induced endocytosis or active penetrations) and damage. In turn, host defense against infecting Candida is mainly presented by the action of macrophages and neutrophiles (reviewed in Moyes et al. 2014; Wilson and Hube 2014). After phagocytosing the fungal cell, neutrophils expose a variety of factors to block hyphae formation and eventually kill the fungus, including nutrient starvation, production of antimicrobial peptides and enzymes (e.g., defensins, lactoferrin, ellastase), oxidative burst, formation of neutrophil extracellular traps (NETs).
As mentioned above, quantities of yeast cells in an infected patient sample are generally very small, which poses many challenges for the analyses (Rosenbach et al. 2010; Bruno et al. 2015). As a consequence, many previous studies have been performed using animal or tissue culture models, where higher loads of the pathogen can be present and larger quantities of tissue are available. One of the first studies using RNA-Seq to decipher host–pathogen interactions of C. albicans was carried out by Tierney et al. (2012). In this study, the authors performed an in vitro timecourse model experiment of interaction between C. albicans and M. musculus bone marrow-derived dendritic cells (BMDCs) with further RNA sequencing and network analysis to identify and predict interspecific interactions. With the aforementioned techniques, the authors predicted and subsequently experimentally verified a mechanism by which C. albicans escapes host immune response mediated by a reorganization of its cell wall, which in turn is triggered by the release of complement-activating and opsonin protein PTX3 from dendritic cells.
In a more recent study, Bruno et al. (2015) used a murine model of vulvovaginal candidiasis (VVC) coupled with RNA-Seq to study the transcriptome and its alterations in mice and C. albicans. This study demonstrated that expression of the NLRP3 inflammasome, which triggers caspases and the maturation of proinflammatory cytokine interleukin 1 beta—the hallmark of VVC immunopathogenesis, was elevated in infected mice. Moreover, Nlrp3 −/− infected mice showed significantly lowered levels of polymorphonuclear leukocytes (PMNs), alarmins, and inflammatory cytokines. These findings suggested an important role of NLRP3 inflammasome in response to C. albicans in VVC. On the other hand, the authors have also attempted to analyze C. albicans response to host; however, the in-depth analysis of C. albicans transcriptome was precluded by a very low amount of fungal reads obtained from infected vaginal samples (on average ~80 thousand reads of C. albicans compared to ~103 million mouse-derived reads). Nevertheless, the analysis of highly expressed C. albicans genes revealed a robust expression of hypha-associated SAP 4, 5, and 6, while mutants of these genes were inducing significantly lower inflammatory response.
Another comprehensive RNA-Seq-based study of C. albicans and host interaction was done by Liu et al. (2015). Here, the authors analyzed host–pathogen interactions in both in vitro and in vivo conditions. In the former case, two human cell cultures were used, namely human endothelial and oral epithelial cell cultures, while in vivo investigation was carried out in both murine model and using real clinical samples from patients. The time series analysis using not only infected samples but also controling on each corresponding time point (non-infected human samples and C. albicans cells in the growth media) allowed the researchers to reveal that, surprisingly, C. albicans showed a minimal transcriptional response to host cells, which was indicated by a low number of DE genes compared to the control samples. This result points out that most of the C. albicans genes involved in host interaction with the studied cell types are also similarly expressed in the growth medium (M199 and DMEM media). Nevertheless, this fact does not lessen the importance of these genes in host–pathogen interaction, but rather once again demonstrates the relevance of careful study design. Thereafter, focusing mainly on the host side the authors demonstrated a distinct transcriptional response of different cell lines to C. albicans. To identify molecular pathways governing host response, the authors used network analysis and identified numerous previously reported up-regulated pathways like MAPK1/3, TLR7, EGF, and novel pathways such as PDGF and NEDD9. Further, in-depth wet-lab analysis has shown that the last two pathways play a crucial role in endocytosis of C. albicans cells in a cadherin-independent manner in cell cultures. Moreover, it was demonstrated that both pathways are also implicated in pathogen interaction in disseminated mouse infection model, while in case of in vivo human infection NEDD9 was intact. Overall, this study is an excellent example where a combination of carefully planned RNA-Seq design and thorough bioinformatical and follow-up wet-lab analysis can successfully reveal novel mechanisms of host–fungal interaction.
A recent study by Niemiec et al. (2017) has evaluated the interaction between C. albicans and human neutrophils by means of RNA-Seq. The authors assessed the transcriptome of neutrophils exposed to the fungus in either yeast or hyphal morphotypes, as well as the transcriptomic response of those morphotypes to intact neutrophils and NETs. The analysis revealed that the core response of neutrophils is largely similar for the two C. albicans morphotypes, with only 11% of DE genes being specific to the interaction with the hyphal morphotype. The core response to fungi included inflammasome induction and release of numerous cytokines, which shows that despite their short life span neutrophils are also orchestrating complex immune response to C. albicans. On the other hand, C. albicans response was also mainly morphotype-independent, while the reaction to either intact neutrophils or NETs was markedly distinct. Overall, fungal response was primarily dominated by metabolic genes, controlled by the regulators of transcription as Tup1p, Cap1p, Hap43p, with the latter being the major regulator in C. albicans of evasion from neutrophils.
As highlighted previously, one of the major limitations of investigating mutual host–fungus interaction (especially in vivo studies) is a very low proportion of fungal cells as compared to host cells (Rosenbach et al. 2010; Bruno et al. 2015). This problem refers not only to RNA-Seq, but to any other high-throughput NGS technique. Subsequent analysis of such kind of a “host-biased” sample generally does not yield enough fungal data for a comprehensive description of the fungal transcriptome. Previous attempts to solve this issue had serious limitations—some of them were altering true gene expression levels (Andes et al. 2005; Thewes et al. 2007), while the others did not provide transcriptome-wide resolution (Geiss et al. 2008). To overcome this issue, Amorim-Vaz et al. (2015) used, for the first time, RNA-Seq coupled with SureSelect targeted RNA enrichment technology. This technology is based on the use of biotinylated oligonucleotide baits directed to target RNA molecules of interest, which are then enriched by probe hybridization and subsequent pool down. Importantly, this enrichment procedure has been shown to not interfere or change the transcriptional profile of the sample. The authors used two animal models of C. albicans infection—murine model of kidney infection and a Galleria mellonella larvae model—and investigated host–pathogen interplay at early and late stages of infection. Consistent with previous studies, the RNA-Seq analysis of the bulk sample showed that barely 0.1–1% of the reads belonged to the pathogen. However, after applying the enrichment approach, the number of reads aligned to C. albicans dramatically increased up to 1670-fold, while biasing the expression levels only for 3% of genes. DGE analysis of C. albicans genes showed consistent results with previously published studies including up-regulation of genes involved in cell host adhesion, hypha formation, and iron acquisition. Moreover, the analyses revealed new, previously uncharacterized targets in both C. albicans and hosts for further exploration. Overall, this study demonstrated that targeted enrichment can be successfully applied for in vivo host–pathogen studies to describe both counterparts in a transcriptome-wide manner.
3.1.1 Non-albicans Candida Species
Other Candida species are less frequently reported than C. albicans in infection cases, but nevertheless they collectively account for ~50% of the cases. After C. albicans, the most widespread species in infections are C. glabrata, C. parapsilosis, and C. tropicalis, generally in this order (Guinea 2014). Despite the importance of these species in fungal infection epidemiology, their virulence mechanisms and host interactions are significantly less studied than those of C. albicans. So far, only a handful of studies have been performed for transcriptional profiling of these species in the context of host–pathogen interactions, and none of them was deeply focused on both counterparts.
Candida glabrata is the second most widespread Candida species that causes human infections. Phylogenetically, this yeast is much closer to S. cerevisiae than to C. albicans, and it does not form true hyphae and has high intrinsic resistance to azole class of antifungal drugs (Gabaldón and Carreté 2016). Rasheed et al. (2018) investigated the role of yapsins (CgYps)—cell surface-associated aspartile proteases of C. glabrata—in the interaction with human THP-1 macrophages and in systemic murine infection. First, to clarify the role of yapsins in fungal homeostasis on gene expression level, the authors performed RNA-Seq of mutant strain Cgyps1–11Δ, which lacks all 11 yapsins, and compared it to the wild-type (WT). Downstream analysis uncovered 35 down- and 89 up-regulated genes in the mutant, with enriched GO categories of “ion transport,” “oxidation-reduction process” and “sterol import,” and “carbohydrate metabolic process,” “fungal-type cell wall organization” and “tricarboxylic acid cycle,” respectively. Using biochemical staining assays, the authors further demonstrated the altered cell wall composition of the Cgyps1–11Δ mutant in β-glucan, chitin, and mannan content, largely caused by the deletion of CgYps1 and CgYps7 yapsins. While the application of RNA-Seq was restricted to the above-mentioned analysis in this study, the authors additionally used microarray technology to describe human THP-1 macrophages response to C. glabrata WT and the forementioned mutant. Broadly, the microarray profiling showed that THP-1 cell line responds differently to WT and mutant C. glabrata strains: In the former case, human DE genes were involved in inflammatory response, chemotaxis, and chemokine-mediated signaling pathways, while in the latter case the cells expressed genes involved in viral response. In addition, the authors elucidated the role of IL-1β in C. glabrata interaction, showing that its production is likely to be deleterious for fungal survival in macrophages and that yapsins play a pivotal role in suppressing the production of host’s IL-1β.
To clarify the role of yapsins in vivo, BALB/c mice were infected with WT and Cgyps1–11Δ mutant. Overall, WT C. glabrata colonized and disseminated in numerous mouse organs, while the mutant strain had a significantly lower survival, demonstrating that yapsins are required for colonization and dissemination of the fungus. Finally, to uncover the roles of each of the yapsins in infection and fungal survival, the authors performed murine infection models with different combinations of single, double, and triple mutants of yapsin genes. Altogether, organ-specific survival effects of different yapsins were identified.
Another recent study (Whaley et al. 2018) focused on C. glabrata addressed the susceptibility mechanisms of the fungus to fluconazole, identifying the gene which negatively regulates the resistance levels. By screening a large collection of single gene mutants, the authors found that the strain with deleted JJJ1 gene (GL0J07370g), increased the minimum inhibitory concentration (MIC) to fluconazole 16-fold as compared to WT. This finding was further supported by deleting this gene in a C. glabrata clinical strain. Since the main mechanism of C. glabrata resistance to azoles is defined by over-expression of the transcription factor PDR1, which directly activates efflux-pump genes such as CDR1, PDH1, and SNQ2, the authors demonstrated that deletion of JJJ1 increased the resistance through Pdr1-dependent up-regulation of CDR1. To further investigate the effect of JJJ1 deletion on the overall transcriptome of C. glabrata, the authors have performed RNA-Seq using Ion Torrent technology. The analysis identified 119 and 149 up and down-regulated genes, respectively, many of which had been previously identified by microarray analysis.
Candida parapsilosis is a member of CTG clade alongside with C. albicans and C. tropicalis. It is considered to be the third most frequent opportunistic Candida pathogen. As for C. glabrata, a restricted number of studies have been performed to clarify host–pathogen mechanisms on transcription level. To our knowledge, the only RNA-Seq-based host–pathogen interaction study with C. parapsilosis was performed recently by Toth et al. (2018), focusing mainly on the fungal side. The authors employed a timecourse in vitro infection model of C. parapsilosis with human THP-1 monocytes with further RNA-Seq of fungal transcriptome to identify potential molecular targets for future antimycotic agents. RNA-Seq analysis revealed 19 highly up-regulated C. parapsilosis genes, which were selected for further investigation. By constructing deletion mutant strains of each of those genes and performing the screening of the mutants for different properties, the authors narrowed the search of virulence factors to three transcriptional regulator genes CPAR2_100540, CPAR2_200390, and CPAR2_303700. Further in-depth analysis demonstrated that these three genes play an important role in nutrient acquisition and alternative carbon source utilization, hyphae and biofilm formation, and sensitivity to low temperatures, respectively.
As for C. tropicalis, two studies have been performed that used RNA-Seq to understand yeast–hyphal transition. Wu et al. (2016) performed RNA sequencing of three C. tropicalis clinical isolates in yeast and filamentous forms. Differential gene expression analysis showed up-regulation of several genes, including SAP2, SAP3, ALS3, LIP1, which have been previously reported to be involved in hyphal transition in C. albicans.
Jiang et al. (2016) have studied 52 clinical isolates of C. tropicalis by estimating different parameters of pathogenicity, such as biofilm formation, hyphal morphology, and hemolytic activities. Based on the ability to form hyphae, two groups of strains (three highly and three lowly hyphae-producing strains) were further selected for performing in vivo murine infection model with subsequent RNA sequencing of C. tropicalis. RNA-Seq analysis between two groups has shown 206 DE genes in highly hyphae-producing strains, enriched in aspartic-type endopeptidase activity, metal homeostasis, and oxidative response. On the other hand, several uncharacterized DE genes were revealed, which might also have an impact on C. tropicalis pathogenicity.
3.2 Aspergillus
Aspergillus is a genus within the Ascomycota phylum that comprises over 300 species (Samson et al. 2014). Aspergillus species have a high and diverse economic and social impact, since they massively spoil food products (Dijksterhuis et al. 2013), serve for various biotechnology productions (Pel et al. 2007), and some are human pathogens (Kwon-Chung and Sugui 2013). From the latter perspective, the most frequent human pathogen is the soil-associated fungus Aspergillus fumigatus, accounting for 90% of Aspergillus-caused infections, which as in the case of Candida affects mainly immunocompromised individuals (Perfect et al. 2001; Paterson and Lima 2017). A. fumigatus produces hydrophobic microscopic spores known as conidia, which are ubiquitous in the environment and are the main cause of infections (Latgé 1999). After being inhaled by immunocompromised individuals, conidia can reach pulmonary alveoli and start germinating, forming hyphae and mycelia. This causes a wide range of nosologies collectively called aspergillosis, with invasive forms reaching 50–95% mortality rates (Abad et al. 2010).
Numerous studies involving transcriptome profiling have been performed to study pathogenic mechanisms of A. fumigatus. Most of them have been carried out in vitro by exposing the fungus to different environments resembling the interaction with the host or to different stresses (Losada et al. 2014; O’Keeffe et al. 2014; Gibbons et al. 2012; Wang et al. 2015b). However, only few recent studies addressed host–pathogen interactions based on RNA-Seq of more realistic in vivo models focusing either on host, pathogen, or both simultaneously.
Using RNA-Seq, Irmer et al. (2015) investigated the response of A. fumigatus exposed to human blood in vitro, mimicking the host environment encountered by the fungus when it germinates and penetrates to blood vessels. The authors took samples at two time points of 30 and 180 min after incubating with either human blood or with minimal medium, as a control. The experiment was performed in duplicate, and all samples were compared to pre-cultured A. fumigatus mycelia. Differential gene expression analysis between pre-cultured fungus and blood-exposed samples revealed 410 up-regulated and 367 down-regulated genes after 30 min of exposure to blood, and 266 up-regulated and 318 down-regulated genes after 180 min. Those numbers of genes were obtained after subtracting the DE genes from the comparison between control samples and fungi grown on minimal media. After differential expression analysis, the authors performed comprehensive GO enrichment analysis. Briefly, four categories of genes were analyzed—early up-regulated genes, early up-regulated and then down-regulated genes, solely late down-regulated genes and late up-regulated genes. Functional analysis of early up-regulated genes showed enrichment in metabolism, cell-rescue, transport, virulence- and protein synthesis-related genes. Genes that were up-regulated after 30 min but down-regulated after 180 min were largely similar to the ones only up-regulated at 30 min or only down-regulated after 180 min, and thus, their functional enrichments were also similar. After 180 min, enrichment categories remained largely the same compared to early up-regulation, but, obviously, were depressed, indicating slow-down of overall fungal metabolism. A modest number of enriched categories was found in late up-regulated genes, including iron starvation, detoxification, and stress. Overall, the gene expression patterns and functional analysis suggested that human blood is not a hostile environment for A. fumigatus, which first senses the environment and then shuts down several important pathways of energy-consuming metabolism after the first hours and thus can not effectively grow in blood.
Kale et al. (2017) performed a time series dual RNA-Seq analysis of two immunosuppressed mice models (one treated with chemotherapy and another one with corticosteroid) challenged with a particular A. fumigatus strain (Cea10) to assess how do host-pathogen interactions vary between two distinct immunocompromised states in pulmonary aspergillosis. Dual RNA-Seq yielded 16-29 mln reads, depending on the sample, from which 98% mapped to the mouse genome and the rest to the fungal counterpart. Further differential expression analysis of the host side revealed that the two immunocompromised models showed distinct patterns of gene expression response to the pathogen, showing that host response to A. fumigatus depends on the type of immunosuppression. Functional enrichment analysis of DE genes showed enrichment in numerous immunological processes related to cytokines, chemokines, and their receptors in the chemotherapeutic model, whereas for the corticosteroid model a limited number of cytokine-related genes were DE. More highlighted differences in functionally enriched categories between the two models were found with regard to metabolic processes—the chemotherapeutic model was enriched with urea cycle, pentose phosphate metabolism, nucleotide and vitamin metabolism, while corticosteroid-treated model in inositol phosphate metabolism, fatty acid oxidation, terpenoid biosynthesis, thiamine biosynthesis, etc. Additionally, the authors identified novel genes from pathogen-sensing gene families of Tlrs, Clecs, and Nlrs, which had not been previously described in pulmonary aspergillosis.
On the fungal side, the comparison of gene expression profiles of A. fumigatus in different mice models has shown a large proportion of similar DE genes (n = 3345), with a restricted number of model- and time-point-specific DE genes (n = 128–204). Nevertheless, the analysis of fungal secreted proteins, which are important for fungal pathogenesis, showed that A. fumigatus has a temporal and model-specific activation of these proteins. However, it has to be noted that the analysis of a large proportion of fungal genes (5175, ~60% of total genes) was precluded due to very low expression values, which once again demonstrates the problem of low concentration of fungal genetic material for in vivo dual RNA-Seq experiments.
Previously, it has been demonstrated that virulence varies among different A. fumigatus strains (Fuller et al. 2016; Kowalski et al. 2016) and moreover that the host immune response against different strains is also distinct (Rizzetto et al. 2013). Thus, opposite to Kale et al., Watkins et al. (Watkins et al. 2018) have recently made RNA-Seq gene expression profiling of two A. fumigatus strains Cea10 and Af293 interacting with human airways cell line A549 to elucidate the differences and commonalities between the virulence mechanisms of aforementioned strains. The experiment comprised two time points (6- and 16-h post-infection) of infected human cells with two fungal strains and time-matched controls for fungal samples (i.e., fungi without host cells). The study focused only on the fungal side, thus the authors did not perform controls for the host counterpart. Since the ratio of fungal and human cells in the infection models was close to 1:1, the RNA-Seq successfully recovered enough amount of fungal reads (53±29.3 million reads per sample) for robust downstream analysis. Differential expression analysis revealed 7888 genes across conditions, and PCA and hierarchical clustering with those genes showed that samples clustered largely according to time points and by strain (on 16-h time point) and that controls were positioned close to infected samples. Taken together, the patterns of sample distribution and clustering highlighted that changes of fungal transcriptional profiles are largely due to growth and metabolism dynamics, rather than to a response to the lung epithelial cells. To dissect the genes that are specifically involved in the infection process, the authors compared transcriptional profiles of time-matched infection and control samples. In this case, a modest response to human cells was found (n = 128–619 DE genes) with 70% of them being strain-specific, indicating the virtual lack of strong conservative response of A. fumigatus strains at least in the analyzed conditions. Nevertheless, a small proportion of genes (n = 47) that were similarly expressed in both strains was found, and the mutants of seven of these genes were shown to have attenuated virulence.
Chen et al. (2015) investigated the interactions of A549 epithelial cell line with A. fumigatus, but unlike in Watkins et al., this study focused on the host side. Here, the authors infected cell cultures with A. fumigatus B5233 for 8 h and performed fungal-free control at similar conditions, with further RNA isolation and sequencing of the host cells. Differential expression analysis between infected and intact cells revealed in total 302 up- and 157 down-regulated genes. GO enrichment analysis showed that down-regulated genes were enriched in ion transport, skeletal system developments, and vascular development. On the other hand, similar to other studies, up-regulated genes were functionally enriched in numerous immune-associated processes such as chemotaxis, inflammatory response, response to bacterium, and also in cytoskeleton remodeling, which also has been reported earlier (Jia et al. 2014). To further investigate the role of specific host genes in fungal response, the authors chose two genes—ARC and EGR1—involved in cytoskeleton rearrangements, since it is known that A. fumigatus conidia are able to internalize in the host cells. Western blotting indicated that corresponding proteins of that genes were up-regulated during the course of infection. Moreover, inhibition of expression of ARC and EGR1 genes by RNAi decreased the internalization rates of conidia by 20 and 40%, respectively.
Another in vivo murine infection model of host–aspergillus interactions was made by Shankar et al. (2018). Here, unlike in the above-mentioned study by Kale et al. (2017), the authors investigated invasive aspergillosis in immunocompetent mice and focused on kidney infection. The study comprised a timecourse infection model at five different time points up to eight days after infection. At all time points and for control animals, infected kidneys were homogenized and subjected to RNA-Seq. Initially, the authors aimed for resolving host–pathogen interaction of both counterparts; however, as usual for in vivo studies, fungal side did not yield interpretable amount of data. Thus, further analysis was focused only on the host transcriptome. Overall, differential expression analysis revealed more than 14,000 DE genes throughout the course of infection in mice. Although notable up-regulation was observable from the first day after infection, functional enrichment was only observed after five days post-infection. Enriched terms included leukocyte aggregation, acute inflammatory responses, positive regulation of chemokines, and other immune response processes. A more in-depth investigation of up-regulated genes showed the activation of several genes such as Ccr5, Cxcr3, Ccr2, or Cxcr4, which are directly involved in activation and recruitment of Th-1 and Th-17 T-helper cells. In turn, after activation of Th cells, the up-regulation of different proinflammatory cytokines such as IFN-c, IL-27, IL-18, IL-24 was detected. Conversely, down-regulated genes were associated with iron and heme binding, electron carrier activity, and aromatase activity. However, in the case of iron-regulation associated genes, the pattern of down-regulation was explained by suppression of P450 related genes, since many other key components of iron homeostasis like Nos1, Nos2, Ltf were systematically up-regulated.
3.3 Cryptococcus
Unlike Candida and Aspergillus, the Cryptococcus genus belongs to the phylum Basidiomycota (http://www.asmscience.org/content/book/10.1128/9781555816858.ch01). There are two main pathogenic Cryptococcus species, Cr. neoformans and Cr. gattii, which are environmental non-host-specific pathogens infecting a wide range of hosts including insects, plants, and mammals. In the case of humans, Cr. neoformans is mainly an opportunistic pathogen, while Cr. gatii can infect immunocompetent individuals [reviewed in (Kwon-Chung et al. 2015)]. In recent decades, the incidence on cryptococcosis has increased drastically which is mainly associated with an emergence of HIV and increasing numbers of organ transplant recipients. The main types cryptococcal infections are cutaneous cryptococcosis, pulmonary cryptococcosis, and cryptococcal meningitis, with the latter being fatal if not treated on initial stages. In the developing part of the world, it has been estimated that these two species cause around one million infections with mortality rates reaching 70% and causing 650,000 deaths per year (Brown et al. 2012; Park et al. 2009). As it is the case of Aspergillus conidia, cryptococcal spores or dried yeast cells enter the host organism through inhalation or through direct interaction in the case of skin-related infections.
As for Candida and Aspergillus, the pathogenicity mechanisms of Cryptococcus species have been more extensively studied in vitro by exposing them to different environmental conditions (O’Meara et al. 2013; Brandão et al. 2018; Zhang et al. 2014). However, in vivo or ex vivo transcriptomic studies of Cryptococcus-host interaction are limited to several recent studies. Moreover, some studies using RNA-Seq were performed for refining the genome and transcriptome annotations of Cryptococcus species (Janbon et al. 2014; Gonzalez-Hilarion et al. 2016; Ferrareze et al. 2017), which are not covered by our review.
The first study investigating transcriptome of Cr. neoformans cells interacting with the host environment was carried out in 2014 by Chen et al. (2014b). The authors performed RNA-Seq of two Cr. neoformans var. grubii strains, G0 and HC1, taken directly from the cerebrospinal fluid (CSF) of two patients with cryptococcal meningitis. Additionally, the same fungal cells were grown in two conditions—ex vivo CSF and YPD, followed by RNA sequencing and comparison with in vivo obtained fungi. Initial analysis showed that gene expression profiles of both strains in each condition were very similar; thus, the strains at the given conditions were considered as biological replicates. Differential gene expression analysis between pairs of conditions identified 129, 45, 256 DE genes when comparing ex vivo versus YPD, in vivo versus YPD, and in vivo versus ex vivo, respectively. This shows that transcriptomes from in vivo and YPD samples are more similar than in ex vivo CSF samples. Compared to ex vivo cells, in vivo samples were enriched with cellular biosynthetic GO terms, indicating that Cr. neoformans cells within the host are transcriptionally more active, which might be explained by interaction with the immune system. On the other hand, as expected, samples exposed to CSF (in both cases) comparing with those with YPD had multiple DE genes previously reported to be important for Cr. neoformans virulence, such as CFO1 (Jung et al. 2009), ENA1 (Idnurm et al. 2009), and RIM101 (O’Meara et al. 2010). The authors also identified 100 strain-specific differentially expressed genes, which were enriched in transporter genes. Additionally, the high sequencing depth allowed the authors to perform variant calling of sequenced strains and compare their genotypes with the reference. Variant calling showed a substantial genomic variation between the analyzed strains and the reference genome—50,155 and 156,880 SNVs were identified in G0 and HC1, respectively, which demonstrates that diverse Cr. neoformans strains have largely similar transcriptomic responses to the host environment. Taking advantage of the high depth and quality of the sequencing data, the authors performed de novo assembly of the transcriptomes of strains identifying novel genes related to transport, localization, and membrane constitution. Taken together, this work was the first study addressing the question of virulence mechanisms of phylogenetically diverse strains of Cr. neoformans obtained directly from host using RNA-Seq and thorough methods of bioinformatics data analysis.
In another study Liu et al. (2014a) compared the transcriptomic profiles of brain tissues in mice, infected by WT Cr. neoformans and double knock-out mutant for the genes of inositol transporters Itr1a and Itr3c, which were previously shown to be involved in fungal virulence through their role in uptaking inositol from the host. Gene expression profiles obtained by RNA-Seq were generated for control mice and were compared in a pairwise manner with those from mice infected by the two fungal strains. Differential expression analysis identified 1133 up- and 1600 down-regulated genes in WT-infected mice, while itr1aΔ itr3cΔ mutant strain showed altered expression of 552 up-regulated and 278 down-regulated genes. Three hundred and seventy-one genes were shared between mice infected by each of the two strains. GO enrichment analysis showed that many enriched functional terms are shared across the two different infections, including cellular death and survival, cell-to-cell interaction, involvement in neurological disease. However, mice infected by the mutant strain extensively activated immune-related responses, such as inflammation, humoral immune response, free radical scavenging. In stark contrast, none of the immune response pathways was significantly enriched in WT-infected mice. Moreover, terms related to cell death and necrosis were enriched only in WT-infected mice. To assess changes in the pathogen that were resulting in different host responses, the authors measured the size of fungal capsule and the secretion of glucuronoxylomannan (GXM)—two important factors for Cr. neoformans virulence. While the capsule size was similar in the two strains, the secretion of GXM was significantly reduced in the itr1aΔ itr3cΔ mutant. This result was also confirmed by immunohistologic staining of GXM in mouse brain tissue, showing that animals affected with mutant had less GXM around brain lessons. Overall, this study demonstrated the role of inositol transporters in host–pathogen interactions, linking their function with the secretion of GXM and altered composition of the fungal capsule, which in turn elicits a highlighted host immune response.
Hu et al. (2014) investigated the ability of environmental Cryptococcus neoformans strains to undergo microevolutionary changes promoting the increase of virulence during serial host passages. The authors used nine haploid serotype A Cr. neoformans strains isolated mainly from soil. Each strain was inoculated into mice sequentially four times over four months. Each following passage to a new mouse was performed using fungal colonies isolated from brains of the precedent mouse. Two strains were revealed with prominent increase of virulence, which in both cases reduced the time of mice death of the first and the last passages by 4-fold (~25 h in the first infected mouse and ~ 6 h in the fourth infected mouse). To disentangle the transcriptomic changes of highly adapted strains compares to their environmental predecessors, the authors performed RNA-Seq of aforementioned two strains and one control strain that did not show virulence changes across the passages. RNA-Seq analysis revealed four genes with significantly higher expression in evolved strains compared to predecessors. One of them, Fre3 (CNAG_06524), was shared between two species. Using RNA interference, the authors identified that Fre3 functions as an iron reductase without copper reductase activity. To confirm the role of the gene in pathogenicity, they over-expressed Fre3 in WT background, which recapitulated the increased adaptive virulence phenotype. Overall, the study shows how RNA-Seq can be used to address the important process of environment-to-mammal transition of Cr. neoformans, identifying the role of iron reductase Fre3 in the adaptation to the host.
4 Emerging Technologies in RNA-Seq
4.1 Single-Cell RNA-Seq
The term RNA-Seq is generally referred to sequencing of RNA, which was isolated from the population of cells (bulk RNA-Seq). Thus, the results obtained from bulk RNA-Seq constitute an averaged signal from the sum of individual cells, while each cell (or a sub-population of cells) might have its own transcriptomic patterns. The limitation of sequencing the bulk RNA was overcome by two major technological advances: efficient cell sorting with single-cell isolation and the availability of efficient protocols for the amplification of minute amounts of RNA from these single cells (Kolodziejczyk et al. 2015). Today, these two methods and their derivatives allow performing single-cell RNA sequencing (scRNA-Seq), disentangling transcriptional profiles of thousands (even hundred of thousands) individual cells (Fan et al. 2015; Zheng et al. 2017; Rosenberg et al. 2018). Despite challenges related to cost, technology, and data analysis (Weinreb et al. 2018; Kolodziejczyk et al. 2015; Stegle et al.2015), scRNA-Seq is now one of the most precise methods in transcriptomics studies. However, as compared to studies on mammals, it has not been used much to study microbial cells (Rosenthal et al. 2017; Kolisko et al. 2014; Wang et al. 2015a) and host-pathogen interaction studies (Avraham et al. 2015; Saliba et al. 2016). One recent advancement in this field was reported in Avital et al. 2017, where the authors developed a method for single-cell dual RNA-Seq for mouse macrophages and Salmonella typhimurium cells during infection, revealing three distinct stages of macrophage response to the pathogen. On the other hand, to our knowledge there are no studies addressing human–fungal interaction on the single-cell level. This technology holds a great potential to unravel specific expression patterns governing the switches between fungal morphotypes, quorum sensing, switches from commensalism to pathogenicity and switches between the stages of infection. Moreover, single-cell transcriptomics approaches can decipher how the host senses and reacts to the pathogen at different infection stages and deconvolve the expression patterns of different cell types, especially in context of in vivo studies.
4.2 Long-Read Sequencing
Today, the dominating sequencing technology “sequencing-by-synthesis” of Illumina, also known as second-generation sequencing, generates relatively short reads (25–300 bp) with a very high throughput and high accuracy. Despite the great advantage of the last two features, short reads are often problematic in some specific tasks, such as assembly of complex and repetitive genomes or accurate reconstruction of transcript isoforms. To overcome this problem, Illumina has recently implemented so-called TruSeq synthetic long-read technology, previously known as Moleculo (McCoy et al. 2014). This experimental and data analysis approach splits the molecule into smaller pieces and uses barcodes to tag the adjacent sequences. Further sequencing and bioinformatics data analysis reassembles the initial sequence, thus allowing to obtain synthetic long reads.
Moreover, in the last decade, the advent of third-generation sequencing has opened new avenues in biomedical research, allowing to sequence much longer reads [up to several hundred kbs (Jain et al. 2018)]. Moreover, their single molecule sequencing technology is PCR free, which eliminates potential PCR amplification biases. However, today long-read sequencing comes with two major disadvantages, which are low throughput and high error rates as compared to sequencing-by-synthesis technology. The two major technologies for long-read sequencing are operated by Oxford Nanopore (ON) and Pacific Biosciences. The details of each technology are reviewed in (Lu et al. 2016) and (Rhoads and Au 2015), respectively. Although long-read sequencing was initially used in genomics field to assemble more contiguous and resolved genomes, today the technologies have also been validated in transcriptomics applications, mainly in transcript discovery (Sharon et al. 2013; Chen et al. 2017; Garalde et al. 2018) and at lesser extent in gene/transcript expression profiling (Byrne et al. 2017). To fill the gap of low throughput of long-read technologies and thus allow reliable expression evaluation, so-called hybrid sequencing can be used, which utilizes both short and long-read sequencing data (Ning et al. 2017; Wang et al. 2018). The third-generation sequencing has already advanced our knowledge about the transcriptomes of even well-studied organisms, identifying numerous previously uncharacterized transcripts and splicing events (Chen et al. 2014a; Byrne et al. 2017; Chen et al. 2017; Sharon et al. 2013; Au et al. 2013). Moreover, it already has been reported that ON can be effectively used in microbial diagnostics (Quick et al. 2015; Mitsuhashi et al. 2017; Schmidt et al. 2017), providing the potential of identifying the pathogen in 2–4 h.
Overall, long-read sequencing technology can dramatically further our knowledge of transcriptomes of poorly studied organisms, as is the case of most of human fungal pathogens. In this case, novel-species-specific transcripts can become promising biomarkers for fungal diagnostics and discovery. On the other hand, when applied to host–pathogen interaction studies, it might allow the precise reconstruction of novel pathogen-specific transcripts, like lncRNAs, in the host side, which have been already shown as immune response regulators (Heward and Lindsay 2014; Ouyang et al. 2016; Jiang et al. 2018).
Nevertheless, the third-generation sequencing is still in its infancy, and further improvements and validations of the technology are necessary in order to make it more versatile and popular in biomedical research.
4.3 Potential Applications of RNA-Seq in Fungal Diagnostics
Next-generation sequencing methods have become increasingly popular in the clinics, especially in the context of diagnosis of cancers (Luthra et al. 2015) and Mendelian diseases (Jamuar and Tan 2015). Moreover, today these techniques have already penetrated to microbiology labs, allowing to achieve high precision of microorganism detection (Turabelidze et al. 2013; Shaw et al. 2016), identify drug resistance (Stoesser et al. 2013; Wain and Mavrogiorgou 2013), control outbreaks (Reuter et al. 2013; Sherry et al. 2013), and study microorganisms that are difficult to grow using conventional culturing methods (Berenguer et al. 1993). However, despite the fact that the incidence of fungal infection is steadily increasing, so far the efforts in applying NGS in microbial diagnostics have been mainly focused on bacteria and viruses. Fungal pathogens possess features that make them difficult for management under the paradigm of traditional microbiology diagnostic methods, such as rapid emergence of antimycotic drug resistance, emergence of new pathogenic species, high biodiversity. Thus, the necessity of novel analytical tools such as NGS in fungal diagnostics becomes inevitable. On the other hand, a distinction of different NGS tools in their applicability for diagnostic purposes has to be done. While DNA sequencing plays a major role for species detection, identification, and characterization, RNA-Seq holds a great potential in identifying biomarkers (in form of novel transcripts) and gene/transcript expression-level signatures specific to different species or for different stages of infection. Nevertheless, to achieve this kind of diagnostics, additional research efforts have to be performed. Precisely, here is where the emerging technologies can immensely further the potential for RNA-Seq diagnostics. For instance, inherent problems such as the low amount of fungal RNA in patient samples can be effectively solved using probe enrichment, while the further identification of novel transcripts is addressable by long-read or hybrid sequencing. Moreover, single-cell RNA-Seq approach could be applied to decipher transcriptomic differences between cell populations, increasing the potential resolution of diagnostics. On the other hand, prices and turn-around time of these technologies are yet to achieve levels that make them suitable for the clinical settings. However, with current trends of diminishing prices, smaller and easier to handle machines, and faster turn-around times. the future of RNAseq-based diagnostics may be approaching. Taken together, RNA-Seq and related methods open promising avenues for fungal diagnostics, but nevertheless still a considerable research and technical developments have to be carried out to truly uncover this potential.
5 Concluding Remarks
In the recent decade, the advent of transcriptome sequencing technologies has opened exciting possibilities for exploring gene regulation and how it varies in different contexts at a level of detail and throughput that surpasses the most optimistic expectations of the previous decade. Many biological disciplines are taking advantage of this new era in transcriptomics, and host-pathogen interaction studies are no exception. As a result, our knowledge about the molecular mechanisms of the interplay between various microbes and their hosts has greatly advanced in this time frame. While the application of RNA-Seq for unraveling human–fungal interactions is just gaining momentum, it is already clear that the use of this technology and its derivatives will be the main trajectory in the field for the coming years. Despite its versatility, today RNA-Seq faces several natural and technical barriers, specifically in human–fungal interaction studies. While in vivo studies are complicated by extremely low amount of pathogen cells, infection models do not entirely reconstitute the whole complexity and peculiarities of human–fungal interactions. On the other hand, RNA-Seq is still relatively expensive and requires specific expertise in study planning, bioinformatics data analysis, and interpretation of results. Moreover, current mainstream technologies are limited in several technical aspects. Nevertheless, the technological advancements in the field are occurring at a fast pace, and they are already partially overcoming most of the aforementioned limitations. We anticipate that dual host–pathogen RNA-Seq analyses in both in vivo models and patients will multiply in the coming years, as current limitations are overcome, and will constitute the basis of key advancements in our understanding of host–pathogen interactions during commensalism and infection. Finally, although there are still many technical and practical impediments for the use of RNA-Seq for diagnostic purposes, we foresee a great potential that may be realized as key biomarker genes of the process of infection are discovered and technical developments enable bringing fast, accurate, and affordable RNA-Seq-based technologies to the clinics.
References
Abad A, Victoria Fernández-Molina J, Bikandi J et al (2010) What makes Aspergillus fumigatus a successful pathogen? Genes and molecules involved in invasive aspergillosis. Rev Iberoam Micol 27:155–182. https://doi.org/10.1016/j.riam.2010.10.003
Adams MD, Kelley JM, Gocayne JD et al (1991) Complementary DNA sequencing: expressed sequence tags and human genome project. Science 252:1651–1656. https://doi.org/10.1126/science.2047873
Alwine JC, Kemp DJ, Stark GR (1977) Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc Natl Acad Sci 74:5350–5354. https://doi.org/10.1073/pnas.74.12.5350
Amorim-vaz S, Tran VDT, Pradervand S et al (2015) RNA enrichment method for quantitative transcriptional analysis of pathogens in vivo applied to the fungus Candida albicans. MBio 6:1–16. https://doi.org/10.1128/mBio.00942-15.Editor
Anders S, Pyl PT, Huber W (2015) HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. https://doi.org/10.1093/bioinformatics/btu638
Andes D, Lepak A, Pitula A et al (2005) A simple approach for estimating gene expression in Candida albicans directly from a systemic infection site. J Infect Dis 192:893–900. https://doi.org/10.1086/432104
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Aprianto R, Slager J, Holsappel S, Veening JW (2016) Time-resolved dual RNA-seq reveals extensive rewiring of lung epithelial and pneumococcal transcriptomes during early infection. Genome Biol 17. https://doi.org/10.1186/s13059-016-1054-5
Au KF, Sebastiano V, Afshar PT et al (2013) Characterization of the human ESC transcriptome by hybrid sequencing. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1320101110
Avital G, Avraham R, Fan A et al (2017) scDual-Seq: Mapping the gene regulatory program of Salmonella infection by host and pathogen single-cell RNA-sequencing. Genome Biol. https://doi.org/10.1186/s13059-017-1340-x
Avraham R, Haseley N, Brown D et al (2015) Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses. Cell. https://doi.org/10.1016/j.cell.2015.08.027
Bainbridge MN, Warren RL, Hirst M et al (2006) Analysis of the prostate cancer cell line LNCaP transcriptome using a sequencing-by-synthesis approach. BMC Genomics 7. https://doi.org/10.1186/1471-2164-7-246
Baruzzo G, Hayer KE, Kim EJ et al (2017) Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat Methods 14:135–139. https://doi.org/10.1038/nmeth.4106
Berenguer J, Buck M, Witebsky F et al (1993) Lysis-centrifugation blood cultures in the detection of tissue-proven invasive candidiasis disseminated versus single-organ infection. Diagn Microbiol Infect Dis 17:103–109. https://doi.org/10.1016/0732-8893(93)90020-8
Binkley J, Arnaud MB, Inglis DO et al (2014) The Candida Genome Database: the new homology information page highlights protein similarity and phylogeny. Nucleic Acids Res 42:D711–D716. https://doi.org/10.1093/nar/gkt1046
Bitar D, Lortholary O, Le Strat Y et al (2014) Population-based analysis of invasive fungal infections, France, 2001–2010. Emerg Infect Dis 20:1149–1155. https://doi.org/10.3201/eid2007.140087
Black MB, Parks BB, Pluta L et al (2014) Comparison of microarrays and RNA-Seq for gene expression analyses of dose-response experiments. Toxicol Sci 137:385–403. https://doi.org/10.1093/toxsci/kft249
Blackwell M (2011) The fungi: 1, 2, 3 … 5.1 million species? Am J Bot 98:426–438. https://doi.org/10.3732/ajb.1000298
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Borodina T, Adjaye J, Sultan M (2011) A Strand-Specific Library Preparation Protocol for RNA Sequencing. In: Methods in enzymology. pp 79–98
Brandão F, Esher SK, Ost KS et al (2018) HDAC genes play distinct and redundant roles in Cryptococcus neoformans virulence. Sci Rep. https://doi.org/10.1038/s41598-018-21965-y
Brandt ME, Lockhart SR (2012) Recent taxonomic developments with candida and other opportunistic yeasts. Curr Fungal Infect Rep 6:170–177. https://doi.org/10.1007/s12281-012-0094-x
Bray NL, Pimentel H, Melsted P, Pachter L (2016) Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34:525–527. https://doi.org/10.1038/nbt.3519
Brown GD, Denning DW, Gow NAR et al (2012) Hidden killers: human fungal infections. Sci. Transl, Med, p 4
Brown NA, Ries LNA, Reis TF et al (2016) RNAseq reveals hydrophobins that are involved in the adaptation of Aspergillus nidulans to lignocellulose. Biotechnol Biofuels 9. https://doi.org/10.1186/s13068-016-0558-2
Bruno VM, Shetty AC, Yano J et al (2015) Transcriptomic Analysis of Vulvovaginal Candidiasis Identifies a Role for the NLRP3 Inflammasome. MBio 6:1–15. https://doi.org/10.1128/mBio.00182-15
Bullard JH, Purdom E, Hansen KD, Dudoit S (2010) Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11:94. https://doi.org/10.1186/1471-2105-11-94
Byrne A, Beaudin AE, Olsen HE et al (2017) Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat Commun. https://doi.org/10.1038/ncomms16027
Campbell JD, Liu G, Luo L et al (2015) Assessment of microRNA differential expression and detection in multiplexed small RNA sequencing data. RNA. https://doi.org/10.1261/rna.046060.114
Castel SE, Levy-Moonshine A, Mohammadi P et al (2015) Tools and best practices for data processing in allelic expression analysis. Genome Biol 16:195. https://doi.org/10.1186/s13059-015-0762-6
Cerqueira GC, Arnaud MB, Inglis DO et al (2014) The Aspergillus Genome Database: multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucleic Acids Res 42:D705–D710. https://doi.org/10.1093/nar/gkt1029
Chalupová J, Raus M, Sedlářová M, Šebela M (2014) Identification of fungal microorganisms by MALDI-TOF mass spectrometry. Biotechnol Adv 32:230–241
Chapman B, Slavin M, Marriott D et al (2017) Changing epidemiology of candidaemia in Australia. J Antimicrob Chemother 72:1103–1108. https://doi.org/10.1093/jac/dkw422
Chen SY, Deng F, Jia X et al (2017) A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing. Sci Rep. https://doi.org/10.1038/s41598-017-08138-z
Chen JJ, Hsueh HM, Delongchamp RR et al (2007) Reproducibility of microarray data: a further analysis of microarray quality control (MAQC) data. BMC Bioinformatics 8. https://doi.org/10.1186/1471-2105-8-412
Chen L, Kostadima M, Martens JHA et al (2014a) Transcriptional diversity during lineage commitment of human blood progenitors. Science. https://doi.org/10.1126/science.1251033
Chen Y, Toffaletti DL, Tenor JL et al (2014b) The Cryptococcus neoformans transcriptome at the site of human meningitis. MBio 5. https://doi.org/10.1128/mbio.01087-13
Chen F, Zhang C, Jia X et al (2015) Transcriptome profiles of human lung epithelial cells A549 interacting with Aspergillus fumigatus by RNA-Seq. PLoS One 10. https://doi.org/10.1371/journal.pone.0135720
Cheon SA, Thak EJ, Bahn YS, Kang HA (2017) A novel bZIP protein, Gsb1, is required for oxidative stress response, mating, and virulence in the human pathogen Cryptococcus neoformans. Sci Rep 7. https://doi.org/10.1038/s41598-017-04290-8
Chhangawala S, Rudy G, Mason CE, Rosenfeld JA (2015) The impact of read length on quantification of differentially expressed genes and splice junction detection. Genome Biol 16:131. https://doi.org/10.1186/s13059-015-0697-y
Chowdhary A, Sharma C, Meis JF (2017) Candida auris: A rapidly emerging cause of hospital-acquired multidrug-resistant fungal infections globally. PLoS Pathog. 13
Cock PJA, Fields CJ, Goto N et al (2009) The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res 38:1767–1771. https://doi.org/10.1093/nar/gkp1137
Conesa A, Madrigal P, Tarazona S et al (2016) A survey of best practices for RNA-seq data analysis. Genome Biol 17:13. https://doi.org/10.1186/s13059-016-0881-8
Cottier F, Tan ASM, Chen J et al (2015) The transcriptional stress response of Candida albicans to weak organic acids. G3 (Bethesda) 5:497–505. https://doi.org/10.1534/g3.114.015941
D’Souza CA, Kronstad JW, Taylor G et al (2011) Genome variation in Cryptococcus gattii, an emerging pathogen of immunocompetent hosts. MBio 2. https://doi.org/10.1128/mbio.00342-10
Dagenais TRT, Keller NP (2009) Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin Microbiol Rev 22:447–465
Dijksterhuis J, Houbraken J, Samson RA (2013) Fungal spoilage of crops and food. In: Agricultural Applications, 2nd Edition. pp 35–56
Dinel S, Bolduc C, Belleau P et al (2005) Reproducibility, bioinformatic analysis and power of the SAGE method to evaluate changes in transcriptome. Nucleic Acids Res 33:1–8. https://doi.org/10.1093/nar/gni025
Dobin A, Davis CA, Schlesinger F et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Dobin A, Gingeras TR (2013) Comment on “TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions” by Kim et al. bioRxiv 000851. https://doi.org/10.1101/000851
Dutton LC, Paszkiewicz KH, Silverman RJ et al (2016) Transcriptional landscape of trans-kingdom communication between Candida albicans and Streptococcus gordonii. Mol Oral Microbiol 31:136–161. https://doi.org/10.1111/omi.12111
Emmert-Streib F, Glazko GV (2011) Pathway analysis of expression data: deciphering functional building blocks of complex diseases. PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1002053
Fan HC, Fu GK, Fodor SPA, Önnerfjord P (2015) Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science. https://doi.org/10.1126/science.1258367
Ferrareze PAG, Streit RSA, dos Santos PR et al (2017) Transcriptional Analysis Allows Genome Reannotation and Reveals that Cryptococcus gattii VGII Undergoes Nutrient Restriction during Infection. Microorganisms 5:49. https://doi.org/10.3390/microorganisms5030049
Flevari A, Theodorakopoulou M, Velegraki A et al (2013) Treatment of invasive candidiasis in the elderly: a review. Clin Interv Aging 8:1199–1208
Francis WR, Christianson LM, Kiko R et al (2013) A comparison across non-model animals suggests an optimal sequencing depth for de novo transcriptome assembly. BMC Genom 14:1–12. https://doi.org/10.1186/1471-2164-14-167
Fuller KK, Cramer RA, Zegans ME et al (2016) Aspergillus fumigatus photobiology illuminates the marked heterogeneity between isolates. MBio 7. https://doi.org/10.1128/mbio.01517-16
Gabaldón T, Carreté L (2016) The birth of a deadly yeast: tracing the evolutionary emergence of virulence traits in Candida glabrata. FEMS Yeast Res. 16
Gabaldón T, Naranjo-Ortíz MA, Marcet-Houben M (2016) Evolutionary genomics of yeast pathogens in the Saccharomycotina. FEMS Yeast Res. 16
Garalde DR, Snell EA, Jachimowicz D et al (2018) Highly parallel direct RN A sequencing on an array of nanopores. Nat Methods. https://doi.org/10.1038/nmeth.4577
Geiss GK, Bumgarner RE, Birditt B et al (2008) Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 26:317–325. https://doi.org/10.1038/nbt1385
Gibbons JG, Beauvais A, Beau R et al (2012) Global transcriptome changes underlying colony growth in the opportunistic human pathogen Aspergillus fumigatus. Eukaryot Cell 11:68–78. https://doi.org/10.1128/EC.05102-11
Gonzalez-Hilarion S, Paulet D, Lee KT et al (2016) Intron retention-dependent gene regulation in Cryptococcus neoformans. Sci Rep. https://doi.org/10.1038/srep32252
Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17:333–351. https://doi.org/10.1038/nrg.2016.49
Griffin AT, Hanson KE (2014) Update on fungal diagnostics. Curr Infect Dis Rep 16. https://doi.org/10.1007/s11908-014-0415-z
Guinea J (2014) Global trends in the distribution of Candida species causing candidemia. Clin Microbiol Infect 20:5–10
Guo Y, Zhao S, Li C-I et al (2014) RNAseqPS: A Web Tool for Estimating Sample Size and Power for RNAseq Experiment. Cancer Inform 13:1–5. https://doi.org/10.4137/CIN.S17688
Haas BJ, Papanicolaou A, Yassour M et al (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512. https://doi.org/10.1038/nprot.2013.084
Hart SN, Therneau TM, Zhang Y et al (2013) Calculating Sample Size Estimates for RNA Sequencing Data. J Comput Biol 20:970–978. https://doi.org/10.1089/cmb.2012.0283
Havlickova B, Czaika VA, Friedrich M (2008) Epidemiological trends in skin mycoses worldwide. Mycoses 51:2–15
Hawksworth DL, Lücking R (2017) Fungal Diversity Revisited: 2.2 to 3.8 Million Species. Microbiol Spectr 5. https://doi.org/10.1128/microbiolspec.funk-0052-2016
Heward JA, Lindsay MA (2014) Long non-coding RNAs in the regulation of the immune response. Trends Immunol
Hu G, Chen SH, Qiu J et al (2014) Microevolution during serial mouse passage demonstrates FRE3 as a virulence adaptation gene in Cryptococcus neoformans. MBio. https://doi.org/10.1128/mBio.00941-14
Hu B, Xie G, Lo C-C et al (2011) Pathogen comparative genomics in the next-generation sequencing era: genome alignments, pangenomics and metagenomics. Brief Funct Genomics 10:322–333. https://doi.org/10.1093/bfgp/elr042
Idnurm A, Walton FJ, Floyd A et al (2009) Identification of ENA1 as a virulence gene of the human pathogenic fungus Cryptococcus neoformans through signature-tagged insertional mutagenesis. Eukaryot Cell. https://doi.org/10.1128/EC.00375-08
Irmer H, Tarazona S, Sasse C et al (2015) RNAseq analysis of Aspergillus fumigatus in blood reveals a just wait and see resting stage behavior. BMC Genom 16:640. https://doi.org/10.1186/s12864-015-1853-1
Jain M, Koren S, Miga KH et al (2018) Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. https://doi.org/10.1038/nbt.4060
Jamuar SS Tan EC (2015) Clinical application of next-generation sequencing for Mendelian diseases. Hum. Genomics
Janbon G, Ormerod KL, Paulet D et al (2014) Analysis of the Genome and Transcriptome of Cryptococcus neoformans var. grubii Reveals Complex RNA Expression and Microevolution Leading to Virulence Attenuation. PLoS Genet 10. https://doi.org/10.1371/journal.pgen.1004261
Jia X, Chen F, Pan W et al (2014) Gliotoxin promotes Aspergillus fumigatus internalization into type II human pneumocyte A549 cells by inducing host phospholipase D activation. Microbes Infect 16:491–501. https://doi.org/10.1016/j.micinf.2014.03.001
Jiang H, Lei R, Ding S-W, Zhu S (2014) Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15:182. https://doi.org/10.1186/1471-2105-15-182
Jiang C, Li Z, Zhang L et al (2016) Significance of hyphae formation in virulence of Candida tropicalis and transcriptomic analysis of hyphal cells. Microbiol Res 192:65–72. https://doi.org/10.1016/j.micres.2016.06.003
Jiang M, Zhang S, Yang Z et al (2018) Self-Recognition of an Inducible Host lncRNA by RIG-I Feedback Restricts Innate Immune Response. Cell. https://doi.org/10.1016/j.cell.2018.03.064
Jung WH, Hu G, Kuo W, Kronstad JW (2009) Role of ferroxidases in iron uptake and virulence of Cryptococcus neoformans. Eukaryot Cell. https://doi.org/10.1128/EC.00166-09
Kale SD, Ayubi T, Chung D et al (2017) Modulation of Immune Signaling and Metabolism Highlights Host and Fungal Transcriptional Responses in Mouse Models of Invasive Pulmonary Aspergillosis. Sci Rep 7. https://doi.org/10.1038/s41598-017-17000-1
Kathiravan, MK, Salake AB, Chothe AS, Dudhe PB, Watode RP, Mukta MS, Gadhwe S (2012). The biology and chemistry of antifungal agents: A review. Bioorganic Med Chem 20(19):5678–5698. https://doi.org/10.1016/j.bmc.2012.04.045
Khot PD, Fredricks DN (2009) PCR-based diagnosis of human fungal infections. Expert Rev Anti Infect Ther 7:1201–1221. https://doi.org/10.1586/eri.09.104
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360. https://doi.org/10.1038/nmeth.3317
Kim D, Pertea G, Trapnell C et al (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. https://doi.org/10.1186/gb-2013-14-4-r36
Kim J, Sudbery P (2011) Candida albicans, a major human fungal pathogen. J Microbiol 49:171–177. https://doi.org/10.1007/s12275-011-1064-7
Klingspor L, Tortorano AM, Peman J et al (2015) Invasive Candida infections in surgical patients in intensive care units: a prospective, multicentre survey initiated by the European Confederation of Medical Mycology (ECMM) (2006-2008). Clin Microbiol Infect 21:87.e1-87.e10. https://doi.org/10.1016/j.cmi.2014.08.011
Kolisko M, Boscaro V, Burki F et al (2014) Single-cell transcriptomics for microbial eukaryotes. Curr, Biol
Kolodziejczyk AA, Kim JK, Svensson V et al (2015) The Technology and Biology of Single-Cell RNA Sequencing. Mol. Cell
Kowalski CH, Beattie SR, Fuller KK et al (2016) Heterogeneity among isolates reveals that fitness in low oxygen correlates with Aspergillus fumigatus virulence. MBio 7. https://doi.org/10.1128/mbio.01515-16
Kozel TR, Wickes B (2014) Fungal diagnostics. Cold Spring Harb Perspect Med 4. https://doi.org/10.1101/cshperspect.a019299
Kwon-Chung KJ, Boekhout T, Wickes BL, Fell JW (2011). Systematics of the genus Cryptococcus and its type species C. neoformans. In Cryptococcus: 3–15. American Society of Microbiology
Kwon-Chung KJ, Fraser JA, Doering TÁL et al (2015) Cryptococcus neoformans and Cryptococcus gattii, the etiologic agents of cryptococcosis. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a019760
Kwon-Chung KJ, Sugui JA (2013) Aspergillus fumigatus-What Makes the Species a Ubiquitous Human Fungal Pathogen? PLoS Pathog 9:1–4. https://doi.org/10.1371/journal.ppat.1003743
Latgé JP (1999) Aspergillus fumigatus and Aspergillosis. Clin Microbiol Rev 12:310–350
Levin JZ, Yassour M, Adiconis X et al (2010) Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 7:709–715. https://doi.org/10.1038/nmeth.1491
Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. https://doi.org/10.1093/bioinformatics/btt656
Lin JQ, Zhao XX, Zhi QQ et al (2013) Transcriptomic profiling of Aspergillus flavus in response to 5-azacytidine. Fungal Genet Biol 56:78–86. https://doi.org/10.1016/j.fgb.2013.04.007
Lister R, O’Malley RC, Tonti-Filippini J et al (2008) Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 133:523–536. https://doi.org/10.1016/j.cell.2008.03.029
Liu Y, Ferguson JF, Xue C et al (2013) Evaluating the Impact of Sequencing Depth on Transcriptome Profiling in Human Adipose. PLoS One 8. https://doi.org/10.1371/journal.pone.0066883
Liu Y, Filler SG (2011) Candida albicans Als3, a multifunctional adhesin and invasin. Eukaryot Cell 10:168–173
Liu Y, Shetty AC, Schwartz JA et al (2015) New signaling pathways govern the host response to C. albicans infection in various niches. Genome Res 125:679–689. https://doi.org/10.1101/gr.187427.114
Liu TB, Subbian S, Pan W et al (2014a) Cryptococcus inositol utilization modulates the host protective immune response during brain infection. Cell Commun Signal 12:1–17. https://doi.org/10.1186/s12964-014-0051-0
Liu Y, Zhou J, White KP (2014b) RNA-seq differential expression studies: more sequence or more replication? Bioinformatics 30:301–304. https://doi.org/10.1093/bioinformatics/btt688
Lockhart DJ, Dong H, Byrne MC et al (1996) Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol 14:1675–1680. https://doi.org/10.1038/nbt1296-1675
Losada L, Barker BM, Pakala S et al (2014) Large-Scale Transcriptional Response to Hypoxia in Aspergillus fumigatus Observed Using RNAseq Identifies a Novel Hypoxia Regulated ncRNA. Mycopathologia 178:331–339. https://doi.org/10.1007/s11046-014-9779-8
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Lowe R, Shirley N, Bleackley M et al (2017) Transcriptomics technologies. PLOS Comput Biol 13:e1005457. https://doi.org/10.1371/journal.pcbi.1005457
Lu H, Giordano F, Ning Z (2016) Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics, Proteomics Bioinforma
Luthra R, Chen H, Roy-Chowdhuri S, Singh RR (2015) Next-generation sequencing in clinical molecular diagnostics of cancer: advantages and challenges. Cancers (Basel)
Marioni JC, Mason CE, Mane SM et al (2008) RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18:1509–1517. https://doi.org/10.1101/gr.079558.108
Martin R, Albrecht-Eckardt D, Brunke S et al (2013) A Core Filamentation Response Network in Candida albicans Is Restricted to Eight Genes. PLoS One 8. https://doi.org/10.1371/journal.pone.0058613
May RC, Stone NRH, Wiesner DL et al (2016) Cryptococcus: from environmental saprophyte to global pathogen. Nat Rev Microbiol 14:106–117
McCoy RC, Taylor RW, Blauwkamp TA et al (2014) Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS ONE. https://doi.org/10.1371/journal.pone.0106689
Metpally RPR, Nasser S, Courtright A et al (2013) Comparison of analysis tools for miRNA high throughput sequencing using nerve crush as a model. Front Genet. https://doi.org/10.3389/fgene.2013.00020
Mitrovich QM, Tuch BB, Guthrie C, Johnson AD (2007) Computational and experimental approaches double the number of known introns in the pathogenic yeast Candida albicans. Genome Res 17:492–502. https://doi.org/10.1101/gr.6111907
Mitsuhashi S, Kryukov K, Nakagawa S et al (2017) A portable system for metagenomic analyses using nanopore-based sequencer and laptop computers can realize rapid on-site determination of bacterial compositions. bioRxiv. https://doi.org/10.1038/s41598-017-05772-5
Mixão V, Gabaldón T (2017) Hybridization and emergence of virulence in opportunistic human yeast pathogens. Yeast. https://doi.org/10.1002/yea.3242
Morrissy AS, Morin RD, Delaney A et al (2009) Next-generation tag sequencing for cancer gene expression profiling. Genome Res 19:1825–1835. https://doi.org/10.1101/gr.094482.109
Moyes DL, Richardson JP, Naglik JR (2014). From: Human Pathogenic Fungi: Molecular Biology and Pathogenic Mechanisms. In: Sullivan DJ, Moran GP (eds). Caister Academic Press, U.K
Moyes DL, Wilson D, Richardson JP et al (2016) Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 532:64–68. https://doi.org/10.1038/nature17625
Naglik JR, Challacombe SJ, Hube B (2003) Candida albicans Secreted Aspartyl Proteinases in Virulence and Pathogenesis. Microbiol Mol Biol Rev 67:400–428. https://doi.org/10.1128/MMBR.67.3.400-428.2003
Nature Microbiology Editorial (2017) Stop neglecting fungi. Nat. Microbiol. 2:17120. https://doi.org/10.1038/nmicrobiol.2017.123
Niemiec MJ, Grumaz C, Ermert D et al (2017) Dual transcriptome of the immediate neutrophil and Candida albicans interplay. BMC Genom 18:696. https://doi.org/10.1186/s12864-017-4097-4
Ning G, Cheng X, Luo P et al (2017) Hybrid sequencing and map finding (HySeMaFi): optional strategies for extensively deciphering gene splicing and expression in organisms without reference genome. Sci Rep. https://doi.org/10.1038/srep43793
Nobile CJ, Nett JE, Andes DR, Mitchell AP (2006) Function of Candida albicans adhesin hwp1 in biofilm formation. Eukaryot Cell 5:1604–1610. https://doi.org/10.1128/EC.00194-06
Nuss AM, Beckstette M, Pimenova M et al (2017) Tissue dual RNA-seq allows fast discovery of infection-specific functions and riboregulators shaping host–pathogen transcriptomes. Proc Natl Acad Sci 114:E791–E800. https://doi.org/10.1073/pnas.1613405114
O’Brien HE, Parrent JL, Jackson JA et al (2005) Fungal Community Analysis by Large-Scale Sequencing of Environmental Samples. Appl Environ Microbiol 71:5544–5550. https://doi.org/10.1128/AEM.71.9.5544
O’Keeffe G, Hammel S, Owens RA et al (2014) RNA-seq reveals the pan-transcriptomic impact of attenuating the gliotoxin self-protection mechanism in Aspergillus fumigatus. BMC Genomics 15. https://doi.org/10.1186/1471-2164-15-894
O’Meara TR, Holmer SM, Selvig K et al (2013) Cryptococcus neoformans Rim101 is associated with cell wall remodeling and evasion of the host immune responses. MBio. https://doi.org/10.1128/mBio.00522-12
O’Meara TR, Norton D, Price MS et al (2010) Interaction of Cryptococcus neoformans Rim101 and protein kinase a regulates capsule. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1000776
O’Neil D, Glowatz H, Schlumpberger M (2013) Ribosomal RNA Depletion for Efficient Use of RNA-Seq Capacity. In: Current Protocols in Molecular Biology. Wiley, Hoboken, NJ, USA, p Unit 4.19
Oren I, Paul M (2014) Up to date epidemiology, diagnosis and management of invasive fungal infections. Clin Microbiol Infect 20:1–4
Otto C, Stadler PF, Hoffmann S (2014) Lacking alignments? The next-generation sequencing mapper segemehl revisited. Bioinformatics 30:1837–1843. https://doi.org/10.1093/bioinformatics/btu146
Ouyang J, Hu J, Chen JL (2016) lncRNAs regulate the innate immune response to viral infection. Wiley Interdiscip Rev RNA 7:129–143. https://doi.org/10.1002/wrna.1321
Papon N, Courdavault V, Clastre M, Bennett RJ (2013) Emerging and Emerged Pathogenic Candida Species: Beyond the Candida albicans Paradigm. PLoS Pathog 9. https://doi.org/10.1371/journal.ppat.1003550
Park BJ, Wannemuehler KA, Marston BJ et al (2009) Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS. https://doi.org/10.1097/QAD.0b013e328322ffac
Parkhomchuk D, Borodina T, Amstislavskiy V et al (2009) Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res 37:e123–e123. https://doi.org/10.1093/nar/gkp596
Patel RK, Jain M (2012) NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7:e30619. https://doi.org/10.1371/journal.pone.0030619
Paterson RRM, Lima N (2017) Filamentous Fungal Human Pathogens from Food Emphasising Aspergillus. Fusarium Mucor Microorganisms 5:44. https://doi.org/10.3390/microorganisms5030044
Patro R, Duggal G, Love MI et al (2017) Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14:417–419. https://doi.org/10.1038/nmeth.4197
Pel HJ, De Winde JH, Archer DB et al (2007) Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol 25:221–231. https://doi.org/10.1038/nbt1282
Perfect JR, Cox GM, Lee JY et al (2001) The impact of culture isolation of Aspergillus species: a hospital-based survey of aspergillosis. Clin Infect Dis 33:1824–1833. https://doi.org/10.1086/323900
Pfaller MA, Diekema DJ (2007) Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20:133–163
Pfaller MA, Diekema DJ (2010) Epidemiology of Invasive Mycoses in North America. Crit Rev Microbiol 36:1–53. https://doi.org/10.3109/10408410903241444
Pfaller MA, Messer SA, Hollis RJ et al (2009) Variation in susceptibility of bloodstream isolates of Candida glabrata to fluconazole according to patient age and geographic location in the United States in 2001 to 2007. J Clin Microbiol 47:3185–3190. https://doi.org/10.1128/JCM.00946-09
Pruitt KD, Tatusova T, Maglott DR (2007) NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35:D61–D65. https://doi.org/10.1093/nar/gkl842
Pryszcz LP, Németh T, Gácser A, Gabaldón T (2014) Genome comparison of candida orthopsilosis clinical strains reveals the existence of hybrids between two distinct subspecies. Genome Biol Evol 6:1069–1078. https://doi.org/10.1093/gbe/evu082
Pryszcz LP, Németh T, Saus E et al (2015) The Genomic Aftermath of Hybridization in the Opportunistic Pathogen Candida metapsilosis. PLoS Genet 11. https://doi.org/10.1371/journal.pgen.1005626
Quick J, Ashton P, Calus S et al (2015) Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol. https://doi.org/10.1186/s13059-015-0677-2
Rapaport F, Khanin R, Liang Y et al (2013) Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol 14:R95. https://doi.org/10.1186/gb-2013-14-9-r95
Rappolee D A, Mark D, Banda MJ, Werb Z (1988) Wound macrophages express TGF-alpha and other growth factors in vivo: analysis by mRNA phenotyping. Science (80-) 241:708–12. https://doi.org/10.1126/science.3041594
Rasheed M, Battu A, Kaur R (2018) Aspartyl proteases in Candida glabrata are required for suppression of the host innate immune response. J Biol Chem jbc.M117.813741. https://doi.org/10.1074/jbc.m117.813741
Reuter S, Ellington MJ, Cartwright EJP et al (2013) Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology. JAMA Intern Med. https://doi.org/10.1001/jamainternmed.2013.7734
Rhoads A, Au KF (2015) PacBio Sequencing and Its Applications. Genomics, Proteomics Bioinforma
Rhodes J, Desjardins CA, Sykes SM et al (2017) Tracing genetic exchange and biogeography of cryptococcus neoformans var. Grubii at the global population level. Genetics 207:327–346. https://doi.org/10.1534/genetics.117.203836
Ritchie ME, Phipson B, Wu D et al (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47–e47. https://doi.org/10.1093/nar/gkv007
Rizzetto L, Giovannini G, Bromley M et al (2013) Strain Dependent Variation of Immune Responses to A. fumigatus: Definition of Pathogenic Species. PLoS One 8. https://doi.org/10.1371/journal.pone.0056651
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Rosenbach A, Dignard D, Pierce JV et al (2010) Adaptations of Candida albicans for growth in the mammalian intestinal tract. Eukaryot Cell 9:1075–1086. https://doi.org/10.1128/EC.00034-10
Rosenberg AB, Roco CM, Muscat RA et al (2018) Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science (80-). https://doi.org/10.1126/science.aam8999
Rosenthal K, Oehling V, Dusny C, Schmid A (2017) Beyond the bulk: Disclosing the life of single microbial cells. FEMS Microbiol, Rev
Saliba AE, Li L, Westermann AJ et al (2016) Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat Microbiol. https://doi.org/10.1038/nmicrobiol.2016.206
Samson RA, Visagie CM, Houbraken J, Hong S-B, Hubka V, Klaassen CHW, Perrones G, Seifert KA, Susca A, Tanney JB, Varga J, Kocsube S, Szigeti G, Yaguchi T, Frisvad JC (2014) Phylogeny, identification and nomenclature of the genus Aspergillus. Stud Mycol 78:343–371. https://doi.org/10.1016/j.simyco.2014.09.001
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467
Sanglard D (2016) Emerging Threats in Antifungal-Resistant Fungal Pathogens. Front Med 3. https://doi.org/10.3389/fmed.2016.00011
Sarma S, Upadhyay S (2017) Current perspective on emergence, diagnosis and drug resistance in Candida auris. Infect Drug Resist 10:155–165
Satoh K, Makimura K, Hasumi Y et al (2009) Candida auris sp. nov., a novel ascomycetous yeast isolated from the external ear canal of an inpatient in a Japanese hospital. Microbiol Immunol 53:41–44. https://doi.org/10.1111/j.1348-0421.2008.00083.x
Saus E, Willis JR, Pryszcz LP et al (2018) nextPARS: parallel probing of RNA structures in Illumina. RNA 24:609–619. https://doi.org/10.1261/rna.063073.117
Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative Monitoring of Gene Expression Patterns with a Complementary DNA Microarray. Science (80-) 270:467–470. https://doi.org/10.1126/science.270.5235.467
Schmidt K, Mwaigwisya S, Crossman LC et al (2017) Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J Antimicrob Chemother. https://doi.org/10.1093/jac/dkw397
Schröder MS, Martinez de San Vicente K, Prandini THR et al (2016) Multiple Origins of the Pathogenic Yeast Candida orthopsilosis by Separate Hybridizations between Two Parental Species. PLoS Genet 12. https://doi.org/10.1371/journal.pgen.1006404
Schulze S, Henkel SG, Driesch D et al (2015) Computational prediction of molecular pathogen-host interactions based on dual transcriptome data. Front Microbiol. https://doi.org/10.3389/fmicb.2015.00065
Schulze S, Schleicher J, Guthke R, Linde J (2016) How to predict molecular interactions between species?. Front, Microbiol
Schurch NJ, Schofield P, Gierliński M et al (2016) How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 22:839–851. https://doi.org/10.1261/rna.053959.115
Seqc/Maqc-Iii Consortium (2014) A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat Biotechnol 32:903–914. https://doi.org/10.1038/nbt.2957
Seyednasrollah F, Laiho A, Elo LL (2015) Comparison of software packages for detecting differential expression in RNA-seq studies. Brief Bioinform 16:59–70. https://doi.org/10.1093/bib/bbt086
Shankar J, Cerqueira GC, Wortman JR et al (2018) RNA-Seq Profile Reveals Th-1 and Th-17-Type of Immune Responses in Mice Infected Systemically with Aspergillus fumigatus. Mycopathologia. https://doi.org/10.1007/s11046-018-0254-9
Sharon D, Tilgner H, Grubert F, Snyder M (2013) A single-molecule long-read survey of the human transcriptome. Nat Biotechnol. https://doi.org/10.1038/nbt.2705
Shaw WH, Lin Q, Muhammad ZZBR et al (2016) Identification of HIV mutation as diagnostic biomarker through next generation sequencing. J Clin Diagnostic Res. https://doi.org/10.7860/JCDR/2016/19760.8140
Sherry NL, Porter JL, Seemann T et al (2013) Outbreak investigation using high-throughput genome sequencing within a diagnostic microbiology laboratory. J Clin Microbiol. https://doi.org/10.1128/JCM.03332-12
Short DP, O’Donnell K, Geiser DM (2014) Clonality, recombination, and hybridization in the plumbing-inhabiting human pathogen Fusarium keratoplasticum inferred from multilocus sequence typing. BMC Evol Biol 14:91. https://doi.org/10.1186/1471-2148-14-91
Smeekens SP, van de Veerdonk FL, Netea MG (2016) An Omics Perspective on Candida Infections: toward Next-Generation Diagnosis and Therapy. Front Microbiol 7:154. https://doi.org/10.3389/fmicb.2016.00154
Soneson C (2013) Delorenzi M (2013) A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinforma 141(14):91. https://doi.org/10.1186/1471-2105-14-91
Soneson C, Love MI, Robinson MD (2015) Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 4:1521. https://doi.org/10.12688/f1000research.7563.2
Stajich JE, Harris T, Brunk BP et al (2012) FungiDB: an integrated functional genomics database for fungi. Nucleic Acids Res 40:D675–D681. https://doi.org/10.1093/nar/gkr918
Stegle O, Teichmann SA, Marioni JC (2015) Computational and analytical challenges in single-cell transcriptomics. Nat. Rev, Genet
Stoesser N, Batty EM, Eyre DW et al (2013) Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J Antimicrob Chemother. https://doi.org/10.1093/jac/dkt180
Subramanian A, Tamayo P, Mootha VK et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.0506580102
Sudbery P, Gow N, Berman J (2004) The distinct morphogenic states of Candida albicans. Trends Microbiol 12:317–324
Sutcliffe JG, Milner RJ, Bloom FE, Lerner RA (1982) Common 82-nucleotide sequence unique to brain RNA. Proc Natl Acad Sci U S A 79:4942–4946. https://doi.org/10.1073/pnas.79.16.4942
Tarazona S, García-Alcalde F, Dopazo J et al (2011) Differential expression in RNA-seq: a matter of depth. Genome Res 21:2213–2223. https://doi.org/10.1101/gr.124321.111
Thänert R, Goldmann O, Beineke A, Medina E (2017) Host-inherent variability influences the transcriptional response of Staphylococcus aureus during in vivo infection. Nat Commun 8. https://doi.org/10.1038/ncomms14268
The Gene Ontology Consortium (2017) Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw1108
Thewes S, Kretschmar M, Park H et al (2007) In vivo and ex vivo comparative transcriptional profiling of invasive and non-invasive Candida albicans isolates identifies genes associated with tissue invasion. Mol Microbiol 63:1606–1628. https://doi.org/10.1111/j.1365-2958.2007.05614.x
Tierney L, Linde J, Müller S et al (2012) An Interspecies Regulatory Network Inferred from Simultaneous RNA-seq of Candida albicans Invading Innate Immune Cells. Front Microbiol 3:85. https://doi.org/10.3389/fmicb.2012.00085
Tóth R, Cabral V, Thuer E et al (2018) Investigation of Candida parapsilosis virulence regulatory factors during host-pathogen interaction. Sci Rep 8:1–14. https://doi.org/10.1038/s41598-018-19453-4
Turabelidze G, Lawrence SJ, Gao H et al (2013) Precise dissection of an escherichia coli o157:H7 outbreak by single nucleotide polymorphism analysis. J Clin Microbiol. https://doi.org/10.1128/JCM.01930-13
Turner SA, Butler G (2014) The Candida pathogenic species complex. Cold Spring Harb Perspect Med 4. https://doi.org/10.1101/cshperspect.a019778
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW (1995) Serial Analysis of Gene Expression. Science (80-) 270:484–487. https://doi.org/10.1126/science.270.5235.484
Wain J, Mavrogiorgou E (2013) Next-generation sequencing in clinical microbiology. Expert Rev. Mol, Diagn
Wan Y, Qu K, Ouyang Z, Chang HY (2013) Genome-wide mapping of RNA structure using nuclease digestion and high-throughput sequencing. Nat Protoc 8:849–869. https://doi.org/10.1038/nprot.2013.045
Wang J, Chen L, Chen Z, Zhang W (2015a) RNA-seq based transcriptomic analysis of single bacterial cells. Integr Biol (United Kingdom). https://doi.org/10.1039/c5ib00191a
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Wang B, Regulski M, Tseng E et al (2018) A comparative transcriptional landscape of maize and sorghum obtained by single-molecule sequencing. Genome Res. https://doi.org/10.1101/gr.227462.117
Wang K, Zhang Z, Chen X et al (2015b) Transcription factor ADS-4 regulates adaptive responses and resistance to antifungal azole stress. Antimicrob Agents Chemother 59:5396–5404. https://doi.org/10.1128/AAC.00542-15
Warner JR (1999) The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 24:437–440
Watkins TN, Liu H, Chung M et al (2018) Comparative transcriptomics of Aspergillus fumigatus strains upon exposure to human airway epithelial cells. Microb Genomics 1–9. https://doi.org/10.1099/mgen.0.000154
Weinreb C, Wolock S, Tusi BK et al (2018) Fundamental limits on dynamic inference from single-cell snapshots. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1714723115
Westermann AJ, Barquist L, Vogel J (2017) Resolving host–pathogen interactions by dual RNA-seq. PLoS Pathog 13:e1006033. https://doi.org/10.1371/JOURNAL.PPAT.1006033
Westermann AJ, Gorski SA, Vogel J (2012) Dual RNA-seq of pathogen and host. Nat Rev Microbiol 10:618–630. https://doi.org/10.1038/nrmicro2852
Whaley SG, Caudle KE, Simonicova L et al (2018) Jjj1 Is a Negative Regulator of Pdr1-Mediated Fluconazole Resistance in Candida glabrata. mSphere 3:1–11. https://doi.org/10.1128/msphere.00466-17
Williams CR, Baccarella A, Parrish JZ, Kim CC (2016) Trimming of sequence reads alters RNA-Seq gene expression estimates. BMC Bioinformatics 17:103. https://doi.org/10.1186/s12859-016-0956-2
Wilson D, Hube B (2014). From: Human Pathogenic Fungi: Molecular Biology and Pathogenic Mechanisms. Sullivan DJ, Moran GP (eds). Caister Academic Press, U.K
Wolf T, Kämmer P, Brunke S, Linde J (2018) Two’s company: studying interspecies relationships with dual RNA-seq. Curr Opin Microbiol 42:7–12. https://doi.org/10.1016/j.mib.2017.09.001
Wu Y, Li Y, Yu S et al (2016) A Genome-Wide Transcriptional Analysis of Yeast-Hyphal Transition in Candida tropicalis by RNA-Seq. PLoS ONE 11:e0166645. https://doi.org/10.1371/journal.pone.0166645
Wu G, Zhao H, Li C et al (2015) Genus-Wide Comparative Genomics of Malassezia Delineates Its Phylogeny, Physiology, and Niche Adaptation on Human Skin. PLoS Genet 11. https://doi.org/10.1371/journal.pgen.1005614
Xie Y, Wu G, Tang J et al (2014) SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30:1660–1666. https://doi.org/10.1093/bioinformatics/btu077
Yang Q, Gao L, Tao M et al (2016) Transcriptomics Analysis of Candida albicans Treated with Huanglian Jiedu Decoction Using RNA-seq. Evidence-based Complement Altern Med 2016. https://doi.org/10.1155/2016/3198249
Yang X, Liu D, Liu F et al (2013) HTQC: a fast quality control toolkit for Illumina sequencing data. BMC Bioinformatics 14:33. https://doi.org/10.1186/1471-2105-14-33
Yu L, Fernandez S, Brock G (2017) Power analysis for RNA-Seq differential expression studies. BMC Bioinformatics 18:234. https://doi.org/10.1186/s12859-017-1648-2
Zhang N, Park YD, Williamson PR (2014) New technology and resources for cryptococcal research. Fungal Genet Biol. https://doi.org/10.1016/j.fgb.2014.11.001
Zhao W, He X, Hoadley KA et al (2014) Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genom 15:419. https://doi.org/10.1186/1471-2164-15-419
Zhao S, Zhang Y, Gordon W et al (2015) Comparison of stranded and non-stranded RNA-seq transcriptome profiling and investigation of gene overlap. BMC Genom 16:675. https://doi.org/10.1186/s12864-015-1876-7
Zheng GXY, Terry JM, Belgrader P et al (2017) Massively parallel digital transcriptional profiling of single cells. Nat Commun. https://doi.org/10.1038/ncomms14049
Zhu YY, Machleder EM, Chenchik A et al (2001) Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques 30:892–897
Zoll J, Snelders E, Verweij PE, Melchers WJG (2016) Next-Generation Sequencing in the Mycology Lab. Curr Fungal Infect Rep 10:37–42. https://doi.org/10.1007/s12281-016-0253-6
Acknowledgements
TG group acknowledges support from the Spanish Ministry of Economy, Industry, and Competitiveness (MEIC) for the EMBL partnership and grants “Centro de Excelencia Severo Ochoa 2013–2017” SEV-2012-0208, and BFU2015-67107 cofounded by European Regional Development Fund (ERDF), from the CERCA Programme/Generalitat de Catalunya, from the Catalan Research Agency (AGAUR) SGR857, and grant from the European Union’s Horizon 2020 research and innovation programme under the grant agreement ERC-2016-724173 the Marie Sklodowska-Curie grant agreement No. H2020-MSCA-ITN-2014-642095.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hovhannisyan, H., Gabaldón, T. (2018). Transcriptome Sequencing Approaches to Elucidate Host–Microbe Interactions in Opportunistic Human Fungal Pathogens. In: Rodrigues, M. (eds) Fungal Physiology and Immunopathogenesis . Current Topics in Microbiology and Immunology, vol 422. Springer, Cham. https://doi.org/10.1007/82_2018_122
Download citation
DOI: https://doi.org/10.1007/82_2018_122
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-30236-8
Online ISBN: 978-3-030-30237-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)