
Abstract
Aberrant pulmonary immune responses are linked to the pathogenesis of multiple human respiratory viral infections. Elevated cytokine and chemokine production “cytokine storm” has been continuously associated with poor clinical outcome and pathogenesis during influenza virus infection in humans and animal models. Initial trials using global immune suppression with corticosteroids or targeted neutralization of single inflammatory mediators proved ineffective to ameliorate pathology during pathogenic influenza virus infection. Thus, it was believed that cytokine storm was either chemically intractable or not causal in the pathology observed. During this review, we will discuss the history of research assessing the roles various cytokines, chemokines, and innate immune cells play in promoting pathology or protection during influenza virus infection. Several promising new strategies modulating lipid signaling have been recently uncovered for global blunting, but not ablation, of innate immune responses following influenza virus infection. Importantly, modulating lipid signaling through various means has proven effective at curbing morbidity and mortality in animal models and may be useful for curbing influenza virus induced pathology in humans. Finally, we highlight future research directions for mechanistically dissecting how modulation of lipid signaling pathways results in favorable outcomes following influenza virus infection.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The morbidity and mortality of severe influenza infections reflects properties intrinsic to the virus strain, including the ability to enter, replicate, and lyse respiratory epithelial cells (Garcia-Sastre 2010; Tscherne and Garcia-Sastre 2011). Host-intrinsic properties reflect both susceptibilities to infection as well as the double-edged sword of host immune responses that may ameliorate or exacerbate both infection and clinical outcome. The correlation of an aggressive immune response and severe disease following influenza virus infection in humans and animal models has been discussed previously (La Gruta et al. 2007). An aggressive innate response, with elevated recruitment of inflammatory leukocytes to lung, likely contributed to the morbidity of the 1918 influenza infection (Ahmed et al. 2007; Kobasa et al. 2007). In fact, lung injury during infection of macaques with the 1918 H1N1 influenza virus strain directly correlated with early dysregulated inflammatory gene expression (Cilloniz et al. 2009; Kobasa et al. 2007). More recently, clinical studies on avian H5N1 infected humans documented a significant association between excessive early cytokine responses and immune cell recruitment as predictive of poor outcome (de Jong et al. 2006). Moreover, an aberrant cytokine/chemokine response was observed in patients with severe disease during the most recent H1N1 pandemic in 2009 (Arankalle et al. 2010).
Thus far, public health approaches to influenza pandemics have relied primarily on preventative vaccine strategies and supportive measures including extensive use of various antiviral therapies. Nevertheless, the speed at which the 2009 H1N1 influenza virus pandemic spread coupled with increased morbidity associated with infection made evident the need to identify additional therapeutic strategies for the amelioration of influenza virus associated pathologies (Openshaw and Dunning 2010). While antiviral drugs that inhibit virus replication run the risk of mutational escape rendering the therapy ineffective, modulating the host immune response is less susceptible to selective pressure and drug resistance. Moreover, the current vaccine platforms require bi- or triennial modification to anticipate newly-emerging viral strains. Thus, uncovering novel therapies that can blunt the pathogenic immune response without compromising viral clearance could save countless lives and economic losses when the next lethal influenza virus pandemic emerges. Because of the strong connection between pathogenic influenza virus infection and excessive cytokine/chemokine production, this review will focus exclusively on the role various cytokines , chemokines, and innate immune cells play in promoting influenza virus protection versus pathogenesis. While it is recognized that some animal models have reported enhanced cytokine responses, control of influenza virus replication and reduced morbidity during influenza virus infection (Maelfait et al. 2012; Strutt et al. 2010), the majority of clinical and animal model studies overwhelmingly support that aberrant immune responses play a commanding role during influenza virus pathogenesis (La Gruta et al. 2007; Salomon and Webster 2009; Tisoncik et al. 2012).
For this reason, the focus of this chapter will be to evaluate the role innate immune cytokines, chemokines, and leukocytes play in protection and pathology during influenza virus infection.
2 Role of Cytokines and Chemokines in Immunopathology Versus Protection
2.1 Interferon
Type I interferon signaling is well-known to inhibit influenza virus replication and spread (Garcia-Sastre and Biron 2006). In fact, a major function of the viral NS1 protein, one of 11 viral proteins, is to inhibit type 1 interferon production and signaling (Hale et al. 2008). Deletion or mutation of the NS1 gene results in significant increases in the levels of type 1 interferon in infected cells and significantly lower virus titers both in vitro and in vivo (Garcia-Sastre et al. 1998; Jiao et al. 2008; Kochs et al. 2007). Despite strong evidence demonstrating extensive antiviral properties of type 1 interferon, several studies also suggest pathogenic roles for IFN-α during influenza virus infection. The production of several proinflammatory cytokines and chemokines are known to be amplified by type I interferon receptor signaling. Moreover, symptom onset correlates directly with the local appearance of IFN-α in respiratory lavage fluid in humans (Hayden et al. 1998; Van Reeth 2000). Thus, type I interferon signaling has dual roles in virus control and pathogenesis. Infection of IFNAR1−/− mice with the PR8 strain of influenza virus resulted in altered recruitment of Ly6Chi versus Ly6Cint monocytes in the lung, translating into increased production of the neutrophil chemoattractant, KC (CXCL8), elevated numbers of neutrophils in the lung and increased morbidity and mortality (Seo et al. 2011). Therefore, modulation of type 1 interferon signaling and production needs to be balanced to have enough to control virus infection but not promote excessive inflammation.
Type II interferon, made up of IFN-γ, is produced throughout influenza virus infection. Early levels of IFN-γ (within the first 3 days of infection) are made by macrophages and natural killer (NK) cells. While later on during infection (days 5–10 postinfection) IFN-γ is produced primarily by antiviral CD4 and CD8 T cells in the lung and secondary lymphoid tissues. Administration of IFN-γ early following influenza virus infection has been demonstrated to exert antiviral properties and is protective (Weiss et al. 2010). The protective role of IFN-γ is supported by the use of proteinase-activated receptor 2 (PAR2) agonist in vivo, which increased IFN-γ production, reduced influenza virus titers, and improved survival (Khoufache et al. 2009). However, a subsequent study demonstrated that following high-dose influenza virus infection, the absence of PAR2 resulted in improved survival (Nhu et al. 2010). Enhanced survival was also observed following lethal influenza virus infection in protease-activated receptor-1 (PAR1)-deficient mice or wild-type mice treated with PAR1 antagonist peptide, which correlated with a decrease in early influenza virus titers though the levels of IFN-γ were not assessed in this study (Khoufache et al. 2013). Protective memory CD4 T cell responses are correlated with the elevated numbers of lung CD4 T cells producing IFN-γ (McKinstry et al. 2012; Teijaro et al. 2010, 2011a). Moreover, IFN-γ-deficient, virus-specific T cells were more pathological than their IFN-γ-sufficient counterparts (Wiley et al. 2001). Antibody neutralization of IFN-γ in vivo affected influenza virus induced humoral and cellular immune responses in the lung (Baumgarth and Kelso 1996). Conversely, several studies reported that IFN-γ-deficient mice have negligible defects in virus clearance or generating efficient immune responses (Bot et al. 1998; Nguyen et al. 2000; Price et al. 2000). These studies suggest that spatial and temporal activity of IFN-γ production and signaling likely play key roles in controlling influenza virus infection and the magnitude of the immune response.
In addition to type I and II interferons, type III interferon, λ-interferon, has been detected in both respiratory epithelial cell cultures as well as mouse lung following influenza virus infection (Crotta et al. 2013; Jewell et al. 2010). Interestingly, λ-interferon did not require type I interferon for its induction and appeared to be sufficient to protect mice in the absence of type 1 interferon signaling during influenza virus challenge (Jewell et al. 2010). Administration of λ-interferon prior to influenza virus infection protected type 1 Interferon receptor knockout mice from infection. However, deletion of IL-28Rα, the receptor that recognizes λ-interferon, rendered mice only slightly more susceptible to influenza virus infection (Mordstein et al. 2008). Thus, it appears that Interferon-λ signaling following influenza virus challenge contributes to antiviral protection similar to type I interferon signaling.
2.2 Interleukin-1 (IL-1) and Tumor Necrosis Factor Alpha (TNF-α)
In addition to endosome (TLR3, 7) and cytosolic (RIG-I) sensors, influenza virus infected cells can activate the NLRP3 inflammasome (Ichinohe et al. 2009, 2010). The end result of inflammasome activation is the cleavage of pro-IL-1β and pro-IL-18 to active proteins. In turn, secreted IL-1β can signal through the IL-1R to induce inflammatory gene production through MyD88 signaling. Signaling through IL-1R has been demonstrated to contribute to both host protection and immune pathology following influenza virus infection. One group found that while IL-1R−/− mice displayed reduced lung inflammatory pathology with reduced neutrophil recruitment, IL-1R−/− mice also displayed reduced anti-influenza virus IgM antibody, along with delayed viral clearance and increased mortality (Schmitz et al. 2005). In contrast, a separate study demonstrated delayed mortality in IL-1R−/− mice following infection with H5N1 influenza virus (Perrone et al. 2010). In fact, elevated levels of IL-1β drive proinflammatory responses in patients with acute respiratory distress syndrome (Pugin et al. 1996). Elevated levels of TNF-α have also been linked to morbidity and mortality following highly pathogenic influenza virus infection in humans and animal models (Szretter et al. 2007). Interestingly, anti-TNF-α antibody therapy in mice resulted in reduced recruitment of inflammatory cells, T cell cytokine production, and morbidity in a mouse model of influenza virus infection (Hussell et al. 2001). Conversely, no difference in disease severity or mortality was observed in TNFR−/− mice following H5N1 infection (Salomon et al. 2007). Interestingly, mice lacking both IL-1R and TNFR displayed reduced morbidity, significantly delayed mortality, suppressed cytokine/chemokine production and reduced numbers of neutrophils and macrophages in the lung compared to wild-type mice (Perrone et al. 2010). Whether the disparate results between these two studies are due to the different viral strains used (H1N1/PR8 versus H5N1), differences in the method of depletion (anti-TNF antibody versus congenital TNFR knock out), or redundancy between the TNFR and IL-1R signaling pathways is still unclear.
2.3 Interleukin-6 (IL-6)
Elevated levels of IL-6 were found in human serum from patients infected with H5N1 and H1N1/2009 infections. Moreover, levels of IL-6 correlate directly with symptom formation in human influenza virus infection (Kaiser et al. 2001; Van Reeth 2000). Infection of macaques with the H1N1/1918/1919 pandemic influenza virus generated increased levels of IL-6 in the serum, suggestive of an aberrant inflammatory response (Kobasa et al. 2007). Despite the strong correlation of IL-6 levels and influenza pathogenesis, genetic depletion of IL-6 did not alter mortality following experimental infection of mice with H5N1 infection (Salomon et al. 2007), suggesting IL-6 may not be causal for influenza virus immune pathogenesis. On the other hand, IL-6 signaling was required to survive sublethal influenza virus infection in a mouse model (Dienz et al. 2012). In this study, the absence of IL-6 signaling led to increased lung pathology and viral titers, however, resulted in significant decreases in neutrophils in the lung, which was shown to be attributable to neutrophil death. Signaling through the IL-6 receptor is also essential for CD4 T follicular helper cell generation, B cell and antibody responses, which may explain the inability of IL-6 deficient mice to control influenza viral titers in the lung. The immune stimulatory antiviral properties of IL-6 may also explain why IL-6 deficient mice succumbed at a similar rate following H5N1 infection (Salomon et al. 2007). Alternatively, the different viral strains used in the above studies could explain the disparate clinical outcomes in mice deficient in IL-6 signaling. Nevertheless, fine-tuning will likely be required during inhibition of IL-6 signaling following influenza virus infection to allow for a sufficient immune response to control virus replication.
2.4 Chemokines
In addition to cytokines , multiple chemotactic molecules are induced following influenza virus infection in humans and animal models. In fact, production of several chemokines, both locally and systemically, correlate with influenza virus pathogenesis (de Jong et al. 2006; Kobasa et al. 2007; Van Reeth 2000). Elevated levels of MCP-1 (CCL2) and IP-10 (CXCL10) were found in the serum of patients infected with H5N1 compared to patients with less virulent strains (de Jong et al. 2006; Peiris et al. 2004). Moreover, elevated levels of RANTES (CCL5) mRNA were detected in human primary macrophages following infection with H5N1 as compared to H1N1 or H3N2 infection (Cheung et al. 2002). Influenza virus infected monocytes also express MIP-1α (Sprenger et al. 1996). While there is a clear correlation between increased production of the above chemokines during lethal influenza virus infection and pathology, a causal relationship between individual chemokines and pathogenesis has been difficult to prove. Infection of CCR2 and CCR5-deficient animals with mouse adapted PR8 H1N1 influenza virus yielded opposing results. CCR5 deficient mice developed a severe pro-inflammatory response, pulmonary cell damage, and decreased survival despite normal control of virus replication (Dawson et al. 2000). In contrast to worse pathology observed in CCR5−/− mice following influenza virus infection, CCR2-deficient mice displayed reduced pulmonary innate immune cell infiltration, decreased pulmonary damage, and increased survival. Interestingly, despite reduced pulmonary immune pathology, influenza virus loads were elevated in CCR2−/− mice (Dawson et al. 2000). Based on the above studies, one might assume that neutralization or deletion of CCL2 (or CCR2) during infection with human pathogenic influenza virus strains would result in a favorable clinical outcome. However, it was later determined that CCL2−/− mice succumbed to H5N1 infection at a similar rate and frequency as CCL2+/+ control mice (Salomon et al. 2007). Again, the discrepancy could be due to the differences in the virus strains used or that other non-CCL2 chemokines that signal through CCR2 are responsible for immune pathology . However, neutralization of CCR2 signaling with antibody, chemotherapeutic, or genetic deletion in animal models has not been tested following infection with H5N1 or other highly pathogenic influenza virus strains. In light of the increased pathology in CCR5−/− mice, it was demonstrated that CXCR3-deficiency (CXCR3 is a receptor for CXCL10) blunted lung pathology and cytokine levels and ultimately restored survival in CCR5−/− mice (Fadel et al. 2008), suggesting that signaling through CXCR3 in the absence of CCR5 may be detrimental during influenza virus infection. Moreover, blockade of CXCR3 with the drug AMG487 reduced symptom formation and delayed mortality in H5N1 infected ferrets (Cameron et al. 2008).
2.5 Negative Regulatory Cytokines , Interleukin-10 (IL-10), and Tumor Growth Factor-Beta (TGF-β)
In addition to initiating immune responses, the host immune system also utilizes multiple anti-inflammatory cytokines to prevent detrimental immune pathology. One major negative immune regulator, IL-10, is a central factor for regulating immune responses to viruses, bacteria, and parasitic infections (Couper et al. 2008). During human pathogenic influenza virus infection, elevated IL-10 levels are found in severely ill patients (de Jong et al. 2006). However, upregulation of IL-10 in these patients was likely a reactionary response to the excessive inflammation generated. One study reported that blockade of IL-10 signaling using an IL-10R neutralizing antibody following sublethal infection of mice resulted in exacerbated morbidity and mortality. The source of the IL-10 was determined to be effector CD8 T cells infiltrating the lung tissue (Sun et al. 2009). In a separate study, IL-10-deficient mice were protected from death following infection with a lethal dose of influenza virus. However, in this model the source of IL-10 was lung effector CD4 T cells (McKinstry et al. 2009). Moreover, a significant increase in the T helper 17 cell subset was detected in IL-10−/− mice following lethal influenza virus infection (McKinstry et al. 2009).
The immune regulatory role of Transforming Growth Factor Beta (TGF-β) has been extensively studied (Li et al. 2006). The activity of TGF-β has been reported to increase following influenza virus infection in mice. In fact, the influenza virus neuraminidase (NA) protein was demonstrated to convert inactive TGF-β into its active form (Schultz-Cherry and Hinshaw 1996). Interestingly, it was also discovered that highly pathogenic strains of H5N1 influenza virus fail to activate TGF-β (Carlson et al. 2010). Thus, one might anticipate that TGF-β activation may be essential for host protection during lethal H5N1 infection. In support of that hypothesis, it was determined that exogenous administration of active TGF-β using an adenovirus vector resulted in reduced viral loads and delayed mortality following lethal H5N1 infection in mice (Carlson et al. 2010). Conversely, neutralization of TGF-β produced higher mortality rates in mice infected with nonlethal doses of influenza virus, suggesting that active TGF-β can serve as a protective factor (Carlson et al. 2010). Despite these interesting results, the degree and mechanisms by which TGF-β regulates pathological immune responses or interferes with virus replication during human pathogenic influenza virus infection requires further studies and is likely to be a fruitful area of investigation. Moreover, a better understanding of these and other negative immune regulatory molecules may lead to treatments that blunt immune pathology following pathogenic influenza virus infection in humans.
3 The Role of Innate Immune Cells in Pathogenesis Versus Protection
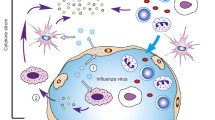
3.1 Macrophages/Monocytes
Experimental infection with H5N1 and 1918 H1N1 influenza viruses results in recruitment of macrophages/monocytic cells into infected lungs. Recruited monocytic cells can serve as reservoirs for influenza virus replication early following infection (Pang et al. 2013) and can be protected from infection via type 1 interferon signaling (Hermesh et al. 2010). Further, this infiltration may contribute to lung pathology and correlates directly with morbidity and mortality. In addition to infiltrating macrophages and monocytes, lung resident alveolar macrophages (AMs) have been demonstrated to play a role in the control of influenza virus clearance (Tumpey et al. 2005). In fact, administration of clodronate containing liposomes prior to infection with influenza viruses containing genes from the 1918–1919 H1N1 pandemic isolate reduced cytokine/chemokine production, however, also led to increased mortality. Moreover, depletion of AMs prior to sublethal influenza virus infection using clondronate liposomes resulted in uncontrolled viral titers and increased mortality despite reduced cytokine and chemokine expression (Tate et al. 2010). In contrast, AM depletion using clodronate liposomes during lethal PR8 infection did not alter the severity of disease or mortality (Tate et al. 2010). These contrasting results highlight the importance of the infectious dose of virus used and the outcome of immune cell depletion. Moreover, it is important to point out that clodronate liposome administration likely eliminates additional non-AM cell populations in the lungs, and a more extensive analysis of the populations depleted in these studies would be beneficial for interpretation. Further, the selective depletion of specific monocyte populations following influenza virus challenge has not been performed to date.
3.2 Neutrophils
Pathology following infection with highly virulent strains of human pathogenic influenza virus correlates with elevated neutrophil recruitment into lung tissue (Perrone et al. 2008). Moreover, the neutrophil responsive chemokine, IL-8, is elevated in patients displaying severe disease during H5N1 infection compared to infection with seasonal strains (de Jong et al. 2006). However, upon closer review of the literature, both protective and pathogenic roles for neutrophils have been reported following influenza virus infection. Complete depletion of neutrophils using antibody methods increased morbidity and mortality that was accompanied by decreased control of virus replication following infection with various strains of influenza virus (Brandes et al. 2013; Tumpey et al. 2005). Increased neutrophil numbers were detected in influenza virus infected CCR2−/− mice, correlating with reduced lung pathology and survival, despite slightly elevated viral loads (Dawson et al. 2000). Interestingly, a recent report demonstrated that attenuation, not depletion, of the neutrophil response correlated with improved survival following infection of mice with the PR8 strain of influenza virus (Brandes et al. 2013). This latter report suggests that too many or too few neutrophils can result in negative pathological outcomes either through exacerbated lung immune pathology or reduced virus control. Collectively, the above studies suggest that there may be a “sweet spot” in the numbers of neutrophils required to allow for the optimal control of both virus replication and immune pathology following influenza virus infection. In light of the above work, a more detailed understanding of the exact mechanisms of neutrophil-mediated protection and pathology will be crucial to designing therapies to quell lung immune pathology while preserving virus control.
3.3 Dendritic Cells
During respiratory virus infection, lung resident dendritic cells (DCs) play important roles in priming naïve T cells in the lung draining mediastinal lymph nodes. Several different subpopulations of these DCs have been identified by phenotypic flow cytometry analysis, with different populations displaying varying abilities to prime virus-specific T cells (Lambrecht and Hammad 2012). This aspect of influenza immune biology will be discussed in more detail in a separate chapter in this volume. Our focus here will be to discuss what is known about the pathogenic roles of DCs during the innate immune response of influenza virus infection. Several studies have linked DC responses with the immune pathology observed following influenza virus infection. Accumulation of CCR2+ monocyte derived DCs in the lung correlates with increased lung pathology, morbidity, and mortality. Importantly, reduced numbers of CCR2+/+ monocyte derived DCs are found in the lung of CCR2−/− mice following influenza challenge, and CCR2−/− mice display reduced morbidity and less mortality (Dawson et al. 2000). A similar study reported that TNF-α and Nitric Oxide Producing DC (TipDC) are found in higher numbers in the lung following infection with highly pathogenic compared to less pathogenic influenza virus strains. Moreover, TipDC recruitment was completely abolished in CCR2−/− mice following PR8 influenza virus infection (Aldridge et al. 2009). Despite reduced TipDC numbers in the lungs in CCR2−/− mice, no differences in morbidity or mortality were observed. The authors further demonstrated that TipDCs were essential for priming virus-specific CD8 T cell responses and conclude that complete depletion of TipDCs in their model may prevent the control of influenza virus replication. Interestingly, using the peroxisome proliferator-activated receptor-γ (PPAR-γ) agonist Pioglitazone prior to influenza virus infection reduced TipDCs, MCP-1, and MCP-3 chemokine production in the lungs, promoting increased survival following influenza virus infection (Aldridge et al. 2009). Collectively, the above studies suggest that a fine balance between protection and pathology exists upon depletion of DC populations during influenza virus infection. Therapeutic modulation of influenza virus induced immune pathology will likely require effective blunting without ablation of specific pulmonary DC subsets.
4 Identifying Therapeutic Interventions to Blunt Immune Pathology
Overabundant innate immune responses correlate with increased morbidity and mortality during infection with multiple pathogenic respiratory viruses (Kobasa et al. 2007; Macneil et al. 2011; Thiel and Weber 2008). In fact, cytokine storm during influenza virus infection is a prospective predictor of morbidity and mortality in humans (de Jong et al. 2006). Despite a strong correlation with pathogenesis, a direct causal link between innate immune responses, morbidity, and mortality has been difficult to prove. While cytokines and chemokines are produced at elevated levels during highly pathogenic compared to less pathogenic human influenza virus infection, inhibition of any single inflammatory mediator only modestly improved morbidity and delayed mortality in animal models (Cameron et al. 2008; Salomon et al. 2007; Salomon and Webster 2009; Szretter et al. 2007). These results indicate that isolated neutralization of single immune signaling molecules is insufficient to curb the resultant immune pathology following highly pathogenic influenza virus infection. However, global immune suppressive strategies such as corticosteroids have proven either ineffective or only modestly altered disease course in humans or animal models (Brun-Buisson et al. 2011; Tisoncik et al. 2012). One critical hurdle to generating effective treatments to curb influenza virus mediated immune pathology is a more detailed understanding of the cellular signaling pathways that elicit cytokine storm and potentiate lung pathology.
The balance between influenza virus immune control and pathology is on a knife’s edge; too much immune modulation results in the loss of virus control and too little can prove ineffective to blunt immune pathology. One must remain in the “goldilocks zone” for the best efficacy. Perhaps it is best to discuss therapies that have proven effective in blunting immune pathology while simultaneously sparing control of influenza virus replication with the hope of gaining insight into effective immune modulatory strategies to curb pathogenesis. Over the past 5 years, several studies have emerged demonstrating efficacious immune modulation of influenza virus replication and immunopathology. One of the more exciting areas of research that has recently developed is the characterization of lipid species following influenza virus infection and how they contribute to pathogenicity or protection. In fact, the majority of effective immune modulatory therapies that suppress morbidity and mortality following influenza virus infection have targeted various pathways that mediate lipid production or signaling. Performing lipidomic profiling following influenza virus infection using viruses with varying pathogenicity, one group identified lipid species that predominated during the pathogenic phase of infection and others that were associated with the resolution phase. Specifically, the authors found 5-Lipoxygenase metabolites in pathogenic influenza virus infection while 12/15-lipoxygenase metabolites were found in less pathogenic influenza infection (Tam et al. 2013). The generation of the omega-3 polyunsaturated fatty acid-derived lipid mediator, Protectin D1, was determined to inhibit influenza virus replication and result in reduced morbidity and mortality following infection (Morita et al. 2013). Another group identified that the oxidized phospholipid OxPAPC (OxPL) was essential for acute lung injury (ALI) following administration of inactivated H5N1 influenza virus to lungs (Imai et al. 2008). Interestingly, OxPL promoted lung injury and cytokine production by lung macrophages, which was dependent on TLR4-TRIF signaling. Moreover, TLR4- or TRIF-deficiency protected mice from ALI following H5N1 stimulation. Complimentary studies confirmed and extended this work, demonstrating that TLR4−/− mice are less susceptible to mortality following infection with a lethal dose of the PR8 strain of influenza virus (Nhu et al. 2012). Moreover, both prophylactic and therapeutic administration of a TLR4 antagonist, Eritoran, protected mice from lethal infection with either PR8 or the A/California/07/2009 H1N1 virus strain (Shirey et al. 2013). Importantly, Eritoran treatment resulted in reduced levels of oxidized phospholipids, suppressed cytokine/chemokine gene expression and ameliorated lung pathology (Shirey et al. 2013). While early viral titers were comparable between control and Eritoran treated mice infected with influenza virus, viral loads were reduced on day 7 postinfection in Eritoran treated mice compared to controls. Despite significant immune modulatory effects, Eritoran displayed no direct antiviral activities. The above reports indicate that a better understanding of pathogenic lipid species following influenza virus infection may lead to superior therapeutic modalities to blunt disease pathogenesis.
The cyclooxygenase enzymes (COX-1 and 2) convert arachidonic acid into prostaglandins and can control various inflammatory processes. The roles these enzymes play in the outcome of influenza virus pathology have been addressed. Deletion or pharmacological inhibition of the COX-1 enzyme was detrimental during influenza virus infection, resulting in enhanced morbidity, mortality, and enhanced cytokine/chemokine production. In contrast, COX-2 deficiency or pharmacological inhibition ameliorated pathology, suppressed inflammation and improved survival despite elevated viral loads (Carey et al. 2005, 2010). Notably, coadministration of the COX-2 inhibitor celecoxib and the antiviral drug zanamivir reduced mortality in mice infected with pathogenic H5N1 virus (Zheng et al. 2008). Thus, inhibiting COX-2 activity can be beneficial during influenza virus infection in the presence of antiviral therapies.
Sphingosine-1-Phosphate (S1P ) is a lipid metabolite synthesized from intracellular ceramide precursors to sphingosine. Sphingosine is subsequently phosphorylated by sphingosine kinase 1 and 2 (Sphk) to bioactive S1P (Chalfant and Spiegel 2005). The levels of bioactive S1P are stringently regulated through the actions of S1P lyases and phosphatases which degrade and dephosphorylate S1P, respectively. Highest levels of S1P are found in the blood and lymph with significantly lower levels maintained in peripheral tissues (Cyster 2005). S1P binds and signals through five G-protein coupled receptors denoted S1PR1–5. The expression of S1P receptors is heterogeneous, being found on various cell types of both hematopoietic and nonhematopoietic lineages and regulation of S1P signaling is mediated primarily through the expression pattern of the five receptors and stringent regulations of S1P levels (Im 2010). The diverse cellular functions associated with S1P receptor signaling is mediated through coupling to multiple heterotrimeric G-proteins adding an additional level of regulation. Binding through these five receptors is known to modulate multiple cellular functions including: cell adhesion, migration, survival, proliferation, endocytosis, barrier function, and cytokine production (Rivera et al. 2008). Since the initial discovery that the immunomodulatory agent, FTY720, induced lymphopenia, investigators sought to understand how S1P signaling affects immune cell function. To this end, genetically altered mice and selective agonists/antagonists have been developed to probe how S1P signaling on different S1P receptors alters immune cell responses (Marsolais and Rosen 2009; Rosen et al. 2008). FTY720 and AAL-R are prodrug promiscuous S1P receptor agonists and once phosphorylated in vivo by SphK-2, bind and signal through S1P1, 3, 4, and 5R. Both FTY720 and AAL-R have been used extensively to characterize the effects of S1P signaling in vivo and have been shown to alter the outcome of autoimmune disease, toxic shock syndrome, and viral infection (Niessen et al. 2008; Oldstone 2013). In addition, several S1P receptor selective agonists have been developed to probe the cellular functions of S1P-receptor signaling. For instance, S1P1R selective agonists (CYM5442, SEW2781 and AUY954) (Gonzalez-Cabrera et al. 2008; Rosen and Liao 2003) and antagonists (W146 and Ex26) (Cahalan et al. 2013; Gonzalez-Cabrera et al. 2008) have been synthesized and have provided insights into how S1P1R signaling affects lymphocyte trafficking and endothelial cell barrier functions (Rosen and Liao 2003; Shea et al. 2010). More recently, an S1P1R-eGFP knock-in mouse has been created, allowing for fluorescent and biochemical detection of functional S1P1R expression (Cahalan et al. 2011). Further, small molecule probes to S1P3 and S1P4 have been developed and are being used to probe the functions of these receptors in modulating DC activation and in turn pathological T cell responses. Most importantly, FTY720 has been approved for use in humans to treat Multiple Sclerosis (MS) and several S1P1R-selective agonists are in phase 2/3 clinical trials for MS and Colitis. Thus, potential of S1P receptor signaling modulation to modulate immune pathology during influenza virus infection wielded great promise.
Studies using the promiscuous S1P receptor agonist, AAL-R, demonstrated significant reductions in the numbers of macrophages and neutrophils in the lung early following infection with a mouse-adapted strain (WSN) (Marsolais et al. 2009). In addition, activation of lung infiltrating macrophage and NK cells, as measured by CD69 expression, was also significantly attenuated by AAL-R treatment following infection with a human virulent strain (pandemic H1N1 2009) of influenza virus (Walsh et al. 2011). Moreover, early administration of AAL-R resulted in significant reductions of multiple proinflammatory cytokines and chemokines following infection with either WSN or human pandemic H1N1 2009 influenza virus (Marsolais et al. 2009; Walsh et al. 2011). Further, AAL-R-mediated reduction of early innate immune cell recruitment and cytokine/chemokine production correlated directly with reduced lung pathology and improved survival during H1N1 2009 influenza virus infection. While AAL-R clearly inhibited innate immune responses, significant inhibition of activated T cell recruitment into the lung at various times postinfection was also observed in both mouse adapted (Marsolais et al. 2008) and human pathogenic strains of influenza virus (Walsh et al. 2011). Thus, whether AAL-R-mediated survival was due to inhibition of innate or adaptive immune responses could not be determined from these studies. The above results were extended using genetic and chemical tools to probe functions of the S1P1 receptor (S1P1 GFP knock-in transgenic mice, S1P1 receptor agonists and antagonists) revealing that pulmonary endothelial cells are major modulators of innate immune cell recruitment and cytokine/chemokine responses early following influenza virus infection (Teijaro et al. 2011b). While S1P1R is expressed on endothelial cells and lymphocytes within lung tissue, an S1P1R-selective agonist suppresses cytokines and innate immune cell recruitment in wild-type and lymphocyte-deficient mice, identifying pulmonary endothelial cells as central players in promoting cytokine storm (Teijaro et al. 2011b). Immune cell infiltration and cytokine production were found to be distinct events, both orchestrated by signaling through the S1P1R located on endothelial cells (Teijaro et al. 2011b). Furthermore, suppression of early innate immune responses through S1P1R signaling results in reduced mortality during infection with human pathogenic strains (H1N1 swine) of influenza virus without compromising the host’s ability to mount a sufficiently effective antiviral immune response to control infection in both mouse and ferret models (Teijaro et al. 2011b, 2014a). Importantly, no differences were observed in T cell functionality or total numbers in the lung during influenza virus infection following S1P1R agonist treatment (Marsolais et al. 2008), suggesting that the protective effect likely occurred primarily through suppression of early innate immune responses. We extended these findings showing that S1P1R agonist treatment suppresses global cytokine amplification. Importantly, S1P1R agonist treatment blunted cytokine/chemokine production and innate immune cell recruitment in the lung independently of endosomal and cytosolic innate sensing pathways (Teijaro et al. 2014b). Further, S1P1R signaling suppression of cytokine amplification was independent of multiple innate signaling adaptor pathways, indicating common pathway inhibition of cytokine storm is likely essential for protection. Moreover, the MyD88 adaptor molecule was determined to be responsible for the majority of cytokine amplification observed following influenza virus challenge (Teijaro et al. 2014b). Collectively, our results suggest that blunting global cytokine and chemokine production and innate immune cell recruitment is likely required for effective host protection from excessive immunopathology. Collectively, the observations that host-generated lipids and lipid signaling pathways can promote or inhibit aberrant immune responses as well as suppress virus replication reveal new targets that may ultimately be utilized to ameliorate disease following pathogenic influenza virus infection.
5 Future Perspectives
Current research is illuminating the cellular and molecular contributions of immunopathology caused by highly virulent influenza viruses that lead to disease. New therapies should focus on blocking inflammation in conjunction with antiviral therapy to reduce morbidity and mortality following infection. Moreover, it is clear from studies performed recently that global blunting, not ablation, of inflammatory mediators will likely be required to ameliorate disease following highly pathogenic influenza virus infections. Thus, future studies should focus on the signaling pathways necessary for cytokine amplification and seek out ways to globally dampen inflammatory immune responses generated through these pathways. Toward this end, it will be essential to understand in greater detail the mechanisms by which the lipid signaling modulators described above improve the outcome during pathogenic influenza virus infection. Additionally, poor outcome during pathogenic influenza virus infection may be predictable by identifying mutations in genetic loci that alter the production of these lipid species.
Despite encouraging reports that differential lipid signaling can affect the outcome of influenza virus infection, more work is necessary to create a thorough understanding of how different host-generated lipids modulate influenza virus generated pathological immune responses. Blockade or deletion of TLR4 signaling significantly improves the clinical outcome of influenza virus infection in mice. Understanding the pulmonary cells that produce oxidized phospholipid species that target TLR4 will be essential. Moreover, the cell types that require TLR4 signaling to amplify cytokine production, innate immune cell recruitment, and the resulting morbidity and mortality will also be informative. The fact that Eritoran is already FDA approved may expedite its use during influenza virus infection in humans. Further investigation into the cellular and molecular sources of these lipids may lead to novel interventions to relieve immune pathology . The detection of 5-Lipoxygenase metabolites in pathogenic influenza virus infection while 12/15-lipoxygenase or docosahexaenoic acid derived metabolites in less pathogenic influenza infection suggests that some lipid species can indicate resolution of infection or exert antiviral protective effects. Again, the sources of these lipids and how they mediate their protective or pathological effects will be important directions for future research. Whether similar lipid profiles are also observed in human pathogenic influenza virus infection would significantly buttress this area of research.
The identification that S1P signaling dampens influenza virus immune pathology provides insights and tools to dissect influenza virus pathogenesis. S1P receptor signaling inhibits inflammation during both the innate and adaptive immune responses to influenza virus infection. Specific targeting of distinct cell populations as well as selective S1P receptors may differentially dampen innate and adaptive inflammatory responses and thus will be important to explore. Further research will focus on pinpointing the S1P receptors and pulmonary cell subsets responsible for blunting innate and adaptive immune responses during influenza virus infection. To this end, more potent receptor selective drugs and knock-in and knock-out mouse models can be used as tools to further understand the mechanisms and signaling pathways involved during the inflammatory response. Moreover, the development and application of potent, selective agonists will likely decrease the potential of adverse side-effects through more precise targeting of specific receptors, modulating distinct immune functions. Additionally, human susceptibilities to inflammatory disease may correlate directly with differential regulation and/or signaling of the S1P receptor cascade. Modulation of inflammation utilizing S1P receptor signaling may be a general phenomenon that occurs in multiple respiratory viral associated maladies and manipulation of this pathway may provide broad anti-inflammatory therapeutic efficacy during multiple respiratory virus infections.
References
Ahmed R, Oldstone MB, Palese P (2007) Protective immunity and susceptibility to infectious diseases: lessons from the 1918 influenza pandemic. Nat Immunol 8:1188–1193
Aldridge JR Jr, Moseley CE, Boltz DA, Negovetich NJ, Reynolds C, Franks J, Brown SA, Doherty PC, Webster RG, Thomas PG (2009) TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Nat Acad Sci U.S.A 106:5306–5311
Arankalle VA, Lole KS, Arya RP, Tripathy AS, Ramdasi AY, Chadha MS, Sangle SA, Kadam DB (2010) Role of host immune response and viral load in the differential outcome of pandemic H1N1 (2009) influenza virus infection in Indian patients. PLoS ONE 5(10):e13099
Baumgarth N, Kelso A (1996) In vivo blockade of gamma interferon affects the influenza virus-induced humoral and the local cellular immune response in lung tissue. J Virol 70:4411–4418
Bot A, Bot S, Bona CA (1998) Protective role of gamma interferon during the recall response to influenza virus. J Virol 72:6637–6645
Brandes M, Klauschen F, Kuchen S, Germain RN (2013) A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154:197–212
Brun-Buisson C, Richard JC, Mercat A, Thiebaut AC, Brochard L, REVA-SRLF A/H1N1v 2009 Registry Group (2011) Early corticosteroids in severe influenza A/H1N1 pneumonia and acute respiratory distress syndrome. Am J Respir Crit Care Med 183:1200–1206
Cahalan SM, Gonzalez-Cabrera PJ, Nguyen N, Guerrero M, Cisar EA, Leaf NB, Brown SJ, Roberts E, Rosen H (2013) Sphingosine 1-phosphate receptor 1 (S1P(1)) upregulation and amelioration of experimental autoimmune encephalomyelitis by an S1P(1) antagonist. Mol Pharmacol 83:316–321
Cahalan SM, Gonzalez-Cabrera PJ, Sarkisyan G, Nguyen N, Schaeffer MT, Huang L, Yeager A, Clemons B, Scott F, Rosen H (2011) Actions of a picomolar short-acting S1P(1) agonist in S1P(1)-eGFP knock-in mice. Nat Chem Biol 7:254–256
Cameron CM, Cameron MJ, Bermejo-Martin JF, Ran L, Xu L, Turner PV, Ran R, Danesh A, Fang Y, Chan PK et al (2008) Gene expression analysis of host innate immune responses during lethal H5N1 infection in ferrets. J Virol 82:11308–11317
Carey MA, Bradbury JA, Rebolloso YD, Graves JP, Zeldin DC, Germolec DR (2010) Pharmacologic inhibition of COX-1 and COX-2 in influenza A viral infection in mice. PLoS ONE 5:e11610
Carey MA, Bradbury JA, Seubert JM, Langenbach R, Zeldin DC, Germolec DR (2005) Contrasting effects of cyclooxygenase-1 (COX-1) and COX-2 deficiency on the host response to influenza A viral infection. J Immunol 175:6878–6884
Carlson CM, Turpin EA, Moser LA, O’Brien KB, Cline TD, Jones JC, Tumpey TM, Katz JM, Kelley LA, Gauldie J et al (2010) Transforming growth factor-beta: activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog 6:e1001136
Chalfant CE, Spiegel S (2005) Sphingosine 1-phosphate and ceramide 1-phosphate: expanding roles in cell signaling. J Cell Sci 118:4605–4612
Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF, Gordon S, Guan Y, Peiris JS (2002) Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360:1831–1837
Cilloniz C, Shinya K, Peng X, Korth MJ, Proll SC, Aicher LD, Carter VS, Chang JH, Kobasa D, Feldmann F et al (2009) Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathog 5:e1000604
Couper KN, Blount DG, Riley EM (2008) IL-10: the master regulator of immunity to infection. J Immunol 180:5771–5777
Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, Staeheli P, Wack A (2013) Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog 9:e1003773
Cyster JG (2005) Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Ann Rev Immunol 23:127–159
Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N (2000) Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol 156:1951–1959
de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC et al (2006) Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12:1203–1207
Dienz O, Rud JG, Eaton SM, Lanthier PA, Burg E, Drew A, Bunn J, Suratt BT, Haynes L, Rincon M (2012) Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol 5:258–266
Fadel SA, Bromley SK, Medoff BD, Luster AD (2008) CXCR3-deficiency protects influenza-infected CCR5-deficient mice from mortality. Eur J Immunol 38:3376–3387
Garcia-Sastre A (2010) Influenza virus receptor specificity: disease and transmission. Am J Pathol 176:1584–1585
Garcia-Sastre A, Biron CA (2006) Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312:879–882
Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, Palese P, Muster T (1998) Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324–330
Gonzalez-Cabrera PJ, Jo E, Sanna MG, Brown S, Leaf N, Marsolais D, Schaeffer MT, Chapman J, Cameron M, Guerrero M et al (2008) Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P-like headgroup interactions. Mol Pharmacol 74:1308–1318
Hale BG, Randall RE, Ortin J, Jackson D (2008) The multifunctional NS1 protein of influenza A viruses. J Gen Virol 89:2359–2376
Hayden FG, Fritz R, Lobo MC, Alvord W, Strober W, Straus SE (1998) Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J Clin Invest 101:643–649
Hermesh T, Moltedo B, Moran TM, Lopez CB (2010) Antiviral instruction of bone marrow leukocytes during respiratory viral infections. Cell Host Microbe 7:343–353
Hussell T, Pennycook A, Openshaw PJ (2001) Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur J Immunol 31:2566–2573
Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A (2009) Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med 206:79–87
Ichinohe T, Pang IK, Iwasaki A (2010) Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 11:404–410
Im DS (2010) Pharmacological tools for lysophospholipid GPCRs: development of agonists and antagonists for LPA and S1P receptors. Acta Pharmacol Sin 31:1213–1222
Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H et al (2008) Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133:235–249
Jewell NA, Cline T, Mertz SE, Smirnov SV, Flano E, Schindler C, Grieves JL, Durbin RK, Kotenko SV, Durbin JE (2010) Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol 84:11515–11522
Jiao P, Tian G, Li Y, Deng G, Jiang Y, Liu C, Liu W, Bu Z, Kawaoka Y, Chen H (2008) A single amino acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J Virol 82:1146–1154
Kaiser L, Fritz RS, Straus SE, Gubareva L, Hayden FG (2001) Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J Med Virol 64:262–268
Khoufache K, Berri F, Nacken W, Vogel AB, Delenne M, Camerer E, Coughlin SR, Carmeliet P, Lina B, Rimmelzwaan GF et al (2013) PAR1 contributes to influenza A virus pathogenicity in mice. J Clin Invest 123:206–214
Khoufache K, LeBouder F, Morello E, Laurent F, Riffault S, Andrade-Gordon P, Boullier S, Rousset P, Vergnolle N, Riteau B (2009) Protective role for protease-activated receptor-2 against influenza virus pathogenesis via an IFN-gamma-dependent pathway. J Immunol 182:7795–7802
Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M et al (2007) Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445:319–323
Kochs G, Garcia-Sastre A, Martinez-Sobrido L (2007) Multiple anti-interferon actions of the influenza A virus NS1 protein. J Virol 81:7011–7021
La Gruta NL, Kedzierska K, Stambas J, Doherty PC (2007) A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 85:85–92
Lambrecht BN, Hammad H (2012) Lung dendritic cells in respiratory viral infection and asthma: from protection to immunopathology. Ann Rev Immunol 30:243–270
Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA (2006) Transforming growth factor-beta regulation of immune responses. Ann Rev Immunol 24:99–146
Macneil A, Nichol ST, Spiropoulou CF (2011) Hantavirus pulmonary syndrome. Virus Res 162:138–147
Maelfait J, Roose K, Bogaert P, Sze M, Saelens X, Pasparakis M, Carpentier I, van Loo G, Beyaert R (2012) A20 (Tnfaip3) deficiency in myeloid cells protects against influenza A virus infection. PLoS Pathog 8:e1002570
Marsolais D, Hahm B, Edelmann KH, Walsh KB, Guerrero M, Hatta Y, Kawaoka Y, Roberts E, Oldstone MB, Rosen H (2008) Local not systemic modulation of dendritic cell S1P receptors in lung blunts virus-specific immune responses to influenza. Mol Pharmacol 74:896–903
Marsolais D, Hahm B, Walsh KB, Edelmann KH, McGavern D, Hatta Y, Kawaoka Y, Rosen H, Oldstone MB (2009) A critical role for the sphingosine analog AAL-R in dampening the cytokine response during influenza virus infection. Proc Nat Acad Sci U.S.A. 106:1560–1565
Marsolais D, Rosen H (2009) Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat Rev Drug Discov 8:297–307
McKinstry KK, Strutt TM, Buck A, Curtis JD, Dibble JP, Huston G, Tighe M, Hamada H, Sell S, Dutton RW et al (2009) IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge. J Immunol 182:7353–7363
McKinstry KK, Strutt TM, Kuang Y, Brown DM, Sell S, Dutton RW, Swain SL (2012) Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J Clin Invest 122:2847–2856
Morita M, Kuba K, Ichikawa A, Nakayama M, Katahira J, Iwamoto R, Watanebe T, Sakabe S, Daidoji T, Nakamura S et al (2013) The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell 153:112–125
Mahlakõiv T, Ritz D, Mordstein M, DeDiego ML, Enjuanes L, Müller MA, Drosten C, Staeheli P et al (2012) Combined action of type I and type III interferon restricts initial replication of severe acute respiratory syndrome coronavirus in the lung but fails to inhibit systemic virus spread. J Gen Virol 2601-2605 doi:10.1099/vir.0.046284-0
Nguyen HH, van Ginkel FW, Vu HL, Novak MJ, McGhee JR, Mestecky J (2000) Gamma interferon is not required for mucosal cytotoxic T-lymphocyte responses or heterosubtypic immunity to influenza A virus infection in mice. J Virol 74:5495–5501
Nhu QM, Shirey K, Teijaro JR, Farber DL, Netzel-Arnett S, Antalis TM, Fasano A, Vogel SN (2010) Novel signaling interactions between proteinase-activated receptor 2 and toll-like receptors in vitro and in vivo. Mucosal Immunol 3:29–39
Nhu QM, Shirey KA, Pennini ME, Stiltz J, Vogel SN (2012) Proteinase-activated receptor 2 activation promotes an anti-inflammatory and alternatively activated phenotype in LPS-stimulated murine macrophages. Innate Immun 18:193–203
Niessen F, Schaffner F, Furlan-Freguia C, Pawlinski R, Bhattacharjee G, Chun J, Derian CK, Andrade-Gordon P, Rosen H, Ruf W (2008) Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature 452:654–658
Oldstone MB (2013) Lessons learned and concepts formed from study of the pathogenesis of the two negative-strand viruses lymphocytic choriomeningitis and influenza. Proc Nat Acad Sci U.S.A 110:4180–4183
Openshaw PJ, Dunning J (2010) Influenza vaccination: lessons learned from the pandemic (H1N1) 2009 influenza outbreak. Mucosal Immunol 3:422–424
Pang IK, Pillai PS, Iwasaki A (2013) Efficient influenza A virus replication in the respiratory tract requires signals from TLR7 and RIG-I. Proc Nat Acad Sci U.S.A 110:13910–13915
Peiris JS, Yu WC, Leung CW, Cheung CY, Ng WF, Nicholls JM, Ng TK, Chan KH, Lai ST, Lim WL et al (2004) Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet 363:617–619
Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM (2008) H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog 4:e1000115
Perrone LA, Szretter KJ, Katz JM, Mizgerd JP, Tumpey TM (2010) Mice lacking both TNF and IL-1 receptors exhibit reduced lung inflammation and delay in onset of death following infection with a highly virulent H5N1 virus. J Infect Dis 202:1161–1170
Price GE, Gaszewska-Mastarlarz A, Moskophidis D (2000) The role of alpha/beta and gamma interferons in development of immunity to influenza A virus in mice. J Virol 74:3996–4003
Pugin J, Ricou B, Steinberg KP, Suter PM, Martin TR (1996) Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin-1. Am J Respir Crit Care Med 153:1850–1856
Rivera J, Proia RL, Olivera A (2008) The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol 8:753–763
Rosen H, Gonzalez-Cabrera P, Marsolais D, Cahalan S, Don AS, Sanna MG (2008) Modulating tone: the overture of S1P receptor immunotherapeutics. Immunol Rev 223:221–235
Rosen H, Liao J (2003) Sphingosine 1-phosphate pathway therapeutics: a lipid ligand-receptor paradigm. Curr Opin Chem Biol 7:461–468
Salomon R, Hoffmann E, Webster RG (2007) Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc Nat Acad Sci U.S.A 104:12479–12481
Salomon R, Webster RG (2009) The influenza virus enigma. Cell 136:402–410
Schmitz N, Kurrer M, Bachmann MF, Kopf M (2005) Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol 79:6441–6448
Schultz-Cherry S, Hinshaw VS (1996) Influenza virus neuraminidase activates latent transforming growth factor beta. J Virol 70:8624–8629
Seo SU, Kwon HJ, Ko HJ, Byun YH, Seong BL, Uematsu S, Akira S, Kweon MN (2011) Type I interferon signaling regulates Ly6C(hi) monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathog 7:e1001304
Shea BS, Brooks SF, Fontaine BA, Chun J, Luster AD, Tager AM (2010) Prolonged exposure to sphingosine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am J Respir Cell Mol Biol 43:662–673
Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, Karp CL, McAlees J, Gioannini TL, Weiss J et al (2013) The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 497:498–502
Sprenger H, Meyer RG, Kaufmann A, Bussfeld D, Rischkowsky E, Gemsa D (1996) Selective induction of monocyte and not neutrophil-attracting chemokines after influenza A virus infection. J Exp Med 184:1191–1196
Strutt TM, McKinstry KK, Dibble JP, Winchell C, Kuang Y, Curtis JD, Huston G, Dutton RW, Swain SL (2010) Memory CD4+ T cells induce innate responses independently of pathogen. Nat Med 16: 558–564 (1p following 564)
Sun J, Madan R, Karp CL, Braciale TJ (2009) Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med 15:277–284
Szretter KJ, Gangappa S, Lu X, Smith C, Shieh WJ, Zaki SR, Sambhara S, Tumpey TM, Katz JM (2007) Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol 81:2736–2744
Tam VC, Quehenberger O, Oshansky CM, Suen R, Armando AM, Treuting PM, Thomas PG, Dennis EA, Aderem A (2013) Lipidomic profiling of influenza infection identifies mediators that induce and resolve inflammation. Cell 154:213–227
Tate MD, Pickett DL, van Rooijen N, Brooks AG, Reading PC (2010) Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J Virol 84:7569–7580
Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL (2011a) Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol 187:5510–5514
Teijaro JR, Verhoeven D, Page CA, Turner D, Farber DL (2010) Memory CD4 T cells direct protective responses to influenza virus in the lungs through helper-independent mechanisms. J Virol 84:9217–9226
Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H (2011b) Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 146:980–991
Teijaro JR, Walsh KB, Long JP, Tordoff KP, Stark GV, Eisfeld AJ, Kawaoka Y, Rosen H, Oldstone MB (2014a) Protection of ferrets from pulmonary injury due to H1N1 2009 influenza virus infection: immunopathology tractable by sphingosine-1-phosphate 1 receptor agonist therapy. Virology 452–453:152–157
Teijaro JR, Walsh KB, Rice S, Rosen H, Oldstone MB (2014) Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc Nat Acad Sci U.S.A 111(10):3799–3804
Thiel V, Weber F (2008) Interferon and cytokine responses to SARS-coronavirus infection. Cytokine Growth Factor Rev 19:121–132
Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG (2012) Into the eye of the cytokine storm. Microbiol Mol Biol Rev (MMBR) 76:16–32
Tscherne DM, Garcia-Sastre A (2011) Virulence determinants of pandemic influenza viruses. J Clin Invest 121:6–13
Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, Schultz-Cherry S, Solorzano A, van Rooijen N, Katz JM et al (2005) Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol 79:14933–14944
Van Reeth K (2000) Cytokines in the pathogenesis of influenza. Vet Microbiol 74:109–116
Walsh KB, Teijaro JR, Wilker PR, Jatzek A, Fremgen DM, Das SC, Watanabe T, Hatta M, Shinya K, Suresh M et al (2011) Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Nat Acad Sci U.S.A 108:12018–12023
Weiss ID, Wald O, Wald H, Beider K, Abraham M, Galun E, Nagler A, Peled A (2010) IFN-gamma treatment at early stages of influenza virus infection protects mice from death in a NK cell-dependent manner. J Interferon Cytokine Res (the official journal of the International Society for Interferon and Cytokine Research) 30:439–449
Wiley JA, Cerwenka A, Harkema JR, Dutton RW, Harmsen AG (2001) Production of interferon-gamma by influenza hemagglutinin-specific CD8 effector T cells influences the development of pulmonary immunopathology. Am J Pathol 158:119–130
Zheng BJ, Chan KW, Lin YP, Zhao GY, Chan C, Zhang HJ, Chen HL, Wong SS, Lau SK, Woo PC et al (2008) Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc Nat Acad Sci U.S.A 105:8091–8096
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Teijaro, J.R. (2014). The Role of Cytokine Responses During Influenza Virus Pathogenesis and Potential Therapeutic Options. In: Oldstone, M., Compans, R. (eds) Influenza Pathogenesis and Control - Volume II. Current Topics in Microbiology and Immunology, vol 386. Springer, Cham. https://doi.org/10.1007/82_2014_411
Download citation
DOI: https://doi.org/10.1007/82_2014_411
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-11157-5
Online ISBN: 978-3-319-11158-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)