
Abstract
Specific IgM, administered together with the antigen it recognizes, enhances primary antibody responses, formation of germinal centers, and priming for secondary antibody responses. The response to all epitopes on the antigen to which IgM binds is usually enhanced. IgM preferentially enhances responses to large antigens such as erythrocytes, malaria parasites, and keyhole limpet hemocyanine. In order for an effect to be seen, antigens must be administered in suboptimal concentrations and in close temporal relationship to the IgM. Enhancement is dependent on the ability of IgM to activate complement, but the lytic pathway is not required. Enhancement does not take place in mice lacking complement receptors 1 and 2 (CR1/2) suggesting that the role of IgM is to generate C3 split products, i.e., the ligands for CR1/2. In mice, these receptors are expressed on follicular dendritic cells (FDCs) and B cells. Optimal IgM-mediated enhancement requires that both cell types express CR1/2, but intermediate enhancement is seen when only FDCs express the receptors and low enhancement when only B cells express them. These observations imply that IgM-mediated enhancement works through several, non-mutually exclusive, pathways. Marginal zone B cells can transport IgM-antigen-complement complexes, bound to CR1/2, from the marginal zone and deposit them onto FDCs. In addition, co-crosslinking of the BCR and the CR2/CD19/CD81 co-receptor complex may enhance signaling to specific B cells, a mechanism likely to be involved in induction of early extrafollicular antibody responses.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1 Introduction
When antibodies are passively administered together with their specific antigen, they can dramatically change the antibody response towards the antigen. The response can be completely suppressed or enhanced by several hundred-fold. This phenomenon is called antibody-mediated feedback regulation. In experimental situations, the regulating antibodies are usually given intravenously within a few hours of the antigen, but feedback regulation also works in a more natural setting with endogenously produced antibodies as regulators. In early studies, reviewed by Uhr and Möller (1968), the source of the regulating antibodies was usually serum from immunized animals, and therefore the effect of individual antibody classes could not be determined. With the arrival of antibody separation techniques and the hybridoma technology, the immunoregulatory effects of different antibody isotypes has been extensively investigated [reviewed in (Heyman 2000; Hjelm et al. 2006; Sörman et al. 2014)]. In studies of antibody-mediated feedback regulation, both antibodies and antigen are administered in physiological salt solutions, i.e., without adjuvants. Although there are exceptions, the regulatory effects are generally antigen but not epitope-specific. This suggests that the response to the entire antigen, captured by the antibody, is affected, regardless of to which epitope the regulating antibody binds.
1.1 IgG-Mediated Feedback Suppression
IgG, administered together with erythrocytes, can completely suppress the antibody response. This is utilized in the clinic to prevent Rh-negative women carrying Rh-positive fetuses from becoming immunized against fetal erythrocytes acquired via transplacental hemorrhage. Maternal IgG crosses the placenta and can damage the erythrocytes of the fetus or newborn. A small dose of preformed IgG anti-Rh, given to the mother during pregnancy or immediately after delivery, prevents hemolytic disease of the newborn (Clarke et al. 1963; Bowman 1988). The mechanism behind IgG-mediated suppression is still not understood. One possibility is that IgG masks the antigen and prevents naïve B cells from binding to it. Another, not mutually exclusive, possibility is that erythrocytes covered with IgG will be rapidly eliminated from the circulation and therefore be unable to stimulate an immune response. Complement activation is not required for IgG-mediated suppression (Heyman et al. 1988b; Bergström and Heyman 2015) and IgG suppresses equally well in the absence of all known Fc-receptors for IgG (Karlsson et al. 1999, 2001; Bernardo et al. 2015; Bergström and Heyman 2015).
1.2 IgG-Mediated Feedback Enhancement
When IgG is administered with soluble protein antigens, it will enhance antibody responses. In fact, the same monoclonal IgG anti-TNP which suppresses responses to SRBC-TNP can enhance responses to KLH-TNP (Enriquez-Rincon and Klaus 1984; Wiersma et al. 1989), illustrating the important role of the type of antigen. Enhancement by the murine subclasses IgG1, IgG2a, and IgG2b requires Fc-receptors for IgG (Wernersson et al. 1999) whereas IgG3 largely operates via complement (Diaz de Ståhl et al. 2003; Zhang et al. 2014). The most likely mechanism for enhancement by the Fc-receptor-dependent subclasses is increased uptake of IgG-antigen by dendritic cells, followed by increased T helper cell induction (Getahun et al. 2004; de Jong et al. 2006; Hamano et al. 2000) whereas IgG3 probably acts by increasing the delivery of antigen to B cell follicles and FDCs (Zhang et al. 2014).
1.3 IgE-Mediated Feedback Enhancement
IgE, administered with soluble protein antigens, will enhance antibody and T helper cell responses (Getahun et al. 2005). This process requires the low affinity receptor for IgE, CD23, and the receptor must be expressed on B cells. The mechanism appears to be that IgE-antigen is captured by recirculating CD23+ B cells which rapidly transport the antigen to B cell follicles (Hjelm 2008). In the spleen, CD11c+ dendritic cells somehow acquire the antigen and present it to T cells which subsequently help B cells to produce antibodies (Henningsson et al. 2011).
1.4 IgM-Mediated Feedback Enhancement
In 1968, Niels Jerne and Claudia Henry published a paper where they dissected the opposing immunoregulatory effects of IgG and IgM on antibody responses to sheep erythrocytes (SRBC) (Henry and Jerne 1968). SRBC-specific IgG (then denoted 7S antigen receptors), SRBC-specific IgM (19S antigen receptors) or a mixture of the two antibodies were administered intravenously to mice. Within one hour, the mice received an intravenous dose of SRBC and a few days later the active IgM-response was measured as hemolytic plaque-forming cells per spleen. With this assay, single plasma cells secreting IgM anti-SRBC can be measured: one hemolytic plaque represents one plasma cell (Jerne and Nordin 1963). Comparisons between groups given SRBC alone and groups given IgM prior to SRBC showed that IgM enhanced the response, provided suboptimal doses of antigen were used. In contrast, IgG suppressed more than 99% of the response and, interestingly, a mixture of IgG and IgM had an intermediate effect.
Thus, by separating antibodies into IgM and IgG instead of using whole serum, Henry and Jerne found that different isotypes could have different regulatory effects (Henry and Jerne 1968). Their paper was the start of modern studies of antibody feedback regulation and was also the foundation for B.H.’s Ph.D. studies in Hans Wigzell’s laboratory in Uppsala in the 1980s. At that time, Niels Jerne’s network theory, based on the notion that antibodies and B cell receptors (BCRs) also constitute antigens, was very much discussed (Jerne 1974). The epitopes of the antigen-binding regions of a certain antibody or BCR is defined as its idiotype. Since our antibody repertoire can recognize all antigens in the universe, an idiotype will be recognized by so called anti-idiotypic antibodies. In the 1970s and 1980s such idiotype/anti-idiotype interactions were thought to be of major importance in regulation of the immune response, and we set out to investigate whether IgM-mediated enhancement could be explained by network regulation. Whereas another laboratory reported that an anti-SRBC response could be generated in mice given IgM anti-SRBC without antigen (Forni et al. 1980), we failed to find evidence of anti-idiotypic regulation by IgM (Heyman et al. 1982).
Today, the most favored explanation for how specific IgM can feedback-enhance antibody responses is that IgM, binding to an antigen, rapidly activates complement thus forming an IgM-antigen-complement complex. This complex can bind to complement receptors 1 and 2 (CR1/2) which are expressed on B cells and FDCs and are known to play an important role in the generation of robust antibody responses [reviewed in (Sörman et al. 2014)]. Binding of immune complexes to these receptors can positively influence the antibody response in at least three different ways, and the relative importance of these is currently not understood. First, co-crosslinking of CR2 and BCR lowers the threshold for B cell activation and an immune complex could serve as the crosslinker (Carter et al. 1988). Second, marginal zone (MZ) B cells shuttle between the MZ and the B cell follicles (Cinamon et al. 2008; Arnon et al. 2013) and because they express high levels of CR1/2, they can transport complement-opsonized immune complexes into the follicle (Youd et al. 2002; Ferguson et al. 2004; Cinamon et al. 2008). Third, the deposition of immune complexes onto FDCs is most likely facilitated by their expression of CR1/2.
Below we will review the experimental observations leading to the hypothesis that antigen-specific IgM enhances antibody responses by activating complement and forming IgM-antigen-complement complexes which bind to CR1/2.
2 Basic Parameters of IgM-Mediated Enhancement
2.1 Antigens
IgM has generally been reported to enhance responses to large antigens such as erythrocytes (Henry and Jerne 1968; Dennert 1971; Wason 1973; Schrader 1973; Heyman et al. 1982; Whited Collisson et al. 1983; Heyman et al. 1985), malaria parasites (Harte et al. 1983), keyhole limpet hemocyanine (KLH) (Ding et al. 2013) and haptens coupled to KLH (Enriquez-Rincon and Klaus 1984; Coulie and Van Snick 1985; Youd et al. 2002) but occasionally IgM enhances responses to small proteins such as ovalbumin (OVA) (Whited Collisson et al. 1983). Notably, also human IgM enhances antibody responses as discovered during studies of Rhesus prophylaxis. Here, IgG anti-Rh suppressed and IgM anti-Rh enhanced the Rhesus-specific antibody responses (Clarke et al. 1963). IgM can only enhance responses to suboptimal doses of antigen (Henry and Jerne 1968; Powell et al. 1982; Lehner et al. 1983).
2.2 The IgM Molecule and Mode of Administration
Not only polyclonal, but also monoclonal IgM antibodies (Heyman et al. 1982; Powell et al. 1982; Harte et al. 1983; Coulie and Van Snick 1985; Heyman et al. 1988a; Youd et al. 2002) can enhance antibody responses. This finding is difficult to reconcile with idiotypic network regulation because a monoclonal IgM antibody cannot be expected to bind to more than a few BCRs and stimulation of only a few B cells would go unnoticed. A wide range of IgM concentrations are able to enhance (Dennert 1971; Heyman et al. 1982) but too high concentrations may lead to suppression (Pearlman 1967; Möller and Wigzell 1965), probably owing to epitope masking. IgM can have dual effects also in other situations. IgM is generally administered within 2 hours of the antigen but delaying IgM-administration until 1–2 days after antigen may result in suppression instead of enhancement (Wason 1973). Moreover, IgM that enhances in vivo can have a suppressive effect in vitro (Schrader 1973) and to our knowledge there are no reports showing that IgM-mediated enhancement works in vitro.
The structural requirements on the IgM molecule for ability to enhance has been scarcely studied. As will be discussed in detail in Sect. 4.1, monoclonal as well as polyclonal IgM with a point mutation in the constant part of the μ heavy chain leading to inability to bind C1q, is unable to feedback enhance antibody responses (Heyman et al. 1988a; Ding et al. 2013). Likewise, monomeric IgM, which cannot activate complement, is unable to enhance (Youd et al. 2002). Hexameric IgM is a more efficient complement activator than pentameric IgM (Davis et al. 1988), but feedback regulation by hexameric IgM has not been studied.
2.3 Primary Antibody Responses
The vast majority of studies demonstrating IgM-mediated enhancement have analyzed primary IgM responses (Henry and Jerne 1968; Dennert 1971; Wason 1973; Schrader 1973; Heyman et al. 1982; Whited Collisson et al. 1983; Heyman and Wigzell 1985) using Jerne’s direct hemolytic plaque-forming cell assay (Jerne and Nordin 1963). However, IgM enhances also primary IgG responses measured either as indirect plaque-forming cells or serum IgG (Heyman et al. 1982; Heyman and Wigzell 1985; Applequist et al. 2000; Rutemark et al. 2012; Ding et al. 2013). All IgG subclasses (Heyman et al. 1985) as well as IgE (Strannegård and Belin 1971) can be enhanced, and the IgG levels remain high during several months (Heyman and Wigzell 1985).
The magnitude of the responses to SRBC and KLH administered together with specific IgM, is similar to that seen with a 10-fold higher dose of antigen alone although the responses to the higher doses of antigen alone peak earlier (Henry and Jerne 1968; Youd et al. 2002).
2.4 Priming for Memory Responses
Mice primed with IgM + antigen and boosted with antigen alone, have an enhanced secondary response (Heyman and Wigzell 1985; Youd et al. 2002). This was most clearly demonstrated in adoptive transfer experiments where spleen cells from the primed mice were transferred to naïve recipients which were “boosted” with antigen (Heyman and Wigzell 1985). Boosting the same mice that had been primed, sometimes concealed the enhanced memory owing to feedback suppression mediated by the higher levels of endogenous IgG anti-SRBC induced in IgM + SRBC-primed mice (Heyman and Wigzell 1985).
2.5 Avidity of the Enhanced Response
The avidity of the response after administration of IgM and antigen has been reported to be either unchanged (Whited Collisson et al. 1984) or enhanced (Corley et al. 2005). In an experimental system where primed mice were challenged with IgM-immune complexes, it was suggested that feedback regulation by endogenous IgM drove affinity maturation (Zhang et al. 2013). When the injected IgM, forming the immune complexes, had a low affinity the endogenous affinity maturation could proceed. When high affinity IgM was injected, the affinity maturation was impaired. These observations are probably due to IgM antibodies masking the antigen that had bound to FDCs.
2.6 Germinal Center Responses
Specific IgM administered together with KLH or SRBC, promotes the formation of germinal centers (Ferguson et al. 2004; Ding et al. 2013). Since germinal centers are crucial for development of memory B cells and longlived plasma cells and for affinity maturation, these results agree well with the observations that these parameters are all enhanced by IgM (Heyman and Wigzell 1985; Youd et al. 2002; Ferguson et al. 2004; Corley et al. 2005).
2.7 Specificity of the Enhanced Antibody Response
IgM-mediated enhancement is antigen specific, but usually IgM specific for one epitope will lead to enhancement also of responses to other epitopes present on the same antigen particle (Henry and Jerne 1968; Heyman et al. 1982; Coulie and Van Snick 1985; Wason 1973; Whited Collisson et al. 1983; Heyman et al. 1988a; Ding et al. 2013). For example, mice immunized with IgM anti-SRBC together with SRBC conjugated to OVA (SRBC-OVA) will have an enhanced antibody response both to SRBC and OVA (Ding et al. 2013). Similarly to the observations that monoclonal IgM antibodies are efficient enhancers, non-epitope-specific enhancement is hard to reconcile with network regulation and suggest involvement of Fc-mediated functions.
2.8 T Cells and IgM-Mediated Enhancement
IgM cannot enhance responses to erythrocytes in mice lacking T cells and thus cannot compensate for T cell help (Whited Collisson et al. 1983; Powell et al. 1982; Lehner et al. 1983; Coutinho and Forni 1981). Very few studies have addressed the question of whether IgM can enhance T cell responses in parallel with antibody responses. In a report using malaria-specific monoclonal IgM, an enhanced induction of T helper cells was seen (Harte et al. 1983). On the other hand, IgM had no effect on proliferation of OVA-specific T cells in mice immunized with IgM anti-SRBC + OVA-SRBC although the antibody response was efficiently enhanced (Ding et al. 2013). To resolve this question, further experiments are required.
3 Complement in Antibody Responses to Uncomplexed Antigen
A still quite unknown function of complement is to facilitate humoral immune responses [reviewed in (Sörman et al. 2014; Carroll 2008)]. This was first demonstrated by the poor antibody responses to SRBC in mice depleted of C3 by treatment with cobra venom factor (Pepys 1974). Subsequently, it was shown that animals and humans lacking C1, C2, C3, C4, or the complement receptors 1 and 2 (CR1/2 or CD35/CD21) have severely impaired antibody responses. The main ligands for CR1/2 are split products of factor C3 of the complement cascade and the phenotype of mice lacking C1, C3, or C4 closely resembles that of mice lacking CR1/2. Therefore, it is generally assumed that the impaired antibody responses seen in C1-, C3-, or C4-deficient animals are explained by their failure to generate ligands for CR1/2. In other words, the effects of these complement factors on antibody responses would all be mediated via CR1/2. In mice, these two receptors are splice variants from the same gene, Cr2, and are expressed on B cells and FDCs. Recent data suggest that FDCs primarily express CR1 while B cells primarily express CR2 (Donius et al. 2013).
4 Complement in Antibody Responses to IgM-Antigen Complexes
4.1 Complement Activation by IgM
IgM is a very efficient activator of the classical pathway and one single IgM molecule can induce lysis of an erythrocyte (Borsos and Rapp 1965). IgM in solution does not bind C1 and it is thought that binding to a multivalent antigen is required to induce the conformation changes that expose the C1 binding sites (Feinstein and Munn 1969; Czajkowsky and Shao 2009).
Regarding antibody-mediated feedback regulation, it is particularly interesting that lack of C1q is associated with severe defects in antibody production against SRBC (Cutler et al. 1998; Rutemark et al. 2011). C1q is required for activation of the classical, but not the alternative or lectin, pathways. In the late 1980s, Marc Shulman generated a series of mutant monoclonal IgM antibodies, one of which (Mutant13) had lost its ability to bind C1q and to initiate hemolysis owing to a point mutation in the μ heavy chain (Shulman et al. 1987). We were interested to find out whether the ability of IgM to enhance antibody responses was related to its ability to activate complement. Therefore, mice were immunized mice with TNP-specific Mutant13 or wild-type IgM together with SRBC-TNP or with antigen alone. The results showed that only wildtype IgM was able to enhance antibody responses (Heyman et al. 1988a). Recently, these findings were confirmed using non-complement activating polyclonal IgM obtained from Cμ13 knock-in mice, which have the same point mutation as Mutant13 (see below) (Ding et al. 2013) (Fig. 1). The importance of complement for IgM-mediated enhancement is further supported by the observations that IgM cannot enhance in C3-depleted mice (Heyman et al. 1988a) and that monomeric IgM, which cannot activate complement, lost its enhancing capacity (Youd et al. 2002). The involvement of complement raises the question whether IgM-mediated lysis of erythrocytes may render them more immunogenic and that this is the mechanism behind the enhancing effect. Two experimental findings argue against this idea. First, response to the protein KLH, which cannot be lysed, is enhanced by IgM (Ding et al. 2013; Youd et al. 2002; Ferguson et al. 2004). Second, IgM enhances responses to SRBC in AKR mice which lack C5 and thereby the lytic pathway (Heyman et al. 1988a).

Mutant IgM from B cell hybridomas and from a knock-in mouse strain. The monoclonal IgM anti-TNP, produced by the B cell hybridoma Mutant13, has a serine - > proline mutation in position 436 of the IgM heavy chain leading to inability to bind C1q and to induce hemolysis (Shulman et al. 1987). The Cμ13 knock-in mouse strain has the same mutation in its genome and all IgM antibodies produced by these mice are unable to bind C1q and to activate the classical pathway (Rutemark et al. 2011)
The complement dependence probably explains why enhancement is generally limited to large antigens such as erythrocytes, malaria parasites, and KLH which are large enough to allow IgM to bind with all five arms and assume the conformation change required for C1 binding (Feinstein and Munn 1969; Czajkowsky and Shao 2009).
4.2 Complement Receptors 1 and 2, CR1/2, in Antibody Responses to IgM-Antigen Complexes
The complement dependence of IgM-mediated enhancement led us to investigate whether CR1/2 were involved in this feedback circle. At that time, there were no CR1/2 knockout mice (Cr2−/−) available, but Taroh Kinoshita had developed a monoclonal antibody, 7G6, which efficiently blocked the ligand-binding sites of both CR1 and CR2 (Kinoshita et al. 1988). Initially, we pretreated mice with 7G6 and then immunized them with IgM anti-SRBC + SRBC + horse erythrocytes (HRBC), or with SRBC + HRBC alone. HRBC was intended as a specificity control, establishing that IgM enhanced only the SRBC response. To our surprise, all mice treated with 7G6 had extremely low antibody responses both to SRBC and HRBC. The conclusion was that CR1/2 were required for all types of antibody responses, and not only for those enhanced by IgM, and led to the first publication of the dramatic role of CR1/2 for antibody responses in vivo (Heyman et al. 1990). Subsequently, other laboratories generated Cr2−/− mice and confirmed the importance of CR1/2 in antibody responses (Molina et al. 1996; Ahearn et al. 1996). Using such mice, we were able to show that IgM could not enhance in the absence of CR1/2 (Applequist et al. 2000; Rutemark et al. 2012) whereas IgE- and IgG2a-mediated enhancement, used as positive controls, remained intact (Applequist et al. 2000). Interestingly, enhancement by murine IgG3 is also dependent on CR1/2 (Diaz de Ståhl et al. 2003; Zhang et al. 2014).
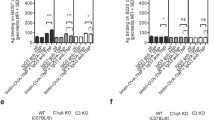
As mentioned above, CR1/2 are expressed on B cells and FDCs in mice. Studies in bone marrow chimeras between CR1/2 knockout and wild-type mice showed that optimal IgM-mediated enhancement required that both B cells and FDCs expressed CR1/2 (Rutemark et al. 2012). However, less pronounced enhancement was also seen when only FDCs or only B cells expressed the receptors (Rutemark et al. 2012) (Fig. 2).

CR1/2 on B cells and FDCs are required for optimal antibody responses to IgM-SRBC complexes. BALB/c and Cr2 −/− mice were irradiated and reconstituted with either BALB/c or Cr2 −/− bone marrow. Six weeks later they were immunized as indicated and screened for IgG anti-SRBC in serum. Two statistical comparisons were made, both using Student’s t-test. First, comparisons between the responses in mice immunized with SRBC alone versus IgM + SRBC (to determine whether IgM enhanced antibody responses significantly); * = p < 0.05; ** = p < 0.01; *** = p < 0.001. Second, comparisons between the responses between various chimeras immunized with IgM + SRBC (to determine whether CR1/2+ B cells contributed significantly to the antibody response to IgM + SRBC in mice with CR1/2+ FDCs (E vs. F) and CR1/2− FDCs (G vs. H); ° = p < 0.05; °° = p < 0.01; °°° = p < 0.001. Non-significant differences are not indicated. Adapted from (Rutemark et al. 2012)
4.3 Cμ13 Knock-in Mice with a Point Mutation in the IgM Heavy Chain Abolishing C1q-Binding
The observation that lack of C1q of the classical pathway leads to impaired primary antibody responses seems paradoxical for two reasons: (i) also alternative and lectin pathway activation would generate the C3 fragments which are the ligands for CR1/2, and (ii) the classical pathway is activated by antibodies binding to their antigens and, in naïve mice, very little specific antibodies would be present at the time of immunization. In 1998, it was found that mice lacking secretory IgM had impaired antibody responses and that the responses could be restored by transfer of non-immune IgM from normal mouse serum (Ehrenstein et al. 1998). This led to a possible explanation to the paradox described above, suggesting that natural IgM, present in naïve mice, would bind antigen with low affinity, activate complement and facilitate an early primary response in the same way as specific IgM does during feedback-enhancement. Once specific IgM is produced, classical IgM-mediated enhancement would ensue thus further potentiating the response.
During B.H.’s sabbatical with Michael Carroll, starting in 2001, we decided to test this hypothesis and generated knock-in mice (Cμ13), carrying the same point mutation in the μ heavy chain as the Mutant13 IgM, known to be unable to activate complement and to enhance antibody responses (Shulman et al. 1987; Heyman et al. 1988a). As a consequence of the mutation, all IgM antibodies produced by these mice, regardless of specificity, are unable to bind C1q. A.S. (neé Bergman) and Christian Rutemark, both junior Ph.D. students in B.H.’s lab at the time, immunized Cμ13 and wild-type animals with KLH or SRBC and compared their antibody responses. In the majority of the experiments, no significant differences between responses in wild-type and Cμ13 mice were seen (Rutemark et al. 2011). Occasionally, the antibody responses in Cμ13 mice were slightly reduced but not nearly to levels as low as those seen in CR1/2 knockout mice (Rutemark et al. 2011). Thus, it appeared that the ability of natural IgM to activate complement did not explain the requirement for classical pathway activation in primary antibody responses. Possibly, the antigen doses that were tested were too high or other C1q-activating substances may play a role. Nevertheless, this was an unexpected result which prompted further investigations described below Sects. (4.4 and 4.6).
4.4 FcμR (Toso/Faim3) and IgM-Mediated Enhancement
In 2012, two research groups published that mice lacking the Fc-receptor for IgM, FcμR (Toso/Faim3), had impaired antibody responses to suboptimal doses of antigen (Ouchida et al. 2012; Honjo et al. 2012). This opened the possibility that FcμR could be involved in IgM-mediated feedback enhancement. The conclusion that IgM-mediated enhancement depends on the ability of IgM to activate complement was based on the loss of enhancing capacity by monoclonal or polyclonal IgM with the same point mutation in the μ heavy chain (Heyman et al. 1988a; Ding et al. 2013) and on the loss of enhancing capacity by monomeric IgM (Youd et al. 2002). Hypothetically, the IgM mutation could also have affected FcμR binding and monomeric IgM may not bind to FcμR. In collaboration with Ji-Yang Wang’s laboratory, we addressed this question and found that IgM from Cμ13 knock-in and wildtype mice bound equally well to cells expressing FcμR (Ding et al. 2013). Thus, since IgM from Cμ13 mice was unable to enhance antibody responses and to activate complement but bound well to FcμR (Ding et al. 2013), the results strongly suggest that complement activation, but not FcμR binding, is required for induction of enhancement.
4.5 Other IgM-Binding Receptors and IgM-Mediated Enhancement
Not only FcμR (Toso/Faim3), but also poly-IgR (pIgR) (Johansen et al. 2000), Fcα-/μR (Ohno et al. 1990; Shibuya et al. 2000), and CD22 (Adachi et al. 2012) are known to bind IgM. However, their involvement in IgM-mediated enhancement has not been studied.
4.6 Specific IgM from Wildtype but not Cμ13 Mice, Causes Rapid Deposition of C3 on SRBC in Vivo
Possible caveats when testing the complement activation by Mutant13 and IgM from Cμ13 mice are that tests are performed in vitro and that guinea pig complement, instead of mouse complement, is used. To compare physiological complement activation by wild-type and Cμ13 IgM in vivo, SRBC-specific IgM of either type was administered intravenously to mice which 30 min later were given SRBC. In blood obtained as early as one minute after the last injection, large amounts of C3 fragments were deposited on SRBC in mice given wild-type but not Cμ13 IgM (Ding et al. 2013). Thus, IgM binds to intravenously administered antigens and activates the classical pathway within seconds, leading to heavy deposition of C3 fragments on the antigen. It is easy to envisage that the complement-opsonized SRBC seen in mice given IgM and SRBC (Ding et al. 2013; Sörman et al. 2014) will bind to CR1/2.
5 Transport of IgM-Antigen Complexes to Splenic B Cell Follicles
Early studies reported a correlation between the degree of IgM-mediated enhancement of the antibody response to SRBC and how much 51Cr-labeled SRBC was trapped in the spleen (Dennert et al. 1971; Dennert 1971). More recently, Richard Corley’s laboratory, using monoclonal IgM anti-NP, found that pentameric, but not monomeric IgM in complex with NP-BSA caused localization of antigen on FDCs in splenic B cell follicles (Youd et al. 2002). In mice lacking CR1/2 or C3, the IgM-antigen complexes were trapped in the MZ and did not move further into follicles. The same pentameric, but not monomeric (non-complement-activating) IgM, enhanced antibody responses to NP-KLH. Enhancement against NP-BSA was not investigated, or at least not reported. The same laboratory later reported that the cells responsible for transport of IgM-NP-BSA complexes from the MZ into the follicle were MZ B cells (Ferguson et al. 2004). Subsequently, Cinamon et al. showed that MZ B cells shuttle between the MZ and the follicle and deliver TNP-Ficoll to FDCs (Cinamon et al. 2008). Intravital imaging of MZ B cells demonstrated that as much as 20% of the cells exchanged compartment every hour (Arnon et al. 2013). Another study where virus-like particles (VLP) were used as antigens, showed that VLP-dimers required specific IgM for transport into follicles, whereas larger VLPs only required natural IgM (Link et al. 2012). In analogy to the studies above, follicular localization generally required CR1/2, C3, and C1q (Link et al. 2012). Thus, although only one study directly correlated IgM-mediated enhancement of antibody responses to antigen localization to the spleen (Dennert 1971), the other studies described above are highly compatible with such a scenario.
6 Summary and Concluding Discussion
The molecular mechanisms behind the onset of an antibody response are complicated and not yet fully understood. A current model, based on recent reviews (Victora et al. 2010; Vinuesa et al. 2010; Chan and Brink 2012; Heesters et al. 2014), is presented in Fig. 3. The question of major interest for the present discussion is how specific IgM can interfere with these processes and cause the enhancement of primary IgM and IgG responses, germinal center formation and induction of memory responses described above.

Schematic overview over generation of antibody responses in the spleen. (1a, b) Antigen enters the splenic B cell follicles. Small antigens can enter via conduits (Nolte et al. 2003) whereas larger antigens, e.g. KLH, bind to MZ B cells via complement receptors (Ferguson et al. 2004) (1a, b). These cells shuttle between the MZ and the follicle and deposit antigen on FDCs (Cinamon et al. 2008) (1a). (2a) In the follicle, antigen is recognized by naïve follicular B cells which migrate towards the T cell zone after antigen encounter. (2b) T cells are simultaneously activated by antigen-presenting cells displaying peptides on their MHC-II. (3) A subgroup of the activated T cells upregulate CXCR5 and down-regulate CCR7, causing them to migrate towards the T-B-cell border where they meet and activate specific B cells. (4) Some of the activated B cells differentiate into short-lived extrafollicular plasma cells, mainly producing IgM (MacLennan et al. 2003). The majority of the activated B cells proliferate and form the dark zone of the germinal center. (5) In the dark zone, B cells undergo somatic hypermutation and then migrate to the light zone. (6) Some of the activated T cells are further triggered by the B cells to upregulate CXCR5 and differentiate into TFH cells and move towards the light zone. (7) Here, B cells meet FDCs that display intact antigens on their dendrites (Heesters et al. 2013). (8) High affinity B cells capture the antigen, process and display it on MHC-II to a limiting number of TFH cells, which provide the B cells with survival signals ensuring that B cells with the highest affinity survive (Schwickert et al. 2011; Shulman et al. 2013; Gitlin et al. 2014). The high affinity B cells subsequently undergo class-switch recombination. (9) Some B cells differentiate into memory B cells or high affinity longlived plasma cells which exit the follicles. Others return to the dark zone for another round of hypermutation. As detailed in the text, experimental observations suggest that specific IgM, through its ability to deposit C3 fragments onto the antigens, can interfere in the generation of antibody responses at several levels: (1a, b) Transport of IgM-antigen-complement complexes by CR1/2+ MZ B cells into the follicle. (2a) Co-crosslinking of BCR and CR2/CD19/CD81 by IgM-antigen-complement complexes leading to facilitated B cell signaling and/or increased B cell activation simply owing to increased levels of antigen. (7) capture of antigen on FDCs for presentation to B cells during the affinity maturation process
A central finding is that IgM must be able to activate complement in order for enhancement to be initiated (Heyman et al. 1988a; Youd et al. 2002; Ding et al. 2013). Studies in mice immunized with IgM anti-SRBC + SRBC show that the SRBC in circulating blood are covered by C3 fragments already 10 s after immunization (Sörman et al. 2014). The role of complement in IgM-mediated enhancement seems to be to opsonize antigen for binding to CR1/2 rather than to increase the immunogenicity of the antigen through hemolysis: IgM enhances in mice lacking C5, a factor required for the lytic pathway (Heyman et al. 1988b) but not in mice lacking CR1/2 (Applequist et al. 2000; Rutemark et al. 2012).
CR1/2 are expressed on B cells and FDCs in mice. The only study which to our knowledge has addressed the question of which of these cells must express CR1/2 in order for IgM to be able to enhance antibody responses, was done in bone marrow chimeras with SRBC as the antigen and measured IgG responses (Rutemark et al. 2012). Expression of CR1/2 on both FDCs and B cells were required for optimal enhancement by IgM. However, expression on FDCs alone resulted in an intermediate enhancement and expression on B cells alone resulted in a weak enhancement (Rutemark et al. 2012) (Fig. 2).
Starting with B cells, at least three mechanisms have been described through which they, via CR1/2, could hypothetically increase antibody responses. In vitro, they can take up and present complement-opsonized antigens to T cells (Thornton et al. 1996; Boackle et al. 1997) and they can co-crosslink the CR2/CD19/CD81 complex and the BCR, lowering the threshold for B cell signaling (Carter et al. 1988; Matsumoto et al. 1993; Dempsey et al. 1996). In vivo, MZ B cells can transport complement-opsonized antigens into the B cell follicles (Youd et al. 2002; Ferguson et al. 2004; Cinamon et al. 2008; Link et al. 2012; Arnon et al. 2013). To date, there is no evidence that antigen presentation to CD4 T cells via increased uptake of IgM-immune complexes by B cells via CR1/2 plays a significant role in vivo but it is noteworthy that the influence of IgM on activation of the T follicular helper cell subset has not been selectively investigated. However, IgM does not enhance activation and proliferation of adoptively transferred transgenic antigen-specific CD4 T cells although the antibody responses were enhanced in the same animal (Ding et al. 2013). Similarly, studies of the role of CR1/2 for in vivo T cell responses to uncomplexed antigen did not reveal a role for these receptors (Gustavsson et al. 1995; Da Costa et al. 1999; Carlsson et al. 2009). Moreover, mice lacking CR1/2, or mice where the receptors were blocked, have poor antibody responses to T cell independent antigens (Thyphronitis et al. 1991; Wiersma et al. 1991; Carlsson et al. 2009). Since such antigens do not need to be processed and presented to T cells in order to induce antibody responses, the observations are hard to reconcile with an in vivo role for CR1/2 in antigen presentation to T cells. This reasoning leaves antigen transport by MZ B cells and facilitated B cell signaling as two non-mutually exclusive mechanisms through which B cells can be involved in IgM-mediated enhancement. The increased availability of antigen as a result of MZ B cell-mediated transport of IgM-complement-opsonized antigens into the follicle could lead to increased deposition of antigen on FDCs (Fig. 31a, 7). It could also lead to increased activation of specific follicular B cells in general and/or to increased B cell signaling caused by complement-opsonized antigens co-crosslinking the BCR and the CR2/CD19/CD81 co-receptor complex (Fig. 31b, 2a). The relative importance of B cell-mediated antigen transport versus B cell signaling is presently unknown. However, since the early IgM responses induced by specific IgM probably represent an extrafollicular response, neither transport of antigen into follicles nor binding to FDCs would be required. Therefore, it seems likely that in this situation co-crosslinking of BCR and CR2/CD19/CD81 plays a significant role.
Not only B cells but also CR1/2+ FDCs are important for optimal IgM-mediated enhancement, and judging from the only direct experiment testing their relative roles, FDCs are the most important cells (Rutemark et al. 2012) (Fig. 2). Complement-opsonized antigen, transported into follicles either via MZ B cells or via other pathways, is likely to be captured by FDCs and presented to B cells competing for antigen after their hypermutation processes (Fig. 31a, 7).
In conclusion, specific IgM must be able to activate complement in order to enhance antibody responses and ligation of CR1/2 on both B cells and FDCs are involved. The ability of specific IgM to enhance antibody responses is likely to play a physiological role in optimizing antibody responses. Since also natural IgM and FcμR influence antibody responses, the relative roles of these components and those of specific IgM and complement is an interesting subject for future research.
References
Adachi T, Harumiya S, Takematsu H, Kozutsumi Y, Wabl M, Fujimoto M, Tedder TF (2012) CD22 serves as a receptor for soluble IgM. Eur J Immunol 42(1):241–247
Ahearn JM, Fischer MB, Croix D, Goerg S, Ma M, Xia J, Zhou X, Howard RG, Rothstein TL, Carroll MC (1996) Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity 4:251–262
Applequist SE, Dahlström J, Jiang N, Molina H, Heyman B (2000) Antibody production in mice deficient for complement receptors 1 and 2 can be induced by IgG/Ag and IgE/Ag, but not IgM/Ag complexes. J Immunol 165:2398–2403
Arnon TI, Horton RM, Grigorova IL, Cyster JG (2013) Visualization of splenic marginal zone B-cell shuttling and follicular B-cell egress. Nature 493(7434):684–688
Bergström JJ, Heyman B (2015) IgG Suppresses Antibody Responses in Mice Lacking C1q, C3, Complement Receptors 1 and 2, or IgG Fc-Receptors. PLoS ONE 10(11):e0143841
Bernardo L, Yu H, Amash A, Zimring JC, Lazarus AH (2015) IgG-mediated immune suppression to erythrocytes by polyclonal antibodies can occur in the absence of activating or inhibitory Fc-gamma receptors in a full mouse model. J Immunol 195(5):2224–2230
Boackle SA, Holers VM, Karp DR (1997) CD21 augments antigen presentation in immune individuals. Eur J Immunol 27(1):122–129
Borsos T, Rapp HJ (1965) Hemolysin titration based on fixation of the activated first component of complement: evidence that one molecule of hemolysin suffices to sensitize an erythrocyte. J Immunol 95:559–566
Bowman JM (1988) The prevention of Rh immunization. Transfus Med Rev 2:129–150
Carlsson F, Getahun A, Rutemark C, Heyman B (2009) Impaired antibody responses but normal proliferation of specific CD4+ T cells in mice lacking complement receptors 1 and 2. Scand J Immunol 70(2):77–84
Carroll MC (2008) Complement and humoral immunity. Vaccine 26(Suppl 8):28–33
Carter RH, Spycher MO, Ng YC, Hoffmann R, Fearon DT (1988) Synergistic interaction between complement receptor type 2 and membrane IgM on B-lymphocytes. J Immunol 141:457–463
Chan TD, Brink R (2012) Affinity-based selection and the germinal center response. Immunol Rev 247(1):11–23
Cinamon G, Zachariah MA, Lam OM, Foss FW Jr, Cyster JG (2008) Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol 9(1):54–62
Clarke CA, Donohoe WTA, Woodrow JC, Finn R, Krevans JR, Kulke W, Lehane D, Sheppard PM (1963) Further experimental studies on the prevention of Rh haemolytic disease. Br Med Journal 1:979–984
Corley RB, Morehouse EM, Ferguson AR (2005) IgM accelerates affinity maturation. Scand J Immunol 62(Suppl 1):55–61
Coulie P, Van Snick J (1985) Enhancement of IgG anti-carrier responses by IgG2-anti-hapten antibodies in mice. Eur J Immunol 15:793–798
Coutinho A, Forni L (1981) The enhancement of antibody response by IgM antibodies is dependent on antigen-specific T helper cells. Immunobiology 158:182–190
Cutler AJ, Botto M, van Essen D, Rivi R, Davies KA, Gray D, Walport MJ (1998) T cell-dependent immune response in C1q-deficient mice: defective interferon g production by antigen-specific T cells. J Exp Med 187:1789–1797
Czajkowsky DM, Shao Z (2009) The human IgM pentamer is a mushroom-shaped molecule with a flexural bias. Proc Natl Acad Sci U S A 106(35):14960–14965
Da Costa XJ, Brockman MA, Alicot E, Ma M, Fischer MB, Zhou X, Knipe DM, Carroll MC (1999) Humoral response to herpes simplex virus is complement-dependent. Proc Natl Acad Sci U S A 96(22):12708–12712
Davis AC, Roux KH, Shulman MJ (1988) On the structure of polymeric IgM. Eur J Immunol 18(7):1001–1008
de Jong JM, Schuurhuis DH, Ioan-Facsinay A, Welling MM, Camps MG, van der Voort EI, Huizinga TW, Ossendorp F, Verbeek JS, Toes RE (2006) Dendritic cells, but not macrophages or B cells, activate major histocompatibility complex class II-restricted CD4+ T cells upon immune-complex uptake in vivo. Immunology 119(4):499–506
Dempsey PW, Allison MED, Akkaraju S, Goodnow CC, Fearon DT (1996) C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 271:348–350
Dennert G (1971) The mechanism of antibody-induced stimulation and inhibition of the immune response. J Immunol 106:951–955
Dennert G, Pohlit H, Rajewsky K (1971) Co-operative antibody: a concentrating device. In: Mäkelä O, Cross A, Kosunen TU (eds) Cell interactions and receptor antibodies in immune responses. Academic Press Limited, London, pp 3–7
Diaz de Ståhl T, Dahlström J, Carroll MC, Heyman B (2003) A role for complement in feedback-enhancement of antibody responses by IgG3. J Exp Med 197:1183–1190
Ding ZJ, Bergman A, Rutemark C, Ouchida R, Ohno H, Wang JY, Heyman B (2013) Complement-activating IgM enhances the humoral but not the T cell immune response in mice. PLoS ONE 8(11):e81299–e81299
Donius LR, Handy JM, Weis JJ, Weis JH (2013) Optimal germinal center B cell activation and T-dependent antibody responses require expression of the mouse complement receptor Cr1. J Immunol 191(1):434–447
Ehrenstein MR, O’Keefe TL, Davies SL, Neuberger MS (1998) Targeted gene disruption reveals a role for natural secretory IgM in the maturation of the primary immune response. Proc Natl Acad Sci U S A 95(17):10089–10093
Enriquez-Rincon F, Klaus GGB (1984) Differing effects of monoclonal anti-hapten antibodies on humoral responses to soluble or particulate antigens. Immunology 52:129–136
Feinstein A, Munn EA (1969) Conformation of the free and antigen-bound IgM antibody molecules. Nature 224(5226):1307–1309
Ferguson AR, Youd ME, Corley RB (2004) Marginal zone B cells transport and deposit IgM-containing immune complexes onto follicular dendritic cells. Int Immunol 16(10):1411–1422
Forni L, Coutinho A, Köhler G, Jerne N (1980) IgM antibodies induce the production of antibodies of the same specificity. Proc Natl Acad Sci USA 77:1125–1128
Getahun A, Dahlström J, Wernersson S, Heyman B (2004) IgG2a-mediated enhancement of Ab- and T-cell responses and its relation to inhibitory and activating FcgRs. J Immunol 172:5269–5276
Getahun A, Hjelm F, Heyman B (2005) IgE enhances antibody and T cell responses in vivo via CD23+ B Cells. J Immunol 175(3):1473–1482
Gitlin AD, Shulman Z, Nussenzweig MC (2014) Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 509(7502):637–640
Gustavsson S, Kinoshita T, Heyman B (1995) Antibodies to murine complement receptor 1 and 2 can inhibit the antibody response in vivo without inhibiting T-helper cell induction. J Immunol 154:6524–6528
Hamano Y, Arase H, Saisho H, Saito T (2000) Immune complex and Fc receptor-mediated augmentation of antigen presentation for in vivo Th cell responses. J Immunol 164(12):6113–6119
Harte PG, Cooke A, Playfair JHL (1983) Specific monoclonal IgM is a potent adjuvant in murine malaria vaccination. Nature 302:256–258
Heesters BA, Chatterjee P, Kim YA, Gonzalez SF, Kuligowski MP, Kirchhausen T, Carroll MC (2013) Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity 38(6):1164–1175
Heesters BA, Myers RC, Carroll MC (2014) Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol 14(7):495–504
Henningsson F, Ding Z, Dahlin JS, Linkevicius M, Carlsson F, Grönvik KO, Hallgren J, Heyman B (2011) IgE-mediated enhancement of CD4+ T cell responses in mice requires antigen presentation by CD11c + cells and not by B cells. PLoS ONE 6(7):e21760
Henry C, Jerne N (1968) Competition of 19S and 7S antigen receptors in the regulation of the primary immune response. J Exp Med 128:133–152
Heyman B (2000) Regulation of antibody responses via antibodies, complement, and Fc receptors. Annu Rev Immunol 18:709–737
Heyman B, Andrighetto S, Wigzell H (1982) Antigen dependent IgM-mediated enhancement of the sheep erythrocyte response in mice. Evidence for induction of B cells with specificities other than that of the injected antibodies. J Exp Med 155:994–1009
Heyman B, Hobbs M, Weigle WO (1985) IgM-mediated enhancement of in vivo anti-sheep erythrocyte antibody responses: Isotype analysis of the enhanced responses. Cell Immunol 92:134–141
Heyman B, Pilström L, Shulman MJ (1988a) Complement activation is required for IgM-mediated enhancement of the antibody response. J Exp Med 167:1999–2004
Heyman B, Wiersma E, Nose M (1988b) Complement activation is not required for IgG-mediated suppression of the antibody response. Eur J Immunol 18:1739–1743
Heyman B, Wiersma EJ, Kinoshita T (1990) In vivo inhibition of the antibody response by a monoclonal complement receptor specific antibody. J Exp Med 172:665–668
Heyman B, Wigzell H (1985) IgM enhances and IgG suppresses immunological memory in mice. Scand J Immunol 21:255–266
Hjelm F, Carlsson F, Getahun A, Heyman B (2006) Antibody-mediated regulation of the immune response. Scand J Immunol 64(3):177–184
Hjelm F, Karlsson MCI, Heyman B (2008) A novel B-cell mediated transport of IgE-immune complexes to the follicle of the spleen. J Immunol 180:6604–6610
Honjo K, Kubagawa Y, Jones DM, Dizon B, Zhu Z, Ohno H, Izui S, Kearney JF, Kubagawa H (2012) Altered Ig levels and antibody responses in mice deficient for the Fc receptor for IgM (FcmuR). Proc Natl Acad Sci U S A 109(39):15882–15887
Jerne NK (1974) Towards a network theory of the immune system. Ann Immunol (Paris) 125C(1–2):373–389
Jerne NK, Nordin AA (1963) Plaque formation in agar by single antibody-producing cells. Science 140:405
Johansen FE, Braathen R, Brandtzaeg P (2000) Role of J chain in secretory immunoglobulin formation. Scand J Immunol 52(3):240–248
Karlsson MCI, Getahun A, Heyman B (2001) FcgRIIB in IgG-mediated suppression of antibody responses: different impact in vivo and in vitro. J Immunol 167:5558–5564
Karlsson MCI, Wernersson S, Diaz de Ståhl T, Gustavsson S, Heyman B (1999) Efficient IgG-mediated suppression of primary antibody responses in Fc-gamma receptor-deficient mice. Proc Natl Acad Sci USA 96:2244–2249
Kinoshita T, Takeda J, Hong K, Kozono H, Sakai H, Inoue K (1988) Monoclonal antibodies to mouse complement receptor type 1 (CR1). Their use in a distribution study showing that mouse erythrocytes and pltelets are CR1-negative. J Immunol 140:3066
Lehner P, Hutchings P, Lydyard PM, Cooke A (1983) Regulation of the immune response by antibody II. IgM-mediated enhancement: dependency on antigen dose, T-cell requirement and lack of evidence for an idiotype-related mechanism. Immunology 50:503–509
Link A, Zabel F, Schnetzler Y, Titz A, Brombacher F, Bachmann MF (2012) Innate immunity mediates follicular transport of particulate but not soluble protein antigen. J Immunol 188(8):3724–3733
MacLennan IC, Toellner KM, Cunningham AF, Serre K, Sze DM, Zuniga E, Cook MC, Vinuesa CG (2003) Extrafollicular antibody responses. Immunol Rev 194:8–18
Matsumoto AK, Martin DR, Carter RH, Klickstein LB, Ahearn JM, Fearon DT (1993) Functional dissection of the CD21/CD19/TAPA-1/Leu-13 complex of B lymphocytes. J Exp Med 178(4):1407–1417
Molina H, Holers VM, Li B, Fang Y-F, Mariathasan S, Goellner J, Strauss-Schoenberger J, Karr RW, Chaplin DD (1996) Markedly impaired humoral immune responses in mice deficient in complement receptors 1 and 2. Proc Natl Acad Sci USA 93:3357–3361
Möller G, Wigzell H (1965) Antibody synthesis at the cellular level. Antibody-induced suppression of 19S and 7S antibody responses. J Exp Med 121:969–989
Nolte MA, Belien JA, Schadee-Eestermans I, Jansen W, Unger WW, van Rooijen N, Kraal G, Mebius RE (2003) A conduit system distributes chemokines and small blood-borne molecules through the splenic white pulp. J Exp Med 198(3):505–512
Ohno T, Kubagawa H, Sanders SK, Cooper MD (1990) Biochemical nature of an Fc mu receptor on human B-lineage cells. J Exp Med 172(4):1165–1175
Ouchida R, Mori H, Hase K, Takatsu H, Kurosaki T, Tokuhisa T, Ohno H, Wang JY (2012) Critical role of the IgM Fc receptor in IgM homeostasis, B-cell survival, and humoral immune responses. Proc Natl Acad Sci U S A 109(40):2699–2706
Pearlman DS (1967) The influence of antibodies on immunologic responses. The effect on the response to particulate antigen in the rabbit. J Exp Med 126:127–148
Pepys MB (1974) Role of complement in induction of antibody production in vivo. Effect of cobra factor and other C3-reactive agents on thymus-dependent and thymus-independent antibody responses. J Exp Med 140:126–145
Powell R, Hutchings P, Cooke A, Lydyard PM (1982) Antibody mediated regulation of immune responses: I. Enhancement of specific antibody responses through IgM antibodies. Immunol Lett 4(5):253–258
Rutemark C, Alicot E, Bergman A, Ma M, Getahun A, Ellmerich S, Carroll MC, Heyman B (2011) Requirement for complement in antibody responses is not explained by the classic pathway activator IgM. Proc Natl Acad Sci U S A 108(43):934–942
Rutemark C, Bergman A, Getahun A, Hallgren J, Henningsson F, Heyman B (2012) Complement receptors 1 and 2 in murine antibody responses to IgM-complexed and uncomplexed sheep erythrocytes. PLoS ONE 7(7):e41968
Schrader JW (1973) Regulation of the immune response by IgM antibody: a paradoxical suppression of the in vitro primary immune response to sheep erythrocytes by IgM. Aust J Exp Biol Med Sci 51:333–346
Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, Dustin ML, Nussenzweig MC (2011) A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J Exp Med 208(6):1243–1252
Shibuya A, Sakamoto N, Shimizu Y, Shibuya K, Osawa M, Hiroyama T, Miyabayashi T, Sakano S, Tsuji T, Nakayama E, Ohillips JH, Lanier LL (2000) Fcα/μ receptor mediates endocytosis of IgM-coated microbes. Nat Immunol 1(5):441–446
Shulman MJ, Collins C, Pennell N, Hozumi N (1987) Complement activation by IgM: evidence for the importance of the third constant domain of the µ heavy chain. Eur J Immunol 17:549–554
Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, Victora GD (2013) T follicular helper cell dynamics in germinal centers. Science 341(6146):673–677
Strannegård Ö, Belin L (1971) Enhancement of reagin formation in rabbits by passively administered 19S antibody. Immunology 20:427–431
Sörman A, Zhang L, Ding Z, Heyman B (2014) How antibodies use complement to regulate antibody responses. Mol Immunol 61(2):79–88
Thornton BP, Vetvicka V, Ross GD (1996) Function of C3 in a humoral response: iC3b/C3dg bound to an immune complex generated with natural antibody and a primary antigen promotes antigen uptake and the expression of co-stimulatory molecules by all B cells, but only stimulates immunoglobulin synthesis by antigen-specific B cells. Clin Exp Immunol 104(3):531–537
Thyphronitis G, Kinoshita T, Inoue K, Schweinle JE, Tsokos GC, Metcalf ES, Finkelman FD, Balow JE (1991) Modulation of mouse complement receptors 1 and 2 suppresses antibody responses in vivo. J Immunol 147:224–230
Uhr JW, Möller G (1968) Regulatory effect of antibody on the immune response. Adv Immunol 8:81–127
Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, Nussenzweig MC (2010) Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143(4):592–605
Vinuesa CG, Linterman MA, Goodnow CC, Randall KL (2010) T cells and follicular dendritic cells in germinal center B-cell formation and selection. Immunol Rev 237(1):72–89
Wason WM (1973) Regulation of the immune response with antigen specific IgM antibody: a dual role. J Immunol 110:1245–1252
Wernersson S, Karlsson M, Dahlström J, Mattsson R, Verbeek JS, Heyman B (1999) IgG-mediated enhancement of Ab responses is low in FcRg chain deficient mice and increased in FcgRII deficient mice. J Immunol 163:618–622
Whited Collisson E, Andersson B, Lamon EW (1984) Avidities of hapten-specific antibodies when the responses are modulated by anti-carrier antibodies. Immunology 53:443–449
Whited Collisson E, Andersson B, Rönnholm M, Lamon E (1983) Potentiation of antibody responses by specific IgM: Specificity and thymus dependency. Cell Immunol 79:44–55
Wiersma EJ, Coulie PG, Heyman B (1989) Dual immunoregulatory effects of monoclonal IgG-antibodies: suppression and enhancement of the antibody response. Scand J Immunol 29:439–448
Wiersma EJ, Kinoshita T, Heyman B (1991) Inhibition of immunological memory and T-independent humoral responses by monoclonal antibodies specific for murine complement receptors. Eur J Immunol 21:2501–2506
Youd ME, Ferguson AR, Corley RB (2002) Synergistic roles of IgM and complement in antigen trapping and follicular localization. Eur J Immunol 32:2328–2337
Zhang L, Ding Z, Xu H, Heyman B (2014) Marginal zone B cells transport IgG3-immune complexes to splenic follicles. J Immunol 193(4):1681–1689
Zhang Y, Meyer-Hermann M, George LA, Figge MT, Khan M, Goodall M, Notley CA, Ehrenstein MR, Kosco-Vilbois M, Toellner KM (2013) Germinal center B cells govern their own fate via antibody feedback. Journal of Experimental Medicine 210(3):457–464
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Sörman, A., Heyman, B. (2017). Specific IgM and Regulation of Antibody Responses. In: Kubagawa, H., Burrows, P. (eds) IgM and Its Receptors and Binding Proteins. Current Topics in Microbiology and Immunology, vol 408. Springer, Cham. https://doi.org/10.1007/82_2017_24
Download citation
DOI: https://doi.org/10.1007/82_2017_24
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64524-7
Online ISBN: 978-3-319-64526-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)