
Abstract
The ultraviolet (UV) radiation contained in sunlight is a powerful immune suppressant. While exposure to UV is best known for its ability to cause skin cancer, it is also associated with protection against a range of autoimmune diseases, particularly multiple sclerosis (MS). Although the precise mechanism by which sunlight affords protection from MS remains to be determined, some have hypothesised that UV immunosuppression explains the “latitude-gradient effect” associated with MS. By stimulating the release of soluble factors in exposed skin, UV activates immune suppressive pathways that culminate in the induction of regulatory cells in distant tissues. Each and every one of the immune suppressive cells and molecules activated by UV exposure are potential targets for treating and preventing MS. A thorough understanding of the mechanisms involved is therefore required if we are to realise the therapeutic potential of photoimmunology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
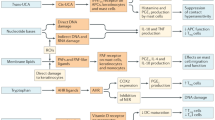
The UV radiation in sunlight can protect us against a variety of autoimmune diseases (Ponsonby et al. 2005), particularly MS (van der Mei et al. 2003). Despite enormous public interest in understanding how UV achieves this protection, the mechanisms involved remain to be determined. Exposure to UV is best known for its contribution to the development of skin cancer, in part through its capacity to damage DNA (Agar et al. 2004). UV is also capable of suppressing adaptive (including anti-tumour) immune responses (Fisher and Kripke 1977) which is required for carcinogenesis. Whether UV-induced immune suppression explains the autoimmune protective effect of sunlight has not been formally tested. Both the UVA (320–400 nm) and UVB (290–320 nm) spectrums of sunlight are immune suppressive (Fig. 1). Far-red/near-infrared wavelengths of sunlight (670 nm) are also immune suppressive (Kandolf-Sekulovic et al. 2003) and have been shown to ameliorate the well-known animal model of MS, experimental autoimmune encephalomyelitis (EAE) (Muili et al. 2012). While the mechanisms involved in long wavelength immune suppression remain to be identified, considerably more progress has been made in our understanding of how UV suppresses immunity. Understanding the mechanisms involved is likely to lead to breakthroughs in the prevention and treatment of MS. This chapter therefore aims to provide a comprehensive overview of the molecular and cellular pathways by which the UV spectrum of sunlight suppresses adaptive immunity.

Immune alterations and events triggered by the different wavelengths in sunlight . An overview of the reported effects caused by different wavelengths of light on distinct immune functions and cells. CGRP calcitonin gene-related peptide; DC dendritic cell; LC Langerhans cell; PAF platelet-activating factor; PGE 2 prostaglandin E2; ROS reactive oxygen species; TNF tumour necrosis factor
2 Increased Exposure to UV Correlates with Lower MS Incidence and Relapse Rates
The incidence of MS increases the further you move away from the equator and is determined by how much UV one receives (van der Mei et al. 2001). This so-called latitude-gradient effect is particularly striking in Australia (McMichael and Hall 1997) where those born and bred in Tasmania (latitude ~43°S) are six times more likely to develop MS than those living in north Queensland (latitude ~19°S). Similar observations have been found across the globe (Simpson Jr et al. 2011). A latitude-gradient effect also exists for other organ-specific autoimmune diseases, including type 1 diabetes (Mohr et al. 2008), Sjögren’s Syndrome (Shapira et al. 2010) and Crohn’s Disease (Armitage et al. 2004). Whether varying levels of UV exposure also explains the susceptibility to these autoimmune diseases remains to be determined. In addition to UV exposure as a neonate, the time of year you are born also impacts on MS incidence and age of onset. Dobson et al. (2013) found the risk of developing MS significantly higher for those born in spring. However, in MS patients, the age of onset is on average 2.8 years earlier for those born in winter (McDowell et al. 2010). MS relapse rates also display a striking correlation with exposure to erythemal ultraviolet radiation , as patients were more likely to relapse in winter and early spring following prolonged periods of insufficient UV exposure (Tremlett et al. 2008). A similar pattern of association was observed between serum 25(OH)D concentrations and active MS lesions (Embry et al. 2000).
3 Is UV -induced Vitamin D Responsible for Sunlight Protection from MS?
Exposure to UVB (those wavelengths between 280 and 320 nm) is the most efficient way to make Vitamin D, which is important for bone health. A Vitamin D deficiency in MS patients (Munger et al. 2006) has prompted a number of trials of Vitamin D supplements to prevent and/or treat MS. However, no difference was found between high- and low-dose treatment groups in one of the first double-blinded, randomised, controlled trials of Vitamin D supplementation to MS patients. On the contrary, MS patients on high-dose Vitamin D had a higher exit expanded disability status scale and were significantly more likely to relapse (Stein et al. 2011). Similar interventions in type 1 diabetics have also been ineffective (Stene et al. 2003). These studies suggest that boosting Vitamin D levels alone may not have the desired therapeutic effect, and that there is something else about UVB exposure that explains the protective properties of sunlight (Hart et al. 2011). Similar to UVB, Vitamin D activates immune suppressive pathways (Damian et al. 2010) that have been utilised for the treatment of inflammatory autoimmune skin diseases (Gruber-Wackernagel et al. 2011). One mechanism may involve Vitamin D3-mediated upregulation of receptor activator of NF-κB (RANK) ligand which in turn leads to systemic increases in CD4+CD25+ T cells (Loser et al. 2006) (Fig. 2). It is not yet clear what this means for MS patients who paradoxically have elevated levels of circulating RANKL (Kurban et al. 2008). Alternatively (or simultaneously), Vitamin D3 may enhance the activity of regulatory T cells (Gorman et al. 2007). Delineating the Vitamin D-mediated effects from the many other factors that are produced following UVB exposure is difficult. However, recent studies have shown that UVB does modulate the immune response independently of Vitamin D in both mice (Schwarz et al. 2012; Gorman et al. 2012) and humans (Milliken et al. 2012). Indeed, UVB-protection of mice from EAE is not due to UVB-induced Vitamin D (Becklund et al. 2010; Wang et al. 2013). In fact, making mice Vitamin D deficient (DeLuca and Plum 2011) or knocking out the Vitamin D receptor (Wang et al. 2012) paradoxically reduces the severity and delays the onset of this autoimmune disease. Thus, another UVB-induced event that is independent of Vitamin D protects the central nervous system (CNS) from immune attack.

Immune suppressive events triggered by UVB. UVB initiates a cascade of molecular and cellular events that culminate in the induction of regulatory cells in distant lymphoid tissues. Many of these events are interactive and additive, leading to large effects on the immune system that are difficult to inhibit due to their redundancy. Many of these cells and molecules are potential therapeutic targets in preventing and treating MS. COX cyclooxygenase; DC dendritic cell; DLN draining lymph node; NKT cells natural killer T cells; PAF platelet-activating factor; PGE 2 prostaglandin E2; RANKL receptor activator of nuclear factor-κB ligand; ST2 IL-33 receptor; TNF tumour necrosis factor; UCA urocanic acid; UVB-B Regs UVB-induced regulatory B cells ; UVB-T Regs UVB-induced regulatory T cells ; VDR Vitamin D receptor
4 How then Does UV Protect Us from MS?
MS is caused by a “perfect storm” of genetic and environmental factors conspiring to initiate and promote damage to the CNS. Work done by The International Multiple Sclerosis Genetics Consortium confirmed that immunologically relevant genes are significantly overrepresented in MS patients (Sawcer et al. 2011). This is important because it means that efforts to interfere with immune-mediated events are amongst the most promising for preventing and ultimately curing MS. This probably explains why some of the most successful MS therapies, including glatiramer acetate, IFNβ, fingolimod (FTY720) and natalizumab, all work by modulating or suppressing the immune system. Exposing the skin to UVB initiates a cascade of molecular and cellular events that also suppresses the immune system (Fig. 2). Whether these events are responsible for the health benefits of sunlight in MS remain to be determined.
5 Molecular Mechanisms of UVB-induced Immune Suppression
5.1 Platelet-activating Factor (PAF) and Serotonin (5-HT) Receptor Agonists
While it is now appreciated that UVB suppresses immunity in internal organs as well as the skin (McGlade et al. 2007; Rana et al. 2011), the trigger must have a cutaneous origin as UVB does not penetrate deep enough to reach internal organs. Two early molecular events following UVB exposure is the release of platelet-activating factor (PAF) from keratinocytes (Barber et al. 1998; Alappatt et al. 2000) and the isomerisation of epidermal trans-urocanic acid (UCA) to cis-UCA (Anglin et al. 1961; Pascher 1962). These are important and relevant because both PAF (Walterscheid et al. 2002) and cis-UCA (De Fabo and Noonan 1983) are potent mediators of UVB immunosuppression. Normal skin has abundant levels of trans-UCA, which is a UVB photoreceptor that gets isomerised to cis-UCA following exposure to UVB (Anglin et al. 1961; Pascher 1962; Kammeyer et al. 1997). By signalling through serotonin (5-HT) 2A receptors (Walterscheid et al. 2006), cis-UCA causes a defect in antigen presentation (Noonan et al. 1988) ultimately leading to systemic suppression of the adaptive immune response (De Fabo and Noonan 1983; el-Ghorr and Norval 1995). While it remains to be determined whether UVB-induced cis-UCA is involved in protection from MS, the fact that patients with relapsing remitting MS have lower plasma levels of cis-UCA compared to healthy controls (Correale and Farez 2013) may indicate a relationship between these two events.
PAF is a plasma membrane phospholipid first discovered in 1972 by studying its influence on the immune system, where it aggregated platelets to release histamine (Benveniste et al. 1972). Exposure to UVB causes PAF and other photo-oxidised cellular phospholipids to be almost immediately released from keratinocytes (Barber et al. 1998; Alappatt et al. 2000). The generation of these PAF receptor agonists (Travers et al. 2010) leads to a positive amplification loop of PAF synthesis and ultimately apoptosis (Marathe et al. 2005; Pei et al. 1998). This cascade of events in turn suppresses the adaptive immune response (Rola-Pleszczynski et al. 1988; Walterscheid et al. 2002). Mice treated with PAF receptor antagonists (Walterscheid et al. 2002) or mice deficient in PAF receptors (Wolf et al. 2006) are resistant to UVB-induced immunosuppression. One mechanism may involve PAF binding to its receptor on keratinocytes leading to the release of tumour necrosis factor-α (TNF) that in turn triggers Langerhans cell migration to the local draining lymph nodes (Fukunaga et al. 2010). Another possibility is that PAF targets PAF receptor+ bone marrow-derived cells to upregulate IL-10 through COX-2-generated prostaglandins (Zhang et al. 2008). More recently, PAF was shown to target dermal mast cells, triggering upregulation of CXCR4 and their migration to CXCL12-expressing local draining lymph nodes (Chacon-Salinas et al. 2014). Using a combination of antagonists and receptor knockout mice we proved that PAF and serotonin receptor signalling were both required for the activation of UVB-induced regulatory B cells (UV-BRegs) (Matsumura et al. 2006).
There are conflicting reports on the role of PAF and serotonin in CNS-targeted autoimmunity. While a pathogenic role for PAF in EAE has been proposed (Kihara et al. 2005), this analysis was restricted to the potential damage caused by PAF in the CNS and not its role in mediating systemic immune suppression . Meanwhile, contrasting studies used specific PAF receptor antagonists to rule out any role for PAF in mediating EAE (Vela et al. 1991). A pathogenic role for serotonin has also been proposed based on studies where serotonin receptor antagonists inhibited the development of EAE (Dietsch and Hinrichs 1989). In contrast, efforts to pharmacologically boost available serotonin shows promise. One way to boost the levels of extracellular free serotonin is through the use of serotonin re-uptake inhibitors (antidepressants). This class of drug boosts IL-10 levels (Kubera et al. 2001) and reduces the formation of new lesions in MS patients (Mostert et al. 2008). Antidepressants can also ameliorate the course of EAE (Vollmar et al. 2009). Whether UVB-induced signalling through PAF and/or serotonin receptors is involved in protection from CNS-targeted autoimmunity remains to be investigated.
5.2 Interleukin-33 (IL-33)
One outcome of the release of PAF receptor agonists by UVB is the production by dermal fibroblasts of cutaneous interleukin-33 (IL-33) (Fig. 2) (Byrne et al. 2011), a member of the IL-1 family of cytokines, which has been found to promote Th2 responses (Schmitz et al. 2005). IL-33 can also expand immunoregulatory myeloid cells and CD4+ Foxp3+ regulatory T cells (Turnquist et al. 2011), suggesting that it may be important for suppressing autoimmunity. We showed that UVB-induced IL-33 was involved in immune suppression because antibodies specific for IL-33 could block the effects of UVB exposure (Byrne et al. 2011). MS patients have elevated levels of this cytokine within the CNS and periphery (Christophi et al. 2012), suggesting IL-33 may play a pathogenic role during this disease. Resting mice also express IL-33 and its receptor (ST2) in the CNS, which is upregulated during EAE. Paradoxically, mice that are deficient in ST2 display exacerbated EAE compared with wild-type mice (Jiang et al. 2012). Furthermore, injecting recombinant IL-33 attenuates EAE severity in wild-type but not ST2 knockout mice, a phenomenon associated with significantly reduced IL-17 and IFN-γ levels. This conflicts with a report by Li and colleagues showing that neutralising IL-33 with monoclonal antibodies suppresses, while injecting recombinant IL-33 exacerbates EAE (Li et al. 2012). Further complicating this story, Oboki and colleagues showed that EAE develops normally in IL-33 deficient mice (Oboki et al. 2010), although this apparent contradiction could be due to cytokine redundancy. At this stage, it is not yet clear what role, if any, UVB-induced IL-33 plays in protection from CNS-targeted autoimmunity.
5.3 Interleukin-4 (IL-4) and IL-13
A major target of UVB-induced IL-33 are likely to be dermal mast cells which are found in close proximity to IL-33-producing fibroblasts in UVB-exposed skin (Byrne et al. 2011) (Fig. 2). Either directly or via mast cells triggered to produce the neutrophil chemoattractant CXCL8 (IL-8) (Allakhverdi et al. 2007; Endoh et al. 2007), and IL-33 may be responsible for recruiting neutrophils to UVB-exposed skin. This is likely to be important for downstream immune suppression because neutrophils are the primary cellular source of immune modulating IL-4 (Fig. 2) (Teunissen et al. 2002). Indeed, wild-type mice treated with anti-IL-4 antibodies (Shreedhar et al. 1998a) or mice deficient in IL-4 (El-Ghorr and Norval 1997) are resistant to UVB-immune suppression . UVB-induced IL-4 is also likely to be a critical differentiation signal for dermal mast cells because degranulation is defective in IL-4-deficient mice exposed to UVB. This defect has a significant impact on downstream UVB-induced immune suppression (Hart et al. 2000).
IL-33-stimulated mast cells also respond by producing anti-inflammatory cytokines such as IL-10 (Allakhverdi et al. 2007) and IL-13 (Sarchio et al. 2012). The significance of IL-13 upregulation following UVB has yet to be explored but will be of much interest to MS researchers because IL-13 can suppress Th1 and Th17 inflammation by regulating the synthesis of IL-6, IFN-γ (Minty et al. 1993) and IL-17-driven autoimmunity (Newcomb et al. 2009), the latter independently from IL-10 (Newcomb et al. 2012). This is important because both EAE (Bettelli et al. 2004; Jäger et al. 2009) and MS (Lock et al. 2002; Tzartos et al. 2008) are caused by a coordinated Th1/Th17 attack (Stromnes et al. 2008; Sawcer et al. 2011).
5.4 Interleukin-10 (IL-10)
A role for IL-10 in mediating UVB-immune suppression is now firmly established. Exposure to UVB results in a cascade of molecular and cellular events that ultimately raises serum IL-10 levels (Hart et al. 2000). IL-10 appears to be a central cytokine involved in mediating the immune suppressive effects of UVB because mice deficient in IL-10 are completely resistant to UVB-induced immune suppression (Beissert et al. 1996) and carcinogenesis (Loser et al. 2007). The triggers and cellular sources of UVB-induced IL-10 are many and varied and will be discussed in detail below. IL-10 is produced by a variety of regulatory cells (Fujio et al. 2010; Mauri and Bosma 2012) and can induce anergy in self-reactive T cells (Groux et al. 1996). Thus, UVB-induced upregulation of IL-10 is a particularly relevant event in MS due to its ability to maintain peripheral tolerance .
5.5 Tumour Necrosis Factor (TNF)
Another early molecular event occurring in UVB-exposed skin is the release of tumour necrosis factor α (TNF) (Skov et al. 1998), most likely produced by degranulating mast cells (Fig. 2) (Walsh 1995). Upregulation of TNF is a major trigger of Langerhans cells migration from the epidermis to the draining lymph node (Moodycliffe et al. 1994) and is required for UVB suppression of skin immunity (Rivas and Ullrich 1994). TNF may be pathogenic in the context of CNS-targeted autoimmunity, as TNF-expressing mast cells are responsible for recruiting neutrophils into the meninges, which in turn alter vascular permeability (Christy et al. 2013). MS patients also have increased levels of TNF within the CNS and cerebrospinal fluid (Hauser et al. 1990), which correlates with disease severity (Sharief and Hentges 1991). Disappointingly, early therapeutic interventions to neutralise TNF in MS patients had to be terminated due to disease exacerbation (Group 1999). These apparent contradictions may be explained in a number of ways including the possibility that TNF has different effects depending on its source and site of production (i.e. skin vs. CNS). Another important consideration is that TNF exerts its effects on target cells by binding to two receptors: TNFR1 (originally TNFR60), which is predominantly activated by soluble TNF; and TNFR2 (originally TNFR80), which is preferentially activated by membrane bound TNF (Grell et al. 1995). Activation of TNFR1 has proinflammatory effects in MS patients (Akassoglou et al. 1998), whereas TNFR2 activation promotes both remyelination and neuroprotection (Arnett et al. 2001; Fontaine et al. 2002). Indeed, only recently it has been empirically confirmed that selective antagonism of TNFR1 receptors attenuates EAE (Williams et al. 2014). This, together with the fact that UVB selectively decreases TNFR1 expression but increases TNFR2 expression in human skin (Barr et al. 1999), suggests that UVB-induced TNF may be promoting remyelination and neuroprotection. This intriguing possibility remains to be explored.
6 Cellular Mechanisms of UVB-induced Immune Suppression
The cascade of molecular events triggered by exposure to UVB leads to suppression of the induction, effector and memory phases of both cell-mediated and humoral immune responses (Fig. 2). UVB affects CD8+ cytotoxic T lymphocyte (CTL) responses (Rana et al. 2011), as well as suppressing CD4+ T helper cell (Th) type 1 (Th1) (Brown et al. 1995), Th2 (McGlade et al. 2007) and Th17 (Singh et al. 2010) responses. UVB suppression of the T cell response is likely to be important in protection from MS because CTLs (Mars et al. 2011) as well as Th1 and Th17 responses are strong drivers of CNS-targeted autoimmunity (Zamvil et al. 1986; Lock et al. 2002; Langrish et al. 2005; Tzartos et al. 2008; Yang et al. 2008; Sweeney et al. 2011; Inoue et al. 2012). T follicular helper cell responses are also significantly suppressed by UVB (Chacon-Salinas et al. 2011). The subsequent inhibition of germinal centre formation leads to significant decrease in high-affinity class-switched antibody production. While this suppression of humoral immunity impacts on the success of vaccination (Cooper et al. 1992; Sleijffers et al. 2002), it may explain the protective effect of UVB in CNS-targeted autoimmunity because myelin-reactive autoantibodies are present in EAE mice (Matsushita et al. 2008) and MS patients (Genain et al. 1999). The broad spectrum of suppressive events initiated and maintained by sunlight makes therapeutic exposure to UVB an attractive option. This has prompted the Australian-based “PhoCIS” randomised controlled clinical trial which will explore whether narrowband UVB therapy decreases the risk of developing multiple sclerosis over 12 months from their first demyelinating event (ANZCTR ID:ACTRN12614000185662).
7 UVB Suppresses Immunity by Activating Regulatory Cells
7.1 UVB-induced Regulatory T Cells (UV-TRegs)
Exposure to UVB ultimately leads to the activation of a number of different types of regulatory cells that suppress inflammation and adaptive immune responses (Fig. 2). The most well known of these is the UVB-activated regulatory T cell (UV-TReg) (Elmets et al. 1983; Shreedhar et al. 1998b). TRegs are a subset of CD4+ T cells responsible for suppressing immunity and maintaining peripheral self-tolerance (Groux et al. 1997). They were originally described as “suppressor T cells” in the 1970s but are now commonly identified as CD4+CD25+ cells (Sakaguchi et al. 1995) that express the transcription factor FoxP3 (Roncador et al. 2005). UV-TRegs are relatively well characterised (Loser and Beissert 2012). They express CD4, CD25, CD62L and the transcription factor FoxP3 (Schwarz 2008; Schwarz et al. 2011) while expression of CD152 (CTLA-4) (Schwarz et al. 2000), GITR (Shimizu et al. 2002) and the putative TReg marker neuropilin-1 (nrp1) (Bruder et al. 2004) is required for their suppression of immunity. This is likely to be important for CNS-targeted autoimmunity because it has been shown that the adoptive transfer of wild type but not nrp1−/− TRegs suppress EAE (Solomon et al. 2011). While the expression of these membrane bound molecules partly explains the mechanism of UV-TReg suppression, their ability to produce immune modulating cytokines, particularly IL-10 (Shreedhar et al. 1998b), is also involved.
A role for UV-TRegs in protecting from MS has yet to be confirmed. Studies in animal models (Stohlman et al. 1999) and MS patients (Tennakoon et al. 2006; Frisullo et al. 2010) have highlighted the important contribution TRegs play in maintaining self-tolerance . Indeed, targeting TRegs is a promising therapeutic strategy. IL-10-producing T cells have been successfully activated in vitro through the combination of Vitamin D and dexamethasone in both humans and mice, which successfully prevented the induction of EAE (Barrat et al. 2002). Treatment of CD4+ T cells with a B7H1(PDL-1)-Ig fusion protein in combination with anti-CD3 activates type 1 TRegs (Tr1), which suppressed the induction of EAE following adoptive transfer 3 days prior to MOG injection (active), as well as during co-injection with MOG-specific T cells (passive) (Ding et al. 2006). Indeed, adoptive transfer of freshly isolated CD4+CD25+ T cells from the lymph nodes of mice were likewise able to prevent the induction of both active and passive forms of EAE, with normal Th1 cell levels but increased MOG-specific Th2 cells (Kohm et al. 2002). While targeting TRegs for MS therapy shows much promise, it is complicated by the fact that a variety of subsets exist with different mechanisms of activation and suppression. Full utilisation of their therapeutic potential awaits further investigations into the most efficient way to activate and amplify TRegs.
7.2 Dendritic Cells (DC)
UV-TRegs are activated in local draining lymph nodes that are not directly exposed to UVB. How then does the suppressive signal generated in the skin reach distant cellular targets? Important cellular messengers include migrating dendritic cells (DC), particularly epidermal Langerhans cells (LC) (Meunier et al. 1995). First, identified in the 1970s by Steinman and Cohn (1973), DC are now well known for their role as antigen-presenting cells (APC) (Green et al. 1980; Steinman et al. 1980; Streilein et al. 1980). Many different blood-borne, cutaneous and lymphoid DC subsets exist with varied functions. Exposure to UVB radiation decreases the antigen-presenting function of dermal DC in humans (Dumay et al. 2001) and induces LC emigration from the epidermis to draining lymph nodes (Meunier et al. 1995). Accumulating evidence now supports a role for these migrating epidermal LC (but not dermal DC) in mediating UVB-induced immune suppression (Fukunaga et al. 2008). DNA-damaged LC will migrate towards the draining lymph nodes following exposure to UVB (Vink et al. 1996) to activate natural killer (NK)T cells (Fukunaga et al. 2010). This subset of CD1d-restricted T cells produces high quantities of immunosuppressive IL-4 and is a major regulatory cell involved in mediating UVB-induced immune suppression (Moodycliffe et al. 2000). Indeed, neither LC-depleted nor NKT cell-deficient (Jα18−/−) mice were susceptible to UVB-induced immune suppression (Fukunaga et al. 2010). While DC are clearly important for suppression of local cutaneous responses, they do not appear to be responsible for suppressing systemic immune responses (Byrne and Halliday 2005, Gorman et al. 2005). More recently, a unique subset of UVB-induced regulatory DC arising from bone marrow precursors has been identified (Ng et al. 2010, 2013a). In response to UVB-induced prostaglandin E2 (PGE2), which itself is immune suppressive (Shreedhar et al. 1998a), very long-lived DC precursors in the bone marrow are imprinted with the capacity to suppress immunity (Fig. 2). These so-called regulatory DC do not express common regulatory molecules such as CCR7, FasL, B7H3 or B7H4. Moreover, their ability to acquire, migrate and present antigens to T cells is normal, and so it is not yet entirely clear how these regulatory DC mediate their immune suppression (Scott et al. 2012). Intriguingly, reduced immunogenicity of these bone marrow-derived DC can be passed from UVB-irradiated mothers to their progeny (Ng et al. 2013b). This has implications for MS, as there are strong epidemiological links between daily ambient UVB radiation in the first trimester of pregnancy and risk of developing MS (Staples et al. 2010). Empirical evidence gathered in animal models suggests that a deficiency in Vitamin D is unlikely to fully explain this effect (Fernandes de Abreu et al. 2010; Gorman et al. 2012; Wang et al. 2012), implying that there is something else about UVB exposure that affords protection to the unborn.
7.3 Mast Cells
Another cell we discovered was responsible for transmitting the immune suppressive signal from UVB-exposed skin to lymphocytes in draining lymph nodes is the mast cell (Byrne et al. 2008). Mast cells are traditionally known for their role in mediating allergic reactions (Oyaizu et al. 1985) whereupon first exposure to an allergen, the immune system is sensitised to produce antigen-specific immunoglobulin (Ig)E. Unlike other antibody classes, IgE can bind to high-affinity Fcε receptors (FcεR1α) on the surface of mast cells in the absence of antigen. Recently, it was shown that dermal mast cells reside along blood vessels to probe and capture IgE antibodies in the circulation (Cheng et al. 2013). In this way, upon re-exposure to the antigen, rapid degranulation of the pre-loaded mast cell occurs with the immediate release of pre-formed effector molecules (Pfeiffer et al. 1985; Rottem et al. 1992; Keown et al. 1998). This may be relevant because IgE-positive cells and mast cells have been found within demyelinated areas (Toms et al. 1990) and plaques of MS patients (Olsson 1974). This is thought to contribute to disease pathogenesis through the production of tryptase within the cerebrospinal fluid (Rozniecki et al. 1995). Others have more recently suggested that multiple sclerosis may be caused by IgE dimer formation on the surface of myelin (Calenoff 2012). The subsequent mast cell degranulation in the CNS is the ultimate mediator of CNS tissue damage. Conflicting reports using a variety of different mast cell-deficient strains suggests that mast cells may be pathogenic (Secor et al. 2000; Sayed et al. 2011), protective (Li et al. 2011; Piconese et al. 2011) or dispensable (Bennett et al. 2009; Feyerabend et al. 2011) for EAE. Thus, the role played by mast cells in CNS autoimmunity is highly controversial and remains unresolved.
In addition to their well-established pro-inflammatory role, mast cells are also capable of regulating immune responses. Indeed, by exposing cKIT-mutant mast cell-deficient mice to UVB, Hart and colleagues were the first to show the immunoregulatory capacity of mast cells (Hart et al. 1998). Mast cell-deficient mice are resistant to UVB immunosuppression , a phenotype that can be restored by reconstituting these mice with wild-type bone marrow-derived mast cells (Hart et al. 1998; Byrne et al. 2008). Grimbaldeston and colleagues discovered that mast cell-derived IL-10 is required to limit the inflammation induced by UVB (Grimbaldeston et al. 2007). It was subsequently shown by this same group that Vitamin D working through its receptor on the surface of mast cells was the molecular trigger for anti-inflammatory IL-10 (Fig. 2) (Biggs et al. 2010). Mast cell-derived IL-10 is also required for UVB-induced immune suppression (Chacon-Salinas et al. 2011).
We provided mechanistic insight into this process by demonstrating that UVB-activated mast cells migrate from the skin to B cells in draining lymph nodes (Byrne et al. 2008). This is important because myelin-derived self-antigens are abundantly expressed in lymph nodes of MS patients (Fabriek et al. 2005), and mast cells can affect B cell activation (Gauchat et al. 1993). Working through PAF (Chacon-Salinas et al. 2014), UVB activates CXCR4+ mast cells to follow a UVB-established CXCL12 chemokine gradient into and away from the skin. Using the highly specific CXCR4 antagonist AMD3100, we blocked cutaneous mast cell trafficking, which revealed the requirement of this migration for UVB suppression of T cell-mediated immunity (Byrne et al. 2008). More recently, we proved the relevance of this by showing that mice treated with AMD3100 developed ~fivefold less UVB-induced skin cancers (Sarchio et al. 2014). UVB significantly upregulates CXCL12 in local draining lymph nodes (Byrne et al. 2008) which may be important for redirecting the polarisation of effector Th1 cells into CD4+CD25−Foxp3−IL-10high autoimmune-protecting TRegs (Meiron et al. 2008). CXCL12 is constitutively expressed in the CNS and the cerebrospinal fluid (Pashenkov et al. 2003) but is higher in active MS lesions (Calderon et al. 2006). Although there are conflicting reports (Kohler et al. 2008), studies in mice show that antagonising CXCR4 with AMD3100 can exacerbate EAE (McCandless et al. 2006), implying that CXCL12 in the brain and spinal cord may not necessarily be responsible for autoimmune pathology. In fact, the location of CXCL12 within the CNS, rather than the total amount expressed, can profoundly affect MS pathogenesis. McCandless and colleagues showed that CXCL12 expression is localised to the parenchymal side of the endothelium in normal healthy brain tissue. In MS lesions, CXCL12 redistributes to the luminal side of the endothelium (McCandless et al. 2008), which is thought to lead to disease progression by allowing the traffic of CXCR4+ cells into and out of the perivascular spaces.
7.4 UVB-induced Regulatory B Cells (UV-BRegs)
In addition to UVB-induced TRegs (Ullrich and Kripke 1984), dendritic cells (Ng et al. 2013b) and mast cells (Grimbaldeston et al. 2007; Byrne et al. 2008; Biggs et al. 2010; Chacon-Salinas et al. 2011), we were the first group to demonstrate that a major way UVB causes immune suppression is via the activation of an IL-10-secreting regulatory B cell . We call these MHC IIhi B220hi cells “UV-BRegs” (Byrne and Halliday 2005; Byrne et al. 2005; Matsumura et al. 2006). While an immunoregulatory role for B cells was first described in the 1970s (Katz et al. 1974), their phenotype and mechanisms of suppression are not as well studied as TRegs. Splenic B cells that are CD5+CD1dhigh and produce large amounts of IL-10 have been termed B10 cells. They are perhaps the most well-known BReg subset having been characterised in mice (Yanaba et al. 2008a), and more recently an equivalent subset has been identified in humans (Iwata et al. 2011). Although IL-10 can be produced by mast cells and UV-TRegs, B cells have been shown to be the most abundant source of this anti-inflammatory cytokine (Madan et al. 2009). Indeed, IL-10-producing B cells have been shown to suppress a range of autoimmune diseases in animal models including type 1 diabetes, rheumatoid arthritis and EAE (Yanaba et al. 2008b). Other BReg subsets have been described, including CD20low tumour-evoked BRegs (tBRegs) (Olkhanud et al. 2011; Bodogai et al. 2013) and IL-35-producing EAE-protecting BRegs (Shen et al. 2014). Whether IL-10-producing UVB-BRegs are related to B10 cells, another BReg subset, or are unique, remains to be determined.
The role of B cells in MS is extremely controversial and somewhat contradictory. It is not yet clear why depletion of CD20+ B cells with Rituximab is therapeutically beneficial, whereas eliminating B cells by targeting B cell growth factors with Atacicept exacerbates MS (Kappos et al. 2014). Studies are therefore urgently needed to identify which B cells provide protection from MS and which are pathogenic. In any event targeting UV-BRegs is an attractive proposition because TReg therapy has currently only been shown to prevent EAE induction (Roncarolo and Battaglia 2007). Similarly, UV-TRegs only suppress the induction of immunity (Glass et al. 1990) and need to be “re-programmed” to suppress established cutaneous responses (Schwarz et al. 2011). In fact, while B cells were EAE protective, the transfer of splenic CD4+ T cells from EAE-regressed donors actually exacerbated EAE (McGeachy et al. 2005). In contrast, artificially induced BRegs suppress both EAE induction and progression of established disease (Rafei et al. 2009; Sun et al. 2012).
8 Conclusion
While MS is a disease that is not restricted by location, genetics or gender, we can learn much by studying these factors as each can influence the overall prevalence of disease. The UV wavelengths contained in sunlight are a particularly strong contributor to the MS latitude-gradient effect, although precisely how increasing the amount of UV one receives leads to protection from an autoimmune attack remains to be determined. While the molecular and cellular mechanisms are extremely complex, much progress has been made in recent decades to unravel the series of events that lead to UV-induced immune suppression . By understanding these pathways, it may be possible to therapeutically target the cells and molecules involved to prevent and treat MS.
References
Agar NS, Halliday GM, Barnetson RS, Ananthaswamy HN, Wheeler M, Jones AM (2004) The basal layer in human squamous tumors harbors more UVBA than UVB fingerprint mutations: a role for UVBA in human skin carcinogenesis. Proc Natl Acad Sci U S A 101:4954–4959
Akassoglou K, Bauer J, Kassiotis G, Pasparakis M, Lassmann H, Kollias G, Probert L (1998) Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: models for multiple sclerosis with primary oligodendrogliopathy. Am J Pathol 153:801–813
Alappatt C, Johnson CA, Clay KL, Travers JB (2000) Acute keratinocyte damage stimulates platelet-activating factor production. Arch Dermatol Res 292:256–259
Allakhverdi Z, Smith DE, Comeau MR, Delespesse G (2007) Cutting edge: the ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol 179:2051–2054
Anglin JH Jr, Bever AT, Everett MA, Lamb JH (1961) Ultraviolet-light-induced alterations in urocanic acid in vivo. Biochim Biophys Acta 53:408–409
Armitage EL, Aldhous MC, Anderson N, Drummond HE, Riemersma RA, Ghosh S, Satsangi J (2004) Incidence of juvenile-onset Crohn’s disease in Scotland: association with northern latitude and affluence. Gastroenterology 127:1051–1057
Arnett HA, Mason J, Marino M, Suzuki K, Matsuchima GK, Ting JP (2001) TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 4:1116–1122
Barber LA, Spandau DF, Rathman SC, Murphy RC, Johnson CA, Kelley SW, Hurwitz SA, Travers JB (1998) Expression of the platelet-activating factor receptor results in enhanced ultraviolet B radiation-induced apoptosis in a human epidermal cell line. J Biol Chem 273:18891–18897
Barr RM, Walker SL, Tsang W, Harrison GI, Ettehadi P, Greaves MW, Young AR (1999) Suppressed alloantigen presentation, increased TNF-alpha, IL-1, IL-1Ra, IL-10, and modulation of TNF-R in UVB-irradiated human skin. J Invest Dermatol 112:692–698
Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, De Waal-Malefyt R, Coffman RL, Hawrylowicz CM, O’Garra A (2002) In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med 195:603–616
Becklund BR, Severson KS, Vang SV, Deluca HF (2010) UVB radiation suppresses experimental autoimmune encephalomyelitis independent of vitamin D production. Proc Natl Acad Sci U S A 107:6418–6423
Beissert S, Hosoi J, Kuhn R, Rajewsky K, Muller W, Granstein RD (1996) Impaired immunosuppressive response to ultraviolet radiation in interleukin-10-deficient mice. J Invest Dermatol 107:553–557
Bennett JL, Blanchet MR, Zhao L, Zbytnuik L, Antignano F, Gold M, Kubes P, McNagny KM (2009) Bone marrow-derived mast cells accumulate in the central nervous system during inflammation but are dispensable for experimental autoimmune encephalomyelitis pathogenesis. J Immunol 182:5507–5514
Benveniste J, Henson PM, Cochrane CG (1972) Leukocyte-dependent histamine release from rabbit platelets. The role of IgE, basophils, and a platelet-activating factor. J Exp Med 136:1356–1377
Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK (2004) Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med 200:79–87
Biggs L, Yu C, Fedoric B, Lopez AF, Galli SJ, Grimbaldeston MA (2010) Evidence that vitamin D(3) promotes mast cell-dependent reduction of chronic UVB-induced skin pathology in mice. J Exp Med 207:455–463
Bodogai M, Lee Chang C, Wejksza K, Lai J, Merino M, Wersto RP, Gress RE, Chan AC, Hesdorffer C, Biragyn A (2013) Anti-CD20 antibody promotes cancer escape via enrichment of tumor-evoked regulatory B cells expressing low levels of CD20 and CD137L. Cancer Res 73:2127–2138
Brown EL, Rivas JM, Ullrich SE, Young CR, Norris SJ, Kripke ML (1995) Modulation of immunity to Borrelia burgdorferi by ultraviolet irradiation: differential effect on Th1 and Th2 immune responses. Eur J Immunol 25:3017–3022
Bruder D, Probst-Kepper M, Westendorf AM, Geffers R, Beissert S, Loser K, Von Boehmer H, Buer J, Hansen W (2004) Neuropilin-1: a surface marker of regulatory T cells. Eur J Immunol 34:623–630
Byrne SN, Ahmed J, Halliday GM (2005) Ultraviolet B but not A radiation activates suppressor B cells in draining lymph nodes. Photochem Photobiol 81:1366–1370
Byrne SN, Beaugie C, O’Sullivan C, Leighton S, Halliday GM (2011) The immune-modulating cytokine and endogenous Alarmin interleukin-33 is upregulated in skin exposed to inflammatory UVB radiation. Am J Pathol 179:211–222
Byrne SN, Halliday GM (2005) B cells activated in lymph nodes in response to ultraviolet irradiation or by interleukin-10 inhibit dendritic cell induction of immunity. J Invest Dermatol 124:570–578
Byrne SN, Limon-Flores AY, Ullrich SE (2008) Mast cell migration from the skin to the draining lymph nodes upon ultraviolet irradiation represents a key step in the induction of immune suppression. J Immunol 180:4648–4655
Calderon TM, Eugenin EA, Lopez L, Kumar SS, Hesselgesser J, Raine CS, Berman JW (2006) A role for CXCL12 (SDF-1alpha) in the pathogenesis of multiple sclerosis: regulation of CXCL12 expression in astrocytes by soluble myelin basic protein. J Neuroimmunol 177:27–39
Calenoff E (2012) Interplaying factors that effect multiple sclerosis causation and sustenance. ISRN Neurol 2012:851541
Chacon-Salinas R, Chen L, Chavez-Blanco AD, Limon-Flores AY, Ma Y, Ullrich SE (2014) An essential role for platelet-activating factor in activating mast cell migration following ultraviolet irradiation. J Leukoc Biol 95:139–148
Chacon-Salinas R, Limon-Flores AY, Chavez-Blanco AD, Gonzalez-Estrada A, Ullrich SE (2011) Mast cell-derived IL-10 suppresses germinal center formation by affecting T follicular helper cell function. J Immunol 186:25–31
Cheng LE, Hartmann K, Roers A, Krummel MF, Locksley RM (2013) Perivascular mast cells dynamically probe cutaneous blood vessels to capture immunoglobulin E. Immunity 38:166–175
Christophi GP, Gruber RC, Panos M, Christophi RL, Jubelt B, Massa PT (2012) Interleukin-33 upregulation in peripheral leukocytes and CNS of multiple sclerosis patients. Clin Immunol 142:308–319
Christy AL, Walker ME, Hessner MJ, Brown MA (2013) Mast cell activation and neutrophil recruitment promotes early and robust inflammation in the meninges in EAE. J Autoimmun 42:50–61
Cooper KD, Oberhelman L, Hamilton TA, Baadsgaard O, Terhune M, Levee G, Anderson T, Koren H (1992) UVB exposure reduces immunization rates and promotes tolerance to epicutaneous antigens in humans: relationship to dose, CD1a-DR+ epidermal macrophage induction, and Langerhans cell depletion. Proc Natl Acad Sci U S A 89:8497–8501
Correale J, Farez MF (2013) Modulation of multiple sclerosis by sunlight exposure: role of cis-urocanic acid. J Neuroimmunol 261:134–140
Damian DL, Kim YJ, Dixon KM, Haliday GM, Javeri A, Mason RS (2010) Topical calcitriol protects from UVB-induced genetic damage but suppresses cutaneous immunity in humans. Exp Dermatol 19:e23–e30
De Fabo EC, Noonan FP (1983) Mechanism of immune suppression by ultraviolet irradiation in vivo. I. Evidence for the existence of a unique photoreceptor in skin and its role in photoimmunology. J Exp Med 158:84–98
DeLuca HF, Plum LA (2011) Vitamin D deficiency diminishes the severity and delays onset of experimental autoimmune encephalomyelitis. Arch Biochem Biophys 513:140–143
Dietsch GN, Hinrichs DJ (1989) The role of mast cells in the elicitation of experimental allergic encephalomyelitis. J Immunol 142:1476–1481
Ding Q, Lu L, Wang B, Zhou Y, Jiang Y, Zhou X, Xin L, Jiao Z, Chou KY (2006) B7H1-Ig fusion protein activates the CD4+ IFN-gamma receptor+ type 1 T regulatory subset through IFN-gamma-secreting Th1 cells. J Immunol 177:3606–3614
Dobson R, Giovannoni G, Ramagopalan S (2013) The month of birth effect in multiple sclerosis: systematic review, meta-analysis and effect of latitude. J Neurol Neurosurg Psychiatry 84:427–432
Dumay O, Karam A, Vian L, Moyal D, Hourseau C, Stoebner P, Peyron JL, Meynadier J, Cano JP, Meunier L (2001) Ultraviolet AI exposure of human skin results in langerhans cell depletion and reduction of epidermal antigen-presenting cell function: partial protection by a broad-spectrum sunscreen. Br J Dermatol 144:1161–1168
El-Ghorr AA, Norval M (1995) A monoclonal antibody to cis-urocanic acid prevents the ultraviolet-induced changes in Langerhans cells and delayed hypersensitivity responses in mice, although not preventing dendritic cell accumulation in lymph nodes draining the site of irradiation and contact hypersensitivity responses. J Invest Dermatol 105:264–268
El-Ghorr AA, Norval M (1997) The role of interleukin-4 in ultraviolet B light-induced immunosuppression. Immunology 92:26–32
Elmets CA, Bergstresser PR, Tigelaar RE, Wood PJ, Streilein JW (1983) Analysis of the mechanism of unresponsiveness produced by haptens painted on skin exposed to low dose ultraviolet radiation. J Exp Med 158:781–794
Embry AF, Snowdon LR, Vieth R (2000) Vitamin D and seasonal fluctuations of gadolinium-enhancing magnetic resonance imaging lesions in multiple sclerosis. Ann Neurol 48:271–272
Endoh I, Di Girolamo N, Hampartzoumian T, Cameron B, Geczy CL, Tedla N (2007) Ultraviolet B irradiation selectively increases the production of interleukin-8 in human cord blood-derived mast cells. Clin Exp Immunol 148:161–167
Fabriek BO, Zwemmer JN, Teunissen CE, Dijkstra CD, Polman CH, Laman JD, Castelijns JA (2005) In vivo detection of myelin proteins in cervical lymph nodes of MS patients using ultrasound-guided fine-needle aspiration cytology. J Neuroimmunol 161:190–194
Fernandes De Abreu DA, Ibrahim EC, Boucraut J, Khrestchatisky M, Feron F (2010) Severity of experimental autoimmune encephalomyelitis is unexpectedly reduced in mice born to vitamin D-deficient mothers. J Steroid Biochem Mol Biol 121:250–253
Feyerabend TB, Weiser A, Tietz A, Stassen M, Harris N, Kopf M, Radermacher P, Moller P, Benoist C, Mathis D, Fehling HJ, Rodewald HR (2011) Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity 35:832–844
Fisher MS, Kripke ML (1977) Systemic alteration induced in mice by ultraviolet light irradiation and its relationship to ultraviolet carcinogenesis. Proc Natl Acad Sci U S A 74:1688–1692
Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U (2002) Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci 22:RC216
Frisullo G, Nociti V, Iorio R, Plantone D, Patanella AK, Tonali PA, Batocchi AP (2010) CD8(+)Foxp3(+) T cells in peripheral blood of relapsing-remitting multiple sclerosis patients. Hum Immunol 71:437–441
Fujio K, Okamura T, Yamamoto K (2010) The family of IL-10-secreting CD4+ T cells. Adv Imunol 105:99–130
Fukunaga A, Khaskhely NM, Ma Y, Sreevidya CS, Taguchi K, Nishigori C, Ullrich SE (2010) Langerhans cells serve as immunoregulatory cells by activating NKT cells. J Immunol 185:4633–4640
Fukunaga A, Khaskhely NM, Sreevidya CS, Byrne SN, Ullrich SE (2008) Dermal dendritic cells, and not Langerhans cells, play an essential role in inducing an immune response. J Immunol 180:3057–3064
Gauchat JF, Henchoz S, Mazzei G, Aubry JP, Brunner T, Blasey H, Life P, Talabot D, Flores-Romo L, Thompson J et al (1993) Induction of human IgE synthesis in B cells by mast cells and basophils. Nature 365:340–343
Genain CP, Cannella B, Hauser SL, Raine CS (1999) Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med 5:170–175
Glass MJ, Bergstresser PR, Tigelaar RE, Streilein JW (1990) UVB radiation and DNFB skin painting induce suppressor cells universally in mice. J Invest Dermatol 94:273–278
Gorman S, Tan JW, Thomas JA, Townley SL, Stumbles PA, Finlay-Jones JJ, Hart PH (2005) Primary defect in UVB-induced systemic immunomodulation does not relate to immature or functionally impaired APCs in regional lymph nodes. J Immunol 174:6677–6685
Gorman S, Kuritzky LA, Judge MA, Dixon KM, McGlade JP, Mason RS, Finlay-Jones JJ, Hart PH (2007) Topically applied 1,25-dihydroxyvitamin D3 enhances the suppressive activity of CD4+CD25+ cells in the draining lymph nodes. J Immunol 179:6273–6283
Gorman S, Scott NM, Tan DH, Weeden CE, Tuckey RC, Bisley JL, Grimbaldeston MA, Hart PH (2012) Acute erythemal ultraviolet radiation causes systemic immunosuppression in the absence of increased 25-hydroxyvitamin D3 levels in male mice. PLoS ONE 7:e46006
Green I, Stingl G, Shevach EM, Katz SI (1980) Antigen presentation and allogeneic stimulation by langerhans cells. J Invest Dermatol 75:44–45
Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P (1995) The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83:793–802
Grimbaldeston MA, Nakae S, Kalesnikoff J, Tsai M, Galli SJ (2007) Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol 8:1095–1104
Group TLMSSGATUOBCMMA (1999) TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and the University of British Columbia MS/MRI Analysis Group. Neurology 53:457–465
Groux H, Bigler M, De Vries JE, Roncarolo MG (1996) Interleukin-10 induces a long-term antigen-specific anergic state in human CD4+ T cells. J Exp Med 184:19–29
Groux H, O’Garra A, Bigler M, Rouleau M, Aantonenko S, De Vries JE, Roncarolo MG (1997) A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 389:737–742
Gruber-Wackernagel A, Bambach I, Legat FJ, Hofer A, Byrne SN, Quehenberger F, Wolf P (2011) Randomized double-blinded placebo-controlled intra-individual trial on topical treatment with a 1,25-dihydroxyvitamin D(3) analogue in polymorphic light eruption. Br J Dermatol 165:152–163
Hart PH, Grimbaldeston MA, Swift GJ, Jaksic A, Noonan FP, Finlay-Jones JJ (1998) Dermal mast cells determine susceptibility to ultraviolet B-induced systemic suppression of contact hypersensitivity responses in mice. J Exp Med 187:2045–2053
Hart PH, Grimbaldeston MA, Jaksic A, Tan JE, Swift GJ, Hosszu EK, Halliday GM, Finlay-Jones JJ (2000) Ultraviolet B-induced suppression of immune responses in interleukin-4−/− mice: relationship to dermal mast cells. J Invest Dermatol 114:508–513
Hart PH, Gorman S, Finlay-Jones JJ (2011) Modulation of the immune system by UVB radiation: more than just the effects of vitamin D? Nat Rev Immunol 11:584–596
Hauser SL, Doolittle TH, Lincoln R, Brown RH, Dinarello CA (1990) Cytokine accumulations in CSF of multiple sclerosis patients: frequent detection of interleukin-1 and tumor necrosis factor but not interleukin-6. Neurology 40:1735–1739
Inoue M, Williams KL, Oliver T, Vandenabeele P, Rajan JV, Miao EA, Shinohara ML (2012) Interferon-beta therapy against eae is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal 5:ra38
Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM, Szabolcs PM, Brenstein SH, Magro CM, Williams AD, Hall RP, St Clair EW, Tedder TF (2011) Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 117:530–541
Jäger A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK (2009) Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol 183:7169–7177
Jiang HR, Milovanovic M, Allan D, Niedbala W, Besnard AG, Fukada SY, Alves-Filho JC, Togbe D, Goodyear CS, Linington C, Xu D, Lukic ML, Liew FY (2012) IL-33 attenuates EAE by suppressing IL-17 and IFN-gamma production and inducing alternatively activated macrophages. Eur J Immunol 42:1804–1814
Kammeyer A, Pavel S, Asghar SS, Bos JD, Teunissen MB (1997) Prolonged increase of cis-urocanic acid levels in human skin and urine after single total-body ultraviolet exposures. Photochem Photobiol 65:593–598
Kappos L, Hartung HP, Freedman MS, Boyko A, Wilhelm Radü E, Mikol DD, Lamarine M, Hyvert Y, Freudensprung U, Plitz T, Van Beek J (2014) Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol 13:353–363
Kandolf-Sekulovic L, Kataranovski M, Pavlovic MD (2003) Immunomodulatory effects of low-intensity near-infrared laser irradiation on contact hypersensitivity reaction. Photodermatol Photoimmunol Photomed 19:203–212
Katz SI, Parker D, Turk JL (1974) B-cell suppression of delayed hypersensitivity reactions. Nature 251:550–551
Keown MB, Henry AJ, Ghirlando R, Sutton BJ, Gould HJ (1998) Thermodynamics of the interaction of human immunoglobulin E with its high-affinity receptor Fc epsilon RI. Biochemistry 37:8863–8869
Kihara Y, Ishii S, Kita Y, Toda A, Shimada A, Shimizu T (2005) Dual phase regulation of experimental allergic encephalomyelitis by platelet-activating factor. J Exp Med 202:853–863
Kohler RE, Comerford I, Townley S, Haylock-Jacobs S, Clark-Lewis I, McColl SR (2008) Antagonism of the chemokine receptors CXCR3 and CXCR4 reduces the pathology of experimental autoimmune encephalomyelitis. Brain Pathol 18:504–516
Kohm AP, Carpentier PA, Anger HA, Miller SD (2002) Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol 169:4712–4716
Kubera N, Lin AH, Kenis G, Bosmans E, Van Bockstaele D, Maes M (2001) Anti-inflammatory effects of antidepressants through suppression of the interferon-gamma/interleukin-10 production ratio. J Clin Psychopharmacol 21:199–206
Kurban S, Akpinar Z, Mehmetoglu I (2008) Receptor activator of nuclear factor kappaB ligand (RANKL) and osteoprotegerin levels in multiple sclerosis. Multiple Scler 14:431–432
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201:233–240
Li HM, Nourbakhsh B, Safavi F, Li K, Xu H, Cullimore M, Zhou F, Zhang GX, Rostami A (2011) Kit (W-sh) mice develop earlier and more severe experimental autoimmune encephalomyelitis due to absence of immune suppression. J Immunol 187:274–282
Li M, Li Y, Liu X, Gao X, Wang Y (2012) IL-33 blockade suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. J Neuroimmunol 247:25–31
Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L (2002) Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 8:500–508
Loser K, Beissert S (2012) Regulatory T cells: banned cells for decades. J Invest Dermatol 132:864–871
Loser K, Mehling A, Loeser S, Apelt J, Kuhn A, Grabbe S, Schwarz T, Penninger JM, Beissert S (2006) Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nat Med 12:1372–1379
Loser K, Apelt J, Voskort M, Mohaupt M, Balkow S, Schwarz T, Grabbe S, Beissert S (2007) IL-10 controls ultraviolet-induced carcinogenesis in mice. J Immunol 179:365–371
Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A, Yogev N, Gu Y, Khodoun M, Hildeman D, Boespflug N, Fogolin MB, Grobe L, Greweling M, Finkleman FD, Cardin R, Mohrs M, Muller W, Waisman A, Roers A, Karp CL (2009) Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J Immunol 183:2312–2320
Marathe GK, Johnson C, Billings SD, Southall MD, Pei Y, Spandau D, Murphy RC, Zimmerman GA, McIntyre TM, Travers JB (2005) Ultraviolet B radiation generates platelet-activating factor-like phospholipids underlying cutaneous damage. J Biol Chem 280:35448–35457
Mars LT, Saikali P, Liblau RS, Arbour N (2011) Contribution of CD8 T lymphocytes to the immuno-pathogenesis of multiple sclerosis and its animal models. Biochim Biophys Acta 1812:151–161
Matsumura Y, Byrne SN, Nghiem DX, Miyahara Y, Ullrich SE (2006) A role for inflammatory mediators in the induction of immunoregulatory B cells. J Immunol 177:4810–4817
Matsushita T, Yanaba K, Bouaziz JD, Fufimoto M, Tedder TF (2008) Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest 118:3420–3430
Mauri C, Bosma A (2012) Immune regulatory function of B cells. Ann Rev Immunol 30:221–241
McCandless EE, Wang Q, Woerner BM, Harper JM, Klein RS (2006) CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J Immunol 177:8053–8064
McCandless EE, Piccio L, Woerner BM, Schmidt RE, Rubin JB, Cross AH, Klein RS (2008) Pathological expression of CXCL12 at the blood-brain barrier correlates with severity of multiple sclerosis. Am J Pathol 172:799–808
McDowell TY, Amr S, Langenberg P, Royal W, Bever C, Culpepper WJ, Bradham DD (2010) Time of birth, residential solar radiation and age at onset of multiple sclerosis. Neuroepidemiology 34:238–244
McGeachy MJ, Stephens LA, Anderton SM (2005) Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol 175:3025–3032
McGlade JP, Gorman S, Zosky GR, Larcombe AN, Sly PD, Finlay-Jones JJ, Turner DJ, Hart PH (2007) Suppression of the asthmatic phenotype by ultraviolet B-induced, antigen-specific regulatory cells. Clin Exp Allergy 37:1267–1276
McMichael AJ, Hall AJ (1997) Does immunosuppressive ultraviolet radiation explain the latitude gradient for multiple sclerosis? Epidemiology 8:642–645
Meiron M, Zohar Y, Anunu R, Wildbaum G, Karin N (2008) CXCL12 (SDF-1alpha) suppresses ongoing experimental autoimmune encephalomyelitis by selecting antigen-specific regulatory T cells. J Exp Med 205:2643–2655
Meunier L, Bata-Csorgo Z, Cooper KD (1995) In human dermis, ultraviolet radiation induces expansion of a CD36+ CD11b+ CD1− macrophage subset by infiltration and proliferation; CD1+ langerhans-like dendritic antigen-presenting cells are concomitantly depleted. J Invest Dermatol 105:782–788
Milliken SV, Wassall H, Lewis BJ, Logie J, Barker RN, MacDonald H, Vickers MA, Ormerod AD (2012) Effects of ultraviolet light on human serum 25-hydroxyvitamin D and systemic immune function. J Allergy Clin Immunol 129:1554–1561
Minty A, Chalon P, Derocq JM, Dumont X, Guillemot JC, Kaghad M, Labit C, Leplatois P, Liauzun P, Miloux B, Minty C, Casellas P, Loison G, Lupker J, Shire D, Ferrara P, Caput D (1993) Interleukin-13 is a new human lymphokine regulating inflammatory and immune-responses. Nature 362:248–250
Mohr SB, Garland CF, Gorham ED, Garland FC (2008) The association between ultraviolet B irradiance, vitamin D status and incidence rates of type 1 diabetes in 51 regions worldwide. Diabetologia 51:1391–1398
Moodycliffe AM, Kimber I, Norval M (1994) Role of tumour necrosis factor-alpha in ultraviolet B light-induced dendritic cell migration and suppression of contact hypersensitivity. Immunology 81:79–84
Moodycliffe AM, Nghiem D, Clydesdale G, Ullrich SE (2000) Immune suppression and skin cancer development: regulation by NKT cells. Nat Immunol 1:521–525
Mostert JP, Admiraal-Behloul F, Hoogduin JM, Luyendijk J, Heersema DJ, Van Buchem MA, De Keyser J (2008) Effects of fluoxetine on disease activity in relapsing multiple sclerosis: a double-blind, placebo-controlled, exploratory study. J Neurol Neurosurg Psychiatry 79:1027–1031
Muili KA, Gopalakrishnan S, Meyer SL, Eells JT, Lyons JA (2012) Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by photobiomodulation induced by 670 nm light. PLoS ONE 7:e30655
Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A (2006) Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. J Am Med Assoc 296:2832–2838
Newcomb DC, Zhou W, Moore ML, Goleniewska K, Hershey GK, Kollis JK, Peebles RS Jr (2009) A functional IL-13 receptor is expressed on polarized murine CD4+ Th17 cells and IL-13 signaling attenuates Th17 cytokine production. J Immunol 182:5317–5321
Newcomb DC, Boswell MG, Huckabee MM, Goleniewska K, Dulek DE, Reiss S, Lukacs NW, Kolls JK, Peebles RS Jr (2012) IL-13 regulates Th17 secretion of IL-17A in an IL-10-dependent manner. J Immunol 188:1027–1035
Ng RL, Bisley JL, Gorman S, Norval M, Hart PH (2010) Ultraviolet irradiation of mice reduces the competency of bone marrow-derived CD11c+ cells via an indomethacin-inhibitable pathway. J Immunol 185:7207–7215
Ng RL, Scott NM, Bisley JL, Lambert MJ, Gorman S, Norval M, Hart PH (2013a) Characterization of regulatory dendritic cells differentiated from the bone marrow of UVB-irradiated mice. Immunology 140:399–412
Ng RL, Scott NM, Strickland DH, Gorman S, Grimbaldeston MA, Norval M, Waithman J, Hart PH (2013b) Altered immunity and dendritic cell activity in the periphery of mice after long-term engraftment with bone marrow from ultraviolet-irradiated mice. J Immunol 190:5471–5484
Noonan FP, De Fabo EC, Morrison H (1988) Cis-urocanic acid, a product formed by ultraviolet B irradiation of the skin, initiates an antigen presentation defect in splenic dendritic cells in vivo. J Invest Dermatol 90:92–99
Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, Nambu A, Abe T, Kiyonari H, Matsumoto K, Sudo K, Okumura K, Saito H, Nakae S (2010) IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A 107:18581–18586
Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, Malchinkhuu E, Wersto RP, Biragyn A (2011) Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res 71:3505–3515
Olsson Y (1974) Mast cells in plaques of multiple sclerosis. Acta Neurol Scand 50:611–618
Oyaizu N, Uemura Y, Izumi H, Morii S, Nishi M, Hioki K (1985) Eosinophilic gastroenteritis. Immunohistochemical evidence for IgE mast cell-mediated allergy. Acta Pathol Jpn 35:759–766
Pascher G (1962) Cis- and trans-urocanic acid as a component of the corneum stratum. Arch Klin Exp Dermatol 214:234–239
Pashenkov M, Söderström M, Link H (2003) Secondary lymphoid organ chemokines are elevated in the cerebrospinal fluid during central nervous system inflammation. J Neuroimmunol 135:154–160
Pei Y, Barber LA, Murphy RC, Johnson CA, Kelley SW, Dy LC, Fertel RH, Nguyen TM, Williams DA, Travers JB (1998) Activation of the epidermal platelet-activating factor receptor results in cytokine and cyclooxygenase-2 biosynthesis. J Immunol 161:1954–1961
Pfeiffer JR, Seagrave JC, Davis BH, Deanin GG, Oliver JM (1985) Membrane and cytoskeletal changes associated with IgE-mediated serotonin release from rat basophilic leukemia cells. J Cell Biol 101:2145–2155
Piconese S, Costanza M, Musio S, Tripodo C, Poliani PL, Gri G, Burocchi A, Pittoni P, Gorzanelli A, Colombo MP, Pedotti R (2011) Exacerbated experimental autoimmune encephalomyelitis in mast-cell-deficient Kit W-sh/W-sh mice. Lab Invest 91:627–641
Ponsonby AL, Lucas RM, Van der Mei IA (2005) UVBR, vitamin D and three autoimmune diseases–multiple sclerosis, type 1 diabetes, rheumatoid arthritis. Photochem Photobiol 81:1267–1275
Rafei M, Hsieh J, Zehntner S, Li M, Forner K, Birman E, Boivin MN, Young YK, Perreault C, Galipeau J (2009) A granulocyte-macrophage colony-stimulating factor and interleukin-15 fusokine induces a regulatory B cell population with immune suppressive properties. Nat Med 15:1038–1045
Rana S, Rogers LJ, Halliday GM (2011) Systemic low-dose UVB inhibits CD8 T cells and skin inflammation by alternative and novel mechanisms. Am J Pathol 178:2783–2791
Rivas JM, Ullrich SE (1994) The role of IL-4, IL-10, and TNF-alpha in the immune suppression induced by ultraviolet radiation. J Leukoc Biol 56:769–775
Rola-Pleszczynski M, Pouloit C, Turcotte S, Pignol B, Braquet P, Bouvbrette L (1988) Immune regulation by platelet-activating factor. I. Induction of suppressor cell activity in human monocytes and CD8+ T cells and of helper cell activity in CD4+ T cells. J Immunol 140:3547–3552
Roncador G, Brown PJ, Maestre L, Hue S, Martinez-Torrecuadrada JL, Ling KL, Pratap S, Toms C, Fox BC, Cerundolo V, Powrie F, Banham AH (2005) Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single-cell level. Eur J Immunol 35:1681–1691
Roncarolo MG, Battaglia M (2007) Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat Rev Immunol 7:585–598
Rottem M, Bbarbieri S, Kinet JP, Metcalfe DD (1992) Kinetics of the appearance of Fc epsilon RI-bearing cells in interleukin-3-dependent mouse bone marrow cultures: correlation with histamine content and mast cell maturation. Blood 79:972–980
Rozniecki JJ, Hauser SL, Stein M, Lincoln R, Theoharides TC (1995) Elevated mast cell tryptase in cerebrospinal fluid of multiple sclerosis patients. Ann Neurol 37:63–66
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M (1995) Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155:1151–1164
Sarchio SN, Kok LF, O’Sullivan C, Halliday GM, Byrne SN (2012) Dermal mast cells affect the development of sunlight-induced skin tumours. Exp Dermatol 21:241–248
Sarchio SN, Scolyer RA, Beaugie C, McDonald D, Marsh-Wakefield F, Halliday GM, Byrne SN (2014) Pharmacologically antagonizing the CXCR4-CXCL12 chemokine pathway with AMD3100 inhibits sunlight-induced skin cancer. J Invest Dermatol 134:1091–1100
Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE et al (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476:214–219
Sayed BA, Walker ME, Brown MA (2011) Cutting edge: mast cells regulate disease severity in a relapsing-remitting model of multiple sclerosis. J Immunol 186:3294–3298
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X et al (2005) IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23:479–490
Schwarz T (2008) 25 years of UVB-induced immunosuppression mediated by T cells-from disregarded T suppressor cells to highly respected regulatory T cells. Photochem Photobiol 84:10–18
Schwarz A, Beissert S, Grosse-Hietmeyer K, Gunzer M, Bluestone JA, Grabbe S, Schwarz T (2000) Evidence for functional relevance of CTLA-4 in ultraviolet-radiation-induced tolerance. J Immunol 165:1824–1831
Schwarz A, Navid F, Sparwasser T, Clausen BE, Schwarz T (2011) In vivo reprogramming of UVB radiation-induced regulatory T-cell migration to inhibit the elicitation of contact hypersensitivity. J Allergy Clin Immunol 128:826–833
Schwarz A, Navid F, Sparwasser T, Clausen BE, Schwarz T (2012) 1,25-dihydroxyvitamin D exerts similar immunosuppressive effects as UVBR but is dispensable for local UVBR-induced immunosuppression. J Invest Dermatol 132:2762–2769
Scott NM, Ng RL, Strickland DH, Bisley JL, Bazely SA, Gorman S, Norval M, Hart PH (2012) Toward homeostasis: regulatory dendritic cells from the bone marrow of mice with inflammation of the airways and peritoneal cavity. Am J Pathol 181:535–547
Secor VH, Secor WE, Gutekunst CA, Brown MA (2000) Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med 191:813–822
Shapira Y, Agmon-Levin N, Shoenfeld Y (2010) Geoepidemiology of autoimmune rheumatic diseases. Nat Rev Rheumatol 6:468–476
Sharief MK, Hentges R (1991) Association between tumor-necrosis-factor-alpha and disease progression in patients with multiple-sclerosis. N Engl J Med 325:467–472
Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, Ries S, Dang VD, Jaimes Y, Daridon C et al (2014) IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507:366–370
Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S (2002) Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol 3:135–142
Shreedhar V, Giese T, Sung VW, Ullrich SE (1998a) A cytokine cascade including prostaglandin E2, IL-4, and IL-10 is responsible for UVB-induced systemic immune suppression. J Immunol 160:3783–3789
Shreedhar VK, Pride MW, Sun Y, Kripke ML, Strickland FM (1998b) Origin and characteristics of ultraviolet-B radiation-induced suppressor T lymphocytes. J Immunol 161:1327–1335
Simpson S Jr, Blizzard L, Otahal P, van der Mei I, Taylor B (2011) Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. J Neurol Neurosurg Psychiatry 82:1132–1141
Singh TP, Schon MP, Wallbrecht K, Michaelis K, Rinner B, Mayer G, Schmidbauer U, Strohmaier H, Wang XJ, Wolf P (2010) 8-methoxypsoralen plus ultraviolet A therapy acts via inhibition of the IL-23/Th17 axis and induction of Foxp3+ regulatory T cells involving CTLA4 signaling in a psoriasis-like skin disorder. J Immunol 184:7257–7267
Skov L, Hansen H, Allen M, Villadsen L, Norval M, Barker JN, Simon J, Baadsgaard O (1998) Contrasting effects of ultraviolet al and ultraviolet B exposure on the induction of tumour necrosis factor-alpha in human skin. Br J Dermatol 138:216–220
Sleijffers A, Garssen J, De Gruijl FR, Boland GJ, Van Hattum J, Van Vloten WA, Van Loveren H (2002) UVB exposure impairs immune responses after hepatitis B vaccination in two different mouse strains. Photochem Photobiol 75:541–546
Solomon BD, Mueller C, Chae WJ, Alabanza LM, Bynoe MS (2011) Neuropilin-1 attenuates autoreactivity in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 108:2040–2045
Staples J, Ponsonby AL, Lim L (2010) Low maternal exposure to ultraviolet radiation in pregnancy, month of birth, and risk of multiple sclerosis in offspring: longitudinal analysis. Br Med J 340:c1640
Stein MS, Liu Y, Gray OM, Baker JE, Kolbe SC, Ditchfield MR, Egan GF, Mitchell PJ, Harrison LC, Butzkueven H et al (2011) A randomized trial of high-dose vitamin D2 in relapsing-remitting multiple sclerosis. Neurology 77:1611–1618
Steinman RM, Cohn ZA (1973) Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med 137:1142–1162
Steinman RM, Witmer MD, Nussenzweig MC, Chen LL, Schlesinger S, Cohn ZA (1980) Dendritic cells of the mouse: identification and characterization. J Invest Dermatol 75:14–16
Stene LC, Joner G, Group NCDS (2003) Use of cod liver oil during the first year of life is associated with lower risk of childhood-onset type 1 diabetes: a large, population-based, case-control study. Am J Clin Nutr 78:1128–1134
Stohlman SA, Pei L, Cua DJ, Li Z, Hinton DR (1999) Activation of regulatory cells suppresses experimental allergic encephalomyelitis via secretion of IL-10. J Immunol 163:6338–6344
Streilein JW, Toews GT, Gilliam JN, Bergstresser PR (1980) Tolerance or hypersensitivity to 2,4-dinitro-1-fluorobenzene: the role of langerhans cell density within epidermis. J Invest Dermatol 74:319–322
Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM (2008) Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med 14:337–342
Sun JB, Czerkinsky C, Holmgren J (2012) B lymphocytes treated in vitro with antigen coupled to cholera toxin B subunit induce antigen-specific Foxp3(+) regulatory T cells and protect against experimental autoimmune encephalomyelitis. J Immunol 188:1686–1697
Sweeney CM, Lonergan R, Basdeo SA, Kinsella K, Dungan LS, Higgins SC, Kelly PJ, Costelloe L, Tubridy N, Mills KH et al (2011) IL-27 mediates the response to IFN-beta therapy in multiple sclerosis patients by inhibiting Th17 cells. Brain Behav Immun 25:1170–1181
Tennakoon DK, Mehta RS, Ortega SB, Bhoj V, Racke MK, Karandikar NJ (2006) Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J Immunol 176:7119–7129
Teunissen MB, Piskin G, Di Nuzzo S, Sylva-Steenland RM, De Rie MA, Bos JD (2002) Ultraviolet B radiation induces a transient appearance of IL-4+ neutrophils, which support the development of Th2 responses. J Immunol 168:3732–3739
Toms R, Weiner HL, Johnson D (1990) Identification of IgE-positive cells and mast cells in frozen sections of multiple sclerosis brains. J Neuroimmunol 30:169–177
Travers JB, Berry D, Yao Y, Yi Q, Konger RL (2010) Ultraviolet B radiation of human skin generates platelet-activating factor receptor agonists. Photochem Photobiol 86:949–954
Tremlett H, van der Mei IA, Pittas F, Blizzard L, Paley G, Mesaros D, Woodbaker R, Nunez M, Dwyer T, Taylor BV, Ponsonby AL (2008) Monthly ambient sunlight, infections and relapse rates in multiple sclerosis. Neuroepidemiology 31:271–279
Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, Wang Z, Lang M, Stolz DB, Zheng XX et al (2011) IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol 187:4598–4610
Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L (2008) Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 172:146–155
Ullrich SE, Kripke ML (1984) Mechanisms in the suppression of tumor rejection produced in mice by repeated UVB irradiation. J Immunol 133:2786–2790
van der Mei IA, Ponsonby AL, Blizzard L, Dwyer T (2001) Regional variation in multiple sclerosis prevalence in Australia and its association with ambient ultraviolet radiation. Neuroepidemiology 20:168–174
van der Mei IA, Ponsonby AL, Dwyer T, Blizzard L, Simmons R, Taylor BV, Butzkueven H, Kilpatrick T (2003) Past exposure to sun, skin phenotype, and risk of multiple sclerosis: case-control study. Br Med J 327:316
Vela L, Garcia Merino A, Fernandez-Gallardo S, Sanchez Crespo M, Lopez Lozano JJ, Saus C (1991) Platelet-activating factor antagonists do not protect against the development of experimental autoimmune encephalomyelitis. J Neuroimmunol 33:81–86
Vink AA, Strickland FM, Bucana C, Cox PA, Roza L, Yarosh DB, Kripke ML (1996) Localization of DNA damage and its role in altered antigen-presenting cell function in ultraviolet-irradiated mice. J Exp Med 183:1491–1500
Vollmar P, Nessler S, KalluriI SR, Hartung HP, Hemmer B (2009) The antidepressant venlafaxine ameliorates murine experimental autoimmune encephalomyelitis by suppression of pro-inflammatory cytokines. Int J Neuropsychopharmacol 12:525–536
Walsh LJ (1995) Ultraviolet B irradiation of skin induces mast cell degranulation and release of tumour necrosis factor-alpha. Immunol Cell Biol 73:226–233
Walterscheid JP, Nghiem DX, Kazimi N, Nutt LK, McConkey DJ, Norval M, Ullrich SE (2006) Cis-urocanic acid, a sunlight-induced immunosuppressive factor, activates immune suppression via the 5-HT2A receptor. Proc Natl Acad Sci U S A 103:17420–17425
Walterscheid JP, Ullrich SE, Nghiem DX (2002) Platelet-activating factor, a molecular sensor for cellular damage, activates systemic immune suppression. PAF can suppress the adaptive immune response. J Exp Med 195:171–179
Wang Y, Marling SJ, Zhu JG, Severson KS, Deluca HF (2012) Development of experimental autoimmune encephalomyelitis (EAE) in mice requires vitamin D and the vitamin D receptor. Proc Natl Acad Sci U S A 109:8501–8504
Wang Y, Marling SJ, McKnight SM, Danielson AL, Severson KS, Deluca HF (2013) Suppression of experimental autoimmune encephalomyelitis by 300–315 nm ultraviolet light. Arch Biochem Biophys 536:81–86
Williams SK, Maier O, Fischer R, Fairless R, Hochmeister S, Stojic A, Pick L, Haar D, Musiol S, Storch MK et al (2014) Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS ONE 9:e90117
Wolf P, Nghiem DX, Walterscheid JP, Byrne S, Matsumura Y, Bucana C, Aanathaswamy HN, Ullrich SE (2006) Platelet-activating factor is crucial in psoralen and ultraviolet A-induced immune suppression, inflammation, and apoptosis. Am J Pathol 169:795–805
Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF (2008a) A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 28:639–650
Yanaba K, Bouaziz JD, Matsushita T, Magro CM, St Clair EW, Tedder TF (2008b) B-lymphocyte contributions to human autoimmune disease. Immunol Rev 223:284–299
Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z et al (2008) Regulation of inflammatory responses by IL-17F. J Exp Med 205:1063–1075
Zamvil SS, Mitchell DJ, Moore AC, Kitamura K, Steinman L, Rothbard JB (1986) T-cell epitope of the autoantigen myelin basic protein that induces encephalomyelitis. Nature 324:258–260
Zhang Q, Yao Y, Konger RL, Sinn AL, Cai S, Pollok KE, Travers JB (2008) UVB radiation-mediated inhibition of contact hypersensitivity reactions is dependent on the platelet-activating factor system. J Invest Dermatol 128:1780–1787
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Marsh-Wakefield, F., Byrne, S.N. (2015). Photoimmunology and Multiple Sclerosis. In: La Flamme, A., Orian, J. (eds) Emerging and Evolving Topics in Multiple Sclerosis Pathogenesis and Treatments. Current Topics in Behavioral Neurosciences, vol 26. Springer, Cham. https://doi.org/10.1007/7854_2014_359
Download citation
DOI: https://doi.org/10.1007/7854_2014_359
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-25541-5
Online ISBN: 978-3-319-25543-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)