
Abstract
Chemokines are a family of small heparin-binding proteins, mostly known for their role in inflammation and immune surveillance, which have emerged as important regulators of angiogenesis. Chemokines influence angiogenesis either through recruitment of pro-angiogenic immune cells and endothelial progenitors to the neo-vascular niche or via direct regulation of endothelial function downstream of activation of G-protein coupled chemokine receptors. The dual function of chemokines in regulating immune response and angiogenesis confers a central role in modulating the tissue microenvironment. Therefore, chemokines may constitute attractive targets for therapeutic intervention in several pathological disorders. This review will summarize the current understanding of the role of chemokines in angiogenesis, and give an overview of angiostatic and angiogenic chemokines and their crosstalk with other angiogenic factors.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Vascular Endothelial Growth Factor
- Chemokine Receptor
- Endothelial Progenitor Cell
- Metastatic Dissemination
- Matrigel Plug
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Angiogenesis is required for embryonic development and physiological functions, but may also affect the outcome of pathological conditions such as cancer, chronic inflammation, and ischemia. Vascular growth and remodeling is a rare event in adults, with the exception of the female menstrual cycle. However, angiogenesis is readily induced when a need for new vasculature arises, for example, during tissue ischemia or wound healing, through a shift in the balance between endogenous pro- and anti-angiogenic factors. For instance, various oxygen-sensing mechanisms directly induce angiogenesis through stabilization of members of the hypoxia-inducible transcription factor (HIF) family that up-regulate expression of pro-angiogenic molecules, including vascular endothelial growth factor (VEGF) (Fraisl et al. 2009). VEGF is critical for embryonic vascular development through binding to its cognate receptor VEGFR2, and is a central mediator of physiological and pathological angiogenesis and vascular function in the adult (Olsson et al. 2006). Using various in vitro and in vivo assays modeling angiogenesis (Table 1), several other pro- and anti-angiogenic factors have been identified, which act in concert in the tight regulation of blood vessel formation in health and disease. In addition, non-endothelial cell types participate in the angiogenic process either through secretion of factors or via stabilization of the growing vasculature.
Blood vessel formation may occur through several distinct mechanisms (Adams and Alitalo 2007). In sprouting angiogenesis, pro-angiogenic growth factors activate endothelial cells in pre-existing vessels and stimulate invasion of endothelial cells into the surrounding matrix through expression of proteases. Endothelial cells then proliferate and migrate towards the growth factor gradient as a growing stalk, guided by a specialized tip-cell that probes the microenvironment using multiple filopodial extensions. Lumenized growing stalks, often surrounded by stabilizing pericytes, then make contacts through tip-cell filopodia and fuse, allowing blood flow in the newly formed vasculature. Alternatively, new vessels may form through pillar formation in intussuceptive growth (splitting of vessels) (Makanya et al. 2009) or through non-angiogenic biomechanical extension of the existing vasculature (Kilarski et al. 2009). Finally, circulating endothelial progenitor cells are mobilized during tissue ischemia and may be recruited to hypoxic tissue, and contribute to neovascularization in a process reminiscent of embryonic vasculogenesis (Jujo et al. 2008).
2 The Chemokine Network
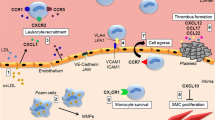
Chemokines are a large family of 8–12 kDa chemoattractant heparin-binding cytokines that may modulate angiogenesis through several distinct mechanisms (Fig. 1). Secreted chemokines accumulate at sites of inflammation through binding to extracellular matrix components and cell-surface carbohydrates, and are presented on the surface of endothelial cells mediating firm adhesion of leukocytes to the vessel wall (Ley et al. 2007; Thelen and Stein 2008). Thereby, chemokines play a central role in the recruitment of immune cells which, in turn, may secrete angiogenic growth factors (Table 2). Also, chemokines regulate recruitment and retention of endothelial progenitor cells that may directly participate in formation of a new vascular plexus (Petit et al. 2007). Importantly, several chemokines influence angiogenesis directly through binding to G-protein-coupled chemokine receptors expressed on endothelial cells, inducing down-stream signaling events that eventually result in enhanced or inhibited formation of new blood vessels (Keeley et al. 2008). The well-established role of chemokines in leukocyte recruitment has been the subject of several excellent reviews (Ley et al. 2007; Thelen and Stein 2008), and, although clearly very important for angiogenesis in health and disease, will not be extensively discussed here. Instead, this review is focused on the crucial role of chemokines as direct regulators of angiogenesis, and on the potential implication of this function in various pathological conditions.

Mechanisms involved in chemokine-mediated regulation of angiogenesis. Chemokines induce angiogenesis through (1) recruitment of pro-angiogenic hematopoietic cells and progenitors, (2) activation of cognate receptors expressed on endothelial cells inducing chemotaxis and tubular morphogenesis, (3) molecular cross-talk with angiogenic growth factor signaling, and (4) direct interaction between chemokine/chemokine receptor complexes and receptor tyrosine kinase receptors. Angiostatic chemokines inhibit angiogenesis through (5) recruitment of T-cells that in turn induce expression of angiostatic chemokines in a positive feedback loop, (6) binding to cognate receptors expressed on endothelial cells inducing apoptosis or regression of vessels, (7) binding of angiogenic growth factors, and (8) inhibition of receptor tyrosine kinase receptor signaling
The human chemokine network involves approximately 20 receptors and 50 ligands, classified according to the spacing of their first cystein residues into four subfamilies designated C, CC, CXC, and CX3C (Taub 2004). Furthermore, the CXC chemokines are divided into two groups depending on the presence or absence of three conserved amino acids (Glu-Leu-Arg; ELR) preceding the first cystein residue in the NH2-terminal domain. The ELR-motif affects the receptor binding specificity and thereby determines biological function, including the promotion or inhibition of angiogenesis (Strieter et al. 1995). Chemokines act through binding to seven-transmembrane G-protein-coupled receptors (GPCR), triggering activation of downstream signaling events typically including release of calcium from intracellular stores (Salanga et al. 2009). Several chemokines can bind multiple chemokine receptors, and many chemokine receptors bind multiple ligands. Adding complexity to the system, chemokines may form homo- or heterodimers or be presented as multimers through binding to glucosaminoglycans, leading to oligomerization of chemokine receptors (Salanga et al. 2009). Interestingly, GPCRs have cross talk with other signaling pathways through multiple mechanisms. This allows chemokine signaling to impinge on, for example, receptor tyrosine kinase pathways, and thereby affect a range of cellular events. As discussed later, many chemokines/chemokine receptor pathways have cross talk with pro-angiogenic VEGF and fibroblast growth factor (FGF) signaling, resulting in differential effects on angiogenesis.
3 Angiostatic Chemokines
Vascular homeostasis requires a strict balance between pro- and anti-angiogenic factors, resulting in restraint of vessel formation denoted angiostasis. Angiogenic stimuli, such as hypoxia, shift the balance of factors towards angiogenesis, initiating formation of vessels. Importantly, during physiological angiogenesis, the process is terminated when adequate vascularization has been achieved, shifting the balance back towards angiostasis. During pathological conditions, such as in tumors or during chronic inflammation, the strict balance between angiogenic and angiostatic (anti-angiogenic) factors is lost, leading to the continued formation of dysfunctional vessels. Notably, the net effect on angiogenesis is equally dependent on the expression levels of pro- and anti-angiogenic factors.
3.1 ELR-Negative CXCR3-Ligands Are Angiostatic
Interferon (IFN)-inducible ELR-negative CXC-family chemokines are potent angiostatic factors that prevent angiogenesis in response to growth factors and angiogenic chemokines (Balestrieri et al. 2008). Among these, CXCL4/platelet factor 4 (PF4) and CXCL10/interferon-γ inducible protein 10 (IP-10) have been most extensively studied, but angiostatic activity has also been noted for CXCL9/monokine induced by gamma (MIG), CXCL11/interferon inducible T-cell alpha chemoattractant (I-TAC), and the CXCL4 analog CXCL4L1 (Strieter et al. 1995; Maione et al. 1990; Angiolillo et al. 1995; Sato et al. 1990; Romagnani et al. 2001; Struyf et al. 2004). These angiostatic chemokines all bind to CXCR3a and CXCR3b receptors, which are splice variants of the same gene, with the exception of CXCL4 that binds CXCR3b exclusively. CXCR3b expressed on the endothelial cell surface mediates the angiostatic activity of ELR-negative chemokines in vitro (Lasagni et al. 2003). Importantly, mice lacking CXCR3 show excessive vessel formation during wound healing, highlighting the importance of ELR-negative CXC-family chemokines in maintaining vascular homeostasis.
Several immune cells express CXCR3 and are consequently recruited and activated by CXCR3 ligands, coupling regulation of immune response with angiostasis. However, CXCL4, CXCL9, CXCL10, and CXCL11 chemokines have been shown to directly inhibit growth factor-induced endothelial chemotaxis and tube formation in vitro and angiogenesis in the chick chorioallantoic membrane (CAM) and matrigel plugs in mice in vivo, implying that the angiostatic function is, at least in certain contexts, cell autonomous (Strieter et al. 1995; Maione et al. 1990; Angiolillo et al. 1995; Sato et al. 1990; Romagnani et al. 2001; Struyf et al. 2004). Interestingly, it was recently demonstrated that CXCL10 induce dissociation and regression of newly formed vessels during wound healing. Endothelial cord dissociation was induced through a CXCR3-dependent activation of µ-calpain, leading to cleavage of the cytoplasmic tail of β3 integrins followed by endothelial apoptosis (Bodnar et al. 2009). This process was equally induced in the presence of angiogenic factors, suggesting a role in pruning of developing vasculature.
3.2 Regulation of Angiostatic Chemokines: Coupling to Immune Response
There is a reciprocal regulation of IFN-inducible ELR-chemokine expression and induction of Th1-type immune response (Balestrieri et al. 2008). CXCR3 ligands activate interleukin secretion from recruited CXCR3-expressing Th1 T-cells, natural killer cells, and mononuclear cells, leading to increased production of IFN-γ. IFN-γ induces expression of CXCL9, CXCL10, and CXCL11, which again enhances recruitment and activation of CXCR3-expressing cells. The direct interaction between monocytes and endothelial cells has also been shown to synergistically induce expression of CXCL10 (Kasama et al. 2002).This positive feedback loop may result in “immunoangiostasis,” in which Type 1 immune response and inhibition of angiogenesis occur simultaneously. Similarly, interleukin (IL)-12, which is a T-cell stimulating factor, induces secretion of angiostatic chemokines from splenocytes, including CXCL10 and CXCL9 (Strasly et al. 2001). Interestingly, IL-12-induced inhibition of FGF-induced angiogenesis is strictly dependent on CXCL10 (Sgadari et al. 1996).
CXCR3 is expressed in a cell-cycle-dependent manner in cultured microvascular endothelial cells, its expression coinciding with that of cyclin A1, in the S-phase of the cell-cycle (Romagnani et al. 2001). This interesting observation suggests that only actively dividing endothelial cells are able to respond to CXCR3 ligands, directly coupling induction of angiogenesis to its potential inhibition. Supporting this notion, CXCL4 was shown to bind specifically to areas of active angiogenesis in vivo (Hansell et al. 1995). Notably, it is not clear to what extent binding to endothelial CXCR3 is required for angiostatic activity of ELR-chemokines in vivo. It has been shown that CXCL4 exerts its inhibitory effect on FGF-induced angiogenesis through direct complex formation with bFGF, which inhibits dimerization, binding, and activation of FGFR2 (Perollet et al. 1998). Similar mechanisms are involved in CXCL4-mediated inhibition of VEGF/VEGFR signaling (Gengrinovitch et al. 1995). CXCL4 may also interact with integrins implicated in angiogenesis, and consequently, soluble CXCL4 inhibits integrin-dependent adhesion of endothelial cells (Aidoudi et al. 2008).
3.3 Angiostatic CXCR3-Ligands in Tumor Growth and Angiogenesis
The concept of chemokine-dependent immunoangiostasis proposes that interferon-inducible ELR-chemokines in combination with Th1-type immune cells may synergistically induce tumor regression. In accordance, neutralization of CXCL10 increases growth, metastasis, and endothelial content in non-small cell lung cancer (NSCLC) implanted in SCID mice (Arenberg et al. 1996a). Viral-mediated transduction of CXCL4 cDNA inhibited angiogenesis and growth of intracerebral gliomas in mice, prolonging survival of treated animals (Tanaka et al. 1997). Interestingly, the CXCL4 analog CXCL4L1 was recently shown to be an even more potent inhibitor of growth and metastasis of melanoma through inhibition of angiogenesis (Struyf et al. 2007). These studies collectively suggest that treatment with angiostatic chemokines may be very beneficial for cancer patients and warrants further research, evaluating this interesting group of chemokines as therapeutic drugs.
4 Angiogenic Chemokines
4.1 ELR-Positive CXC-Family Chemokines Induce Angiogenesis
A subgroup of the CXC family chemokines with an ELR-motif in the N-terminal region has the potential to increase angiogenesis via the recruitment of polymorphonuclear neutrophils into inflamed tissue or through direct modulation of endothelial function. CXCL8/IL-8 was the first discovered chemokine to exhibit angiogenic activity, initially found to induce neo-vascularization in vivo using the rabbit corneal pocket model and then identified as a macrophage-derived factor that enhances angiogenesis through induction of proliferation and chemotaxis of endothelial cells (Strieter et al. 1992; Koch et al. 1992). Subsequently, several related ELR-positive chemokines, including CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, and CXCL7, were found to directly induce endothelial migration in vitro and cornea neovascularization in vivo (Strieter et al. 1995). Although ELR-positive chemokines mediate angiogenesis in the absence of preceding inflammation in the rat cornea model, CXCL1, CXCL2, and CXCL8 failed to induce angiogenesis in matrigel plugs in neutropenic mice, suggesting that additional neutrophil-derived pro-angiogenic factors are required for efficient vascularization in this setting (Benelli et al. 2002). The importance of the presence of an ELR-motif has been elegantly proven by mutating the ELR-sequence in CXCL8, which completely switched the function of the protein to inhibition of angiogenesis (Strieter et al. 1995). Similarly, the introduction of an ELR-motif converted CXCL9 to a pro-angiogenic protein. There is an important exception to this rule, as discussed later, namely the pro-angiogenic ELR-negative chemokine CXCL12/stromal-derived factor (SDF)-1.
There is ample proof that CXCR2 is the common receptor mediating the pro-angiogenic effects of ELR+ chemokines. Importantly, aside from CXCL8 and CXCL6, which also bind to CXCR1, all angiogenic ELR+ chemokines are exclusive ligands for CXCR2 (Keeley et al. 2008). A functional role of CXCR2 in angiogenesis was convincingly demonstrated through antibody-mediated inhibition of CXCR2, resulting in blocked chemotaxis induced by ELR+ chemokines and perturbed angiogenesis in rat corneal micropockets (Addison et al. 2000). Consistent with this, ELR+ chemokines failed to induce angiogenesis in corneas of CXCR2-deficient mice. There is also a delay in wound healing in mice lacking CXCR2, which was associated with decreased neovascularization of the wound tissue (Devalaraja et al. 2000). However, the inhibition of angiogenesis in this setting may be partially due to perturbed recruitment of neutrophils, precluding conclusions to be drawn regarding a direct function of CXCR2 in neovascularization during wound healing.
4.2 Pro-Angiogenic ELR-Positive Chemokines Are Induced Downstream of NF-kB Activation
CXCR2 ligands have been shown to be pro-angiogenic downstream of several distinct pathways, typically involving activation of NF-κB transcription factors. Chemokines are direct targets for NF-κB signaling pathways, which are central mediators of inflammatory signaling and endothelial activation downstream of tumor necrosis factor (TNF)-α and IL-1β stimulation. For instance, CXCL2 was up-regulated in IL-1β-expressing lewis lung carcinoma cells, and induced tumor angiogenesis through binding to CXCR2 (Saijo et al. 2002). The NF-κB-pathway is also induced during hypoxia downstream of HIF-1a induction. Constitutive activation of HIF-1 in keratinocytes enhanced NF-κB activation and induced transcription of CXCL1, CXCL2, and CXCL3 in a transgenic mouse model (Scortegagna et al. 2008). However, HIF-1-independent up-regulation of CXCL8 in response to hypoxia has also been described, suggesting that multiple pathways may lead to chemokine activation during low oxygen pressure (Mizukami et al. 2005). Tissue injury also induces chemokine expression through alternative pathways. For instance, CXCL1 is induced by thrombin in endothelial cells and tumor cells, and plays a pivotal role in thrombin-induced angiogenesis through up-regulation of pro-angiogenic molecules, including its receptor CXCR2 (Caunt et al. 2006).
CXCL8 interacts with the VEGF-signaling pathway in several ways. Autocrine VEGF stimulation was shown to induce CXCL8 secretion in tumor-associated brain endothelial cells (Charalambous et al. 2005). Conversely, CXCL8 has recently been shown to induce VEGF expression through Carma3/Bcl10/Malt1-dependent activation of NF-κB, leading to autocrine stimulation of VEGFR2 (Martin et al. 2009). A direct interaction between CXCL8 and VEGF-signaling pathways has also been described. Petreaca and co-authors showed that CXCL8 induced phosphorylation of VEGFR2 in a VEGF-independent manner, and that transactivation of VEGFR2 was required for CXCL8-induced endothelial permeability (Petreaca et al. 2007). Treatment with recombinant CXCL8 induced complex formation between CXCR1/CXCR2 and VEGFR2 in a Src-dependent manner, leading to phosphorylation of VEGFR2 and endothelial gap formation downstream of RhoA-activation.
4.3 The CXCR2L/CXCR2 Axis in Tumor Vascularization
Interestingly, oncogenes and growth factors may directly induce chemokines, coupling tumorigenesis to inflammation and angiogenesis. Overexpression of Bcl-2, a pro-survival protein up-regulated in many tumors, induces CXCL1 and CXCL8 in microvascular endothelial cells through activation of NF-κB (Karl et al. 2005). Similarly, EGF-induced up-regulation of Bcl-XL induces sprouting angiogenesis in a CXCR2-dependent fashion, involving an ERK-dependent increase in VEGF and a subsequent rise in CXCL1 and CXCL8 downstream of Bcl-2 (Karl et al. 2007). CXCL8 stimulation has in turn been shown to increase survival of endothelial cells through up-regulation of Bcl-2 and inhibition of Bax, suggesting the existence of a synergistic feedback loop between chemokines and Bcl-2 family signaling pathways (Li et al. 2003). Semaphorin 3B, previously described as a tumor suppressor, increases metastatic dissemination in experimental tumor models through up-regulation of CXCL8 downstream of neuropilin-1-mediated p38-mitogen-activated protein kinase activation (Rolny et al. 2008). Notably, CXCL8 is a transcriptional target for oncogenic RAS-signaling required for initiation of tumor-associated inflammation and neovascularization in RAS-expressing tumors (Sparmann and Bar-Sagi 2004). In a recent report, p53 was implicated in ID4-mediated stabilization of CXCL1 and CXCL8 mRNA, offering a new level of regulation of chemokine expression by oncogenes (Fontemaggi et al. 2009).
The role of CXCL8 in tumor angiogenesis is undisputed. Aside from its direct role in enhancing survival, chemotaxis, and tubular morphogenesis of endothelial cells, CXCL8 is a potent chemoattractant for monocytes and neutrophils, which in turn provide angiogenic growth factors and MMPs. The important function of CXCL8 in tumor angiogenesis was first suggested by the observation that angiogenic activity of bronchogenic carcinoma tumor cell homogenates was blocked through inhibition of CXCL8 (Smith et al. 1994). Consistent with this, neutralizing antibodies to CXCL8 or its cognate receptor CXCR2 inhibited tumorigenesis in human NSCLC in SCID mice, associated with reduced angiogenic activity and decreased vascular density (Arenberg et al. 1996b; Numasaki et al. 2005). Furthermore, CXCR2-deficient mice show decreased growth of renal cell carcinoma (RENCA), associated with decreased angiogenesis, reduced metastatic dissemination, and increased necrosis (Mestas et al. 2005). Tumor growth, angiogenesis, and metastasis of CXCL8-expressing human melanoma were also reduced in CXCR2-deficient nude mice, in association with decreased neutrophil infiltration, underscoring an important role of host CXCR2 in tumorigenesis (Singh et al. 2009). Transgene expression of CXCR2 in endothelial cells enhanced angiogenesis and tumor growth of melanocytes expressing a functional homologue to human CXCL8/CXCL1, macrophage inflammatory protein (MIP)-1, providing additional evidence for a direct role of CXCR ligands on endothelial cell function in vivo (Horton et al. 2007). Conversely, endothelial expression of the chemokine decoy receptor DARC decreased tumor vascular density and growth in the same model, while increasing trafficking of leukocytes.
4.4 CXCL12/CXCR4 Signaling in Recruitment of Pro-Angiogenic Bone Marrow Derived Cells
CXCL12/stromal cell-derived factor (SDF)-1 is an ELR-negative chemokine that acts as a potent inducer of angiogenesis. While angiostatic ELR-negative chemokines bind CXCR3, CXCL12 is a ligand for CXCR4 and the newly discovered receptor CXCR7 (Petit et al. 2007; Burns et al. 2006). CXCL12 is constitutively expressed in many different cell types, and expression is further induced upon ischemia or tissue injury. In the bone marrow, where the oxygen pressure is lower than in peripheral blood, CXCL12 is constitutively secreted by stromal cells and presented on endothelial cells (Petit et al. 2007; Mendez-Ferrer and Frenette 2007; Burger and Burkle 2007). Thereby, CXCL12 regulates homing and retention of CXCR4-expressing hematopoietic stem and progenitor cells in the marrow.
CXCL12 may enhance angiogenesis through several distinct mechanisms. First, CXCL12 can act directly through CXCR4 expressed on endothelial cells, inducing endothelial chemotaxis, tubular morphogenesis, and endothelial sprouting from rat aortic rings in vitro (Gupta et al. 1998; Salcedo et al. 1999; Deshane et al. 2007). Recently, the signaling pathway regulating CXCL12-induced endothelial migration has been elucidated and shown to be dependent on JNK3 activation, which occur downstream of eNOS-induced nitrosylation of MKP7 (Pi et al. 2009). CXCL12/CXCR4 activation is an important mediator of VEGF- and FGF-induced angiogenic programs. Indeed, blocking either CXCL12 or CXCR4 inhibits VEGF and FGF-induced tubular morphogenesis of endothelial cells (Salvucci et al. 2002; Salcedo et al. 2003). Conversely, the CXCL12/CXCR4 signaling pathway has been implicated in regulating the angiogenic switch in prostate cancer through down-regulation of phosphoglycerate kinase, leading to the production of VEGF and CXCL8 and decreasing expression of anti-angiogenic angiostatin (Wang et al. 2007). Second, CXCL12 induces adhesion, chemotaxis, and homing of circulating pro-angiogenic CXCR4+ hemotopoetic cells and endothelial progenitor cells to neo-vascular niches, contributing to angiogenesis in ischemic tissue and tumors (Petit et al. 2007). The recruitment of pro-angiogenic bone marrow-derived cells to wound tissue may be induced by platelet release of CXCL12 in response to vessel injury (Jin et al. 2006). Hypoxic gradients directly induce CXCL12 expression through activation of HIF-1α transcription factors in endothelial cells, leading to mobilization of bone marrow-derived progenitors (Schioppa et al. 2003; Ceradini et al. 2004). Endothelial progenitor cells express CXCR4 and have been suggested to contribute to CXCL12-induced angiogenesis. Indeed, CXCL12 increases the number of circulating endothelial progenitor cells during ischemia, and induce formation of tubular structures of co-injected c-kit+ progenitor cells in matrigel plugs in mice (De Falco et al. 2004). However, the relative contribution of endothelial progenitor cells to angiogenesis in various pathological conditions is still controversial (Ahn and Brown 2009; Pearson 2009). Finally, CXCL12 is important for retention of recruited bone marrow-derived cells close to the developing neo-vasculature, allowing continuous secretion of angiogenic growth factors during wound healing (Grunewald et al. 2006).
CXCL12 is well established as a potent inducer of tumor growth and metastatic dissemination through induction of angiogenesis (Guleng et al. 2005; Orimo et al. 2005; Singh et al. 2007; Yoon et al. 2007; Li and Ransohoff 2009). However, the pro-angiogenic role of CXCL12 may be dependent on the tumor type, as neutralizing antibodies to CXCL12 blocked tumor growth and metastasis of NSCLC in SCID mice without affecting tumor angiogenesis (Phillips et al. 2003). The molecular mechanisms that regulate CXCL12/CXCR4-induced angiogenesis in vivo are still not fully elucidated, and are likely to be at least partially dependent on recruitment of pro-angiogenic bone marrow-derived cells. Interestingly, mice lacking either CXCL12 or CXCR4 reveal vascular abnormalities, strongly supporting an important role of CXCL12/CXCR4 signaling in embryonic vasculogenesis (Tachibana et al. 1998; Ara et al. 2005). CXCL12 does not induce signaling through CXCR7, but CXCR4/CXCR7 heterodimers enhance CXCL12 signaling (Sierro et al. 2007). This appears to be specifically important in cardiac development, as endothelial-specific deletion of CXCR7 resulted in ventricular septal defects and heart valve malformations (Sierro et al. 2007).
4.5 CCL-Family Chemokines as Inducers of Angiogenesis
The CCL family is the largest chemokine subgroup involved in both inflammatory response during infection and tissue injury, immune surveillance, and lymphocyte homing. Several CCL family members promote an angiogenic program inducing migration, invasion, and tubular morphogenesis of endothelial cells in vitro, but it is not clear as to what extent the angiogenic function of these chemokines in vivo depend on recruitment of leukocytes. Interestingly, release of CCL-family chemokines from tumor neovessels and recruitment of CCR2- and CCR5-expressing progenitors has been described as a late event in carcinogenesis, correlating with mobilization of circulating endothelial progenitor cells (Spring et al. 2005).
CCL1 participates in the recruitment of monocytes and Th2-cells, and has been shown to induce angiogenesis in the rabbit cornea micropocket and CAM assays (Bernardini et al. 2000). CCL1 binds to CCR8, expressed on endothelial cells, and induces migration, invasion, and tubular morphogenesis in vitro. However, CCL1-induced recruitment of pro-angiogenic leukocytes is likely to at least partially contribute to angiogenesis in vivo. The eosinophil chemoattractant CCL11/eotaxin has angiogenic effects in vivo and induces chemotaxis of endothelial cells in vitro through binding to CCR3 (Salcedo et al. 2001). Blood vessel formation in vivo in the CAM and matrigel plug assay in mice following CCL11-stimulation was accompanied by infiltration of eosinophils, which may in turn release pro-angiogenic factors. Importantly, the observation of robust endothelial sprouting from rat aortic rings, in the absence of eosinophils, supports a direct pro-angiogenic function of CCL11. Likewise, CCL3 is an indirect inducer of angiogenesis during inflammation through recruitment of macrophages (Barcelos et al. 2009; Wu et al. 2008), but has also been implicated in autocrine stimulation of neovessel proliferation in a murine model of hepatocellular carcinoma through binding to its cognate receptor CCR5 (Ryschich et al. 2006).
CCL15, CCL16, and CCL23 induce migration and tube formation of endothelial cells in vitro, and induce neo-vascularization in the CAM assay through binding to CCR1 (Hwang et al. 2004, 2005; Strasly et al. 2004). In the case of CCL15, this effect was partially dependent on CCR3 binding, evidenced by a complete block in migration requiring treatment with neutralizing antibodies towards both CCR1 and CCR3 (Hwang et al. 2004). Interestingly, CCL16 signaling has cross talk with other pro-angiogenic pathways through increased production of CXCL8 and CCL2, and primes endothelial cells to mitogen signals by VEGF (Strasly et al. 2004). Because of the common binding and activation of CCR1, it is likely that similar pathways are induced by CCL15 and CCL23.
4.5.1 CCL2 in Arteriogenesis and Angiogenesis
CCL2/monocyte chemoattractant protein (MCP)-1 has an important role in arteriogenesis, mainly due to its role in recruitment of monocytes. Monocyte recruitment plays an important role during arteriogenesis, especially during collateral growth in response to vessel occlusion and tissue ischemia. CCL2 has a nonredundant function in monocyte recruitment through binding to its cognate receptor CCR2, evidenced by the impaired ability of CCL2−/− or CCR2−/− mice to recruit monocytes to affected tissues during inflammation (Charo and Taubman 2004).
CCL2 treatment induces chemotaxis of endothelial cells expressing CCR2, increases sprouting of rat aortic rings, and enhances angiogenesis in the CAM assay and in matrigel plugs in mice (Weber et al. 1999; Salcedo et al. 2000). Although the in vitro data support a direct role of CCL2 in modulation of endothelial function, the in vivo effects of CCL2 on vessel formation may be partially due to recruitment of monocytes secreting pro-angiogenic factors. CCL2-induced angiogenesis involves activation of Ets-1 and MCPIP transcription factors and up-regulation of MTI-MMP, and can be attenuated through blocking of either of these events (Galvez et al. 2005; Stamatovic et al. 2006; Niu et al. 2008).
CCL2 has a functional role in angiogenesis and arteriogenesis downstream of cytokine and growth factor signaling. CCL2 up-regulation is critical for VEGF-induced tubular morphogenesis and permeability (Yamada et al. 2003; Marumo et al. 1999). TGF-β up-regulates CCL2 directly through SMAD-signaling, which leads to angiogenesis and smooth muscle cell migration dependent on CCL2 (Ma et al. 2007). bFGF enhances CCL2 production from mesenchymal cells, which in turn improves arteriogenesis during hind limb ischemia in mice (Fujii et al. 2006).
4.6 CX3CL/CX3CR Axis in Angiogenesis
CX3CL/Fractalkine is the only known member of the CX3C-subfamily of chemokines, and is expressed as a transmembrane protein that can also be cleaved to a soluble variant. CX3CL induces chemotaxis and tubular morphogenesis of endothelial cells and increases sprouting in rat aortic rings in vitro, and enhances angiogenesis in the CAM assay, rabbit corneal micropocket neovascularization assay, and mouse matrigel plugs in vivo (Volin et al. 2001; Ryu et al. 2008; You et al. 2007). The pro-angiogenic effects of CX3CL are due to induction of HIF-1α transcription factors, leading to increased VEGF-levels (Ryu et al. 2008). Consequently, CX3CL-induced angiogenesis is blocked by inhibition of VEGFR2. CXC3L levels are increased in vitreous fluid in diabetic retinopathy patients and in synovial fluid from rheumatoid arthritis patient, and may be a target for anti-angiogenic treatment of these diseases (Volin et al. 2001; You et al. 2007). However, CX3CL may induce opposite effects in different microenvironmental settings. While CX3CL injection improved the condition of hind limb ischemia through pro-angiogenic effects, it reduced alkali-induced corneal neovascularization through production of anti-angiogenic factors in CX3CR expressing macrophages (Ryu et al. 2008; Lu et al. 2008).
4.7 KSHV-Encoded and CMV-Encoded Chemokines in Angiogenesis
Virally encoded chemokines and chemokine receptors that induce angiogenesis have been described and may influence human disease. A constitutively active GPCR of Kaposis sarcoma-associated herpesvirus (KSHV), with homology to chemokines receptors, has been shown to induce an angiogenic switch mediated by VEGF (Bais et al. 1998). Conditioned media from transfected NIH3T3 fibroblasts induced HUVEC proliferation and tubular morphogenesis in a VEGF-dependent fashion. The KSHV-encoded chemokines vMIP-11 is a CCR4 agonist that has been shown to stimulate angiogenesis in the CAM assay and attract Th2-cells (Stine et al. 2000). Finally, the CMV-encoded chemokine receptor US28 promotes angiogenesis and tumorigenesis via NFκB-driven VEGF-induction of COX-2 (Maussang et al. 2006, 2009).
5 The Chemokines and Their Receptors as Future Therapeutic Targets
Angiogenesis and inflammation are linked processes that affect the outcome of many pathological conditions. Chemokines are central regulators of both these processes, suggesting that targeting chemokines or chemokine receptors may be beneficial in a wide range of diseases (Ley et al. 2007; Thelen and Stein 2008) (Table 3). Several anti-angiogenic drugs, many of which disrupt VEGF/VEGFR activation, have been approved and are successfully used in the treatment of, for example, age-related macula degeneration and various types of cancer (Olsson et al. 2006; Jain et al. 2006). Combining anti-angiogenic therapy with chemotherapy is beneficial in cancer treatment, presumably due to normalization of the tumor vasculature and increased delivery of drugs into the tumor. However, applying anti-angiogenic therapy does not cure the disease. In fact, aggressive anti-angiogenic therapy has recently been associated with increased metastatic dissemination in animal models, although this has yet to be confirmed in human patients (Ebos et al. 2009; Paez-Ribes et al. 2009). Moreover, tumor resistance to anti-angiogenic therapy is still an unresolved clinical problem (Kerbel 2008). One mechanism of refractoriness to anti-VEGF therapy is through recruitment of pro-angiogenic myeloid cells (Shojaei et al. 2007). Drugs targeting chemokines may simultaneously block angiogenesis and inhibit immune cell recruitment, thereby reducing the risk of relapse. Targeting chemokines may also decrease the rate of metastatic dissemination as cancer cells are believed to utilize similar pathways for extravasation as do hematopoietic cells. Indeed, treatment with angiostatic chemokines or inhibition of angiogenic chemokines/chemokine receptors reduces angiogenesis, inhibits tumor growth, and reduces metastasis in many different tumor models. The observation that chemokines frequently have cross talk with several angiogenic factors suggests that combined targeting of chemokines and growth factors may have synergistic effects. Importantly, chemokines have been proposed as therapeutic targets for chronic inflammatory disorders, myocardial ischemia, artherosclerosis, and pulmonary fibrosis, and play an important role during wound healing (Keeley et al. 2008; Strieter et al. 2007). Strategies to inhibit or augment chemokine signaling may therefore be of general importance, and would potentially be utilized in the clinical treatment of several conditions. However, the central role of chemokines in the immune system and the extensive redundancy of chemokine/chemokine receptor signaling necessitate further investigation regarding the putative benefit in intervening with chemokine signaling in pathological disorders.
References
Adams RH, Alitalo K (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8:464–478
Addison CL, Daniel TO, Burdick MD, Liu H, Ehlert JE, Xue YY, Buechi L, Walz A, Richmond A, Strieter RM (2000) The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol 165:5269–5277
Ahn GO, Brown JM (2009) Role of endothelial progenitors and other bone marrow-derived cells in the development of the tumor vasculature. Angiogenesis 12:159–164
Aidoudi S, Bujakowska K, Kieffer N, Bikfalvi A (2008) The CXC-chemokine CXCL4 interacts with integrins implicated in angiogenesis. PLoS One 3:e2657
Angiolillo AL, Sgadari C, Taub DD, Liao F, Farber JM, Maheshwari S, Kleinman HK, Reaman GH, Tosato G (1995) Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med 182:155–162
Ara T, Tokoyoda K, Okamoto R, Koni PA, Nagasawa T (2005) The role of CXCL12 in the organ-specific process of artery formation. Blood 105:3155–3161
Arenberg DA, Kunkel SL, Polverini PJ, Morris SB, Burdick MD, Glass MC, Taub DT, Iannettoni MD, Whyte RI, Strieter RM (1996a) Interferon-gamma-inducible protein 10 (IP-10) is an angiostatic factor that inhibits human non-small cell lung cancer (NSCLC) tumorigenesis and spontaneous metastases. J Exp Med 184:981–992
Arenberg DA, Kunkel SL, Polverini PJ, Glass M, Burdick MD, Strieter RM (1996b) Inhibition of interleukin-8 reduces tumorigenesis of human non-small cell lung cancer in SCID mice. J Clin Invest 97:2792–2802
Bais C, Santomasso B, Coso O, Arvanitakis L, Raaka EG, Gutkind JS, Asch AS, Cesarman E, Gershengorn MC, Mesri EA (1998) G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 391:86–89
Balestrieri ML, Balestrieri A, Mancini FP, Napoli C (2008) Understanding the immunoangiostatic CXC chemokine network. Cardiovasc Res 78:250–256
Barcelos LS, Coelho AM, Russo RC, Guabiraba R, Souza AL, Bruno-Lima G Jr, Proudfoot AE, Andrade SP, Teixeira MM (2009) Role of the chemokines CCL3/MIP-1 alpha and CCL5/RANTES in sponge-induced inflammatory angiogenesis in mice. Microvasc Res 78:148–154
Benelli R, Morini M, Carrozzino F, Ferrari N, Minghelli S, Santi L, Cassatella M, Noonan DM, Albini A (2002) Neutrophils as a key cellular target for angiostatin: implications for regulation of angiogenesis and inflammation. FASEB J 16:267–269
Bernardini G, Spinetti G, Ribatti D, Camarda G, Morbidelli L, Ziche M, Santoni A, Capogrossi MC, Napolitano M (2000) I-309 binds to and activates endothelial cell functions and acts as an angiogenic molecule in vivo. Blood 96:4039–4045
Bodnar RJ, Yates CC, Rodgers ME, Du X, Wells A (2009) IP-10 induces dissociation of newly formed blood vessels. J Cell Sci 122:2064–2077
Burger JA, Burkle A (2007) The CXCR4 chemokine receptor in acute and chronic leukaemia: a marrow homing receptor and potential therapeutic target. Br J Haematol 137:288–296
Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ, Wei K, McMaster BE, Wright K, Howard MC, Schall TJ (2006) A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 203:2201–2213
Caunt M, Hu L, Tang T, Brooks PC, Ibrahim S, Karpatkin S (2006) Growth-regulated oncogene is pivotal in thrombin-induced angiogenesis. Cancer Res 66:4125–4132
Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC (2004) Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10:858–864
Charalambous C, Pen LB, Su YS, Milan J, Chen TC, Hofman FM (2005) Interleukin-8 differentially regulates migration of tumor-associated and normal human brain endothelial cells. Cancer Res 65:10347–10354
Charo IF, Taubman MB (2004) Chemokines in the pathogenesis of vascular disease. Circ Res 95:858–866
De Falco E, Porcelli D, Torella AR, Straino S, Iachininoto MG, Orlandi A, Truffa S, Biglioli P, Napolitano M, Capogrossi MC, Pesce M (2004) SDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells. Blood 104:3472–3482
Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Li Calzi S, Lach R, Hock TD, Chen B, Hill-Kapturczak N, Siegal GP, Dulak J, Jozkowicz A, Grant MB, Agarwal A (2007) Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J Exp Med 204:605–618
Devalaraja RM, Nanney LB, Du J, Qian Q, Yu Y, Devalaraja MN, Richmond A (2000) Delayed wound healing in CXCR2 knockout mice. J Invest Dermatol 115:234–244
Dirkx AE, Oude Egbrink MG, Wagstaff J, Griffioen AW (2006) Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. J Leukoc Biol 80:1183–1196
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS (2009) Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15:232–239
Fontemaggi G, Dell'Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F, Terrenato I, Mottolese M, Muti P, Domany E, Del Bufalo D, Strano S, Blandino G (2009) The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol 16:1086–1093
Fraisl P, Mazzone M, Schmidt T, Carmeliet P (2009) Regulation of angiogenesis by oxygen and metabolism. Dev Cell 16:167–179
Fujii T, Yonemitsu Y, Onimaru M, Tanii M, Nakano T, Egashira K, Takehara T, Inoue M, Hasegawa M, Kuwano H, Sueishi K (2006) Nonendothelial mesenchymal cell-derived MCP-1 is required for FGF-2-mediated therapeutic neovascularization: critical role of the inflammatory/arteriogenic pathway. Arterioscler Thromb Vasc Biol 26:2483–2489
Galvez BG, Genis L, Matias-Roman S, Oblander SA, Tryggvason K, Apte SS, Arroyo AG (2005) Membrane type 1-matrix metalloproteinase is regulated by chemokines monocyte-chemoattractant protein-1/ccl2 and interleukin-8/CXCL8 in endothelial cells during angiogenesis. J Biol Chem 280:1292–1298
Gengrinovitch S, Greenberg SM, Cohen T, Gitay-Goren H, Rockwell P, Maione TE, Levi BZ, Neufeld G (1995) Platelet factor-4 inhibits the mitogenic activity of VEGF121 and VEGF165 using several concurrent mechanisms. J Biol Chem 270:15059–15065
Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Jung S, Chimenti S, Landsman L, Abramovitch R, Keshet E (2006) VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell 124:175–189
Guleng B, Tateishi K, Ohta M, Kanai F, Jazag A, Ijichi H, Tanaka Y, Washida M, Morikane K, Fukushima Y, Yamori T, Tsuruo T, Kawabe T, Miyagishi M, Taira K, Sata M, Omata M (2005) Blockade of the stromal cell-derived factor-1/CXCR4 axis attenuates in vivo tumor growth by inhibiting angiogenesis in a vascular endothelial growth factor-independent manner. Cancer Res 65:5864–5871
Gupta SK, Lysko PG, Pillarisetti K, Ohlstein E, Stadel JM (1998) Chemokine receptors in human endothelial cells. Functional expression of CXCR4 and its transcriptional regulation by inflammatory cytokines. J Biol Chem 273:4282–4287
Hansell P, Maione TE, Borgstrom P (1995) Selective binding of platelet factor 4 to regions of active angiogenesis in vivo. Am J Physiol 269:H829–836
Horton LW, Yu Y, Zaja-Milatovic S, Strieter RM, Richmond A (2007) Opposing roles of murine duffy antigen receptor for chemokine and murine CXC chemokine receptor-2 receptors in murine melanoma tumor growth. Cancer Res 67:9791–9799
Hwang J, Kim CW, Son KN, Han KY, Lee KH, Kleinman HK, Ko J, Na DS, Kwon BS, Gho YS, Kim J (2004) Angiogenic activity of human CC chemokine CCL15 in vitro and in vivo. FEBS Lett 570:47–51
Hwang J, Son KN, Kim CW, Ko J, Na DS, Kwon BS, Gho YS, Kim J (2005) Human CC chemokine CCL23, a ligand for CCR1, induces endothelial cell migration and promotes angiogenesis. Cytokine 30:254–263
Jain RK, Duda DG, Clark JW, Loeffler JS (2006) Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol 3:24–40
Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, Hooper AT, Amano H, Avecilla ST, Heissig B, Hattori K, Zhang F, Hicklin DJ, Wu Y, Zhu Z, Dunn A, Salari H, Werb Z, Hackett NR, Crystal RG, Lyden D, Rafii S (2006) Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med 12:557–567
Jujo K, Ii M, Losordo DW (2008) Endothelial progenitor cells in neovascularization of infarcted myocardium. J Mol Cell Cardiol 45:530–544
Karl E, Warner K, Zeitlin B, Kaneko T, Wurtzel L, Jin T, Chang J, Wang S, Wang CY, Strieter RM, Nunez G, Polverini PJ, Nor JE (2005) Bcl-2 acts in a proangiogenic signaling pathway through nuclear factor-kappaB and CXC chemokines. Cancer Res 65:5063–5069
Karl E, Zhang Z, Dong Z, Neiva KG, Soengas MS, Koch AE, Polverini PJ, Nunez G, Nor JE (2007) Unidirectional crosstalk between Bcl-xL and Bcl-2 enhances the angiogenic phenotype of endothelial cells. Cell Death Differ 14:1657–1666
Kasama T, Muramatsu M, Kobayashi K, Yajima N, Shiozawa F, Hanaoka R, Miwa Y, Negishi M, Ide H, Adachi M (2002) Interaction of monocytes with vascular endothelial cells synergistically induces interferon gamma-inducible protein 10 expression through activation of specific cell surface molecules and cytokines. Cell Immunol 219:131–139
Keeley EC, Mehrad B, Strieter RM (2008) Chemokines as mediators of neovascularization. Arterioscler Thromb Vasc Biol 28:1928–1936
Kerbel RS (2008) Tumor angiogenesis. N Engl J Med 358:2039–2049
Kilarski WW, Samolov B, Petersson L, Kvanta A, Gerwins P (2009) Biomechanical regulation of blood vessel growth during tissue vascularization. Nat Med 15:657–664
Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM (1992) Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 258:1798–1801
Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, Sagrinati C, Mazzinghi B, Orlando C, Maggi E, Marra F, Romagnani S, Serio M, Romagnani P (2003) An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med 197:1537–1549
Ley K, Laudanna C, Cybulsky MI, Nourshargh S (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7:678–689
Li M, Ransohoff RM (2009) The roles of chemokine CXCL12 in embryonic and brain tumor angiogenesis. Semin Cancer Biol 19:111–115
Li A, Dubey S, Varney ML, Dave BJ, Singh RK (2003) IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol 170:3369–3376
Lu P, Li L, Kuno K, Wu Y, Baba T, Li YY, Zhang X, Mukaida N (2008) Protective roles of the fractalkine/CX3CL1-CX3CR1 interactions in alkali-induced corneal neovascularization through enhanced antiangiogenic factor expression. J Immunol 180:4283–4291
Ma J, Wang Q, Fei T, Han JD, Chen YG (2007) MCP-1 mediates TGF-beta-induced angiogenesis by stimulating vascular smooth muscle cell migration. Blood 109:987–994
Maione TE, Gray GS, Petro J, Hunt AJ, Donner AL, Bauer SI, Carson HF, Sharpe RJ (1990) Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. Science 247:77–79
Maione TE, Gray GS, Hunt AJ, Sharpe RJ (1991) Inhibition of tumor growth in mice by an analogue of platelet factor 4 that lacks affinity for heparin and retains potent angiostatic activity. Cancer Res 51:2077–2083
Makanya AN, Hlushchuk R, Djonov VG (2009) Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis 12:113–123
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23:549–555
Martin D, Galisteo R, Gutkind JS (2009) CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem 284:6038–6042
Marumo T, Schini-Kerth VB, Busse R (1999) Vascular endothelial growth factor activates nuclear factor-kappaB and induces monocyte chemoattractant protein-1 in bovine retinal endothelial cells. Diabetes 48:1131–1137
Maussang D, Verzijl D, van Walsum M, Leurs R, Holl J, Pleskoff O, Michel D, van Dongen GA, Smit MJ (2006) Human cytomegalovirus-encoded chemokine receptor US28 promotes tumorigenesis. Proc Natl Acad Sci USA 103:13068–13073
Maussang D, Langemeijer E, Fitzsimons CP, Stigter-van Walsum M, Dijkman R, Borg MK, Slinger E, Schreiber A, Michel D, Tensen CP, van Dongen GA, Leurs R, Smit MJ (2009) The human cytomegalovirus-encoded chemokine receptor US28 promotes angiogenesis and tumor formation via cyclooxygenase-2. Cancer Res 69:2861–2869
Mendez-Ferrer S, Frenette PS (2007) Hematopoietic stem cell trafficking: regulated adhesion and attraction to bone marrow microenvironment. Ann N Y Acad Sci 1116:392–413
Mestas J, Burdick MD, Reckamp K, Pantuck A, Figlin RA, Strieter RM (2005) The role of CXCR2/CXCR2 ligand biological axis in renal cell carcinoma. J Immunol 175:5351–5357
Mizukami Y, Jo WS, Duerr EM, Gala M, Li J, Zhang X, Zimmer MA, Iliopoulos O, Zukerberg LR, Kohgo Y, Lynch MP, Rueda BR, Chung DC (2005) Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat Med 11:992–997
Murdoch C, Muthana M, Coffelt SB, Lewis CE (2008) The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 8:618–631
Niu J, Azfer A, Zhelyabovska O, Fatma S, Kolattukudy PE (2008) Monocyte chemotactic protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein (MCPIP). J Biol Chem 283:14542–14551
Numasaki M, Watanabe M, Suzuki T, Takahashi H, Nakamura A, McAllister F, Hishinuma T, Goto J, Lotze MT, Kolls JK, Sasaki H (2005) IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol 175:6177–6189
Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L (2006) VEGF receptor signalling – in control of vascular function. Nat Rev Mol Cell Biol 7:359–371
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121:335–348
Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O (2009) Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15:220–231
Pearson JD (2009) Endothelial progenitor cells – hype or hope? J Thromb Haemost 7:255–262
Perollet C, Han ZC, Savona C, Caen JP, Bikfalvi A (1998) Platelet factor 4 modulates fibroblast growth factor 2 (FGF-2) activity and inhibits FGF-2 dimerization. Blood 91:3289–3299
Petit I, Jin D, Rafii S (2007) The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol 28:299–307
Petreaca ML, Yao M, Liu Y, Defea K, Martins-Green M (2007) Transactivation of vascular endothelial growth factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for IL-8/CXCL8-induced endothelial permeability. Mol Biol Cell 18:5014–5023
Phillips RJ, Burdick MD, Lutz M, Belperio JA, Keane MP, Strieter RM (2003) The stromal derived factor-1/CXCL12-CXC chemokine receptor 4 biological axis in non-small cell lung cancer metastases. Am J Respir Crit Care Med 167:1676–1686
Pi X, Wu Y, Ferguson JE 3rd, Portbury AL, Patterson C (2009) SDF-1alpha stimulates JNK3 activity via eNOS-dependent nitrosylation of MKP7 to enhance endothelial migration. Proc Natl Acad Sci USA 106:5675–5680
Rolny C, Capparuccia L, Casazza A, Mazzone M, Vallario A, Cignetti A, Medico E, Carmeliet P, Comoglio PM, Tamagnone L (2008) The tumor suppressor semaphorin 3B triggers a prometastatic program mediated by interleukin 8 and the tumor microenvironment. J Exp Med 205:1155–1171
Romagnani P, Annunziato F, Lasagni L, Lazzeri E, Beltrame C, Francalanci M, Uguccioni M, Galli G, Cosmi L, Maurenzig L, Baggiolini M, Maggi E, Romagnani S, Serio M (2001) Cell cycle-dependent expression of CXC chemokine receptor 3 by endothelial cells mediates angiostatic activity. J Clin Invest 107:53–63
Ryschich E, Lizdenis P, Ittrich C, Benner A, Stahl S, Hamann A, Schmidt J, Knolle P, Arnold B, Hammerling GJ, Ganss R (2006) Molecular fingerprinting and autocrine growth regulation of endothelial cells in a murine model of hepatocellular carcinoma. Cancer Res 66:198–211
Ryu J, Lee CW, Hong KH, Shin JA, Lim SH, Park CS, Shim J, Nam KB, Choi KJ, Kim YH, Han KH (2008) Activation of fractalkine/CX3CR1 by vascular endothelial cells induces angiogenesis through VEGF-A/KDR and reverses hindlimb ischaemia. Cardiovasc Res 78:333–340
Saijo Y, Tanaka M, Miki M, Usui K, Suzuki T, Maemondo M, Hong X, Tazawa R, Kikuchi T, Matsushima K, Nukiwa T (2002) Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: in vivo analysis of tumor-stromal interaction. J Immunol 169:469–475
Salanga CL, O'Hayre M, Handel T (2009) Modulation of chemokine receptor activity through dimerization and crosstalk. Cell Mol Life Sci 66:1370–1386
Salcedo R, Wasserman K, Young HA, Grimm MC, Howard OM, Anver MR, Kleinman HK, Murphy WJ, Oppenheim JJ (1999) Vascular endothelial growth factor and basic fibroblast growth factor induce expression of CXCR4 on human endothelial cells: In vivo neovascularization induced by stromal-derived factor-1alpha. Am J Pathol 154:1125–1135
Salcedo R, Ponce ML, Young HA, Wasserman K, Ward JM, Kleinman HK, Oppenheim JJ, Murphy WJ (2000) Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood 96:34–40
Salcedo R, Young HA, Ponce ML, Ward JM, Kleinman HK, Murphy WJ, Oppenheim JJ (2001) Eotaxin (CCL11) induces in vivo angiogenic responses by human CCR3+ endothelial cells. J Immunol 166:7571–7578
Salcedo R, Zhang X, Young HA, Michael N, Wasserman K, Ma WH, Martins-Green M, Murphy WJ, Oppenheim JJ (2003) Angiogenic effects of prostaglandin E2 are mediated by up-regulation of CXCR4 on human microvascular endothelial cells. Blood 102:1966–1977
Salvucci O, Yao L, Villalba S, Sajewicz A, Pittaluga S, Tosato G (2002) Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood 99:2703–2711
Sato Y, Abe M, Takaki R (1990) Platelet factor 4 blocks the binding of basic fibroblast growth factor to the receptor and inhibits the spontaneous migration of vascular endothelial cells. Biochem Biophys Res Commun 172:595–600
Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, Mantovani A, Melillo G, Sica A (2003) Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med 198:1391–1402
Scortegagna M, Cataisson C, Martin RJ, Hicklin DJ, Schreiber RD, Yuspa SH, Arbeit JM (2008) HIF-1alpha regulates epithelial inflammation by cell autonomous NFkappaB activation and paracrine stromal remodeling. Blood 111:3343–3354
Sgadari C, Angiolillo AL, Tosato G (1996) Inhibition of angiogenesis by interleukin-12 is mediated by the interferon-inducible protein 10. Blood 87:3877–3882
Shojaei F, Ferrara N (2008) Refractoriness to antivascular endothelial growth factor treatment: role of myeloid cells. Cancer Res 68:5501–5504
Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N (2007) Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol 25:911–920
Shojaei F, Zhong C, Wu X, Yu L, Ferrara N (2008) Role of myeloid cells in tumor angiogenesis and growth. Trends Cell Biol 18:372–378
Sierro F, Biben C, Martinez-Munoz L, Mellado M, Ransohoff RM, Li M, Woehl B, Leung H, Groom J, Batten M, Harvey RP, Martinez AC, Mackay CR, Mackay F (2007) Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci USA 104:14759–14764
Singh S, Sadanandam A, Singh RK (2007) Chemokines in tumor angiogenesis and metastasis. Cancer Metastasis Rev 26:453–467
Singh S, Varney M, Singh RK (2009) Host CXCR2-dependent regulation of melanoma growth, angiogenesis, and experimental lung metastasis. Cancer Res 69:411–415
Smith DR, Polverini PJ, Kunkel SL, Orringer MB, Whyte RI, Burdick MD, Wilke CA, Strieter RM (1994) Inhibition of interleukin 8 attenuates angiogenesis in bronchogenic carcinoma. J Exp Med 179:1409–1415
Sparmann A, Bar-Sagi D (2004) Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 6:447–458
Spring H, Schuler T, Arnold B, Hammerling GJ, Ganss R (2005) Chemokines direct endothelial progenitors into tumor neovessels. Proc Natl Acad Sci USA 102:18111–18116
Stamatovic SM, Keep RF, Mostarica-Stojkovic M, Andjelkovic AV (2006) CCL2 regulates angiogenesis via activation of Ets-1 transcription factor. J Immunol 177:2651–2661
Staton CA, Reed MW, Brown NJ (2009) A critical analysis of current in vitro and in vivo angiogenesis assays. Int J Exp Pathol 90:195–221
Stine JT, Wood C, Hill M, Epp A, Raport CJ, Schweickart VL, Endo Y, Sasaki T, Simmons G, Boshoff C, Clapham P, Chang Y, Moore P, Gray PW, Chantry D (2000) KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood 95:1151–1157
Strasly M, Cavallo F, Geuna M, Mitola S, Colombo MP, Forni G, Bussolino F (2001) IL-12 inhibition of endothelial cell functions and angiogenesis depends on lymphocyte-endothelial cell cross-talk. J Immunol 166:3890–3899
Strasly M, Doronzo G, Cappello P, Valdembri D, Arese M, Mitola S, Moore P, Alessandri G, Giovarelli M, Bussolino F (2004) CCL16 activates an angiogenic program in vascular endothelial cells. Blood 103:40–49
Strieter RM, Kunkel SL, Elner VM, Martonyi CL, Koch AE, Polverini PJ, Elner SG (1992) Interleukin-8. A corneal factor that induces neovascularization. Am J Pathol 141:1279–1284
Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Marriott D et al (1995) The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem 270:27348–27357
Strieter RM, Gomperts BN, Keane MP (2007) The role of CXC chemokines in pulmonary fibrosis. J Clin Invest 117:549–556
Struyf S, Burdick MD, Proost P, Van Damme J, Strieter RM (2004) Platelets release CXCL4L1, a nonallelic variant of the chemokine platelet factor-4/CXCL4 and potent inhibitor of angiogenesis. Circ Res 95:855–857
Struyf S, Burdick MD, Peeters E, Van den Broeck K, Dillen C, Proost P, Van Damme J, Strieter RM (2007) Platelet factor-4 variant chemokine CXCL4L1 inhibits melanoma and lung carcinoma growth and metastasis by preventing angiogenesis. Cancer Res 67:5940–5948
Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N, Nishikawa S, Kishimoto T, Nagasawa T (1998) The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393:591–594
Tanaka T, Manome Y, Wen P, Kufe DW, Fine HA (1997) Viral vector-mediated transduction of a modified platelet factor 4 cDNA inhibits angiogenesis and tumor growth. Nat Med 3:437–442
Taub DD (2004) Cytokine, growth factor, and chemokine ligand database. Curr Protoc Immunol Chapter 6: Unit 6 29
Thelen M, Stein JV (2008) How chemokines invite leukocytes to dance. Nat Immunol 9:953–959
Volin MV, Woods JM, Amin MA, Connors MA, Harlow LA, Koch AE (2001) Fractalkine: a novel angiogenic chemokine in rheumatoid arthritis. Am J Pathol 159:1521–1530
Wang J, Dai J, Jung Y, Wei CL, Wang Y, Havens AM, Hogg PJ, Keller ET, Pienta KJ, Nor JE, Wang CY, Taichman RS (2007) A glycolytic mechanism regulating an angiogenic switch in prostate cancer. Cancer Res 67:149–159
Weber KS, Nelson PJ, Grone HJ, Weber C (1999) Expression of CCR2 by endothelial cells: implications for MCP-1 mediated wound injury repair and In vivo inflammatory activation of endothelium. Arterioscler Thromb Vasc Biol 19:2085–2093
Wu Y, Li YY, Matsushima K, Baba T, Mukaida N (2008) CCL3-CCR5 axis regulates intratumoral accumulation of leukocytes and fibroblasts and promotes angiogenesis in murine lung metastasis process. J Immunol 181:6384–6393
Yamada M, Kim S, Egashira K, Takeya M, Ikeda T, Mimura O, Iwao H (2003) Molecular mechanism and role of endothelial monocyte chemoattractant protein-1 induction by vascular endothelial growth factor. Arterioscler Thromb Vasc Biol 23:1996–2001
Yoon Y, Liang Z, Zhang X, Choe M, Zhu A, Cho HT, Shin DM, Goodman MM, Chen ZG, Shim H (2007) CXC chemokine receptor-4 antagonist blocks both growth of primary tumor and metastasis of head and neck cancer in xenograft mouse models. Cancer Res 67:7518–7524
You JJ, Yang CH, Huang JS, Chen MS, Yang CM (2007) Fractalkine, a CX3C chemokine, as a mediator of ocular angiogenesis. Invest Ophthalmol Vis Sci 48:5290–5298
Zumsteg A, Christofori G (2009) Corrupt policemen: inflammatory cells promote tumor angiogenesis. Curr Opin Oncol 21:60–70
Acknowledgments
Owing to space limitations, I have not been able to cite all relevant work, and in some cases, reviews are cited rather than specific papers. I apologize to the authors of original work who were not cited. Supported by grants from the Swedish Research Council, the Swedish Cancer Society, the Åke Wiberg foundation, the Magnus Bergvall foundation, and the Swedish Society of Medicine.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Dimberg, A. (2010). Chemokines in Angiogenesis. In: Bruserud, O. (eds) The Chemokine System in Experimental and Clinical Hematology. Current Topics in Microbiology and Immunology, vol 341. Springer, Berlin, Heidelberg. https://doi.org/10.1007/82_2010_21
Download citation
DOI: https://doi.org/10.1007/82_2010_21
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-12638-3
Online ISBN: 978-3-642-12639-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)