
Abstract
Bipolar disorder (BD) is a severe, debilitating psychiatric condition with onset in adolescence or young adulthood and often follows a relapsing and remitting course throughout life. The concept of neuroprogression in BD refers to the progressive path with an identifiable trajectory that takes place with recurrent mood episodes, which eventually leads to cognitive, functional, and clinical deterioration in the course of BD. Understanding the biological basis of neuroprogression helps to explain the subset of BD patients who experience worsening of their disorder over time. Additionally, the study of the neurobiological mechanisms underpinning neuroprogression will help BD staging based on systems biology. Replicated epidemiological studies have suggested inflammatory mechanisms as primary contributors to the neuroprogression of mood disorders. It is known that dysregulated inflammatory/immune pathways are often associated with BD pathophysiology. Hence, in this chapter, we focus on the evidence for the involvement of inflammation and immune regulated pathways in the neurobiological consequences of BD neuroprogression. Herein we put forth the evidence of immune markers from autoimmune disorders, chronic infections, and gut-brain axis that lead to BD neuroprogression. Further, we highlighted the peripheral and central inflammatory components measured along with BD progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Bipolar disorder (BD) is a severe episodic mental illness with chronic and accelerating course, composed of mood swings ranging from extreme elation, called mania, to extreme lows, called depression, which may sometimes occur together (Association AP 2013). BD is classified as BD-1 (which represents classic manic depressive disorder), BD-2 (represented by the manifestation of hypomanic and depressive symptoms), and cyclothymic disorder. In BD, the obligatory symptoms (gate A criteria) are hypomanic or manic episodes. Lifetime prevalence is about 1.0% for BD-1, 1.1% for BD-2, and 2.4% for sub-threshold BD (Merikangas et al. 2007). Among mental disorders, the absolute risk of suicide was found to be the highest for men with BD (7.77%), while for women, it was 4.78% (Nordentoft et al. 2011). There was a reported increase in the suicide risk due to comorbid occurrence of substance abuse and unipolar affective disorders, while the co-occurrence of deliberate self-harm generally doubled the suicide risk (Nordentoft et al. 2011). Growing evidence has suggested that BD, like many other chronic illnesses, may have a progressive course with functional impairment and neuroanatomical changes.
The current clinical focus is mainly on stabilizing the acute mood episodes and preventing their recurrence in BD patients leading to negligence of the need to promote functional recovery (Kapczinski et al. 2017). Clinical, neuroimaging, and neurocognitive studies further support the progressive nature of BD. In a large cross-sectional study, it was found that 42% of the euthymic BD patients had poor overall functioning (Samalin et al. 2016). Also, it was found that episode density, level of residual depressive symptoms, estimated verbal intelligence, and inhibitory control are the risk factors predictive of poor functional outcome of BD (Reinares et al. 2013). Clinical staging model, which is used in oncology and medicine for a long time and recently introduced in psychiatry, proposes that there is a stepwise progression through a series of identifiable steps, which have characteristic features and potential treatment implications (Berk et al. 2007). A large study has reported progressive functional impairment from stage I to stage IV of BD on clinical staging (Rosa et al. 2014). More specifically, stage I includes individuals who exhibit the same status in the interepisodic period as they did before the onset of BD (i.e., premorbid status); stage II includes individuals whose interepisodic period is characterized by psychiatric comorbidities or residual symptoms that require changes in pharmacological treatment, but who are able to maintain daily activities; stage III includes individuals who require occupational and social rehabilitation and face difficulties in their daily activities; and stage IV includes individuals who are unable to maintain personal self-care and to live autonomously.
Various studies have described that the presence of widespread structural brain abnormalities in BD is associated with the incidence of manic episodes and higher illness burden – which points to neuroprogression (Abe et al. 2015; Mwangi et al. 2016). A study found that reduced hippocampal volume and severe cognitive impairment were associated with the increased number of manic episodes and hospitalizations in BD patients (Cao et al. 2016), while another study reported decreased posterior corpus callosum volume in women with late-stage BD (Lavagnino et al. 2015). Reduced hippocampal volume has also been seen in particular viral infections pointing towards an association between inflammation and structural changes seen in severe BD (Almanzar et al. 2005). Besides, a longitudinal study found reduced frontal cortex volume (dorsolateral prefrontal and inferior frontal cortex) in patients who had at least one manic episode (Abe et al. 2015). Similar findings were described in a study that having larger lateral ventricles was associated with a higher number of prior manic episodes in patients with BD (Strakowski et al. 2002). Given these findings, the concept of neuroprogression was postulated to encompass the progressive functional impairment and neuroanatomical changes in BD presentation (Grande et al. 2016).
Neuroprogression has thus been proposed as the pathological alterations in the brain that take place simultaneously with the clinical and neurocognitive deterioration in the course of BD (Berk et al. 2011). However, the clinical implications and molecular foundations of neuroprogression remain incompletely understood. It seems that changes in some peripheral biomarkers from oxidative, inflammatory, and neurotrophic pathways are associated with neuroprogression. Hence, in this book chapter, we will highlight the association of various immune pathways and also the gut microbiota with neuroprogression in a subset of more severe patients with BD. Light will also be shed on the association of neuroprogression and the peripheral biomarker changes in BD.
2 Evidence of Inflammatory and Infectious Diseases in BD Neuroprogression
Even though BD is thought to be a neuroprogressive disorder (Berk et al. 2011), evidence suggests that disruption in neurodevelopmental pathways may play a pivotal role in the etiopathology of this illness (Savitz et al. 2014). Neurodevelopment can be disrupted by several pathways, causing the mood dysregulations seen in BD (Harrison 2016). A multiple-hit model has been postulated as a series of three factors, with hit 1 being a genetic predisposition to BD, hit 2 being the perinatal environment, which gives rise to phenotypes of vulnerability, and hit 3 is the later life experiences and exposures (Daskalakis et al. 2013). Although the mechanism is still unclear, this multiple-hit model has been suggested to dysregulate the homeostasis chronically in a process that is thought to involve immune dysfunction (Leboyer et al. 2016).
2.1 Maternal Immune Activation as a Risk Factor for BD Development and Neuroprogression
Epidemiologic evidence has repeatedly suggested that prenatal environmental influences, such as maternal immune activation (MIA), are involved in the pathophysiology of neuropsychiatric disorders (Brown and Derkits 2010; Estes and McAllister 2016). Prenatal infections and inflammation are potential risk factors associated with schizophrenia (Brown and Derkits 2010; Estes and McAllister 2016), autism spectrum disorders (Canetta et al. 2014), and BD (Brown 2015; Canetta et al. 2014). Animal models of MIA have also demonstrated behavioral, chemical, anatomical, and physiologic disturbances in the CNS of the progeny (Meyer 2014; Meyer et al. 2009). While the role of prenatal and postnatal infections as a risk factor for schizophrenia has been studied extensively (Debnath et al. 2015; Khandaker et al. 2012), not many studies have evaluated the role of infection during the prenatal period as a risk factor for BD (Canetta et al. 2014). For instance, multiple studies have reported an association between serologically documented maternal influenza infection and increased risk of BD in the offspring (Canetta et al. 2014; Parboosing et al. 2013). A case–control study has demonstrated a nearly four times increased risk of BD in adult offspring after maternal influenza infection at any time during pregnancy (Parboosing et al. 2013). Another study of T2-weighted magnetic resonance imaging (MRI) findings described that in BD patients three times more periventricular white matter hyperintensities were seen as compared to controls (Altshuler et al. 1995). Increased number of BD patients showing deep subcortical white matter lesions were born during the winter (Moore et al. 2001) when influenza incidence is high (Kilbourne 1987). These results suggest that structural and functional abnormalities are induced in the CNS of the offspring by maternal infections or inflammation, which might be responsible for neuroprogression of BD in later life.
Although animal models of MIA have not explicitly been explored for their validity as a BD model, some of the experimentally induced phenotypes may be considered for this disorder. Deficits in sensorimotor gating, as present in various rodent MIA models (Estes and McAllister 2016; Meyer 2014; Meyer et al. 2009), are also seen in acute mania (Perry et al. 2001) and euthymic BD patients (Giakoumaki et al. 2007). Besides, several animal studies have reported depression-like behaviors in offspring exposed to MIA (Khan et al. 2014; Ronovsky et al. 2016). The latter phenotypes may not only be seen in unipolar depression, but also depressive episodes in BD. Evaluation of other core behavioral symptoms of BD, such as poor decision-making, altered risk-taking behavior, impulsivity, and loss of inhibitory control, remain unexplored in MIA models.
2.2 Role of Infectious Disease as a Trigger to Develop BD Neuroprogression
Infections with several pathogenic agents have been studied as risk factors for BD (Stich et al. 2015; Yolken and Torrey 2008). Herpes virus can lead to latent and lytic infection in the brain, and it has been associated with memory impairment (Kapur et al. 1994), mania (Koehler and Guth 1979), and psychosis (Schlitt et al. 1985). A study reported that serologic evidence of herpes simplex virus type 1 (HSV-1) infection was an independent predictor of low cognitive functioning in BD patients (Dickerson et al. 2004). Similar results were reported in another study showing a negative association between HSV-1 infection and cognitive performance in both BD patients and controls (Yolken 2011), suggesting that HSV-1 infection is associated with worse functioning in BD patients. Rising evidence also suggests a possible association between cytomegalovirus (CMV) and BD. A study described that CMV IgG levels are associated with a reduction in hippocampal volume and worse episodic verbal memory in BD patients (Houenou et al. 2014). The association between CMV latent infection and lower cognitive functioning is thought to be mediated by a chronic inflammatory response and subsequent reduced hippocampal volume (Almanzar et al. 2005). On the other hand, the hippocampal volume is usually normal in BD and reported to be reduced only in the most severe forms of BD (Strasser et al. 2005). Hence, this evidence suggests that exposure to CMV infection may contribute to the pathological rewiring of neurons and neuroprogression of BD.
A case–control study showed that BD patients had an increased seroprevalence for Toxoplasma gondii compared to controls (Tedla et al. 2011). Indeed, T. gondii is a neurotropic protozoan with high seropositivity rates globally. Two separate studies have reported that BD patients with manic episodes had elevated T. gondii IgM antibody levels as compared to healthy controls. Also, these studies reported a significant negative correlation between T. gondii IgM antibody levels and cognitive scores in both controls and BD patients (Dickerson et al. 2014a, b). The negative association between the IgM antibody load and cognitive functioning points towards the worsening of BD with exposure to T. gondii infection. Evidence also shows that the cognitive deterioration index (DI) in BD patients correlated to high IL-6 mRNA expression only among T. gondii seropositive group (Hamdani et al. 2015), asserting that inflammatory pathways may be involved in the cognitive dysfunction caused by the infection.
Various infections activate a common inflammatory pathway within the cell. Infectious agents are recognized by pattern-recognition receptors (PRRs), which are critical components of the innate immune system (Mook-Kanamori et al. 2011; Sellner et al. 2010). Activation of PRRs causes the release of mediators, such as pro-inflammatory cytokines and chemokines, that propagate and regulate the immune response necessary to remove invaded microorganisms (Iwasaki and Medzhitov 2010). Some of the pro-inflammatory cytokines that are produced during infections, like TNF-α, IL-1β, and IL-6, are increased in BD patients compared to healthy controls (Dong and Zhen 2015; Munkholm et al. 2013; Soderlund et al. 2011), providing a potential pathophysiological mechanism linking infections to neuroprogression of BD.
2.3 Autoimmune Disorders and Their Association with BD Neuroprogression
Growing evidence has described an association between BD and various autoimmune disorders. Multiple epidemiologic studies have pointed towards an association between autoimmune diseases, autoantibodies, and BD (Rosenblat and McIntyre 2015). The most common associations established over the years have been those with autoimmune thyroiditis (strongest association), multiple sclerosis, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), autoimmune hepatitis, inflammatory bowel disease, and Guillain barre syndrome (Hillegers et al. 2007; Hsu et al. 2014; Marrie et al. 2018a, b; Tiosano et al. 2017).
Several mechanisms have been suggested through which the autoimmune diseases have been hypothesized to affect the neuroprogression of BD. Benros et al. proposed a correlation between autoantibodies and psychiatric symptoms (Benros et al. 2013). The most well-accepted hypothesis has been synthesized from the observation of psychiatric symptoms in the autoimmune encephalitis. In autoimmune encephalitis, IgG autoantibodies are formed against the NMDA (N-methyl-D-aspartate receptor) receptors (NR1 subunit) (Vitaliani et al. 2005). As the NMDA receptor is a primary receptor for the excitatory neurotransmitter glutamate, the activation of the pathway will lead to an increase in excitatory neurotransmission. When the NMDA receptor is activated, it will lead to a cascade of activation of various components such as protein kinases. One of the protein kinases (PKC) that is significant in the presentation of a manic episode is also activated via this pathway (Traynelis et al. 2010). In a similar study, it was observed that the levels of autoantibodies against the NR2 subunit of NMDA were high during a manic episode in BD and schizoaffective disorder (Dickerson et al. 2012). Astrocytes are responsible for the clearance of glutamate, by converting it into glutamine. However, due to the interaction of astrocytes with pro-inflammatory cytokines, the glutamate clearance is hampered (Zou et al. 2010). This increased glutamate receptor activation causes an increase in the levels of calcium in the mitochondria. These changes cause neuroplastic alterations and excitotoxicity (Berk et al. 2000; Kato 2007).
Another mechanism that can affect the neuroprogression of BD involves the activation of the tryptophan–kynurenine pathway by the systemic pro-inflammatory cytokines. NMDAR and tryptophan–kynurenine pathways are responsible for the regulation of serotonin (directly) and dopamine levels (indirectly) (Dantzer et al. 2008). Some of the pro-inflammatory cytokines that are observed to be increased in BD patients are C-reactive protein (CRP), interleukin (IL)-1 beta, soluble IL-2 receptor, IL-4, IL-6, tumor necrosis factor-alpha (TNF-α), and soluble receptor of TNF-α type 1 (sTNFR1) (Barbosa et al. 2014a; Brietzke et al. 2009a, b; Modabbernia et al. 2013). Serum levels of these cytokines have been seen to be mood dependent. In this regard, high serum concentrations of IL-4, IL-6, IL-RA, TNF-α, sTNFR1, CXCL10, and CXCL11 are seen in the manic phases. Similarly, elevated serum concentrations of IL-6, IL-1β, CRP, TNF-α, sTNFR1, and CXCL10 are also seen in the depressed period (Barbosa et al. 2014a, b; Rosenblat et al. 2014). IL-6 and TNF-α levels correlate directly with the severity of the disease (Kauer-Sant'Anna et al. 2009). Increased levels of these cytokines lead to neuroplastic changes in the brain. According to one hypothesis, high levels of TNF-α could lead to reduced expression of muscarinic acetylcholine receptor (M2 receptors) in the cortex (Gibbons et al. 2009). The M2 receptors are observed to be reduced in the cortex of patients with major depressive disorder and BD.
Autoimmune diseases have a baseline inflammatory condition. Inflammation causes an increase in the permeability of the blood–brain barrier (BBB). In this instance, the pro-inflammatory cytokines and other components of the inflammatory pathways, such as autoantibodies, can enter the CSF directly and cause various psychiatric symptoms (Modabbernia et al. 2013). The baseline chronic inflammatory state in autoimmune disorders can lead to excessive microglial activation (Frick et al. 2013). The microglial activation can cause detrimental changes in some of the neuronal circuits associated with mood and cognitive functions. Another effect of the microglial overactivation would be a surge in the levels of reactive oxygen species (ROS), which can lead to oxidative stress and further damage to the neuronal circuits, and hence, the neuroprogression of the BD (Frick et al. 2013; Stertz et al. 2013).
2.4 Role of the Gut–Brain Axis on BD Neuroprogression
In the last decade, there has been mounting evidence suggesting that gut microbiota makes a substantial contribution to mental health and, subsequently, to the neuroprogression of various neuropsychiatric disorders such as BD, depression, and anxiety (Cryan and Dinan 2012; Forsythe et al. 2010; Painold et al. 2019). The gut–brain axis refers to a bidirectional communication between the intestine (enteric nervous system, gut microbiota, and metabolites of gut microbiome) and the brain (Carabotti et al. 2015). Multiple studies have shown that the diversity of the gut bacteria is inversely linked to the illness duration of BD (Carabotti et al. 2015; Painold et al. 2019). However, the diversity of the gut bacteria is not solely responsible for the severity of the BD. It is one of the contributing factors to the overall disease state. Patients suffering from BD have decreased levels of Faecalibacterium sp. and of an unknown bacterium of the Ruminococcaceae family (Bengesser et al. 2019; Carabotti et al. 2015; Evans et al. 2017; Huang et al. 2019). Notably, Ruminococcus species are related to the synthesis of butyrate, which presents anti-inflammatory and mood regulatory activities (Hwang et al. 2017). The Enterobacteriaceae family was found in higher fractions in BD patients suffering from depressive symptoms (Carabotti et al. 2015; Evans et al. 2017; Huang et al. 2019). A high proportion of genus Lactobacillus and genus Streptococcus in the gut was associated with higher levels of pro-inflammatory cytokines such as IL-6 (Painold et al. 2019) suggesting activation of inflammatory pathways by these bacteria. A low-grade inflammatory state is seen in a subgroup of BD patients (Bechter 2013; Fillman et al. 2014; Miller et al. 2011). Over the years, various studies have suggested that the cause of this inflammation might be related to the dysbiosis of the gut microbiome. The concept of microbial translocation (gut bacteria leaking into the circulation due to changes in the permeability of the intestinal lumen) plays a central role in this hypothesis. Microbial translocation has been measured by markers such as soluble CD14 (sCD14) and fungal antibodies. The inflammatory reaction mounted in response to the gut bacteria releases mediators in circulation that, in turn, have effects on the behavioral and cognitive patterns (Dickerson et al. 2017). It can be postulated that the chronic inflammatory state produced by long-term changes in the gut microbiota diversity might cause activation of microglia. This may cause a detrimental effect on the neuronal networks directly and indirectly (via the formation of ROS damaging DNA, and proteins). In animal models, it has been shown that the gut flora is responsible for regulating serotonin levels in plasma (Collins and Bercik 2009). The inflammatory state produced by the gut bacteria upregulates the indoleamine 2,3 deoxygenase (IDO) enzyme. The IDO enzyme upregulation causes increased degradation of serotonin. This pathway plays a vital role in the acute manic episodes in BD (Myint et al. 2007).
According to the monoamine hypothesis, the decreased levels of monoamines play a role in the pathophysiology of depressive symptoms. Building on this hypothesis, Bengessar et al. showed that the change in gut microbiota diversity affects the CpG methylation status of the clock gene of aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL), which plays a vital role in the neuroprogression of BD (Bengesser et al. 2018, 2019). The ARNTL gene codes for one of the transcription factors of the monoamine oxidase A (MAO-A) enzyme (responsible for the degradation of monoamine neurotransmitters). The gut bacteria diversity is negatively correlated to the methylation status of the ARNTL gene. Hence, a decrease in gut bacteria diversity would lead to more methylation of the ARNTL gene and decreased expression of the MAO-A transcription factor. This cascade will lead to a reduced breakdown of the monoamine neurotransmitters. The increased levels of monoamine neurotransmitters would have a pro-manic effect, according to this model. However, this hypothesis has loopholes as well since the mechanism of action of mood stabilizers does not support this hypothesis as the MAO-A levels in the brain do not correlate with the mechanisms of these drugs.
3 Inflammatory and Oxidative Mechanisms in BD Neuroprogression
3.1 Mechanisms of Inflammation and Their Contribution to BD Neuroprogression
Many experimental facts solidify the link between BD and inflammation such as an increase in inflammatory biomarker levels seen in both manic and depressive phases of BD, pro-inflammatory cytokine infusion being the best experimental model of depression, epidemiologic evidence of increased rates of inflammatory medical comorbidities in BD associated with an upsurge in the levels of inflammatory mediators, and most importantly anti-inflammatory agents being considered as the novel therapeutic agents in BD trials (Ayorech et al. 2015; Barbosa et al. 2014b; Goldstein et al. 2009; Sayana et al. 2017; Wadee et al. 2002).
Neuroprogression in BD is evident in the form of brain structural changes, neuronal and glial cell abnormalities, biochemical alterations that comprise of inflammatory cytokines, neurotrophins including brain-derived neurotrophic factor (BDNF), oxidative stress, autoimmunity, mitochondrial and endoplasmic reticulum stress, dopaminergic and glutamatergic system alterations, kynurenine pathway imbalance, and involvement of epigenetic changes such as histone and DNA methylation leading to gene expression variations (Berk et al. 2008, 2011; Grayson et al. 2010; Post 2007; Wadee et al. 2002). BD has also been linked to changes in neuroplasticity and neuronal survival that are determined by neurotransmitters, hormones, neurotrophins, and inflammatory biomarkers such as cytokines and chemokines; and acute phase proteins such as immunoglobulins, complement proteins, factor B and high-sensitivity CRP (Brietzke et al. 2009b; Cunha et al. 2008).
The cytokines influence the BD course via their direct actions on the immune system as well as their effect on neurotransmitter and neuropeptide systems. Cytokine production is controlled by phosphoinositides, the arachidonic acid (AA) cascade, adenyl cyclase, tyrosine phosphorylation, and the protein kinase C systems. Similar to neurotransmitters and hormones, cytokines act through the hypothalamus-pituitary-adrenal (HPA) axis to maintain stress response, alter serotonin–catecholamine associated pathways in the brain, and cause mood changes (Ortiz-Dominguez et al. 2007). Studies have demonstrated increased levels of circulating pro-inflammatory cytokines in various phases of BD that lead to activation of neutrophils, the proliferation of B cells, synthesis of acute-phase proteins, and an increase in vascular permeability (Bai et al. 2014).
Pro-inflammatory cytokines, such as IL-1, IL-6, and TNF-α, are involved in neural processes like the induction of long-term potentiation, neurogenesis, survival regulation of neurons, and the development of astrocytes, impacting on several cognitive functions in normal states. However, cytokines may contribute to the neurodegenerative process in neurotoxic and stress-induced states of BD (Eyre and Baune 2012; Khairova et al. 2009). For example, enhanced TNF-α levels seem to be involved in neuronal death via the activation of caspases and apoptotic machinery in BD cases (Cacci et al. 2005).
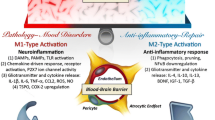
Cyclo-oxygenase 2 inhibitor (celecoxib), an anti-inflammatory drug, showed better improvement of depressive symptoms compared to the standard treatment. Other immune-based treatments in vogue for BD are anti-inflammatory drugs (aspirin, statins), immune-based drugs (minocycline), and an anti-TNF-α monoclonal antibody, infliximab (Austin and Tan 2012; Berk et al. 2013; Elisa and Beny 2010; Nery et al. 2008; Savitz et al. 2012). Thus, the immune basis in BD seems to be a promising novel therapeutic target for BD patients, and the direct and indirect effects of inflammation help us to understand the behavioral modification by the immunological system. The Fig. 1 illustrates the details of inflammation mediated bipolar neuroprogression.

Inflammation as a mechanism of bipolar disorder neuroprogression. Infection, maternal immune activation, dysbiosis, and autoimmune diseases can activate the host immune response increasing the inflammatory mediators in the bloodstream, subsequently triggering cell damage and mitochondrial dysfunction. The inflammatory mediators and damage-associated molecular patterns (DAMPs) released from the mitochondrial dysfunction can increase the blood-brain barrier (BBB) permeability inducing microglial activation leading to bipolar disorder (BD) neuroprogression
3.2 Oxidative Stress and Mitochondrial Dysfunction Associated with BD Neuroprogression
There is ample proof in the literature linking BD and impairment in oxidative metabolism. Increase in the generation of brain energy, basal metabolic rate, resting energy expenditure, maximum oxygen uptake, independent of total consumed calories is seen in the BD-manic phase, whereas in the depressed phase, there is a decrease in energy generation (Baxter Jr. et al. 1985; Caliyurt and Altiay 2009). Brain metabolic rates and energy production showed a gradual ascent from depression to the mixed state to euthymia to manic phase (Baxter Jr. et al. 1985). Oxidative stress biomarkers are encountered in different types (Type I and Type II), stages (early and late), and periods (manic, depressive, and euthymic) of BD (Panizzutti et al. 2015).
Mitochondria are double membrane-bound cytoplasmic organelles that aid in the production of ATP, amino acid, lipid, and steroid metabolism. It also involves activating apoptosis and in the uptake of calcium ions, and it is a significant source of intracellular free radicals. Mitochondrial dysfunction in BD is evidenced by impaired energy metabolism in the brain detected by magnetic resonance spectroscopy (Kato 2007).
BD patients also show discrepancies in mitochondrial electron transport chain (ETC) complex I, where electron escape leads to the formation of superoxide anion from molecular oxygen, which is the precursor for most ROS (Guo et al. 2018). Studies on BD patients showed a reduction in ETC complex 1 activity in prefrontal postmortem brain tissue and administration of lithium increased the activity of mitochondrial complexes I/II and II/III in the brain, altered expression of ETC complex 1 subunits, linking BD to chromosome 19p13 that has multiple ETC complex 1 subunit genes (Cheng et al. 2006; Konradi et al. 2004; Maurer et al. 2009; Sun et al. 2006). In the literature, there are alterations in antioxidant enzymes in BD, such as increased activity of SOD during manic and depressive phases, reduced activity of catalase during the euthymic period, and increased activity of both enzymes in unmedicated manic patients (Andreazza et al. 2007; Machado-Vieira et al. 2007). N-acetyl cysteine (NAC), which is a free radical scavenger and glutathione precursor, when used in BD reduced the depressive symptoms and presented overall functional improvement (Berk et al. 2011). Experimental evidence has proven that oxidative parameters have shown a stage-dependent pattern, where glutathione reductase and glutathione s-transferase (GST) are increased during late-stage, suggesting a failure of compensatory mechanisms during BD progression (Andreazza et al. 2009). Overall oxidative stress, mitochondrial dysfunction, and antioxidant enzyme alterations can cause neuronal cell death via apoptosis or aggregated antioxidants that may result in impairment of mood-stabilizing mechanisms.
3.3 Peripheral Inflammatory Mediators as a Trigger or Accelerator of BD Neuroprogression
In chronic BD, mood relapses lead to neuroprogression with higher frequencies and rapid cycling, resulting in worst outcomes (Berk et al. 2011). One of the crucial mechanisms accountable for neuroprogression in BD is the aberrant exacerbation of the inflammatory mediators during mood episodes (mania and depression) in BD (Sayana et al. 2017).
Inflammation appears to be phase dependent on BD. On marker analysis, manic patients experienced an elevation in peripheral TNF-α and IL-4 along with a reduction in IL-1β and IL-2 levels, while depressed patients showed elevated levels of IL-6 and TNF-α and decreased IL-2 levels (Ortiz-Dominguez et al. 2007). Another study showed an increase in IL-2, IL-4, and IL-6 in mania and IL-6 in depression (Brietzke et al. 2009b). Kim and his team showed an increase in IL-4, IFN-γ, TNF-α, IL-6, IFN-γ/transforming growth factor (TGF)β1, IL-4/TGF-β1, IL-6/IL-4, TNF-α/IL-4, IL-2/IL-4, and IFN-γ/IL-4 ratios and low levels of TGF-β1 in manic episodes (Kim et al. 2004, 2007). Data from the available literature suggest that hsCRP levels were significantly elevated in mania compared to euthymic, depressed phase or controls, and they positively correlated with Bech Rafaelson Manic Rating Scale (BRMRS) and Young Mania Rating Scale (YMRS). The levels of acute-phase reactants, notably CRP, demonstrated an elevation in depressive phase compared to controls and positively correlated with Hamilton Rating Scale for Depression (HAM-D) scores in one study, whereas in another study they did not show any association with bipolar depression or HAM-D scores (Cunha et al. 2008; De Berardis et al. 2008; Dickerson et al. 2007).
When the early and late stages of BD were compared, IL-6 and TNF-α were elevated in both groups, while IL-10 levels were higher in the early stages. However, TNF-α was more elevated in late stages than in early (Kauer-Sant'Anna et al. 2009). In addition to these cytokines, eotaxin/CCL11, a chemokine, was also shown to increase in late-stage euthymic BD compared to controls, suggesting a link between pathological aging, eosinophil function marker CCL11, and BD neuroprogression (Panizzutti et al. 2015). Also, damage-associated molecular patterns (DAMPs) such as circulating nuclear DNA, HSP70, and HSP90α that bind to Toll-like receptors (TLRs) cause systemic toxicity via immune activation in BD patients, as TLRs activate the signaling pathways of immune system triggering inflammatory pathways. The DAMPs activation of TLR signaling cascades can explain how initial insults such as drugs, stress, and relapses can cause systemic inflammation (Kapczinski et al. 2017).
The inflammatory pathways play a crucial role in progressive cognitive impairment in BD. Peripherally measured markers such as TNF-α, hsCRP, sCD40L, IL-1Ra, and sTNFR1 seem to influence cognitive performance in BD (Barbosa et al. 2012; Chung et al. 2013; Hope et al. 2015; Hoseth et al. 2016). The circulating levels of TNF-α correlated with inhibitory control part of executive dysfunction in BD patients, impairment of which is regarded as a cognitive endophenotype of BD (Barbosa et al. 2012). Increased CRP levels are associated with cognitive decline, as evidenced by the low Repeatable Battery for the Assessment of the Neuropsychological Status (RBANS) (Dickerson et al. 2013). Also raised serum hsCRP levels are negatively correlated with the volume of the orbitofrontal cortex, which is, in turn, associated with the poor cognitive performance (Chung et al. 2013). IL-1 receptor antagonist (IL-1Ra) and sTNFR1 levels were associated with the worst performance on the Global Assessment of Functioning (GAF) scale (Hope et al. 2015; Hoseth et al. 2016). The pro-inflammatory profile characterized by activation of cell-mediated immunity, systemic inflammation, phase, and stage related changes in BD with deleterious clinical, cognitive, and neurological consequences, seem to act as a significant player in disease neuroprogression.
3.4 Cerebrospinal Fluid (CSF) System Inflammatory Markers in BD Neuroprogression
BD patients present with higher CSF concentrations of markers of neuroinflammation, glial activation, and neuronal injury compared to controls (Isgren et al. 2017). In this regard, CSF studies demonstrated an increased CSF/serum albumin ratio indicating increased BBB permeability, elevated CSF cell count, IgG index, oligoclonal bands, IL-1β, IL-6, and IL-8 suggesting inflammation and intrathecal immunoglobulin production (Orlovska-Waast et al. 2019).
Studies revealed elevation of inflammatory CSF markers such as IL-8, monocyte activation marker, monocyte chemoattractant protein 1 (MCP-1; also known as CCL-2), glial activation marker, chitinase 3 like protein 1 (CHI3L1; also known as YKL-40) and axonal damage marker, neurofilament light chain (NFL) in BD patients compared to controls. The IL-8 showed a positive association with lithium and antipsychotics, whereas NFL to atypical antipsychotic drugs (Isgren et al. 2015; Jakobsson et al. 2014, 2015). On the evaluation of cognitive decline in BD patients, CSF biomarkers, especially microglial marker, YKL-40 showed a significant impairment in executive functions for euthymic BD patients compared to controls, which is independent of patient age, medication, disease status, and type of BD (Rolstad et al. 2015a). Moreover, the marker, NFL concentrations showed a negative association with verbal function and working memory (Rolstad et al. 2015b). Previously assessed CSF proteins may be involved in adaptive immune processes or may reflect immune aberrations or a state of vulnerability for BD rather than being of predictive value for disease progression.
3.5 Postmortem Inflammatory Markers in BD Neuroprogression
The role of neuroinflammation in BD and alterations in microglial, astrocyte, and oligodendrocyte markers are evident in postmortem BD studies (Giridharan et al. 2019). The innate immune cells that contribute to the neuroinflammation are microglia, astrocytes, macrophages, natural killer (NK) cells, mast cells, as well as oligodendrocytes and neurons (Stephenson et al. 2018). Postmortem BD studies also revealed increased neuroinflammation with decreased anti-inflammatory marker levels in the frontal cortex (Bezchlibnyk et al. 2001; Rao et al. 2010). Accurately, increased protein and mRNA levels of IL-1β, IL-1R, and myeloid differentiation primary response 88 (MyD88) were described, as well as upregulation of nuclear factor kappa B (NF-kB) transcription factor and its subunits (p50 and p65), and astroglial and microglial markers (GFAP, inducible nitric oxide synthase (iNos), c-fos and CD11b) in the pre frontal cortex of BD (Rao et al. 2010).
The neurodegenerative process mediated by TNF-α may result in the volumetric reduction and hypoactivation of frontal lobes in BD patients, along with the disinhibition of limbic structures (Brooks 3rd et al. 2009; Kupferschmidt and Zakzanis 2011). A study examining TNF parameters in BA 24 and BA 46 demonstrated that BD patients presented increased transmembrane TNF-α (tmTNF-α) protein level in the anterior cingulate cortex (ACC; BA 24), and decreased TNFR2 protein levels in the dorso lateral PFC (BA 46). Peripheral tissue inflammation notably increased TNF-α levels, leads to reduced expression of muscarinic M2 receptors in the cortex of MDD and BD, and ultimately results in cognitive deficits in BD (Gibbons et al. 2009; Haddad et al. 1996; Jones et al. 2004).
On the evaluation of kynurenine pathway metabolites, an increase in quinolinic acid (QUIN) expression and QUIN-immunopositive microglia have been observed in the subgenual and supracallosal anterior cingulate cortex (ACC) in depressed patients (Steiner et al. 2011). Overall, the changes in QUIN levels signify the importance of NMDA-R signaling, glutamate transmission, and mononuclear phagocyte system in BD depression. The ratio of kynurenic acid (KA) to kynurenine was lower in the BD group than in the control group, and KA levels were unchanged. There was also an elevation in the density and intensity of both TDO (Tryptophan-2,3-dioxygenase) 2-positive white matter glia and TDO2-positive gray matter glia in the BD group (Miller et al. 2006). Overall, postmortem studies showed that BD patients presented increased markers of neuroinflammation and decreased anti-inflammatory markers that lead to neuroprogression in the form of neuroanatomical changes, neurotransmitter imbalance, cognitive decline, and progressive deterioration of mental health.
4 Conclusions and Future Directions
In summary, we are now beginning to understand the underlying processes of neuroprogression in BD that include the involvement of inflammatory cytokines, neurotrophins, and epigenetic effects. Infection and other inflammatory processes have clearly shown to be associated not only with increased risk of developing BD but also with worsening cognitive impairment and structural changes indicative of neuroprogression in BD. Various cytokines and chemokines activate multitudes of immune pathways leading to mitochondrial dysfunction, oxidative stress generation, and activation of microglial cells in the CNS, leading to worsening of this illness. Cross-sectional studies suggest that inflammatory markers can be used as both a biomarker of illness activity and stage of the disease. Longitudinal studies are needed to clarify the exact role of inflammation and neuroinflammation in BD. Thus prevention of infections with neurotropic pathogens in pregnant women as well as later in life can be one of the strategies implemented to prevent the development and progression of BD in genetically predisposed individuals. Also, specific steps in the molecular pathways of neuroprogression in BD patients may provide new targets for further research to develop new therapeutic drugs for this chronic mental disorder.
Change history
02 February 2021
The original version of this chapter is updated with the below mentioned updates as per the author’s request: Tatiana Barichello, Vijayasree V. Giridharan, Gursimrat Bhatti, Pavani Sayana, Tejaswini Doifode, Danielle Macedo, and Joao Quevedo
References
Abe C, Ekman CJ, Sellgren C, Petrovic P, Ingvar M, Landen M (2015) Manic episodes are related to changes in frontal cortex: a longitudinal neuroimaging study of bipolar disorder 1. Brain 138(Pt 11):3440–3448
Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Wurzner R et al (2005) Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J Virol 79(6):3675–3683
Altshuler LL, Curran JG, Hauser P, Mintz J, Denicoff K, Post R (1995) T2 hyperintensities in bipolar disorder: magnetic resonance imaging comparison and literature meta-analysis. Am J Psychiatry 152(8):1139–1144
Andreazza AC, Cassini C, Rosa AR, Leite MC, de Almeida LM, Nardin P et al (2007) Serum S100B and antioxidant enzymes in bipolar patients. J Psychiatr Res 41(6):523–529
Andreazza AC, Kapczinski F, Kauer-Sant'Anna M, Walz JC, Bond DJ, Goncalves CA et al (2009) 3-Nitrotyrosine and glutathione antioxidant system in patients in the early and late stages of bipolar disorder. J Psychiatry Neurosci 34(4):263–271
Association AP (2013) Diagnostic and statistical manual of mental disorders, 5th edn. American Psychiatric Publishing, Arlington, pp 123–154
Austin M, Tan YC (2012) Mania associated with infliximab. Aust N Z J Psychiatry 46(7):684–685
Ayorech Z, Tracy DK, Baumeister D, Giaroli G (2015) Taking the fuel out of the fire: evidence for the use of anti-inflammatory agents in the treatment of bipolar disorders. J Affect Disord 174:467–478
Bai YM, Su TP, Tsai SJ, Wen-Fei C, Li CT, Pei-Chi T et al (2014) Comparison of inflammatory cytokine levels among type I/type II and manic/hypomanic/euthymic/depressive states of bipolar disorder. J Affect Disord 166:187–192
Barbosa IG, Rocha NP, Huguet RB, Ferreira RA, Salgado JV, Carvalho LA et al (2012) Executive dysfunction in euthymic bipolar disorder patients and its association with plasma biomarkers. J Affect Disord 137(1–3):151–155
Barbosa IG, Bauer ME, Machado-Vieira R, Teixeira AL (2014a) Cytokines in bipolar disorder: paving the way for neuroprogression. Neural Plast 2014:360481
Barbosa IG, Machado-Vieira R, Soares JC, Teixeira AL (2014b) The immunology of bipolar disorder. Neuroimmunomodulation 21(2–3):117–122
Baxter LR Jr, Phelps ME, Mazziotta JC, Schwartz JM, Gerner RH, Selin CE et al (1985) Cerebral metabolic rates for glucose in mood disorders. Studies with positron emission tomography and fluorodeoxyglucose F 18. Arch Gen Psychiatry 42(5):441–447
Bechter K (2013) Updating the mild encephalitis hypothesis of schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry 42:71–91
Bengesser SA, Reininghaus EZ, Lackner N, Birner A, Fellendorf FT, Platzer M et al (2018) Is the molecular clock ticking differently in bipolar disorder? Methylation analysis of the clock gene ARNTL. World J Biol Psychiatry 19(Supp 2):S21–Ss9
Bengesser SA, Morkl S, Painold A, Dalkner N, Birner A, Fellendorf FT et al (2019) Epigenetics of the molecular clock and bacterial diversity in bipolar disorder. Psychoneuroendocrinology 101:160–166
Benros ME, Waltoft BL, Nordentoft M, Ostergaard SD, Eaton WW, Krogh J et al (2013) Autoimmune diseases and severe infections as risk factors for mood disorders: a nationwide study. JAMA Psychiat 70(8):812–820
Berk M, Plein H, Belsham B (2000) The specificity of platelet glutamate receptor supersensitivity in psychotic disorders. Life Sci 66(25):2427–2432
Berk M, Hallam KT, McGorry PD (2007) The potential utility of a staging model as a course specifier: a bipolar disorder perspective. J Affect Disord 100(1–3):279–281
Berk M, Ng F, Dean O, Dodd S, Bush AI (2008) Glutathione: a novel treatment target in psychiatry. Trends Pharmacol Sci 29(7):346–351
Berk M, Kapczinski F, Andreazza AC, Dean OM, Giorlando F, Maes M et al (2011) Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev 35(3):804–817
Berk M, Dean O, Drexhage H, McNeil JJ, Moylan S, O’Neil A et al (2013) Aspirin: a review of its neurobiological properties and therapeutic potential for mental illness. BMC Med 11:74
Bezchlibnyk YB, Wang JF, McQueen GM, Young LT (2001) Gene expression differences in bipolar disorder revealed by cDNA array analysis of post-mortem frontal cortex. J Neurochem 79(4):826–834
Brietzke E, Kauer-Sant’Anna M, Teixeira AL, Kapczinski F (2009a) Abnormalities in serum chemokine levels in euthymic patients with bipolar disorder. Brain Behav Immun 23(8):1079–1082
Brietzke E, Stertz L, Fernandes BS, Kauer-Sant'anna M, Mascarenhas M, Escosteguy Vargas A et al (2009b) Comparison of cytokine levels in depressed, manic and euthymic patients with bipolar disorder. J Affect Disord 116(3):214–217
Brooks JO 3rd, Wang PW, Bonner JC, Rosen AC, Hoblyn JC, Hill SJ et al (2009) Decreased prefrontal, anterior cingulate, insula, and ventral striatal metabolism in medication-free depressed outpatients with bipolar disorder. J Psychiatr Res 43(3):181–188
Brown AS (2015) The Kraepelinian dichotomy from the perspective of prenatal infectious and immunologic insults. Schizophr Bull 41(4):786–791
Brown AS, Derkits EJ (2010) Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry 167(3):261–280
Cacci E, Claasen JH, Kokaia Z (2005) Microglia-derived tumor necrosis factor-alpha exaggerates death of newborn hippocampal progenitor cells in vitro. J Neurosci Res 80(6):789–797
Caliyurt O, Altiay G (2009) Resting energy expenditure in manic episode. Bipolar Disord 11(1):102–106
Canetta SE, Bao Y, Co MD, Ennis FA, Cruz J, Terajima M et al (2014) Serological documentation of maternal influenza exposure and bipolar disorder in adult offspring. Am J Psychiatry 171(5):557–563
Cao B, Passos IC, Mwangi B, Bauer IE, Zunta-Soares GB, Kapczinski F et al (2016) Hippocampal volume and verbal memory performance in late-stage bipolar disorder. J Psychiatr Res 73:102–107
Carabotti M, Scirocco A, Maselli MA, Severi C (2015) The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol 28(2):203–209
Cheng R, Juo SH, Loth JE, Nee J, Iossifov I, Blumenthal R et al (2006) Genome-wide linkage scan in a large bipolar disorder sample from the National Institute of Mental Health genetics initiative suggests putative loci for bipolar disorder, psychosis, suicide, and panic disorder. Mol Psychiatry 11(3):252–260
Chung KH, Huang SH, Wu JY, Chen PH, Hsu JL, Tsai SY (2013) The link between high-sensitivity C-reactive protein and orbitofrontal cortex in euthymic bipolar disorder. Neuropsychobiology 68(3):168–173
Collins SM, Bercik P (2009) The relationship between intestinal microbiota and the central nervous system in normal gastrointestinal function and disease. Gastroenterology 136(6):2003–2014
Cryan JF, Dinan TG (2012) Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 13(10):701–712
Cunha AB, Andreazza AC, Gomes FA, Frey BN, da Silveira LE, Goncalves CA et al (2008) Investigation of serum high-sensitive C-reactive protein levels across all mood states in bipolar disorder. Eur Arch Psychiatry Clin Neurosci 258(5):300–304
Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9(1):46–56
Daskalakis NP, Bagot RC, Parker KJ, Vinkers CH, de Kloet ER (2013) The three-hit concept of vulnerability and resilience: toward understanding adaptation to early-life adversity outcome. Psychoneuroendocrinology 38(9):1858–1873
De Berardis D, Conti CM, Campanella D, Carano A, Scali M, Valchera A et al (2008) Evaluation of C-reactive protein and total serum cholesterol in adult patients with bipolar disorder. Int J Immunopathol Pharmacol 21(2):319–324
Debnath M, Venkatasubramanian G, Berk M (2015) Fetal programming of schizophrenia: select mechanisms. Neurosci Biobehav Rev 49:90–104
Dickerson FB, Boronow JJ, Stallings C, Origoni AE, Cole S, Krivogorsky B et al (2004) Infection with herpes simplex virus type 1 is associated with cognitive deficits in bipolar disorder. Biol Psychiatry 55(6):588–593
Dickerson F, Stallings C, Origoni A, Boronow J, Yolken R (2007) Elevated serum levels of C-reactive protein are associated with mania symptoms in outpatients with bipolar disorder. Prog Neuro-Psychopharmacol Biol Psychiatry 31(4):952–955
Dickerson F, Stallings C, Vaughan C, Origoni A, Khushalani S, Yolken R (2012) Antibodies to the glutamate receptor in mania. Bipolar Disord 14(5):547–553
Dickerson F, Stallings C, Origoni A, Vaughan C, Khushalani S, Yolken R (2013) Elevated C-reactive protein and cognitive deficits in individuals with bipolar disorder. J Affect Disord 150(2):456–459
Dickerson F, Stallings C, Origoni A, Katsafanas E, Schweinfurth L, Savage C et al (2014a) Antibodies to Toxoplasma gondii and cognitive functioning in schizophrenia, bipolar disorder, and nonpsychiatric controls. J Nerv Ment Dis 202(8):589–593
Dickerson F, Stallings C, Origoni A, Vaughan C, Katsafanas E, Khushalani S et al (2014b) Antibodies to Toxoplasma gondii in individuals with mania. Bipolar Disord 16(2):129–136
Dickerson F, Severance E, Yolken R (2017) The microbiome, immunity, and schizophrenia and bipolar disorder. Brain Behav Immun 62:46–52
Dong XH, Zhen XC (2015) Glial pathology in bipolar disorder: potential therapeutic implications. CNS Neurosci Ther 21(5):393–397
Elisa B, Beny L (2010) Induction of manic switch by the tumour necrosis factor-alpha antagonist infliximab. Psychiatry Clin Neurosci 64(4):442–443
Estes ML, McAllister AK (2016) Maternal immune activation: implications for neuropsychiatric disorders. Science 353(6301):772–777
Evans SJ, Bassis CM, Hein R, Assari S, Flowers SA, Kelly MB et al (2017) The gut microbiome composition associates with bipolar disorder and illness severity. J Psychiatr Res 87:23–29
Eyre H, Baune BT (2012) Neuroplastic changes in depression: a role for the immune system. Psychoneuroendocrinology 37(9):1397–1416
Fillman SG, Sinclair D, Fung SJ, Webster MJ, Shannon WC (2014) Markers of inflammation and stress distinguish subsets of individuals with schizophrenia and bipolar disorder. Transl Psychiatry 4:e365
Forsythe P, Sudo N, Dinan T, Taylor VH, Bienenstock J (2010) Mood and gut feelings. Brain Behav Immun 24(1):9–16
Frick LR, Williams K, Pittenger C (2013) Microglial dysregulation in psychiatric disease. Clin Dev Immunol 2013:608654
Giakoumaki SG, Roussos P, Rogdaki M, Karli C, Bitsios P, Frangou S (2007) Evidence of disrupted prepulse inhibition in unaffected siblings of bipolar disorder patients. Biol Psychiatry 62(12):1418–1422
Gibbons AS, Scarr E, McLean C, Sundram S, Dean B (2009) Decreased muscarinic receptor binding in the frontal cortex of bipolar disorder and major depressive disorder subjects. J Affect Disord 116(3):184–191
Giridharan VV, Sayana P, Pinjari OF, Ahmad N, da Rosa MI, Quevedo J et al (2019) Postmortem evidence of brain inflammatory markers in bipolar disorder: a systematic review. Mol Psychiatry:1–20
Goldstein BI, Kemp DE, Soczynska JK, McIntyre RS (2009) Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. J Clin Psychiatry 70(8):1078–1090
Grande I, Berk M, Birmaher B, Vieta E (2016) Bipolar disorder. Lancet 387(10027):1561–1572
Grayson DR, Kundakovic M, Sharma RP (2010) Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol 77(2):126–135
Guo R, Gu J, Zong S, Wu M, Yang M (2018) Structure and mechanism of mitochondrial electron transport chain. Biom J 41(1):9–20
Haddad EB, Rousell J, Lindsay MA, Barnes PJ (1996) Synergy between tumor necrosis factor alpha and interleukin 1beta in inducing transcriptional down-regulation of muscarinic M2 receptor gene expression. Involvement of protein kinase A and ceramide pathways. J Biol Chem 271(51):32586–32592
Hamdani N, Daban-Huard C, Lajnef M, Gadel R, Le Corvoisier P, Delavest M et al (2015) Cognitive deterioration among bipolar disorder patients infected by Toxoplasma gondii is correlated to interleukin 6 levels. J Affect Disord 179:161–166
Harrison PJ (2016) Molecular neurobiological clues to the pathogenesis of bipolar disorder. Curr Opin Neurobiol 36:1–6
Hillegers MH, Reichart CG, Wals M, Verhulst FC, Ormel J, Nolen WA et al (2007) Signs of a higher prevalence of autoimmune thyroiditis in female offspring of bipolar parents. Eur Neuropsychopharmacol 17(6–7):394–399
Hope S, Hoseth E, Dieset I, Morch RH, Aas M, Aukrust P et al (2015) Inflammatory markers are associated with general cognitive abilities in schizophrenia and bipolar disorder patients and healthy controls. Schizophr Res 165(2–3):188–194
Hoseth EZ, Westlye LT, Hope S, Dieset I, Aukrust P, Melle I et al (2016) Association between cytokine levels, verbal memory and hippocampus volume in psychotic disorders and healthy controls. Acta Psychiatr Scand 133(1):53–62
Houenou J, d'Albis MA, Daban C, Hamdani N, Delavest M, Lepine JP et al (2014) Cytomegalovirus seropositivity and serointensity are associated with hippocampal volume and verbal memory in schizophrenia and bipolar disorder. Prog Neuro-Psychopharmacol Biol Psychiatry 48:142–148
Hsu CC, Chen SC, Liu CJ, Lu T, Shen CC, Hu YW et al (2014) Rheumatoid arthritis and the risk of bipolar disorder: a nationwide population-based study. PLoS One 9(9):e107512
Huang TT, Lai JB, Du YL, Xu Y, Ruan LM, Hu SH (2019) Current understanding of gut microbiota in mood disorders: an update of human studies. Front Genet 10:98
Hwang N, Eom T, Gupta SK, Jeong SY, Jeong DY, Kim YS et al (2017) Genes and gut bacteria involved in luminal butyrate reduction caused by diet and loperamide. Genes (Basel) 8(12)
Isgren A, Jakobsson J, Palsson E, Ekman CJ, Johansson AG, Sellgren C et al (2015) Increased cerebrospinal fluid interleukin-8 in bipolar disorder patients associated with lithium and antipsychotic treatment. Brain Behav Immun 43:198–204
Isgren A, Sellgren C, Ekman CJ, Holmen-Larsson J, Blennow K, Zetterberg H et al (2017) Markers of neuroinflammation and neuronal injury in bipolar disorder: relation to prospective clinical outcomes. Brain Behav Immun 65:195–201
Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 327(5963):291–295
Jakobsson J, Bjerke M, Ekman CJ, Sellgren C, Johansson AG, Zetterberg H et al (2014) Elevated concentrations of neurofilament light chain in the cerebrospinal fluid of bipolar disorder patients. Neuropsychopharmacology 39(10):2349–2356
Jakobsson J, Bjerke M, Sahebi S, Isgren A, Ekman CJ, Sellgren C et al (2015) Monocyte and microglial activation in patients with mood-stabilized bipolar disorder. J Psychiatry Neurosci 40(4):250–258
Jones KA, Porjesz B, Almasy L, Bierut L, Goate A, Wang JC et al (2004) Linkage and linkage disequilibrium of evoked EEG oscillations with CHRM2 receptor gene polymorphisms: implications for human brain dynamics and cognition. Int J Psychophysiol 53(2):75–90
Kapczinski NS, Mwangi B, Cassidy RM, Librenza-Garcia D, Bermudez MB, Kauer-Sant'anna M et al (2017) Neuroprogression and illness trajectories in bipolar disorder. Expert Rev Neurother 17(3):277–285
Kapur N, Barker S, Burrows EH, Ellison D, Brice J, Illis LS et al (1994) Herpes simplex encephalitis: long term magnetic resonance imaging and neuropsychological profile. J Neurol Neurosurg Psychiatry 57(11):1334–1342
Kato T (2007) Mitochondrial dysfunction as the molecular basis of bipolar disorder: therapeutic implications. CNS Drugs 21(1):1–11
Kauer-Sant'Anna M, Kapczinski F, Andreazza AC, Bond DJ, Lam RW, Young LT et al (2009) Brain-derived neurotrophic factor and inflammatory markers in patients with early- vs. late-stage bipolar disorder. Int J Neuropsychopharmacol 12(4):447–458
Khairova RA, Machado-Vieira R, Du J, Manji HK (2009) A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int J Neuropsychopharmacol 12(4):561–578
Khan D, Fernando P, Cicvaric A, Berger A, Pollak A, Monje FJ et al (2014) Long-term effects of maternal immune activation on depression-like behavior in the mouse. Transl Psychiatry 4:e363
Khandaker GM, Zimbron J, Dalman C, Lewis G, Jones PB (2012) Childhood infection and adult schizophrenia: a meta-analysis of population-based studies. Schizophr Res 139(1–3):161–168
Kilbourne ED (1987) Influenza. Plenum Medical Book, New York
Kim YK, Myint AM, Lee BH, Han CS, Lee SW, Leonard BE et al (2004) T-helper types 1, 2, and 3 cytokine interactions in symptomatic manic patients. Psychiatry Res 129(3):267–272
Kim YK, Jung HG, Myint AM, Kim H, Park SH (2007) Imbalance between pro-inflammatory and anti-inflammatory cytokines in bipolar disorder. J Affect Disord 104(1–3):91–95
Koehler K, Guth W (1979) The mimicking of mania in “benign” herpes simplex encephalitis. Biol Psychiatry 14(2):405–411
Konradi C, Eaton M, MacDonald ML, Walsh J, Benes FM, Heckers S (2004) Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry 61(3):300–308
Kupferschmidt DA, Zakzanis KK (2011) Toward a functional neuroanatomical signature of bipolar disorder: quantitative evidence from the neuroimaging literature. Psychiatry Res 193(2):71–79
Lavagnino L, Cao B, Mwangi B, Wu MJ, Sanches M, Zunta-Soares GB et al (2015) Changes in the corpus callosum in women with late-stage bipolar disorder. Acta Psychiatr Scand 131(6):458–464
Leboyer M, Oliveira J, Tamouza R, Groc L (2016) Is it time for immunopsychiatry in psychotic disorders? Psychopharmacology 233(9):1651–1660
Machado-Vieira R, Andreazza AC, Viale CI, Zanatto V, Cereser V Jr, da Silva VR et al (2007) Oxidative stress parameters in unmedicated and treated bipolar subjects during initial manic episode: a possible role for lithium antioxidant effects. Neurosci Lett 421(1):33–36
Marrie RA, Hitchon CA, Walld R, Patten SB, Bolton JM, Sareen J et al (2018a) Increased burden of psychiatric disorders in rheumatoid arthritis. Arthritis Care Res (Hoboken) 70(7):970–978
Marrie RA, Walld R, Bolton JM, Sareen J, Walker JR, Patten SB et al (2018b) Physical comorbidities increase the risk of psychiatric comorbidity in immune-mediated inflammatory disease. Gen Hosp Psychiatry 51:71–78
Maurer IC, Schippel P, Volz HP (2009) Lithium-induced enhancement of mitochondrial oxidative phosphorylation in human brain tissue. Bipolar Disord 11(5):515–522
Merikangas KR, Akiskal HS, Angst J, Greenberg PE, Hirschfeld RM, Petukhova M et al (2007) Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey replication. Arch Gen Psychiatry 64(5):543–552
Meyer U (2014) Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry 75(4):307–315
Meyer U, Feldon J, Fatemi SH (2009) In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev 33(7):1061–1079
Miller CL, Llenos IC, Dulay JR, Weis S (2006) Upregulation of the initiating step of the kynurenine pathway in postmortem anterior cingulate cortex from individuals with schizophrenia and bipolar disorder. Brain Res 1073-1074:25–37
Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B (2011) Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry 70(7):663–671
Modabbernia A, Taslimi S, Brietzke E, Ashrafi M (2013) Cytokine alterations in bipolar disorder: a meta-analysis of 30 studies. Biol Psychiatry 74(1):15–25
Mook-Kanamori BB, Geldhoff M, van der Poll T, van de Beek D (2011) Pathogenesis and pathophysiology of pneumococcal meningitis. Clin Microbiol Rev 24(3):557–591
Moore PB, El-Badri SM, Cousins D, Shepherd DJ, Young AH, McAllister VL et al (2001) White matter lesions and season of birth of patients with bipolar affective disorder. Am J Psychiatry 158(9):1521–1524
Munkholm K, Vinberg M, Vedel KL (2013) Cytokines in bipolar disorder: a systematic review and meta-analysis. J Affect Disord 144(1–2):16–27
Mwangi B, Wu MJ, Cao B, Passos IC, Lavagnino L, Keser Z et al (2016) Individualized prediction and clinical staging of bipolar disorders using neuroanatomical biomarkers. Biol Psychiatry Cogn Neurosci Neuroimag 1(2):186–194
Myint AM, Kim YK, Verkerk R, Park SH, Scharpe S, Steinbusch HW et al (2007) Tryptophan breakdown pathway in bipolar mania. J Affect Disord 102(1–3):65–72
Nery FG, Monkul ES, Hatch JP, Fonseca M, Zunta-Soares GB, Frey BN et al (2008) Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum Psychopharmacol 23(2):87–94
Nordentoft M, Mortensen PB, Pedersen CB (2011) Absolute risk of suicide after first hospital contact in mental disorder. Arch Gen Psychiatry 68(10):1058–1064
Orlovska-Waast S, Kohler-Forsberg O, Brix SW, Nordentoft M, Kondziella D, Krogh J et al (2019) Cerebrospinal fluid markers of inflammation and infections in schizophrenia and affective disorders: a systematic review and meta-analysis. Mol Psychiatry 24(6):869–887
Ortiz-Dominguez A, Hernandez ME, Berlanga C, Gutierrez-Mora D, Moreno J, Heinze G et al (2007) Immune variations in bipolar disorder: phasic differences. Bipolar Disord 9(6):596–602
Painold A, Morkl S, Kashofer K, Halwachs B, Dalkner N, Bengesser S et al (2019) A step ahead: exploring the gut microbiota in inpatients with bipolar disorder during a depressive episode. Bipolar Disord 21(1):40–49
Panizzutti B, Gubert C, Schuh AL, Ferrari P, Bristot G, Fries GR et al (2015) Increased serum levels of eotaxin/CCL11 in late-stage patients with bipolar disorder: an accelerated aging biomarker? J Affect Disord 182:64–69
Parboosing R, Bao Y, Shen L, Schaefer CA, Brown AS (2013) Gestational influenza and bipolar disorder in adult offspring. JAMA Psychiat 70(7):677–685
Perry W, Minassian A, Feifel D, Braff DL (2001) Sensorimotor gating deficits in bipolar disorder patients with acute psychotic mania. Biol Psychiatry 50(6):418–424
Post RM (2007) Kindling and sensitization as models for affective episode recurrence, cyclicity, and tolerance phenomena. Neurosci Biobehav Rev 31(6):858–873
Rao JS, Harry GJ, Rapoport SI, Kim HW (2010) Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry 15(4):384–392
Reinares M, Papachristou E, Harvey P, Mar Bonnin C, Sanchez-Moreno J, Torrent C et al (2013) Towards a clinical staging for bipolar disorder: defining patient subtypes based on functional outcome. J Affect Disord 144(1–2):65–71
Rolstad S, Jakobsson J, Sellgren C, Ekman CJ, Blennow K, Zetterberg H et al (2015a) Cognitive performance and cerebrospinal fluid biomarkers of neurodegeneration: a study of patients with bipolar disorder and healthy controls. PLoS One 10(5):e0127100
Rolstad S, Jakobsson J, Sellgren C, Isgren A, Ekman CJ, Bjerke M et al (2015b) CSF neuroinflammatory biomarkers in bipolar disorder are associated with cognitive impairment. Eur Neuropsychopharmacol 25(8):1091–1098
Ronovsky M, Berger S, Molz B, Berger A, Pollak DD (2016) Animal models of maternal immune activation in depression research. Curr Neuropharmacol 14(7):688–704
Rosa AR, Magalhaes PV, Czepielewski L, Sulzbach MV, Goi PD, Vieta E et al (2014) Clinical staging in bipolar disorder: focus on cognition and functioning. J Clin Psychiatry 75(5):e450–e456
Rosenblat JD, McIntyre RS (2015) Are medical comorbid conditions of bipolar disorder due to immune dysfunction? Acta Psychiatr Scand 132(3):180–191
Rosenblat JD, Cha DS, Mansur RB, McIntyre RS (2014) Inflamed moods: a review of the interactions between inflammation and mood disorders. Prog Neuro-Psychopharmacol Biol Psychiatry 53:23–34
Samalin L, de Chazeron I, Vieta E, Bellivier F, Llorca PM (2016) Residual symptoms and specific functional impairments in euthymic patients with bipolar disorder. Bipolar Disord 18(2):164–173
Savitz J, Preskorn S, Teague TK, Drevets D, Yates W, Drevets W (2012) Minocycline and aspirin in the treatment of bipolar depression: a protocol for a proof-of-concept, randomised, double-blind, placebo-controlled, 2x2 clinical trial. BMJ Open 2(1):e000643
Savitz JB, Price JL, Drevets WC (2014) Neuropathological and neuromorphometric abnormalities in bipolar disorder: view from the medial prefrontal cortical network. Neurosci Biobehav Rev 42:132–147
Sayana P, Colpo GD, Simoes LR, Giridharan VV, Teixeira AL, Quevedo J et al (2017) A systematic review of evidence for the role of inflammatory biomarkers in bipolar patients. J Psychiatr Res 92:160–182
Schlitt M, Lakeman FD, Whitley RJ (1985) Psychosis and herpes simplex encephalitis. South Med J 78(11):1347–1350
Sellner J, Tauber MG, Leib SL (2010) Pathogenesis and pathophysiology of bacterial CNS infections. Handb Clin Neurol 96:1–16
Soderlund J, Olsson SK, Samuelsson M, Walther-Jallow L, Johansson C, Erhardt S et al (2011) Elevation of cerebrospinal fluid interleukin-1ss in bipolar disorder. J Psychiatry Neurosci 36(2):114–118
Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z et al (2011) Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation 8:94
Stephenson J, Nutma E, van der Valk P, Amor S (2018) Inflammation in CNS neurodegenerative diseases. Immunology 154(2):204–219
Stertz L, Magalhaes PV, Kapczinski F (2013) Is bipolar disorder an inflammatory condition? The relevance of microglial activation. Curr Opin Psychiatry 26(1):19–26
Stich O, Andres TA, Gross CM, Gerber SI, Rauer S, Langosch JM (2015) An observational study of inflammation in the central nervous system in patients with bipolar disorder. Bipolar Disord 17(3):291–302
Strakowski SM, DelBello MP, Zimmerman ME, Getz GE, Mills NP, Ret J et al (2002) Ventricular and periventricular structural volumes in first- versus multiple-episode bipolar disorder. Am J Psychiatry 159(11):1841–1847
Strasser HC, Lilyestrom J, Ashby ER, Honeycutt NA, Schretlen DJ, Pulver AE et al (2005) Hippocampal and ventricular volumes in psychotic and nonpsychotic bipolar patients compared with schizophrenia patients and community control subjects: a pilot study. Biol Psychiatry 57(6):633–639
Sun X, Wang JF, Tseng M, Young LT (2006) Downregulation in components of the mitochondrial electron transport chain in the postmortem frontal cortex of subjects with bipolar disorder. J Psychiatry Neurosci 31(3):189–196
Tedla Y, Shibre T, Ali O, Tadele G, Woldeamanuel Y, Asrat D et al (2011) Serum antibodies to Toxoplasma gondii and Herpesvidae family viruses in individuals with schizophrenia and bipolar disorder: a case-control study. Ethiop Med J 49(3):211–220
Tiosano S, Nir Z, Gendelman O, Comaneshter D, Amital H, Cohen AD et al (2017) The association between systemic lupus erythematosus and bipolar disorder - a big data analysis. Eur Psychiatry 43:116–119
Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK et al (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62(3):405–496
Vitaliani R, Mason W, Ances B, Zwerdling T, Jiang Z, Dalmau J (2005) Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol 58(4):594–604
Wadee AA, Kuschke RH, Wood LA, Berk M, Ichim L, Maes M (2002) Serological observations in patients suffering from acute manic episodes. Hum Psychopharmacol 17(4):175–179
Yolken RHDF (2011) Neurotropic viruses and cognitive impairment. Schizophr Bull 37:235–236
Yolken RH, Torrey EF (2008) Are some cases of psychosis caused by microbial agents? A review of the evidence. Mol Psychiatry 13(5):470–479
Zou J, Wang YX, Dou FF, Lu HZ, Ma ZW, Lu PH et al (2010) Glutamine synthetase down-regulation reduces astrocyte protection against glutamate excitotoxicity to neurons. Neurochem Int 56(4):577–584
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Barichello, T. et al. (2020). Inflammation as a Mechanism of Bipolar Disorder Neuroprogression. In: Young, A.H., Juruena, M.F. (eds) Bipolar Disorder: From Neuroscience to Treatment. Current Topics in Behavioral Neurosciences, vol 48. Springer, Cham. https://doi.org/10.1007/7854_2020_173
Download citation
DOI: https://doi.org/10.1007/7854_2020_173
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-72142-8
Online ISBN: 978-3-030-72143-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)