
Abstract
Covalent targeting has experienced a revival in the last decade, especially in the area of protein kinase inhibitor development. Generally, covalent inhibitors make use of an electrophilic moiety often termed “warhead” to react with a nucleophilic amino acid, most frequently a cysteine. High efficacy and excellent selectivity in the kinome have been achieved by addressing poorly conserved, non-catalytic cysteine residues with so-called targeted covalent inhibitors (TCIs). Despite the challenges associated with covalent modifiers, application of the TCI approach for the discovery of new treatments has been very successful with six covalent kinase inhibitors having gained approval in the last few years. A multitude of reactive chemical probes and tool compounds has further been developed. Beside cysteine, other nucleophilic amino acids including tyrosine and lysine have also been addressed with suitable electrophiles and covalent-reversible chemistry has recently complemented our toolbox for designing covalent kinase inhibitors. Covalent ligands have also been used in the framework of chemical-genetics approaches or to tackle allosteric pockets, which are often difficult to address.
This chapter aims at providing a general introduction to covalent kinase inhibitors and an overview of the current state of research highlighting major targeting strategies, developments, and advances in this field. More detailed information on certain targets and approaches can be found in dedicated chapters of this book.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Chemical probes
- Electrophilic warheads
- Kinase inhibitors
- Structure-based drug design
- Targeted covalent inhibitors
1 Introduction
Covalent inhibitors have a long history in medicinal chemistry and various covalent modifiers, such as aspirin, β-lactam antibiotics or omeprazole, to name just a few, have been among the most frequently used drugs for decades [1]. However, many of the drug classes acting via a covalent mechanism have been discovered serendipitously. Due to concerns about their potential for haptenization and idiosyncratic toxicity as well as side effects or toxicity arising from irreversible off-target labeling, reactive compounds have long been regarded with skepticism by pharmaceutical companies [2, 3]. Since the beginning of the twenty-first century, however, we have seen a resurgence of covalent targeting strategies in medicinal chemistry. Targeted covalent inhibitors (TCIs) which have been defined as “inhibitors bearing a bond-forming functional group of low reactivity that, following binding to the target protein, is positioned to react rapidly with a specific non-catalytic residue at the target site” [1] are now becoming more and more common especially in the field of protein kinase drug discovery [4,5,6,7].
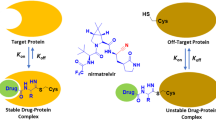
The renewed interest in covalent inhibitors is based on the growing awareness that a well-designed TCI can offer a variety of benefits over classical, non-covalently binding molecules (an excellent review summarizing the opportunities and pitfalls associated with the development of covalent-modifier drugs has recently been provided by De Cesco et al. [8]). Currently, most TCIs address non-catalytic cysteine residues with an electrophilic headgroup termed “warhead” [9], which is highlighted in red throughout this chapter. Thereby, TCIs can make use of the combined specificity of two orthogonal selectivity filters: (1) reversible recognition and (2) the covalent bond-forming reaction. Consequently (and counterintuitively), covalent targeting can increase selectivity provided that the intrinsic reactivity of the warhead is low enough to hit only residues juxtaposed by the reversible binding event [3, 7]. This second selectivity filter is particularly useful when targeting the protein kinases’ ATP pocket, which features a high overall similarity throughout the kinome but a relatively low degree of conservation in the so-called cysteinome (i.e., the entire set of cysteines present in the kinome) [7]. In addition, irreversible covalent binding eliminates the competition with natural ligands, substrates or co-factors (i.e., ATP in the case of kinases) thereby increasing overall efficacy [7]. Given the low millimolar ATP concentrations in cells, the lack of competitivity after covalent bond formation is a key advantage. Due to their time-dependent binding behavior, efficient irreversible inhibitors can achieve full target occupancy even at very low concentrations, provided that the exposure time is long enough. Thus, lower doses may be sufficient to achieve equivalent therapeutic efficacy compared to reversibly binding drugs. At the same time, the target protein remains inhibited until its function is restored by de novo synthesis. If the re-synthesis rate is not too high, persistent target engagement leads to a decoupling of pharmacokinetics (PK) from pharmacodynamics (PD) which can translate into prolonged dosage intervals even for high-clearance compounds and a lower side effect burden due to a decreased overall exposure [2]. Since covalent bond formation can be used as a powerful promoter of potency and selectivity, it can also enable the reduction of molecule size and lipophilicity. Thereby, molecular obesity [10] may be prevented, and physicochemical properties be improved.
Covalent-reversible targeting strategies can be a viable alternative when off-target labeling, GSH-mediated clearance, or haptenization is an issue, when sustained target engagement causes mechanism-based side effects, but also when high turnover targets need to be addressed. Ideally, the unmodified inhibitor dissociates after protein degradation to be “recycled” by engaging with a newly translated target protein. Moreover, this approach offers the potential to benefit from advantages of irreversible covalent inhibitors (e.g., prolonged target occupancy, increased potency, and selectivity) at a decreased risk of drug safety issues [11]. Remarkably, the target residence times of covalent-reversible inhibitors can be fine-tuned by both warhead chemistry and stabilization of the complex via non-covalent interactions, thus providing tailor-made solutions for the desired application. However, although these features hold the promise of enabling the design of better and safer kinase-targeted drugs, no covalent-reversible kinase inhibitors have been approved so far and critical evaluation of the benefit–risk balance in comparison to traditional non-reactive ligands will still be necessary.
As mentioned above, irreversible covalent inhibition is a non-equilibrium process. Due to its time-dependent nature, it can be accurately described neither by the equilibrium dissociation constant Ki (= koff/kon) nor by IC50 values [1]. TCI binding usually involves two steps (Fig. 1). In the first step, the inhibitor reversibly binds the target while covalent bond formation takes place in the second step. Importantly, only the first of these two steps is ATP-competitive. For irreversible covalent binders, the (reversible) binding affinity is described by the constant KI being the inhibitor concentration required to achieve the half-maximal rate of covalent inactivation (= kinact/2). It should be noted that KI does not equal Ki although these values converge for kinact << koff. The first-order rate constant kinact defines the maximal potential rate of covalent inactivation, i.e., the rate of covalent bond formation at full occupancy with the reversibly bound ligand. Accordingly, kinact represents a measure for the efficiency of the covalent inactivation step. kinact depends on several factors including the intrinsic reactivity of the warhead and the target nucleophile, but also the accurate positioning of both reactive groups in terms of distance and angle to favor the reaction. The appropriate measure for the overall efficiency of the two-step binding event is the quotient kinact/KI that is the second-order rate constant of covalent target inactivation [12]. The above descriptions refer to (quasi)-irreversible binders where the rate constant of covalent bond cleavage (denoted here as krev) approximates zero. However, krev can vary over a large range for covalent-reversible ligands, whose binding kinetics shall not be further discussed here. An in-depth description of the binding kinetics of both covalent-reversible and irreversible ligands is beyond the scope of this chapter and can be found elsewhere [8, 12, 13]. However, it should be noted that although kinact/KI is the recommended potency measure for irreversible covalent inhibitors, the bulk of literature in the protein kinase field still relies on IC50 data which is much easier to obtain. Comparing covalent inhibitors on the basis of apparent IC50 values can be useful to deduce early SAR within a series provided that all compounds were tested in the same assay system under identical conditions. Nevertheless, it should be kept in mind that apparent IC50 values, which are determined at a single point in time, do neither provide a quantitative picture of a covalent inhibitor’s overall efficiency nor allow for the deconvolution of contributions from reversible and covalent binding. Thus, caution should be exercised when comparing covalent inhibitors merely on the basis of IC50 data [13].

(a) General mechanism of irreversible and reversible covalent target engagement by TCIs. An initial reversible binding event takes place and positions the reactive warhead (red) close to its target amino acid (dark blue). Covalent trapping represents the second step described by the rate constant kinact. The rate of the reverse reaction (defined by krev) is negligible for (quasi)-irreversible inhibitors while being significant for covalent-reversible inhibitors. (b) General mechanism of one-step non-covalent (reversible), covalent-irreversible and covalent-reversible binding
As mentioned before, the by-far most common residues to be addressed by TCIs are non-catalytic cysteines. The cysteine side chain (pKa ≈ 8.5) [14] can readily be deprotonated to form a strongly nucleophilic thiolate which has a much higher reactivity than the neutral thiol form. Notably, cysteine has the strongest intrinsic nucleophilicity among the proteinogenic amino acids except the rare selenocysteine. While catalytic cysteines, that occur for example in cysteine proteases or phosphates, are pKa-depressed to favor the more nucleophilic deprotonated form, the neutral thiol is often dominant for non-catalytic cysteines at physiological pH making them more difficult to address.
Recent computational studies have suggested that most cysteine residues in kinases’ active sites have even higher pKa’s than the aforementioned reference value rendering those moieties comparably weak nucleophiles [14]. Nevertheless, even cysteines with a very high predicted pKa of >20 (e.g., Cys814 in PDGFRα or Cys788 in c-KIT [14]) have been amenable to covalent targeting [15]. It should also be taken into account that the kinase conformation but also the presence of the ligand itself can influence a cysteine’s pKa and thereby its reactivity [16]. Moreover, not only the distance but also the orientation between the electrophilic warhead and the nucleophilic amino acid as well as the flexibility of the latter two are important determinants for the efficiency of covalent bond formation and may require further consideration in the design process [17, 18].
The prototypical warhead type for targeting non-catalytic cysteine residues are acrylamides and analogous attenuated Michael acceptors (vide infra). Usually, rational TCI design starts from an appropriate non-reactive ligand which is equipped with a warhead to target a proximal nucleophilic amino acid residue [19]. The design process is normally guided by structural information from X-ray crystallography allowing for the rational selection of suitable linkers and attachment points to install the reactive group. On the other hand, the off-target profiles of known covalent ligands can be used to identify starting points for re-design and optimization [6]. Alternative TCI discovery strategies that have recently been pursued include fragment-based approaches employing electrophiles of low structural complexity [20,21,22], which can then be optimized to become potent and specific TCIs. Moreover, DNA-encoded libraries featuring reactive compounds may be screened [23, 24]. A schematic overview of these strategies is depicted in Fig. 2.

Typical strategies for the discovery of TCIs
In order to be useful for TCI design, warheads must fulfill several criteria. Ideally, reactivity is just sufficient to ensure rapid, proximity-driven covalent bond formation with the targeted residue while being too low for promiscuous bonding to other physiological nucleophiles. Since the requirements vary between different targets, the reactivity of the employed functional groups should be tunable over a wide range. An appropriate balance between target engagement and promiscuity needs to be found for each individual application. Moreover, warheads (and their metabolites) should be non-toxic and sufficiently stable against metabolic degradation. In some cases, however, rapid warhead depletion may be beneficial to make use of kinetic selectivity while minimizing off-target modification, especially if more reactive electrophiles are employed. As mentioned, α,β-unsaturated amides have been most frequently used as cysteine-targeted warheads since they feature a relatively low intrinsic reactivity, which can be adjusted by the addition of steric bulk or by tuning the electronic properties of the amide N-substituents [25]. Such moieties rapidly react with cysteine thiol(ate)s (Scheme 1a), and less frequently with other nucleophilic amino acids, e.g., lysine [9], if the latter are located nearby in the binding pocket and possess sufficient nucleophilicity and conformational flexibility to hit the electrophilic β-position via a favorable trajectory. Non-specific reaction with ubiquitous thiols, e.g., in glutathione (GSH) or cysteines from other proteins must be much slower to prevent premature inhibitor depletion and off-target-mediated side effects. Nucleophilic addition of the cysteines’ thiol group leads to β-thioether adducts, which are generally stable with respect to the half-life of most target proteins.

Mechanisms of covalent cysteine modification by acrylamide-derived Michael acceptors. (a) (Quasi)-irreversible reaction of regular acrylamides with cysteine side chains via thiol-Michael addition. (b) Reversible reaction of α-cyanoacrylamides with cysteine side chains. The reverse β-elimination reaction is facilitated by the increased acidity of the Cα-proton arising from the additional electron-withdrawing Cα-substituent (exemplified by a nitrile group)
An interesting recent development are TCIs with reversibly binding warheads [26]. For addressing cysteines transiently, α-cyanoacrylamides and analogous “hyper-activated” Michael acceptors have proven suitable. Although the dual activation by two electron-withdrawing group increases the intrinsic reactivity of such Michael acceptor systems, it also causes a thermodynamic destabilization of the addition product along with an increased α-CH-acidity promoting reversibility by favoring the elimination of the corresponding thiolate anion (Scheme 1b) [27, 28]. Hence, the half-life of the covalent adduct largely depends on stabilizing interactions with the protein’s binding pocket and steric shielding of the Cα-proton. Ideally, off-targets, GSH, or the degraded target protein would not provide sufficient thermodynamic stabilization of the covalent complex, thus rapidly liberating the unmodified ligand while the ligand would bind the intact target in a quasi-irreversible fashion. Reactivity and dissociation rates of such dually activated Michael acceptors can readily be tuned by adjusting the activating group (e.g., acrylonitriles equipped with amides and similar –M-substituents or with electron-deficient heterocycles) [28] and by modulating the steric bulk at the β-position [11]. Other covalent-reversible warhead types employed to address kinases include, for example, cysteine-targeted cyanamides [17] or aldehydes [29] and carboxylate-targeted boronic acids [30]. Moreover, chlorofluoroacetamides have very recently been shown to react with cysteines to form products that can be readily hydrolyzed to the corresponding (hydrated) glyoxamides [31].
The growing toolbox of novel or re-purposed warheads for targeting cysteines and other residues has recently been reviewed [9]. A representative selection of such headgroups is depicted in Fig. 3.

Selected examples of reversibly and irreversibly binding warheads for TCIs
2 Covalent Protein Kinase Inhibitors
2.1 The Protein Kinases’ Cysteinome
Protein kinases feature a deep and highly conserved ATP binding cleft. This pocket is perfectly ligandable and designing potent (typically low nanomolar), ATP-competitive kinase inhibitors is not considered a major challenge anymore. In contrast, achieving selectivity within the kinome, which includes more than 500 different protein kinases, can be very difficult due to the conserved nature of the ATP binding pocket [32]. Classical strategies for obtaining selective active site ligands, which are discussed by Knapp and co-workers in a dedicated chapter of this book, exploit subpockets that are not addressed by ATP (such as the hydrophobic clefts often referred to as hydrophobic regions I and II [33, 34]) since those regions are considerably less conserved. Moreover, inactive kinase conformations that are unique by themselves or that expose non-conserved regions can be addressed [35, 36]. Although poorly conserved allosteric pockets can also be targeted, this strategy is challenging due to the comparably shallow topology and the plasticity of these binding sites. Since most protein kinase inhibitors address the ATP pocket, therapeutic targeting is complicated by the high intracellular ATP concentrations (generally in the low millimolar range [37]) competing with the ligand. Covalent inhibitors hold the potential to address the aforementioned challenges as they combine the use of an additional selectivity filter with limited ATP-competitivity.
Protein kinases possess neither a catalytic cysteine residue nor any other highly activated nucleophile in the active site. Instead, they feature only non-catalytic cysteines that may have regulatory functions but are not directly involved in phosphate transfer. Collectively, these cysteine residues have been denominated the protein kinases’ cysteinome [5, 6]. Cysteine residues are distributed at various locations inside and around the ATP pocket that could potentially be tackled by TCIs. According to a recent analysis by the groups of Koch, Laufer, and Knapp, there are at least 18 positions or subsites (a subsite includes several spatially similar positions) inside or proximal to the ATP pocket harboring cysteines with a high probability of being addressable by electrophilic inhibitors (see Fig. 4) [7]. Additional cysteine positions become accessible in inactive conformations (e.g., DFG-out or αC-out) [15]. Cysteines are very unevenly distributed among these locations (see Table 1). For example, over 80 protein kinases feature a cysteine at the hinge region’s H2 position (see Fig. 4 and Table 1) while only five kinases with a cysteine at the neighboring H1 position were identified [7]. Moreover, there is a bias in the availability of inhibitors targeting individual positions or subsites. While several locations have remained completely unaddressed so far, 8 out of 11 kinases with a cysteine at the F2 position (αD − 1) were targeted with covalent inhibitors when this manuscript was written (February 2019). In the following sections, representative inhibitors engaging cysteine residues at the respective locations will be highlighted and the underlying design principles be discussed.

Cysteine positions potentially amenable to covalent targeting according to Chaikuad et al. [7]. Cysteines were mutated in silico into the X-ray crystal structure of cAMP-dependent protein kinase catalytic subunit α in complex with ATP (PDB-code: 1ATP) and depicted as spheres with the side chains highlighted as sticks. Colors and indentifiers were assigned according to the regions where the cysteines are located. Orange glycine-rich loop/P-loop, pink gatekeeper residue, lime hinge region, red front region, magenta pre-DFG motif, salmon backpocket, brown roof region, cyan activation segment, blue outside the ATP-pocket, dark-green/deep-teal positions exposed in inactive kinase conformations and additional positions (non-exhaustive). Positions are labeled according to the nomenclature used by Chaikuad et al [7]. Position A1 has been complemented according to ref. [38]
2.2 Approved Covalent Protein Kinase Inhibitors
So far, the bulk of efforts in covalent kinase inhibitor discovery has been directed toward kinases involved in cancer [39, 40]. The tremendous interest in covalent kinase targeting is highlighted by over 60 patents disclosed only between 2010 and 2013 [41]. Currently, six covalent kinase inhibitors are approved by the FDA (Fig. 5): afatinib (BIBW-2992, 1), dacomitinib (PF-00299804, 2), neratinib (HKI-272, 3), osimertinib (AZD9291, 4), ibrutinib (PCI-32765, 5), and acalabrutinib (ACP-196, 6). Among these, the first one to gain FDA approval (07/2013) was afatinib, a gefitinib (7)-derived second-generation EGFR/ErbB(HER) family kinase inhibitor (vide supra) developed by Boehringer Ingelheim, which is used in the therapy of metastatic non-small-cell lung cancer (NSCLC) driven by activating EGFR-mutations [42]. This compound hits a cysteine in the F2 position of the EGFR kinase domain (Cys797), which is also present in ErbB2 and ErbB4 but not in the pseudokinase ErbB3. Later in 2013, ibrutinib, an inhibitor addressing an equivalently positioned cysteine in Bruton’s tyrosine kinase (BTK), was approved for the treatment of mantle cell lymphoma. The marketing authorization was later expanded to other conditions including chronic lymphocytic leukemia (2014) and chronic graft versus host disease (2017) [43] making ibrutinib one of the few small molecule kinase inhibitors therapeutically used for the modulation of immune response in a non-oncology setting. Osimertinib, a third-generation mutant-selective EGFR inhibitor, was approved in 2015 for metastatic NSCLC haboring the EGFR-T790M resistance mutation [44]. In July 2017, the FDA granted approval to neratinib, a quinoline-derived pan-ErbB-inhibitor for adjuvant treatment of early stage HER2-positive breast cancer. In October 2017, acalabrutinib, an ibrutinib-derived second-generation BTK inhibitor featuring a but-2-yne amide warhead, gained marketing authorization for the treatment of mantle cell lymphoma [45]. The sixth and most recently approved covalent kinase inhibitor is dacomitinib, a close structural analog of afatinib developed by Pfizer and employed in the treatment of metastatic NSCLC with activating EGFR mutations (L858R) or exon 19 deletions [46].

Currently approved covalent kinase inhibitors. Gefitinib (7), a non-covalent EGFR inhibitor, is depicted as the structural template for the design of covalent (second generation) ErbB family kinase inhibitors
2.3 Natural Products as Covalent Protein Kinase Inhibitors
Natural products have been a rich source of covalent ligands [47] and such compounds have been known as covalent kinase inhibitors for more than two decades [48,49,50]. For example, hypothemycin (8, Fig. 6) and analogous resorcylic acid lactones (RALs) incorporating an endocyclic cis-enone motif engage cysteine residues preceding the conserved DFG motif (D1 position, e.g., Cys166 in ERK2 or Cys174 in TAK1). Equivalent cysteine residues are present in about 50 kinases from several distinct families [5,6,7] many of which are covalently inhibited by hypothemycin [51, 52]. Modification occurs via hypothemycin’s reactive enone moiety while the epoxide, which is also present, behaves as a bystander electrophile in this context. LL-Z1640-2 (FR148083, 5Z-7-oxozeaenol, 9), a close analog of hypothemycin binds kinases such as ERK1/2 and TAK via the expected cysteine residues in the D1 position [53]. Despite the presence of a D1 cysteine, a distinct binding mode is observed for mitogen-activated protein kinase kinase MKK7 (MAP2K7). Here, a cysteine in the F2 position of the solvent-exposed front region is modified (see the overlay in Fig. 7a, b) [54]. LL-Z1640-2 was further developed to clinical candidate E6201 (10), a potent MEK/FLT3 inhibitor, by researchers from Eisai [55, 56]. The compound was efficacious in in vivo models and moved to phase II clinical studies for the treatment of psoriasis (NCT01268527, NCT00539929), hematologic malignancies with FLT3 and/or Ras mutations (NCT02418000, terminated), and recently for melanoma CNS metastases (NCT03332589). Further natural products that are known to covalently engage protein kinases via cysteine adduction include, for example, cyclopentenone prostaglandins (e.g., PGA1, 11) targeting the activation loop cysteine (Cys178/179) in IKKα/β [57] or pyranonaphthoquinones (e.g., lactoquinomycin, 12) targeting Cys296 and Cys310 (O2-position) in the activation loop of AKT1 [58].

Selected natural products or natural product derivatives covalently targeting kinases. The reactive moieties involved in covalent bond formation are highlighted in red

Overlay of the X-ray crystal structures of 9 covalently bound to MKK7 (gray, both cysteine side chains depicted in the ball and stick representation; PDB: 3WZU) and TAK1 (cyan, cysteine side chain shown as sticks; PDB: 4GS6). While the cysteine moitey at the D1 position, which is common to both kinases is adresssed in TAK1, an alternative binding mode engaging a distinct cysteine in the front region of the ATP cleft (F2 position) is observed in MKK7. (a) Top view. (b) Front view
Wortmannin (13), a natural product belonging to the viridin furanosteroids is a potent inhibitor of the lipid kinases from the phosphoinositide 3-kinases (PI3Ks) family. In contrast to the cysteine-targeted enones discussed above, this and analogous compounds covalently modify the conserved catalytic lysine (Lys833 in PI3Kγ) [48]. Attack by the lysine’s ε-amino group occurs at the unsubstituted carbon atom of the dually activated furan ring [59]. The furan undergoes a ring opening, presumably via an intermediate enolate, to form a conjugated enamine (see the mechanism and binding mode in Fig. 8a, b). Opening the ring with diallylamine furnished PX-866 (14), an analog with improved PK properties that moved to clinical trials. In contrast to the addition products of primary amines, which feature an inactive Z-configured enamine that is stabilized by an intramolecular hydrogen bond with the lactone’s carbonyl oxygen atom, secondary amine-derived adducts retain inhibitory activity. The latter undergo exchange reactions with primary amines while the inverse reaction could not be observed [60]. Compound 14 alone or in combination was tested in phase II clinical trials for the treatment of different cancer types including prostate cancer (NCT01331083), glioblastoma (NCT01259869), and non-small cell lung cancer (NSCLC) (NCT01204099).

Wortmannin (13), a lysine-targeted PI3 kinase inhibitor. (a) Mechanism suggested for the reaction with PI3Kγ-Lys833. (b) Binding mode of 13 in the ATP-pocket of PI3Kγ confirming the ring opening and the covalent binding to the Lys833 ε-amino group (PDB: 1E7U)
2.4 Development of Inhibitors Targeting Cysteines in the Front Region
2.4.1 Inhibitors Targeting the F2 Position
So far, the most extensive research efforts have been directed toward kinases featuring a cysteine residue at the F2 (αD − 1) position. Being located in the solvent-exposed front region, F2 cysteines are common to 11 kinases, namely the ErbB family members EGFR, ErbB2, and ErbB4 (but not the pseudokinase ErbB3), the TEC family kinases (TEC, BTK, ITK, BMX, and TXK), the SRC kinase BLK, the Janus kinase JAK3, and the MAP kinase kinase MKK7. The most considerable drug discovery efforts have been aimed toward Cys797 of the EGFR receptor tyrosine kinase. As a member of the ErbB family, EGFR (also referred to as HER1 or ErbB1) transduces growth signals either as a homodimer or as a heterodimer with other ErbB family members. Aberrant EGFR activity is a common driver of non-small cell lung cancer (NSCLC) and patients with certain activating EGFR mutations (e.g., L858R or exon 19 deletions) showed impressive response rates to first-generation (reversible) EGFR inhibitors such as gefitinib (see Fig. 5) or erlotinib. Unfortunately, secondary resistance mutations, most notably the T790M mutation of the gatekeeper residue, appeared rapidly and rendered first-generation EGFR inhibitors virtually inactive in approximately half of the responder population. In this light, second- and third-generation EGFR inhibitors were developed to exploit the nucleophilic nature of Cys797.
Pioneering work was reported already in the late 1990s by Singh, Fry, and colleagues from Parke-Davis. They characterized 2′-thioadenosine (15, Fig. 9a) [61] and the 4-aminoquinazoline-derived acrylamide PD-168393 (16) [62] as covalent modifiers of EGFR-Cys797 and the analogous Cys805 in ErbB2 (and presumably also Cys803 in ErbB4). Improved efficacy of 16 compared to the biochemically almost equipotent propionamide analog was observed in a xenograft model highlighting the potential pharmacological advantages of covalent inhibitors. Further development of analogous compounds led to canertinib (CI-1033, 17), the first irreversible pan-ErbB inhibitor entering clinical trials [63]. The development of canertinib, however, was recently discontinued. Afatinib (1), the first approved covalent ErbB inhibitor features a very similar structure. Besides modifications at the quinazoline’s C7 substituent, a key difference consists in the replacement of canertinib’s acrylamide warhead by an analogous 4-(dialkylamino) crotonamide to assist the deprotonation of the cysteine’s thiol group (see the mechanism in Fig. 9b). Although afatinib showed high efficacy in the treatment of NSCLC with activating EGFR mutations, the drug suffered from dose-limiting side effects hampering its use for overcoming the T790M resistance. Approval of the structural analog dacomitinib (2) was delayed until recently since early trails did not prove superiority compared to first generation EGFR inhibitors.

(a) Early covalent ErbB family kinase inhibitors targeting the cysteine in the F2 position. (b) Mechanism of 4-(dialkylamino) crotonamide-assisted cysteine deprotonation/nucleophilic attack
On the basis of the related quinoline scaffold, compounds featuring a carbonitrile group at the C3-position of the heterocyclic core were developed in order to increase ErbB2 inhibitory activity, the latter being frequently overexpressed in breast cancer [64]. These efforts culminated in the recent approval of neratinib (3).
Third-generation EGFR inhibitors were designed to target resistant/activating EGFR-mutants (e.g., EGFR L858R/T790M) while sparing the wild-type kinase to decrease dose-limiting side effects, which constitute a key liability of the second-generation drugs. The structure of such compounds does not rely on the prototypical 4-aminoquinazoline scaffold. WZ4002 (18, Fig. 10), the first third-generation irreversible ErbB kinase inhibitor, was reported in 2009 by researchers at the Dana-Farber Cancer Institute [65]. This 2-aminopyrimidine derivative features a moderate selectivity for EGFR with an activating L858R plus the aforementioned T790M resistance mutation. An X-ray crystal structure of the compound bound to the EGFR T790M mutant shows the formation of two hydrogen bonds between the aminopyrimidine and the hinge region and confirms labeling of Cys797 by the acrylamide warhead (PDB: 3IKA). Moreover, a hydrophobic contact between the pyrimidine’s 5-chloro substituent and the sulfur atom of the gatekeeper methionine residue is observed. The latter interaction, which also can be interpreted within the conceptual framework of halogen bonding [66, 67], is thought to contribute to the observed selectivity for the T790M mutant. Rociletinib (CO1686, 19), a structurally related inhibitor developed by Clovis Oncology and tested in clinical trials up to phase III (NCT02322281) was dropped in May 2016 for several reasons including lower response rates and a less favorable benefit–risk profile compared to osimertinib [68], which was simultaneously developed by AstraZeneca. Osimertinib successfully gained fast track approval in 2015/16 [69].

Third-generation and more recent covalent EGFR/ErbB kinase inhibitors
Besides escape pathways (e.g., HER2 and MET amplification or PI3K/AKT/mTOR activation), resistance to third-generation EGFR inhibitors is frequently driven by mutation of the reactive cysteine to serine. The common L858R/T790M/C797S triple mutant, for example, precludes covalent binding and is resistant against all EGFR inhibitor generations. A new EGFR inhibitor type (exemplified by GM-597, 20) that covalently binds gefitinib-resistant double mutants while maintaining nanomolar reversible activity against EGFR L858R/T790M/C797S has recently been described by Günther et al. [70, 71].
The second class of clinically successful covalent kinase inhibitors addresses Bruton’s tyrosine kinase (BTK), with two compounds being currently approved by the FDA (vide supra). BTK is a cytoplasmic tyrosine kinase belonging to the TEC family and plays a key role in B-cell receptor signaling. Due to its essential function in B-cell development, BTK has been selected as a target not only for the treatment of B-cell malignancies, but also for inflammatory and autoimmune disorders. Like the aforementioned ErbB family kinases, BTK features a cysteine residue in the F2 position (Cys481). In 2006, the first covalent BTK inhibitors were reported by researchers from Celera Genomics [72] and subsequently employed as tool compounds [73]. A structure-based design approach was used to transform the potent reversible BTK inhibitor 21 (Fig. 11) into reactive analogs addressing Cys481. Key compound 5-rac, an inhibitor with subnanomolar potency featuring an acrylamide warhead attached to a piperidin-3-yl residue, was capable of covalently modifying BTK according to washout and MS-experiments. In this context, it is worth mentioning that aliphatic amine-derived acrylamides typically feature lower intrinsic reactivities compared to analogous anilides [74] which might translate into decreased promiscuity. Compound 5-rac was effective in an arthritis mouse model and the more potent R-enantiomer now known as ibrutinib (5, see Fig. 5) was further developed by Pharmacyclics and Johnson & Johnson. Interestingly, ibrutinib was later shown to potently inhibit most other kinases with an equivalently positioned cysteine [45].

Development of irreversible BTK inhibitors from reversible inhibitor 21. The R-enantiomer of compound 5-rac equals ibrutinib
Acalabrutinib (6, see Fig. 5), an approved second-generation covalent BTK inhibitor, features a related imidazo[1,5-a]pyrazine core [45]. In this case, a but-2-ynamide warhead linked via the nitrogen atom of an S-configured pyrrolidin-2-yl substituent was employed. While propiolamides are more reactive than analogous acrylamides, but-2-ynamides are slightly less reactive [74] making acalabrutinib more stable toward GSH when compared to ibrutinib (t1/2 = 5.5 h vs. 1.9 h). Acalabrutinib (IC50BTK = 5.1 nM, kinact/KI = 3.1 × 104 M−1 s−1) was shown to be slightly less potent than ibrutinib (IC50BTK = 1.5 nM, kinact/KI = 4.8 × 105 M−1 s−1) but relatively selective against most kinases harboring an F2 cysteine with only ErbB4 (IC50 = 16 nM), BMX (IC50 = 46 nM), and TEC (IC50 = 126 nM) being significantly hit (MKK7 was not tested). The compound features a clean kinome profile, oral availability, and durable target engagement in vivo (99% after 4 and 12 h, 100 mg p.o.) although being rapidly eliminated thus highlighting the disconnection between PD and PK, a key feature of many irreversible inhibitors. Acalabrutinib showed a tolerable adverse effect profile and gained accelerated approval by the FDA as second-line therapy for mantle cell lymphoma (MLL) [75]. Several phase III studies for the treatment of chronic lymphocytic leukemia (CLL) are ongoing. However, BTK C481S mutation renders both ibrutinib and acalabrutinib ineffective at the recommended dosage since the decreased potency in combination with the relatively fast clearance of these compounds precludes sustained reversible target engagement [76].
Besides the two aforementioned drugs, many other irreversible BTK inhibitors with distinct chemotypes and good or excellent selectivity profiles (e.g., CHMFL-BTK-11 [77], branebrutinib [78], spebrutinib [45], or poseltinib [79], 22–25, Fig. 12a) have been investigated in preclinical and clinical studies. A covalent-reversible approach was chosen by researchers from the Taunton group and Principia Biopharma [11], who generated α-cyanoacrylamide derivatives of ibrutinib to engage BTK Cys481. Target residence times could be modulated as a function of the β-substituent. For example, compound 26a (Fig. 12b) bearing a bulky tert-butyl group in the β-position retained >50% cellular target occupancy 20 h after washout. In contrast, occupancy of methyl-capped analog 26b was negligible under the same conditions. The extended target residence time of compound 26a may be rationalized by additional thermodynamic stabilization of the covalent complex via hydrophobic interactions with the bulky tert-butyl moiety, which simultaneously induces a conformation with decreased α-CH-acidity while also shielding the Cα-proton from water thereby impeding its abstraction (see PDB: 4YHF). Further optimization with special emphasis on improving solubility and PK properties furnished a compound series exemplified by 27a and 27b. While 27a (IC50 = 0.7 nM, kinact/KI = 1.9 × 103 M−1 s−1) had a residence time of 34 h, key compound 27b (IC50 = 1.9 nM, kinact/KI = 4.3 × 102 M−1 s−1) behaved quasi-irreversible with a residence time of approx. 1 week. In accordance with the applied design concept, binding was rapidly reversible after proteolysis. Compound 27b showed sustained target occupancy after clearance in vivo (as determined with a fluorescent covalent probe) and had a relatively clean profile in a panel of 254 kinases. Nevertheless, five other kinases sharing the combination of an F2 cysteine and a threonine gatekeeper residue were strongly hit at 1 μM including BMX, which was even inhibited >90% at 100 nM. The discussed compound series was further developed to drug candidate PRN1008 (28, IC50 = 1.3 nM) [80], an orally available, covalent-reversible BTK inhibitor with a slow dissociation rate that recently entered phase III clinical trials for the treatment of pemphigus (NCT03762265).

(a) Selected examples of irreversible BTK inhibitors. (b) α-Cyanoacrylamide-based covalent-reversible BTK inhibitors. (c) Cyanamide-based covalent-reversible BTK inhibitor PF-303. (d) Mechanism of the reversible cysteine addition to cyanamides
PF-303 (29, Fig. 12c), a covalent-reversible BTK inhibitor from Pfizer, addresses the same cysteine residue via a cyanamide group [81]. Cyanamides, a warheads class known from cysteine protease inhibitors [82, 83], react with cysteines at similar rates as acrylamides [74]. This Pinner-type addition results in the reversible formation of isothiourea products (Fig. 12d). PF-303, which can be regarded as an ibrutinib bioisostere, is a very potent BTK inhibitor (IC50 = 0.64 nM, kinact/KI = 1.44 × 105 M−1 s−1) with a moderate dissociation rate (t1/2 = 5 h). While being highly selective against JAK3, ITK (both >10,000-fold), and several related kinases, PF303 inhibits TEC and BMX at similar potency as BTK. The compound efficiently blocked anti-IgM F(ab’)2-mediated proliferation of murine B-cells (IC50 = 2 nM) and showed in vivo activity in mice upon oral dosing.
Interleukin-2 inducible T-cell kinase (ITK) is closely related to BTK and features an equivalently positioned cysteine (Cys422) that was suggested to possess a lower nucleophilicity due to a pKa increase promoted by the proximal Asp445 [84]. As a mediator of T-cell receptor signaling, ITK plays an important role in T-cell development, differentiation, and function. It is considered a promising target in the treatment of inflammatory and autoimmune disorders as well as T-cell malignancies. Based on the fact that ibrutinib and similar BTK inhibitors show significant off-target activity on ITK, researchers from Pfizer developed analogous compounds to address this kinase. A structure-based design approach furnished acrylamide 30a (PF-06465469, Fig. 13a), a highly potent ITK inhibitor (IC50 = 2 nM at 1 mM ATP, kinact/KI = 1.6 × 104 M−1 s−1) [84]. Several alternative warheads were tested, however, only the more reactive propiolamide 30b showed similar potency. Compound 30a was potent in a human whole blood assay and showed sustained activity in cells. Covalent engagement of Cys442 was confirmed for several analogs by X-ray crystallography (PDB: 4HCT, 4HCV, 4HCU). However, 30a was also shown to be an equipotent inhibitor of BTK. Selectivity against BTK seems to be difficult to achieve not only because of the lower acidity of ITK’s F2 cysteine but also due to its higher affinity toward ATP. Nevertheless, one compound with a moderate selectivity for ITK could be obtained (30c) albeit at the expense of potency (IC50ITK = 60 nM vs. IC50BTK = 1,050 nM).

Covalent ITK inhibitors from (a) Pfizer and (b) GSK
Another series of covalent ITK inhibitors exemplified by compound 31 (Fig. 13b) was reported by researchers at GSK [85]. This key compound possessed high potency (IC50 = 5 nM at 1 mM ATP), a more pronounced selectivity against BTK and a higher kinact/KI ratio (5.2 × 105 M−1 s−1) compared to the aforementioned inhibitors. Covalent binding was verified by jump-dilution, cellular washout, and X-ray crystallography using a close analog (PDB: 4KIO). Compound 31 demonstrated low reactivity toward GSH, favorable PK properties for inhaled dosing, and prevented anti-CD3-induced T-cell activation in rat lung tissue (p.i.). In human PBMCs, the compound further suppressed the production of TH1, TH2, and TH17 cytokines.
Other kinases with a cysteine at the F2 position that have been addressed by rationally designed covalent inhibitors include JAK3, BMX, and MKK7. Efforts toward highly isoform-selective JAK3 inhibitors culminated in the development of the clinical candidate PF-06651600 (32, Fig. 14a) and have been summarized in a separate chapter of this book and a recent review [86]. Notably, efforts published very recently from researchers at Pfizer showed that JAK3 Cys909 is also amenable to covalent-reversible targeting with cyanamides (e.g., compound 33a) [17]. The latter compound features excellent isoform selectivity (>245-fold vs. other JAKs at 1 mM ATP), high potency (IC50 = 11 nM at 1 mM ATP), and efficient inactivation kinetics (kinact/KI = 1.9 × 105 M−1 s−1) while being reasonably stable against GSH. X-ray crystallography (e.g., with the analog 33b) unambiguously confirmed isothiourea formation (Fig. 15a).

(a) Examples of irreversible and covalent-reversible JAK3 inhibitors. (b) Irreversible covalent BMX inhibitors. (c) Irreversible covalent MKK7 inhibitors. (d) Disulfide-tagged covalent PDK1-E166C inhibitor 39

(a) Binding mode of cyanamide-based covalent-reversible JAK3 inhibitor 33b (PDB: 6DB4). The formed isothiourea is involved in several direct and water-mediated hydrogen bonds contributing to the stabilization of the covalent complex. In the hinge region and the P-loop region, side chains were omitted for clarity. (b) Binding modes of α-cyanoacrylamide-based covalent-reversible JAK3 inhibitor 34b (PDB: 5LWN). The simultaneous presence of both, the covalently and the non-covalently bound compound (highlighted in cyan and yellow, respectively) is observed
A distinct series of α-cyanoacrylamide-based covalent-reversible JAK3 inhibitors (exemplified by 34a and b) with excellent isoform and kinome selectivity has been developed by Forster et al. [87, 88]. Remarkably, both the covalent and the non-covalent complexes coexist in the X-ray crystal structure of 34b bound to JAK3 (Fig. 15b). The nitrile substituent of these compounds opens up a rare induced-fit pocket formed by Arg911, Arg953, and Asp912 which constitutes an additional selectivity filter contributing to the excellent selectivity of this compound class in the kinome.
Covalent inhibitors for other kinases with an F2 cysteine have also been reported. Inhibitors that target the kinase BMX include, for example, the dual BMX/BTK inhibitor BMX-IN-1 (35, Fig. 14b) [89], or the type II inhibitor CHMFL-BMX-078 (36) [90] featuring increased selectivity against BTK. As mentioned before, the MAP kinase kinase MKK7, one of the two activators of the c-Jun N-terminal kinases (JNKs) features several cysteines in the active site: one at the F2 position (Cys218), one at the D1 position (Cys276), one at the P3 position (Cys147), and one at the O3 subsite (Cys296). The F2 cysteine, which is also addressed by LL-Z1640-2 (9, see Figs. 6 and 7), has recently been targeted by indazole-derived inhibitors exemplified by 37 (MKK7-COV-2, Fig. 14c) discovered in a virtual screening campaign using DOCKovalent [91, 92]. Even more recently, ibrutinib-derived 1,2,3-triazoles (e.g., 38) addressing MKK7-Cys218 have been reported by the Rauh group [93].
An engineered cysteine at an equivalent position in PDK1 (E166C) has been addressed by Erlanson et al. using an acrylamide-based “extender” (39, Fig. 14d) equipped with a (di)sulfide-tag. A subsequent fragment-based disulfide tethering screen enabled the generation of potent reversible PDK1 inhibitors [94]. Moreover, the introduction of analogous engineered cysteines has been used in the context of chemical-genetics approaches to generate probes for kinases such as Aurora kinase [95] and c-SRC [96].
2.4.2 Inhibitors Targeting the F3 Position
The three c-Jun N-terminal kinases (JNK1–3) are the only kinases known to feature an accessible cysteine in the F3 position of the αD-helix (αD + 2), eight amino acids after the gatekeeper residue. This cysteine has been targeted by imatinib-derived covalent inhibitors serendipitously discovered by Gray and co-workers [97]. These researchers initially aimed to address cysteines located in the catalytic loop of the PDGFR and c-KIT receptor tyrosine kinases being only accessible in the DFG-out conformation (subsite I1, see Fig. 4 and Table 1). However, the designed compound JNK-IN-1 (40, Fig. 16a) showed strong off-target activity on JNKs [15]. Structure-guided optimization of this initial hit furnished the potent pan-JNK inhibitor JNK-IN-8 (41, IC50 = 4, 19, and 1 nM for JNK1–3, respectively), a compound with an excellent selectivity in the kinome and reasonable cellular potency. Covalent modification of Cys154 (JNK3 numbering) was shown for some close analogs by MS and X-ray crystallography (see PDB: 3V6R and 3V6S).

(a) Imatinib-derived covalent JNK inhibitors. (b) Pyridinylimidazole-derived covalent JNK inhibitors
Structurally distinct pyridinylimidazole-derived covalent JNK inhibitors (e.g., 42a and b, Fig. 16a) were developed from reversible p38α MAP kinase inhibitors in the groups of Koch and Laufer using a structure-based approach [98]. Both compounds potently block JNK3 enzymatic activity with IC50 values bordering the picomolar range. The inhibitors covalently labeled JNK3 as confirmed by MS experiments while leaving the C154A mutant unmodified. However, while the tetra-substituted imidazole 42a retained significant (reversible) inhibitory activity on p38α (IC50 = 36 nM), N-desmethyl analog 42b displayed an approx. 1,000-fold selectivity window over the latter enzyme. Both compounds showed a relatively clean profile in a panel of 410 kinases. More detailed information on covalent and non-covalent JNK inhibitors can be found in a dedicated chapter of this book and in recent reviews [99, 100].
2.4.3 Inhibitors Targeting the F1 and the F4 Position
Further cysteines in the front region of protein kinases that have proven amenable to covalent targeting are located at the F1 (αD − 2) and the F4 (αD + 6) positions. According to the underlying analysis, an F1 cysteine occurs in the three kinases EphB3, LKB1, and PINK1 but it has only been addressed in the EphB3 receptor tyrosine kinase so far. In a series of 4-aminoquinazoline-derived inhibitors evaluated by Kung et al., different electrophiles were tested and chloroacetamide 43a (Fig. 17a) potently blocked Eph3B kinase activity (IC50 = 55 nM). Compound 43a was devoid of significant inhibitory activity on the EphB3 C717S mutant and the related kinases EphA4 and EphB4 [101]. The analogous α-chloromethyl ketone 43b showed an even higher apparent potency (IC50 = 6 nM), presumably due to the increased intrinsic reactivity of this electrophile, and both compounds were active in cells. In contrast, analog 43c lacking a leaving group showed only negligible inhibitory activity (IC50 > 10 μM). Covalent modification of Cys717 was confirmed by washout experiments, mass spectrometry, and X-ray crystallography (PDB: 5L6P and 5L6O). However, compound 43a inhibited EGFR more potently than EphB3. The optimized analog 44 at 50 nM possessed a clean profile in a panel of 98 kinases while fully abrogating EphB3 activity. No substantial inhibition of LKB1 also possessing an F1 cysteine was observed. In this context, it is worth mentioning that although cysteines in LBK1 and PINK1 are positioned at F1 (i.e., two residues before the start of the αD helix), the hinge region of these kinases is shortened by one amino acid (GK + 5 position vs. GK + 6 in EphB3). Consequently, these cysteine residues adopt different orientations in the latter two kinases. A clickable analog of compound 44 suggested cellular target engagement and limited off-target modification at concentrations below 100 nM. Noteworthily, inhibitor 44 was used as a covalent probe for EphB1 in a chemical-genetics-based study where EphB1 was engineered to feature an equivalent cysteine (G703C mutant). An analogous approach was also applicable to the kinases FGFR4, ABL, and RAF [102].

(a) Covalent Eph3B kinase inhibitors. (b) Covalent PI3Kα inhibitor 45
Being among the few kinases known to harbor a cysteine residue in the F4 (αD + 6) position, the lipid kinase PI3Kα was addressed with inhibitor CNX-1351 (45, Fig. 17b) [103]. This compound potently hits PI3Kα (6.8 nM) with good selectivity against some other PI3Ks. It caused prolonged inhibition of PI3Kα signaling in cells and target engagement was demonstrated in vivo. Despite being equipped with an enone warhead, inhibitor 45 possessed only low reactivity toward GSH and several plasma proteins, showcasing that the β,β-dimethyl substitution efficiently attenuates the enone’s intrinsic reactivity [25]. Covalent modification of Cys862 was demonstrated by MS and X-ray crystallography (PDB: 3ZIM). Due to the cysteine being located relatively far outside the ATP pocket, a long spacer was required. Remarkably, the morpholine oxygen atom acts as the non-canonical hinge-binding anchor of this compound.
2.5 Development of Inhibitors Targeting Cysteines around the P-Loop and in the Roof Region
2.5.1 Inhibitors Targeting the R1 Subsite
ErbB3 (HER3), a pseudokinase which forms catalytically active heterodimers with other ErbB kinases, is lacking the F2 cysteine residue common to the remaining ErbB family members. However, this protein has recently been addressed via Cys721, a cysteine located at the R1 subsite at the roof of the ATP binding cleft [104]. R1-cysteines are only known in four other kinases (WNK1–4) [7]. In the WNK family kinases, however, these moieties are located two positions further C-terminally (β3 + 3 in ErbB3 vs. β3 + 5 in WNKs) making the positioning of Cys271 unique. A non-reactive screening hit was developed to the initial lead compound TX1-85-1 (46, Fig. 18), a covalent ErbB3 ligand with nanomolar potency. Unfortunately, 46 was inefficient in suppressing the proliferation of ErbB3-addicted cell lines and ErbB3-dependent downstream signaling at concentrations that would fully label the target protein. However, transforming 46 into (partial) ErbB3-degrader TX2-121-1 (47) by employing a hydrophobic tagging approach resulted in a more pronounced reduction of ErbB3-dependent signal transduction and proliferation. These effects were not observed for the ErbB3 C721S mutant. Consistently, an analogous non-reactive degrader proved to be less efficient. In contrast to these data, covalent binding was recently found to impair PROTAC-promoted degradation of BTK [105].

Covalent ErbB3 ligand 46 and hydrophobically tagged degrader 47
2.5.2 Inhibitors Targeting the Positions or Subsites P1–P4 and A1
Another series of cysteines amenable to covalent targeting is located in the glycine-rich loop (P-loop) or the adjacent β-sheets (P1–P4). An additional cysteine (subsite A1; not included in earlier analyses [5,6,7, 15, 18]) can also be covalently trapped despite its orientation outward the ATP pocket as demonstrated by a recent study (vide infra) [38].
In their pioneering work, Taunton and co-workers developed fluoromethylketone-based inhibitor 48 (FMK, Fig. 19a) to address the C-terminal kinase domain (CTD) of p90 ribosomal protein S6 kinases (RSKs) via a cysteine located at position P4 [106]. The RSKs and the closely related mitogen- and stress-activated protein kinases (MSKs) possess two functional kinase domains i.e. the aforementioned CTD and an additional N-terminal kinase domain (NTD), with the P4 cysteine being only present in the CTD. Besides the four RSKs (RSK1–4), only 7 other kinases (PLK1–3, NEK2, MSK1/2, and MEKK1) are known to possess an analogous cysteine. In this approach, the relatively small threonine gatekeeper residue, which among these kinases is only shared by the CTDs of RSK1, 2, and 4 (RSK3 has a methionine gatekeeper), was used as an additional selectivity filter. Fluoromethylketone 48 is a potent RSK2-CTD inhibitor (IC50 = 15 nM) with high selectivity against the RSK2 C436V and the T493M gatekeeper mutant. In cells, the compound inactivated both RSK1 and RSK2 and cellular selectivity was demonstrated with a clickable probe in a subsequent study [107]. The same scaffold was also used in Taunton’s seminal work on dually activated Michael acceptors as tunable electrophiles for reversible cysteine targeting [27]. Replacement of the above α-fluoromethylketone by a β-linked α-cyanoacrylamide moiety furnished 49a, a covalent-reversible inhibitor with a dissociation half-life of 245 min. As predicted, protein labeling was rapidly reversible upon unfolding. Besides the RSK2-CTD (IC50 = 5 nM), compound 49a potently inhibited RSK1 and 4 (CTDs), while the RSK2 C436V mutation conferred resistance. In a large kinase panel, the compound showed high selectivity and sustained RSK1 and RSK2 occupancy was observed in cells. Covalent engagement of Cys436 was demonstrated for a close analog by X-ray crystallography (PDB: 4D9U) while no adducts could be detected in MS experiments. Modification of a second accessible cysteine (Cys560) located in the DFG-1 position was not observed. In a subsequent study, the above targeting concept was extended to (hetero)aryl-activated acrylonitriles exemplified by inhibitors 49b and 49c (IC50RSK2-CTD = 47 and 38 nM, respectively) [28]. The β-elimination rates of such compounds spanned three orders of magnitude. It is worth mentioning that Taunton and co-workers also identified α-cyanoacrylamide-based inhibitors for other kinases harboring the P4 cysteine such as MSK1-CTD, NEK2, or PLK1 [91, 108].

(a) Irreversible and covalent-reversible RSK inhibitors. (b) Covalent NEK2 inhibitors featuring a 6-ethynyl purine warhead. (c) Irreversible NEK2 inhibitor 51. (d) Electron-deficient heteroarenes suggested to target PLK1 via reversible Meisenheimer complex formation
Interestingly, Cys22, the equivalent cysteine in NEK2, was recently shown to react with 6-ethynyl purines (e.g., compound 50, Fig. 19b) to form vinyl thioether adducts [109, 110]. Furthermore, NEK2 has been covalently targeted by propiolamide JH295 (51, Fig. 19c) [111]. PLK1 has also been addressed non-canonically using electron-deficient heteroarenes exemplified by 52 (Fig. 19d) [112]. These compounds, which resulted from a virtual screening campaign aiming to identify non-covalent PLK1 inhibitors, presumably bind PLK1 Cys67 via the reversible formation of a stable Meisenheimer complex.
While cysteines located at the tip of the glycine-rich loop (P3 position) have not been deliberately addressed at the time of writing, a cysteine close to the end of the β1-sheet (P1 subsite) has been shown to be trapped by ibrutinib in the MAP kinase kinase kinase ZAK [113]. As mentioned above, a very recent study proved the accessibility of a cysteine one position further N-terminally on the β1-sheet (A1) present in few kinases including the non-receptor tyrosine kinase FAK (focal adhesion kinase). The A1 position has not been part of recent analyses presumably due to its unfavorable orientation pointing from the top of the β1-sheet away from the ATP binding site. Design started from the structure of known reversible FAK inhibitor TAE226 (53, PDB: 2JKK) and led to subnanomolar inhibitor 54 (Fig. 20a) [38]. The key to achieve covalent engagement of the cysteine residue was the employment of a squaryldiamide linker forming hydrogen bonds with the side chain of Arg426 and the backbone of Ile428 directing the acrylamide warhead toward Cy427 (Fig. 20b).

Covalent FAK inhibitor 54. (a) Design from unreactive reversible inhibitor TAE226. (b) Binding mode of compound 54 covalently linked to FAK (PDB: 6GCR). Hydrogen bonds between the backbone of Ile428 and the Arg426 side chain and the squaryldiamide linker favor an orientation facilitating covalent bond formation
Clinical candidates have arisen from targeting fibroblast growth factor receptor tyrosine kinases (FGFR1–4) all sharing a cysteine residue on the N-terminal side of the P-loop’s tip (P2 position). An analogous cysteine has been identified in six other kinases (SCR, LIMK1, TNK1, FGR, YES, and SgK069/SBK2). FGFR tyrosine kinases represent promising targets for the treatment of different cancer types. The prototype covalent pan-FGFR inhibitor FIIN-1 (56, Fig. 21a) was designed in the Gray lab from the reversible inhibitor PD173074 (55) [114]. However, mutation of the gatekeeper valine moiety conferred resistance to this and other first-generation FGFR inhibitors. Efficacy could be restored by second-generation compounds exemplified by FIIN-2 (57) [115]. It is worth mentioning that the latter bind FGFR4 in a P-loop-induced DFG-out conformation. The same scaffold has been developed to phase I clinical candidate PRN1371 (58a; NCT02608125) by researchers from Principia Biopharma [116] and a similar approach furnished α-cyanoacrylamide 58b, a related covalent-reversible FGFR inhibitor. Moreover, structurally distinct irreversible FGFR inhibitors such as TAS-120 (59, Fig. 21b) [117], which is currently in phase I/II clinical studies (NCT02052778), have been reported.

(a) Irreversible and covalent-reversible FGFR kinase inhibitors derived from unreactive inhibitor PD173074. (b) 1H-pyrazolo[3,4-d]pyrimidine-derived covalent FGFR inhibitors exemplified by clinical candidate TAS-120. (c) Covalent c-SRC kinase inhibitors
Covalent inhibitors of the proto-oncogene c-SRC have been generated, for example, by Kwarcinski et al. via attaching electrophilic moieties to promiscuous kinase inhibitor scaffolds [118]. These compounds exemplified by 60a and 60b (Fig. 21c; IC50 = 91 and 93 nM; kinact/KI = 1.7 × 103 M−1s−1 and 4.0 × 103 M−1s−1) displayed good selectivity against the homologous kinases c-ABL and HCK both lacking the P2 cysteine (Cys277 in c-SRC). As expected, 60a/b were found to strongly hit the c-ABL Q252C mutant where an equivalent cysteine had been introduced. Remarkably, compound 60b inactivated c-ABL Q252C and also c-YES with a roughly two times higher kinact compared to c-SRC which was attributed to a kinked P-loop conformation in the former two kinases positioning the cysteine in closer proximity of the warhead. Similar inhibitors were designed from the scaffold of the c-SRC/BCR-ABL inhibitor dasatinib [118]. The exemplary vinyl sulfonamide 61 is a potent inhibitor of the dasatinib-resistant c-SRC T338M mutant (IC50 = 44 nM; kinact/KI = 4.0 × 104 M−1s−1) with good cellular potency and increased selectivity compared to the relatively promiscuous template dasatinib.
2.6 Development of Inhibitors Targeting Cysteines in the Hinge Region
2.6.1 Inhibitors Targeting the H1 Position and the H3 Subsite
Among the four FGF receptor tyrosine kinases, FGFR4 stands out as the only family member harboring a cysteine at the H2 (GK + 2) position in the middle of the hinge region. Equivalent cysteines have only been identified in four other kinases, namely TTK, MAPKAPK2, MAPKAPK3, and p70S6Kβ. This cysteine (Cys552) was first covalently addressed by the inhibitor BLU9931 from Blueprint Medicines (62a, Fig. 22), another compound related to the FIIN series albeit with the warhead attached via the quinazoline C2-position [119]. Being a potent FGFR4 inhibitor (IC50 = 3 nM; kinact/KI = 0.6 × 105 M−1 s−1), BLU9931 showed high selectivity within the FGFR family, but also against several kinases with an equivalently positioned cysteine and the remaining kinome. It further demonstrated activity in cells and antitumor activity in hepatocellular carcinoma (HCC) xenograft models. Close analog BLU554 (62b) and the distantly related inhibitor H3B-6527 (63) from Eisai [120] entered phase I clinical trials (NCT02508467, NCT02834780) as potential oral treatments for patients with FGF19/FGFR4-driven HCC. These compounds feature an interesting binding mode with a U-shaped arrangement between the hinge-binding motif, the spacer moiety, and the warhead (Fig. 23a, b) directing the electrophile toward the cysteine sulfur atom. This conformation is stabilized by a bidentate hydrogen bond between the two ortho-amino groups of the 1,2-phenylenediamine spacer and the backbone carbonyl oxygen of Ala553. In the case of BLU9931, an additional ortho-methyl group forcing an “out-of-plane” conformation of the phenyl ring is required to obtain an optimal balance of FGFR4 potency and selectivity against FGFR1–3 [119]. A water-mediated hydrogen bond of the acrylamide oxygen atom with Arg483 may further assist in orienting the warhead to be attacked by Cys552.

Selected irreversible FGFR4 inhibitors

(a) Schematic binding mode of prototypical covalent FGFR4 inhibitor BLU9931 (62a) deduced from the X-ray crystal structure of the covalent complex (PDB: 4XCU). (b) X-ray crystal structure of 62a covalently bound to the FGFR4 ATP pocket. A U-shaped conformation is adopted to hit Cys552 located in the hinge region. A water-mediated H-bond of the inhibitor’s amide oxygen atom and Arg483 was omitted for clarity
Non-canonical approaches to address FGFR4-Cys552 have been reported by Fairhurst and colleagues from Novartis [121]. In an HTS campaign, they identified 6-chloro-3-nitropyridine 64 (Fig. 24a) as a potent inhibitor of FGFR4 (IC50 = 32 nM; kinact/KI = 3.0 × 104 M−1 s−1) sparing FGFR1–3. The compound binds Cys552 via SNAr-displacement of the chloride from the pyridine C6-atom as shown by X-ray crystallography (PDB: 5NUD). The same screening campaign identified 2-formylquinoline amide 65 (Fig. 24b) as an FGFR4 inhibitor (IC50 = 65 nM) engaging Cys552 by highly reversible hemithioacetal formation. The latter compound was further developed to clinical candidate FGF401 (66), an orally available inhibitor with a favorable preclinical profile currently undergoing phase I/II studies (NCT02325739) as a potential treatment of FGFR4/β-klotho-positive HCC and solid tumors.

(a) Irreversible FGFR4 inhibitor 64 engaging FGFR4-Cys552 via an SNAr-electrophile. (b) Covalent-reversible FGFR4 inhibitors relying on an aldehyde warhead. (c) Covalent FLT3 inhibitor FF-10101
No reports describing the covalent targeting of other kinases with an H1 cysteine could be identified at the time of writing. Similarly, cysteines in the H2 position (GK + 3), which are present in a large set of kinases, have remained unaddressed so far. This is presumably owed to the H2 cysteines’ limited flexibility and their location underneath the region that is usually occupied by the hinge-binding motif, which is difficult to reach with typical type I inhibitor scaffolds. However, a cysteine located at the H3 subsite (GK + 4/5) has recently been engaged in Fms-like tyrosine kinase 3 (FLT3) by 4-(dialkylamino)crotonamide-based inhibitor FF-10101 (67, Fig. 24c) [122]. Notably, this kinase representing a validated target in the treatment of acute myeloid leukemia features two other cysteines (D1 and H2 position) which could potentially be addressed. FF-10101 potently inhibits wild-type FLT3 (IC50 = 0.2 nM) and shows a high activity against a variety of leukemia cell lines including those harboring quizartinib-resistance mutations. Along with several other kinases, the compound potently hit the two related kinases KIT and FMS (IC50 = 0.9 nM and 2.0 nM, respectively) and, to a lesser extent, the non-receptor tyrosine kinase FGR (IC50 = 12 nM), all sharing a H3-GK + 4 cysteine. Binding to Cys695 was demonstrated by X-ray crystallography (PDB: 5X02) and confirmed by a set of experiments using the FLT3 C695S mutant and an unreactive analog. FF-10101 showed promising effects in preclinical models and entered phase I/II clinical trials as a potential treatment for patients with relapsed or refractory hematological malignancies (NCT03194685 and NCT02193958).
2.7 Development of Inhibitors Targeting Cysteines Around the DFG Motif and in the Activation Segment
The activation segment is located between the conserved DFG and APE motifs and contains the activation loop and the P + 1 loop. It represents a very flexible region and is often poorly resolved in X-ray crystal structures. Thus, exhaustive analysis of cysteine positioning and accessibility is complicated and structure-based design approaches can be difficult to accomplish. A large set of kinases possesses cysteine moieties in direct neighborhood to the DFG motif, either at the O2 subsite (DFG + 1/2) or in the D1 position (DFG-1). Another set of cysteines can be found at the activation segment’s O3 subsite, frequently two residues after the phosphorylation site. Further accessible cysteines are located in the activation loop. Examples are Cys178 and 179 in IKKα and β, which have been identified as the target of natural products and endogenous ligands like PGA1 (vide supra) [5]. However, proper assignment of the activation loop cysteines is impeded by the highly plastic nature and the variable length of this region [5].
A cysteine located at O2 (Cys296, DFG + 2 position) in the protein kinases B (PKBs, also known as AKTs) is addressed by allosteric inhibitors (vide infra) and presumably also by pyranonaphthoquinones (vide supra) while many kinases with a D1 cysteine are targeted by the aforementioned resorcylic acid lactones like hypothemycin, LL-Z1640-2, and analogs thereof (see Fig. 6), albeit with limited specificity. D1 cysteine residues have also been tackled by rational design efforts. Although there are many kinases (≈ 10% of the kinome) featuring such a cysteine, these targets are distributed over almost all kinase families suggesting that distinct structural features may readily be exploited as additional selectivity filters. For example, Ward and co-workers from AstraZeneca reported on covalent ERK1/2 inhibitors derived from several reversible inhibitor classes using a structure-based approach [123]. The most promising series was based on a 2,4-diaminopyrimidine scaffold (exemplified by 68a and 68b, Fig. 25a). The study identified highly potent compounds covalently engaging Cys166 in ERK2 (see, for example, PDB: 4ZZO) with good selectivity against several other kinases of this subset and in the kinome. CC-90003 (69) [124], a structurally related covalent ERK1/2 inhibitor from Celgene, was investigated in phase I clinical trials (NCT02313012) which were, however, terminated in 2016 due to lack of response, an inappropriate PK-profile, and neurotoxicity [125]. Analogous probe 70 (see also PDB: 5LCJ) enabled imaging of ERK1/2 in living cells by employing trans-cyclooctene (TCO)/tetrazine (Tz) click chemistry [126].

(a) Irreversible ERK1/2 inhibitors including clickable TCO-probe 70. (b and c) Covalent TAK1, dual EGFR/VEGFR2, and GSK-3β/CK-1δ inhibitors
Covalent TAK1 targeting was accomplished by Gray and co-workers with closely related inhibitors such as 71a (IC50 = 5.1 nM) and 71b (IC50 = 50 nM; kinact/KI = 6.9 × 103 M−1 s−1) shown in Fig. 25b. Inhibitor 71b hit multiple kinases in a panel among those being 10 kinases with a cysteine at the D1 position. However, both compounds possessed a moderate selectivity against MEK1 and ERK2 also sharing this cysteine. Interestingly, replacement of the acrylamide warhead by a shorter chloroacetamide moiety led to a strong increase in selectivity against the latter two kinases, which was accompanied by a rearrangement of the DFG motif in TAK as observed by X-ray crystallography (see, for example, the PDB structures 5JH6 vs. 5E7R).
Various warhead chemistries were evaluated by Yang et al. to covalently trap GSK-3β [127]. Out of their series, one of the most promising inhibitors was fluoroacetamide 72 (Fig. 25c; IC50 = 17 nM). Labeling of Cys199 was suggested by experiments with a fluorescent competitor probe and by digestion/MS using an acrylamide-based analog. In contrast, two derivatives equipped with 2-chloropyridine moieties as SNAr-type warheads proved to be reversible inhibitors albeit with similar biochemical potency. Another interesting study was conducted by Wissner et al. who installed two independent warheads on a quinazoline scaffold to generate dual irreversible inhibitors of EGFR and vascular endothelial growth factor receptor 2 (VEGFR2, also termed KDR) as exemplified by compound 73 [128]. While the 4-(dimethylamino)crotonamide residue was directed toward the aforementioned F2 cysteine in EGFR, the substituted benzoquinone moiety was meant to target Cys1045 located at the D1 position of VEGFR2, presumably via a radical-based reductive addition mechanism. Very recently, α-cyanoacrylamide 73 has been described as dual GSK-3β/CK-1δ inhibitor [129]. While being only moderately potent, the compound covalently modified GSK-3β as shown by X-ray crystallography (PDB ID: 6H0U) while no covalent interaction can be assumed for CK-1δ, which is devoid of cysteine moieties in the active site.
2.8 Development of Inhibitors Targeting Remote Cysteines or Inactive Conformations
Remote cysteines are located outside of the regular kinase domain. Such cysteines may also be amenable to covalent targeting with ATP pocket ligands, provided that the cysteine of interest is positioned in spatial proximity to ATP binding cleft. A first study employing this approach was reported in 2014 by researches from the Dana-Farber Cancer Institute and MIT [130]. A screening campaign identified THZ1 (75a, Fig. 26a) as low nanomolar inhibitor of the cyclin-dependent kinase CDK7. Since saturation of the reactive α,β-unsaturated amide moiety led to a strong loss in anti-proliferative and biochemical activity, a covalent binding mode was assumed, which was confirmed with a biotinylated analog. Subsequent MS experiments confirmed Cys312, a residue located in a C-terminal extension traversing the ATP pocket, as the site of modification. As expected, mutation of the target cysteine to serine prevented labeling. Besides several kinases that were hit reversibly, higher inhibitor concentrations also affected CDK12 and CDK13 possessing cysteines that occupy spatially similar locations. Optimization toward these kinases furnished THZ531 (75b), a CDK12/13 inhibitor with IC50 values in the mid-nanomolar range and good selectivity against CDK7 and CDK9 [131]. The compound demonstrated high specificity in a cellular KiNativ screen which was corroborated in cell lysates by means of a biotin-labeled probe. In contrast, several off-targets were hit in a large kinase panel. Remarkably, potent binding to JNKs was observed. JNKs feature a cysteine in the F3 position on the first turn of the αD-helix which can probably be reached by the same warhead/spacer combination. As confirmed by X-ray crystallography and MS, covalent modification of CDK12 occurred at Cys1039 while the CDK12-C1039S mutant resisted covalent labeling in cells. Interestingly, two different binding modes were observed in which both, the loop bearing Cys1039 and the piperidine-linked amide moiety, adopted distinct conformations to enable covalent bond formation (Fig. 26b). Compound 75b strongly inhibited the expression of DNA damage response (DDR) and super-enhancer genes in cells and induced apoptosis. Since resistance to the THZ-series of compounds was acquired by up-regulation of ABC-transporters, the structurally unrelated follow-up CDK12 inhibitor E9 (76) was developed to escape ABC-transporter-mediated efflux [132].

(a) Covalent CDK inhibitors targeting remote cysteines. (b) Binding modes observed in the X-ray crystal structure of THZ531 covalently bound to CDK12 (PDB: 5ACB)
Plasticity is a key regulatory feature of the kinases’ catalytic domain. However, only few cysteines accessible in inactive conformations have been targeted to date. In their seminal work analyzing cysteines available in DFG-out and αC-helix-out conformations, Leproult et al. also designed imatinib-derived electrophilic inhibitors to target the kinases KIT and PDGFRα (platelet-derived growth factor receptor α) via cysteine moieties in the catalytic loop (Cys788 and Cys814, respectively) [15]. Despite its relatively weak apparent potency, key compound 77 (Fig. 27) labeled the predicted cysteines as shown by MS and hit only seven other targets (PDGFRβ, JNK 1–3, DDR1, BRAFV600E, and CSF1R) in a panel of 440 kinases. Notably, the panel contained 17 out of 20 kinases sharing an equivalently positioned cysteine of which (besides KIT and PDGFRα) only PDGFRβ and CSF1R were potently bound.

Covalent PDGFR/KIT inhibitor 77
2.9 Development of Inhibitors Targeting Cysteines in Allosteric Pockets
Besides ligands addressing the ATP pocket, covalent kinase inhibitors engaging allosteric sites have also been identified. As an example, covalent allosteric inhibition of RSKs and MSKs has been suggested to contribute to the biological activity of dimethyl fumarate (78, Fig. 28), a reactive low molecular weight drug used in the treatment of psoriasis and multiple sclerosis [133]. The complex natural product (+)-ainsliadimer A (79) featuring two Michael acceptor moieties has also been shown to act as a covalent kinase inhibitor targeting IKKα and IKKβ via a (putative) allosteric pocket involving Cys46 [134]. Recently discovered acrylamide-based covalent allosteric inhibitors include the AKT inhibitor borussertib (80) [135] and the fragment 81 addressing CDK2 [136]. Moreover, chemical-genetics approaches have been used to enable covalent targeting of non-canonical binding sites via the introduction of cysteines. For example, Bührmann et al. recently identified probes covalently binding to a lipid pocket of p38α MAP kinase cysteine mutants (S251C and S252C) without labeling the wild-type enzyme [137]. It should, however, be noted that ligands addressing allosteric- and other non-canonical binding sites are often identified serendipitously since such pockets tend to be highly plastic and are frequently not observed in the apo-structures.

Examples of covalent kinase inhibitors targeting allosteric pockets
2.10 Development of Inhibitors Targeting Lysine or Tyrosine
As mentioned before (see Sect. 2.3), natural products covalently engaging kinases via lysine residues are well-known. In addition, lysine and tyrosine moieties have been addressed with synthetic small molecules. For example, activated esters have been employed to target the catalytic lysine in PI3Kδ by researchers at GSK [138]. Since the sulfonamide substituent of the reversible PI3Kδ inhibitor GSK2292767 (82, Fig. 29a) interacts with the ε-amino group of PI3Kδ-Lys779, replacement of this moiety by activated phenolic esters was considered as a strategy to address the amino nucleophile. Biochemical potencies of the generated compounds roughly correlated with the leaving group properties of the corresponding phenolates reaching down to the subnanomolar range for the most activated derivative (83a, IC50 = 0.6 nM; kinact/KI = 1.9 × 105 M−1 s−1). Interestingly, analysis of binding kinetics revealed that kinact was relatively constant in the series while differences in the overall potency seemed to be mainly driven by KI. This finding was attributed to a more complex reaction mechanism as compared to the canonical two-step model (see Fig. 1). Notably, the analogous non-activated methyl ester bound only reversibly to the enzyme. Key compound 83b showed high activity in the enzymatic assay (IC50 = 8 nM; kinact/KI = 2.1 × 104 M−1 s−1) and in human whole blood (IC50 = 13 nM) while being selective against other PI3K isoforms (ca. three orders of magnitude against PI3Kα/β/γ) and in a larger kinase panel. A clean chemoproteomic profile was shown for an analogous azide-labeled probe. Exclusive labeling of Lys779 was further demonstrated by MS analysis and X-ray crystallography (see Fig. 31a). The inhibitor also showed a selectivity window in which PI3Kδ was covalently inhibited while PI3Kα and β engagement was reversible.

Examples of lysine-targeted covalent kinase inhibitors. (a) Design of activated esters addressing the catalytic lysine in PI3Kδ. (b) Vinyl sulfones targeting the solvent-exposed Lys89 in CDK2
Michael-acceptor chemistry was used by Anscombe et al. to obtain an irreversible CDK2 inhibitor [139]. Starting from non-covalently binding sulfonamide NU6102 (84a, Fig. 29b), replacement of this functional group by an electrophilic vinyl sulfone furnished covalent analog NU6300 (84b). In this context, it is worth mentioning that previous model experiments by Dahal et al. demonstrated vinyl sulfones and vinyl sulfonamides to have a certain preference to react with lysine over cysteine [9, 140]. Covalent binding of compound 84b to Lys89 located in the front region of the ATP pocket was suggested by MS experiments and confirmed by X-ray crystallography (PDB: 5CYI). The inhibitor, however, featured only moderate reversible binding affinity (Ki = 1.31 μM) and showed relatively slow inactivation kinetics, presumably due to the solvent-exposed nature of the target lysine favoring the protonated state.
Sulfonyl fluorides have frequently been used to address the lysines [9]. For example, ATP-derived probe 5’-O-4-((fluorosulfonyl)benzoyl)adenosine (p-FSBA, 85, Fig. 30a) acts as a non-selective covalent kinase inhibitor typically modifying the catalytic lysine. While investigating the reactivity of sulfur(VI) fluorides, Mukherjee and colleagues from AstraZeneca tested meta-substituted FSBA analogs (m-FSBAs, general structure 86) with a variety of additional substituents in the 4-position of the phenyl ring (−R, ortho to the sulfonyl fluoride warhead) as inhibitors of the kinases FGFR1 and SYK. Covalent binding of the two analogs 86a and 86b to Lys514 in FGFR1 was shown by X-ray crystallography (PDB: 5O49A and 5O4A) and only a single product was observed in MS. The rate of covalent complex formation correlated reasonably with the σp− parameter of the substituent −R and also with the reactivity data previously determined using model amino acids. In contrast, negligible covalent modification was observed for two analogous aryl fluorosulfates derived from m- and p-FSBA, respectively, which is in line with the very low intrinsic reactivity described for this electrophile.

Lysine and tyrosine-targeted sulfonyl fluorides. (a) p-/m-FSBA and analogs. (b) Broad-spectrum kinase probe XO44. (c) Tyrosine-targeted SRPK1/2 inhibitor SRPKIN-1
An interesting application for sulfonyl fluoride-based probes was recently reported by Zhao et al. [141]. Pyrimidinyl 3-aminopyrazole XO44 (87, Fig. 30b) was designed as a clickable broad-spectrum kinase ligand enabling chemoproteomic selectivity profiling. As predicted, the compound specifically labeled the catalytic lysine (Lys295; see Fig. 31b) in the model kinase c-SRC. A similar binding mode was observed in the EGFR kinase domain (PDB: 5U8L). At a concentration of 1 μM, XO44 inhibited 219 of 375 protein kinases in a panel (≥50%) and captured 133 kinases in Jurkat T-cells. Although many non-kinase off-targets were also modified, kinases accounted for most of the signal intensity in MS experiments. Using the poorly selective reversible kinase inhibitor dasatinib as a competitor, compound 87 was validated as a probe enabling cellular selectivity profiling.

(a) Inhibitor 83b covalently bound to Lys779 of PI3Kδ (PDB: 6EYZ). (b) Probe XO44 covalently bound to chicken c-SRC-Lys295 (PDB: 5K9I)
A very recent example of a tyrosine-targeted covalent kinase inhibitor was provided by Hatcher et al. [142]. After discovering that the approved reversible anaplastic lymphoma kinase (ALK) inhibitor alectinib (88a, Fig. 30c) strongly inhibits the SR-protein kinase SRPK1 (IC50 = 11 nM), an m-phenylsulfonyl fluoride moiety was introduced to address Tyr227, a unique tyrosine adjacent to the solvent-exposed front region of the SRPK1 active site. The obtained inhibitor SRPKIN-1 (88b) showed similar SRPK1 activity (IC50 = 36 nM) while retaining only moderate potency on ALK (IC50 = 195 nM). No kinetic analysis was provided to deduce the contribution of covalent bond formation to the observed activity. However, washout experiments supported a covalent binding mode and MS experiments confirmed the predicted labeling of Tyr227. Cellular profiling revealed good selectivity for SRPK1 and SRPK2 and the inhibitor suppressed neovascularization in a choroidal neovascularization (CNV) model.
3 Summary and Outlook
The field of covalent protein kinase inhibitor research has matured over the last years. Covalent targeting strategies have enabled the design of chemical probes with excellent selectivity in the kinome and the benefits of TCIs have successfully been implemented in drug discovery. Covalent approaches have been used, for example, to boost potency and break resistance (e.g., with covalent EGFR inhibitors), to achieve durable target occupancy (e.g., with covalent BTK inhibitors), or to promote excellent selectivity over closely related enzymes (e.g., with covalent JAK3 or FGFR4 inhibitors). The recent FDA approval of six irreversible kinase inhibitors impressively highlights the utility of covalent design strategies for the discovery of new medicines. Although covalent kinase inhibitors have almost exclusively been approved for cancer treatment so far, substantial efforts are currently being made to develop such drugs for non-oncology indications, especially for the treatment of inflammatory and autoimmune disorders.
In the current practice, covalent kinase targeting largely relies on limited warhead chemistry. Generally, attenuated Michael acceptors such as α,β-unsaturated amides are used to address non-catalytic cysteines in a (quasi)-irreversible fashion. However, more diverse warhead types are increasingly receiving attention [9]. Most notably, covalent-reversible targeting approaches relying, for example, on dually activated Michael acceptors, but also on other electrophiles such as cyanamides or aldehydes, are gaining importance as highlighted by the inhibitors PRN1008 and FGF401 (see Figs. 12b and 24b) which are currently under clinical investigation. Furthermore, attempts to address other nucleophilic amino acids inside or proximal to the ATP binding pocket (e.g., Lys and Tyr) or cysteine moieties at allosteric sites have recently been successful and such approaches are expected to gain traction in the near future. Although the structure-guided modification of known reversible inhibitors, which is facilitated by the steeply increasing amount of publicly available X-ray crystal structures, is still by far the most common strategy for the discovery of kinase TCIs, alternative concepts such as the screening of reactive fragments or DNA-encoded libraries show great promise. Methods for estimating the extent of specific and non-specific labeling by irreversible covalent inhibitors in living systems have also advanced. Especially MS-based chemoproteomic approaches are now enabling a more complete understanding of the cellular labeling profiles of covalent inhibitors while biotinylated or fluorescent covalent probes have also been used as valuable tools to monitor target occupancy in cells or in vivo.
Nevertheless, we still face many challenges in the realm of covalent kinase inhibitor discovery, especially, when aiming for clinical applications. A general limitation of the TCI approach, especially in oncology, consists in its vulnerability to resistance development by mutation of the nucleophilic amino acid (e.g., cysteine to serine). This is an intrinsic liability since TCIs target non-conserved residues, which are typically not crucial for protein integrity and function. On the other hand, when aiming for other therapeutic areas beyond oncology where resistance mutations are less likely, an even more stringent benefit/risk assessment is required. The latter, however, is currently complicated by a lack of reliable models to predict toxicity and immune-mediated adverse events including idiosyncratic drug reactions, which may only become apparent in late stage clinical trials or even after long-term therapeutic application. Beside the frequently discussed issue of permanent off-target modification, we also lack a comprehensive understanding of how reversible interactions with unintended targets contribute to the biological profiles of reactive inhibitors, a fact that is rarely alluded to in the current literature. Similarly, covalent modification of non-protein off-targets (e.g., DNA or RNA) by kinase TCIs remains widely unexplored.
As mentioned in the introduction, a detailed analysis of target-binding kinetics is still not performed by default. Thus, the contribution of the covalent binding event to the observed overall biological effects often remains incompletely understood [13]. Further exploration of warhead chemistry to enable the generation of inhibitors with tailored reactivities specifically matching the requirements of the target of interest represents another future challenge [9]. There is also an increasing awareness of cysteine oxidation as a posttranslational modification, which may regulate kinase function and impair covalent binding [13]. The influence of the “cysteine redoxome” and its dynamics on the efficacy covalent inhibitors clearly merits further investigations which may have an important impact on our perspective on covalent cysteine targeting. Finally, and despite the success of covalent approaches in the development of chemical probes and drugs, the coverage of the kinases’ cysteinome by suitable inhibitors is still low and several of the presented cysteine locations (see Fig. 4 and Table 1) have not yet been targeted at all. While a thorough examination of the protein kinases’ lysinome and tyrosinome remains to be performed, it also unclear, how many ligandable cysteine locations, especially in inactive states or induced/allosteric pockets have not been captured in the published analyses. The latter may open up new avenues for covalent ligand design.
Despite the many challenges associated with covalent inhibitor development, a bright future can be expected for protein kinase TCIs. Since many kinases are still understudied and lack suitable chemical probes required for investigation of their function, there is a large and untapped potential for covalent kinase inhibitor discovery, which will inspire continuing research efforts. Moreover, a multitude of covalent protein kinase inhibitors are currently in clinical development for application in cancer therapy and beyond. Therefore, we can expect many more success stories from this field lying ahead of us.
References
Singh J, Petter RC, Baillie TA, Whitty A (2011) The resurgence of covalent drugs. Nat Rev Drug Discov 10:307–317. https://doi.org/10.1038/nrd3410
Bauer RA (2015) Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov Today 20:1061–1073. https://doi.org/10.1016/j.drudis.2015.05.005
Baillie TA (2016) Targeted covalent inhibitors for drug design. Angew Chem Int Ed 55:13408–13421. https://doi.org/10.1002/anie.201601091
Singh J, Petter RC, Kluge AF (2010) Targeted covalent drugs of the kinase family. Curr Opin Chem Biol 14:475–480. https://doi.org/10.1016/j.cbpa.2010.06.168
Barf T, Kaptein A (2012) Irreversible protein kinase inhibitors: balancing the benefits and risks. J Med Chem 55:6243–6262. https://doi.org/10.1021/jm3003203
Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS (2013) Developing irreversible inhibitors of the protein kinase Cysteinome. Chem Biol 20:146–159. https://doi.org/10.1016/j.chembiol.2012.12.006
Chaikuad A, Koch P, Laufer SA, Knapp S (2018) The Cysteinome of protein kinases as a target in drug development. Angew Chem Int Ed 57:4372–4385. https://doi.org/10.1002/anie.201707875
de Cesco S, Kurian J, Dufresne C, Mittermaier AK, Moitessier N (2017) Covalent inhibitors design and discovery. Eur J Med Chem 138:96–114. https://doi.org/10.1016/j.ejmech.2017.06.019
Gehringer M, Laufer SA (2019) Emerging and re-emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J Med Chem 62:5673–5724. https://doi.org/10.1021/acs.jmedchem.8b01153
Hann MM (2011) Molecular obesity, potency and other addictions in drug discovery. Med Chem Commun 2:349–355. https://doi.org/10.1039/C1MD00017A
Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, Romanov S, Finkle D, Shu J, Patel V, Ton T, Li X, Loughhead DG, Nunn PA, Karr DE, Gerritsen ME, Funk JO, Owens TD, Verner E, Brameld KA, Hill RJ, Goldstein DM, Taunton J (2015) Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat Chem Biol 11:525–531. https://doi.org/10.1038/nchembio.1817
Strelow JM (2017) A perspective on the kinetics of covalent and irreversible inhibition. SLAS Discov 22:3–20. https://doi.org/10.1177/1087057116671509
Schwartz PA, Kuzmic P, Solowiej J, Bergqvist S, Bolanos B, Almaden C, Nagata A, Ryan K, Feng J, Dalvie D, Kath JC, Xu M, Wani R, Murray BW (2014) Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc Natl Acad Sci 111:173–178. https://doi.org/10.1073/pnas.1313733111
Awoonor-Williams E, Rowley CN (2018) How reactive are druggable cysteines in protein kinases? J Chem Inf Model 58:1935–1946. https://doi.org/10.1021/acs.jcim.8b00454
Leproult E, Barluenga S, Moras D, Wurtz J-M, Winssinger N (2011) Cysteine mapping in conformationally distinct kinase nucleotide binding sites: application to the design of selective covalent inhibitors. J Med Chem 54:1347–1355. https://doi.org/10.1021/jm101396q
Onufriev AV, Alexov E (2013) Protonation and pK changes in protein–ligand binding. Q Rev Biophys 46:181–209. https://doi.org/10.1017/S0033583513000024
Casimiro-Garcia A, Trujillo JI, Vajdos F, Juba B, Banker ME, Aulabaugh A, Balbo P, Bauman J, Chrencik J, Coe JW, Czerwinski R, Dowty M, Knafels JD, Kwon S, Leung L, Liang S, Robinson RP, Telliez J-B, Unwalla R, Yang X, Thorarensen A (2018) Identification of Cyanamide-based Janus kinase 3 (JAK3) covalent inhibitors. J Med Chem. https://doi.org/10.1021/acs.jmedchem.8b01308
Zhao Z, Liu Q, Bliven S, Xie L, Bourne PE (2017) Determining Cysteines available for covalent inhibition across the human Kinome. J Med Chem 60:2879–2889. https://doi.org/10.1021/acs.jmedchem.6b01815
Lonsdale R, Ward RA (2018) Structure-based design of targeted covalent inhibitors. Chem Soc Rev 47:3816–3830. https://doi.org/10.1039/C7CS00220C
Backus KM, Correia BE, Lum KM, Forli S, Horning BD, González-Páez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF (2016) Proteome-wide covalent ligand discovery in native biological systems. Nature 534:570–574. https://doi.org/10.1038/nature18002
Kathman SG, Xu Z, Statsyuk AV (2014) A fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. J Med Chem 57:4969–4974. https://doi.org/10.1021/jm500345q
Jöst C, Nitsche C, Scholz T, Roux L, Klein CD (2014) Promiscuity and selectivity in covalent enzyme inhibition: a systematic study of electrophilic fragments. J Med Chem 57:7590–7599. https://doi.org/10.1021/jm5006918
Zimmermann G, Rieder U, Bajic D, Vanetti S, Chaikuad A, Knapp S, Scheuermann J, Mattarella M, Neri D (2017) A specific and covalent JNK-1 ligand selected from an encoded self-assembling chemical library. Chem Eur J 23:8152–8155. https://doi.org/10.1002/chem.201701644
Chan AI, McGregor LM, Jain T, Liu DR (2017) Discovery of a covalent kinase inhibitor from a DNA-encoded small-molecule library × protein library selection. J Am Chem Soc 139:10192–10195. https://doi.org/10.1021/jacs.7b04880
Jackson PA, Widen JC, Harki DA, Brummond KM (2017) Covalent modifiers: a chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-Michael addition reactions. J Med Chem 60:839–885. https://doi.org/10.1021/acs.jmedchem.6b00788
Bandyopadhyay A, Gao J (2016) Targeting biomolecules with reversible covalent chemistry. Curr Opin Chem Biol 34:110–116. https://doi.org/10.1016/j.cbpa.2016.08.011
Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frödin M, Taunton J (2012) Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat Chem Biol 8:471–476. https://doi.org/10.1038/nchembio.925
Krishnan S, Miller RM, Tian B, Mullins RD, Jacobson MP, Taunton J (2014) Design of reversible, cysteine-targeted Michael acceptors guided by kinetic and computational analysis. J Am Chem Soc 136:12624–12630. https://doi.org/10.1021/ja505194w
Knoepfel T, Furet P, Mah R, Buschmann N, Leblanc C, Ripoche S, Graus-Porta D, Wartmann M, Galuba I, Fairhurst RA (2018) 2-formylpyridyl ureas as highly selective reversible-covalent inhibitors of fibroblast growth factor receptor 4. ACS Med Chem Lett 9:215–220. https://doi.org/10.1021/acsmedchemlett.7b00485
Müller J, Kirschner RA, Geyer A, Klebe G (2019) Conceptional design of self-assembling bisubstrate-like inhibitors of protein kinase A resulting in a Boronic acid glutamate linkage. ACS Omega 4:775–784. https://doi.org/10.1021/acsomega.8b02364
Shindo N, Fuchida H, Sato M, Watari K, Shibata T, Kuwata K, Miura C, Okamoto K, Hatsuyama Y, Tokunaga K, Sakamoto S, Morimoto S, Abe Y, Shiroishi M, Caaveiro JMM, Ueda T, Tamura T, Matsunaga N, Nakao T, Koyanagi S, Ohdo S, Yamaguchi Y, Hamachi I, Ono M, Ojida A (2019) Selective and reversible modification of kinase cysteines with chlorofluoroacetamides. Nat Chem Biol 15:250. https://doi.org/10.1038/s41589-018-0204-3
Zhang J, Yang PL, Gray NS (2009) Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 9:28–39. https://doi.org/10.1038/nrc2559
Traxler P (1998) Tyrosine kinase inhibitors in cancer treatment (part II). Expert Opin Ther Pat 8:1599–1625. https://doi.org/10.1517/13543776.8.12.1599
Traxler P, Furet P (1999) Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol Ther 82:195–206. https://doi.org/10.1016/S0163-7258(98)00044-8
Backes A, Zech B, Felber B, Klebl B, Müller G (2008) Small-molecule inhibitors binding to protein kinase. Part II: the novel pharmacophore approach of type II and type III inhibition. Exp Opin Drug Discovery 3:1427–1449. https://doi.org/10.1517/17460440802580106
Liao JJ-L (2007) Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J Med Chem 50:409–424. https://doi.org/10.1021/jm0608107
Yoshida T, Kakizuka A, Imamura H (2016) BTeam, a novel BRET-based biosensor for the accurate quantification of ATP concentration within living cells. Sci Rep 6:39618. https://doi.org/10.1038/srep39618
Yen-Pon E, Li B, Acebrón-Garcia-de-Eulate M, Tomkiewicz-Raulet C, Dawson J, Lietha D, Frame MC, Coumoul X, Garbay C, Etheve-Quelquejeu M, Chen H (2018) Structure-based design, synthesis, and characterization of the first irreversible inhibitor of focal adhesion kinase. ACS Chem Biol 13:2067–2073. https://doi.org/10.1021/acschembio.8b00250
Ferguson FM, Gray NS (2018) Kinase inhibitors: the road ahead. Nat Rev Drug Discov 17:353–377. https://doi.org/10.1038/nrd.2018.21
Zhao Z, Bourne PE (2018) Progress with covalent small-molecule kinase inhibitors. Drug Discov Today 23:727–735. https://doi.org/10.1016/j.drudis.2018.01.035
Gilbert AM (2014) Recent advances in irreversible kinase inhibitors. Pharm Patent Anal 3:375–386. https://doi.org/10.4155/ppa.14.24
Sanderson K (2013) Irreversible kinase inhibitors gain traction. Nat Rev Drug Discov 12:649–651. https://doi.org/10.1038/nrd4103
Miklos D, Cutler CS, Arora M, Waller EK, Jagasia M, Pusic I, Flowers ME, Logan AC, Nakamura R, Blazar BR, Li Y, Chang S, Lal I, Dubovsky J, James DF, Styles L, Jaglowski S (2017) Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood 130:2243–2250. https://doi.org/10.1182/blood-2017-07-793786
Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T, Okamoto I, Zhou C, Cho BC, Cheng Y, Cho EK, Voon PJ, Planchard D, Su W-C, Gray JE, Lee S-M, Hodge R, Marotti M, Rukazenkov Y, Ramalingam SS (2017) Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. N Engl J Med 378:113–125. https://doi.org/10.1056/NEJMoa1713137
Barf T, Covey T, Izumi R, van de Kar B, Gulrajani M, van Lith B, van Hoek M, de Zwart E, Mittag D, Demont D, Verkaik S, Krantz F, Pearson PG, Ulrich R, Kaptein A (2017) Acalabrutinib (ACP-196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J Pharmacol Exp Ther 363:240–252. https://doi.org/10.1124/jpet.117.242909
Shirley M (2018) Dacomitinib: first global approval. Drugs. https://doi.org/10.1007/s40265-018-1028-x
Lagoutte R, Winssinger N (2017) Following the lead from nature with covalent inhibitors. CHIMIA Int J Chem 71:703–711. https://doi.org/10.2533/chimia.2017.703
Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, Panayotou G (1996) Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol 16:1722–1733. https://doi.org/10.1128/MCB.16.4.1722
Zhao A, Lee SH, Moiena M, Jenkins RG, Patrick DR, Huber HE, Goetz MA, Hensens OD, Zink DL, Vilella D, Dombrowski AW, Lingham RB, Huang L (1999) Resorcylic acid lactones: naturally occurring potent and selective inhibitors of MEK. J Antibiot 52:1086–1094. https://doi.org/10.7164/antibiotics.52.1086
Williams DH, Wilkinson SE, Purton T, Lamont A, Flotow H, Murray EJ (1998) Ro 09-2210 exhibits potent anti-proliferative effects on activated T cells by selectively blocking MKK activity. Biochemistry 37:9579–9585. https://doi.org/10.1021/bi972914c
Rastelli G, Rosenfeld R, Reid R, Santi DV (2008) Molecular modeling and crystal structure of ERK2–hypothemycin complexes. J Struct Biol 164:18–23. https://doi.org/10.1016/j.jsb.2008.05.002
Schirmer A, Kennedy J, Murli S, Reid R, Santi DV (2006) Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. PNAS 103:4234–4239. https://doi.org/10.1073/pnas.0600445103
Wu J, Powell F, Larsen NA, Lai Z, Byth KF, Read J, Gu R-F, Roth M, Toader D, Saeh JC, Chen H (2013) Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)-7-Oxozeaenol. ACS Chem Biol 8:643–650. https://doi.org/10.1021/cb3005897
Sogabe Y, Matsumoto T, Hashimoto T, Kirii Y, Sawa M, Kinoshita T (2015) 5Z-7-Oxozeaenol covalently binds to MAP2K7 at Cys218 in an unprecedented manner. Bioorg Med Chem Lett 25:593–596. https://doi.org/10.1016/j.bmcl.2014.12.011
Shen Y, Boivin R, Yoneda N, Du H, Schiller S, Matsushima T, Goto M, Shirota H, Gusovsky F, Lemelin C, Jiang Y, Zhang Z, Pelletier R, Ikemori-Kawada M, Kawakami Y, Inoue A, Schnaderbeck M, Wang Y (2010) Discovery of anti-inflammatory clinical candidate E6201, inspired from resorcylic lactone LL-Z1640-2, III. Bioorg Med Chem Lett 20:3155–3157. https://doi.org/10.1016/j.bmcl.2010.03.087
Borthakur G, Gao C, Chen Y, Lan YS, Ruvolo VR, Nomoto K, Zhao N, Konopleva M, Andreeff M (2013) Study of activity of E6201, a dual FLT3 and MEK inhibitor, in acute Myelogenous leukemia with FLT3 or RAS mutation. Blood 122:2683–2683
Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG (2000) Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature 403:103–118. https://doi.org/10.1038/47520
Toral-Barza L, Zhang W-G, Huang X, McDonald LA, Salaski EJ, Barbieri LR, Ding W-D, Krishnamurthy G, Hu YB, Lucas J, Bernan VS, Cai P, Levin JI, Mansour TS, Gibbons JJ, Abraham RT, Yu K (2007) Discovery of lactoquinomycin and related pyranonaphthoquinones as potent and allosteric inhibitors of AKT/PKB: mechanistic involvement of AKT catalytic activation loop cysteines. Mol Cancer Ther 6:3028–3038. https://doi.org/10.1158/1535-7163.MCT-07-0211
Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL (2000) Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell 6:909–919. https://doi.org/10.1016/S1097-2765(05)00089-4
Norman BH, Shih C, Toth JE, Ray JE, Dodge JA, Johnson DW, Rutherford PG, Schultz RM, Worzalla JF, Vlahos CJ (1996) Studies on the mechanism of phosphatidylinositol 3-kinase inhibition by wortmannin and related analogs. J Med Chem 39:1106–1111. https://doi.org/10.1021/jm950619p
Singh J, Dobrusin EM, Fry DW, Haske T, Whitty A, McNamara DJ (1997) Structure-based design of a potent, selective, and irreversible inhibitor of the catalytic domain of the erbB receptor subfamily of protein tyrosine kinases. J Med Chem 40:1130–1135. https://doi.org/10.1021/jm960380s
Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, Hook KE, Keller PR, Leopold WR, Loo JA, McNamara DJ, Nelson JM, Sherwood V, Smaill JB, Trumpp-Kallmeyer S, Dobrusin EM (1998) Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. PNAS 95:12022–12027. https://doi.org/10.1073/pnas.95.20.12022
Smaill JB, Gonzales AJ, Spicer JA, Lee H, Reed JE, Sexton K, Althaus IW, Zhu T, Black SL, Blaser A, Denny WA, Ellis PA, Fakhoury S, Harvey PJ, Hook K, McCarthy FOJ, Palmer BD, Rivault F, Schlosser K, Ellis T, Thompson AM, Trachet E, Winters RT, Tecle H, Bridges A (2016) Tyrosine kinase inhibitors. 20. Optimization of substituted Quinazoline and Pyrido[3,4-d]pyrimidine derivatives as orally active, irreversible inhibitors of the epidermal growth factor receptor family. J Med Chem 59:8103–8124. https://doi.org/10.1021/acs.jmedchem.6b00883
Tsou H-R, Overbeek-Klumpers EG, Hallett WA, Reich MF, Floyd MB, Johnson BD, Michalak RS, Nilakantan R, Discafani C, Golas J, Rabindran SK, Shen R, Shi X, Wang Y-F, Upeslacis J, Wissner A (2005) Optimization of 6,7-Disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor Receptor-2 kinase activity. J Med Chem 48:1107–1131. https://doi.org/10.1021/jm040159c
Zhou W, Ercan D, Chen L, Yun C-H, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong K-K, Eck MJ, Gray NS, Jänne PA (2009) Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462:1070–1074. https://doi.org/10.1038/nature08622
Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM (2013) Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem 56:1363–1388. https://doi.org/10.1021/jm3012068
Boeckler FM, Zimmermann M. Personal communication
Van Der Steen N, Caparello C, Rolfo C, Pauwels P, Peters GJ, Giovannetti E (2016) New developments in the management of non-small-cell lung cancer, focus on rociletinib: what went wrong? Onco Targets Ther 9:6065–6074. https://doi.org/10.2147/OTT.S97644
Yver A (2016) Osimertinib (AZD9291) – a science-driven, collaborative approach to rapid drug design and development. Ann Oncol 27:1165–1170. https://doi.org/10.1093/annonc/mdw129
Günther M, Juchum M, Kelter G, Fiebig H, Laufer S (2016) Lung cancer: EGFR inhibitors with low nanomolar activity against a therapy-resistant L858R/T790M/C797S mutant. Angew Chem Int Ed 55:10890–10894. https://doi.org/10.1002/anie.201603736
Günther M, Lategahn J, Juchum M, Döring E, Keul M, Engel J, Tumbrink HL, Rauh D, Laufer S (2017) Trisubstituted Pyridinylimidazoles as potent inhibitors of the clinically resistant L858R/T790M/C797S EGFR mutant: targeting of both hydrophobic regions and the phosphate binding site. J Med Chem 60:5613–5637. https://doi.org/10.1021/acs.jmedchem.7b00316
Pan Z, Scheerens H, Li S-J, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KCK, Grothaus PG, Jeffery DA, Spoerke JM, Honigberg LA, Young PR, Dalrymple SA, Palmer JT (2006) Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2:58–61. https://doi.org/10.1002/cmdc.200600221
Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ (2010) The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. PNAS 107:13075–13080. https://doi.org/10.1073/pnas.1004594107
Flanagan ME, Abramite JA, Anderson DP, Aulabaugh A, Dahal UP, Gilbert AM, Li C, Montgomery J, Oppenheimer SR, Ryder T, Schuff BP, Uccello DP, Walker GS, Wu Y, Brown MF, Chen JM, Hayward MM, Noe MC, Obach RS, Philippe L, Shanmugasundaram V, Shapiro MJ, Starr J, Stroh J, Che Y (2014) Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J Med Chem 57:10072–10079. https://doi.org/10.1021/jm501412a
Markham A, Dhillon S (2018) Acalabrutinib: first global approval. Drugs 78:139–145. https://doi.org/10.1007/s40265-017-0852-8
Buhimschi AD, Armstrong HA, Toure M, Jaime-Figueroa S, Chen TL, Lehman AM, Woyach JA, Johnson AJ, Byrd JC, Crews CM (2018) Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry 57:3564–3575. https://doi.org/10.1021/acs.biochem.8b00391
Wu H, Huang Q, Qi Z, Chen Y, Wang A, Chen C, Liang Q, Wang J, Chen W, Dong J, Yu K, Hu C, Wang W, Liu X, Deng Y, Wang L, Wang B, Li X, Gray NS, Liu J, Wei W, Liu Q (2017) Irreversible inhibition of BTK kinase by a novel highly selective inhibitor CHMFL-BTK-11 suppresses inflammatory response in rheumatoid arthritis model. Sci Rep 7:466. https://doi.org/10.1038/s41598-017-00482-4
Watterson SH, Liu Q, Beaudoin Bertrand M, Batt DG, Li L, Pattoli MA, Skala S, Cheng L, Obermeier MT, Moore R, Yang Z, Vickery R, Elzinga PA, Discenza L, D’Arienzo C, Gillooly KM, Taylor TL, Pulicicchio C, Zhang Y, Heimrich E, McIntyre KW, Ruan Q, Westhouse RA, Catlett IM, Zheng N, Chaudhry C, Dai J, Galella MA, Tebben AJ, Pokross M, Li J, Zhao R, Smith D, Rampulla R, Allentoff A, Wallace MA, Mathur A, Salter-Cid L, Macor JE, Carter PH, Fura A, Burke JR, Tino JA (2019) Discovery of branebrutinib (BMS-986195): a strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton’s tyrosine kinase (BTK). J Med Chem 62:3228–3250. https://doi.org/10.1021/acs.jmedchem.9b00167
Park JK, Byun J-Y, Park JA, Kim Y-Y, Lee YJ, Oh JI, Jang SY, Kim YH, Song YW, Son J, Suh KH, Lee Y-M, Lee EB (2016) HM71224, a novel Bruton’s tyrosine kinase inhibitor, suppresses B cell and monocyte activation and ameliorates arthritis in a mouse model: a potential drug for rheumatoid arthritis. Arthritis Res Ther 18:91. https://doi.org/10.1186/s13075-016-0988-z
Smith PF, Krishnarajah J, Nunn PA, Hill RJ, Karr D, Tam D, Masjedizadeh M, Funk JO, Gourlay SG (2017) A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br J Clin Pharmacol 83:2367–2376. https://doi.org/10.1111/bcp.13351
Benson MJ, Rodriguez V, von Schack D, Keegan S, Cook TA, Edmonds J, Benoit S, Seth N, Du S, Messing D, Nickerson-Nutter CL, Dunussi-Joannopoulos K, Rankin AL, Ruzek M, Schnute ME, Douhan J (2014) Modeling the clinical phenotype of BTK inhibition in the mature murine immune system. J Immunol 193:185. https://doi.org/10.4049/jimmunol.1302570
Falgueyret J-P, Oballa RM, Okamoto O, Wesolowski G, Aubin Y, Rydzewski RM, Prasit P, Riendeau D, Rodan SB, Percival MD (2001) Novel, nonpeptidic cyanamides as potent and reversible inhibitors of human Cathepsins K and L. J Med Chem 44:94–104. https://doi.org/10.1021/jm0003440
Lainé D, Palovich M, McCleland B, Petitjean E, Delhom I, Xie H, Deng J, Lin G, Davis R, Jolit A, Nevins N, Zhao B, Villa J, Schneck J, McDevitt P, Midgett R, Kmett C, Umbrecht S, Peck B, Davis AB, Bettoun D (2011) Discovery of novel Cyanamide-based inhibitors of Cathepsin C. ACS Med Chem Lett 2:142–147. https://doi.org/10.1021/ml100212k
Zapf CW, Gerstenberger BS, Xing L, Limburg DC, Anderson DR, Caspers N, Han S, Aulabaugh A, Kurumbail R, Shakya S, Li X, Spaulding V, Czerwinski RM, Seth N, Medley QG (2012) Covalent inhibitors of interleukin-2 inducible T cell kinase (Itk) with nanomolar potency in a whole-blood assay. J Med Chem 55:10047–10063. https://doi.org/10.1021/jm301190s
Harling JD, Deakin AM, Campos S, Grimley R, Chaudry L, Nye C, Polyakova O, Bessant CM, Barton N, Somers D, Barrett J, Graves RH, Hanns L, Kerr WJ, Solari R (2013) Discovery of novel irreversible inhibitors of interleukin (IL)-2-inducible tyrosine kinase (Itk) by targeting cysteine 442 in the ATP pocket. J Biol Chem 288:28195–28206. https://doi.org/10.1074/jbc.M113.474114
Forster M, Gehringer M, Laufer SA (2017) Recent advances in JAK3 inhibition: isoform selectivity by covalent cysteine targeting. Bioorg Med Chem Lett 27:4229–4237. https://doi.org/10.1016/j.bmcl.2017.07.079
Forster M, Chaikuad A, Bauer SM, Holstein J, Robers MB, Corona CR, Gehringer M, Pfaffenrot E, Ghoreschi K, Knapp S, Laufer SA (2016) Selective JAK3 inhibitors with a covalent reversible binding mode targeting a new induced fit binding pocket. Cell Chem Biol 23:1335–1340. https://doi.org/10.1016/j.chembiol.2016.10.008
Forster M, Chaikuad A, Dimitrov T, Döring E, Holstein J, Berger B-T, Gehringer M, Ghoreschi K, Müller S, Knapp S, Laufer SA (2018) Development, optimization, and structure–activity relationships of covalent-reversible JAK3 inhibitors based on a tricyclic Imidazo[5,4-d]pyrrolo[2,3-b]pyridine scaffold. J Med Chem 61:5350–5366. https://doi.org/10.1021/acs.jmedchem.8b00571
Liu F, Zhang X, Weisberg E, Chen S, Hur W, Wu H, Zhao Z, Wang W, Mao M, Cai C, Simon NI, Sanda T, Wang J, Look AT, Griffin JD, Balk SP, Liu Q, Gray NS (2013) Discovery of a selective irreversible BMX inhibitor for prostate cancer. ACS Chem Biol 8:1423–1428. https://doi.org/10.1021/cb4000629
Liang X, Lv F, Wang B, Yu K, Wu H, Qi Z, Jiang Z, Chen C, Wang A, Miao W, Wang W, Hu Z, Liu J, Liu X, Zhao Z, Wang L, Zhang S, Ye Z, Wang C, Ren T, Wang Y, Liu Q, Liu J (2017) Discovery of 2-((3-Acrylamido-4-methylphenyl)amino)-N-(2-methyl-5-(3,4,5-trimethoxybenzamido)phenyl)-4-(methylamino)pyrimidine-5-carboxamide (CHMFL-BMX-078) as a highly potent and selective type II irreversible bone marrow kinase in the X Chromosome (BMX) kinase inhibitor. J Med Chem 60:1793–1816. https://doi.org/10.1021/acs.jmedchem.6b01413
London N, Miller RM, Krishnan S, Uchida K, Irwin JJ, Eidam O, Gibold L, Cimermančič P, Bonnet R, Shoichet BK, Taunton J (2014) Covalent docking of large libraries for the discovery of chemical probes. Nat Chem Biol 10:1066–1072. https://doi.org/10.1038/nchembio.1666
Shraga A, Olshvang E, Davidzohn N, Khoshkenar P, Germain N, Shurrush K, Carvalho S, Avram L, Albeck S, Unger T, Lefker B, Subramanyam C, Hudkins RL, Mitchell A, Shulman Z, Kinoshita T, London N (2018) Covalent docking identifies a potent and selective MKK7 inhibitor. Cell Chem Biol. https://doi.org/10.1016/j.chembiol.2018.10.011
Wolle P, Hardick J, Cronin SJF, Engel J, Baumann M, Lategahn J, Penninger JM, Rauh D (2019) Targeting the MKK7–JNK (mitogen-activated protein kinase kinase 7–c-Jun N-terminal kinase) pathway with covalent inhibitors. J Med Chem 62:2843–2848. https://doi.org/10.1021/acs.jmedchem.9b00102
Erlanson DA, Arndt JW, Cancilla MT, Cao K, Elling RA, English N, Friedman J, Hansen SK, Hession C, Joseph I, Kumaravel G, Lee W-C, Lind KE, McDowell RS, Miatkowski K, Nguyen C, Nguyen TB, Park S, Pathan N, Penny DM, Romanowski MJ, Scott D, Silvian L, Simmons RL, Tangonan BT, Yang W, Sun L (2011) Discovery of a potent and highly selective PDK1 inhibitor via fragment-based drug discovery. Bioorg Med Chem Lett 21:3078–3083. https://doi.org/10.1016/j.bmcl.2011.03.032
Koch A, Rode HB, Richters A, Rauh D, Hauf S (2012) A chemical genetic approach for covalent inhibition of analogue-sensitive Aurora kinase. ACS Chem Biol 7:723–731. https://doi.org/10.1021/cb200465c
Blair JA, Rauh D, Kung C, Yun C-H, Fan Q-W, Rode H, Zhang C, Eck MJ, Weiss WA, Shokat KM (2007) Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat Chem Biol 3:229–238. https://doi.org/10.1038/nchembio866
Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, Laughlin JD, Park H, LoGrasso PV, Patricelli M, Nomanbhoy TK, Sorger PK, Alessi DR, Gray NS (2012) Discovery of potent and selective covalent inhibitors of JNK. Chem Biol 19:140–154. https://doi.org/10.1016/j.chembiol.2011.11.010
Muth F, El-Gokha A, Ansideri F, Eitel M, Döring E, Sievers-Engler A, Lange A, Boeckler FM, Lämmerhofer M, Koch P, Laufer SA (2017) Tri- and tetrasubstituted pyridinylimidazoles as covalent inhibitors of c-Jun N-terminal kinase 3. J Med Chem 60:594–607. https://doi.org/10.1021/acs.jmedchem.6b01180
Koch P, Gehringer M, Laufer SA (2015) Inhibitors of c-Jun N-terminal kinases: an update. J Med Chem 58:72–95. https://doi.org/10.1021/jm501212r
Gehringer M, Muth F, Koch P, Laufer SA (2015) c-Jun N-terminal kinase inhibitors: a patent review (2010–2014). Expert Opin Ther Patents 25:849–872. https://doi.org/10.1517/13543776.2015.1039984
Kung A, Chen Y-C, Schimpl M, Ni F, Zhu J, Turner M, Molina H, Overman R, Zhang C (2016) Development of specific, irreversible inhibitors for a receptor tyrosine kinase EphB3. J Am Chem Soc 138:10554–10560. https://doi.org/10.1021/jacs.6b05483
Kung A, Schimpl M, Ekanayake A, Chen Y-C, Overman R, Zhang C (2017) A chemical-genetic approach to generate selective covalent inhibitors of protein kinases. ACS Chem Biol 12:1499–1503. https://doi.org/10.1021/acschembio.6b01083
Nacht M, Qiao L, Sheets MP, St. Martin T, Labenski M, Mazdiyasni H, Karp R, Zhu Z, Chaturvedi P, Bhavsar D, Niu D, Westlin W, Petter RC, Medikonda AP, Singh J (2013) Discovery of a potent and isoform-selective targeted covalent inhibitor of the lipid kinase PI3Kα. J Med Chem 56:712–721. https://doi.org/10.1021/jm3008745
Xie T, Lim SM, Westover KD, Dodge ME, Ercan D, Ficarro SB, Udayakumar D, Gurbani D, Tae HS, Riddle SM, Sim T, Marto JA, Jänne PA, Crews CM, Gray NS (2014) Pharmacological targeting of the pseudokinase Her3. Nat Chem Biol 10:1006–1012. https://doi.org/10.1038/nchembio.1658
Tinworth CP, Lithgow H, Dittus L, Bassi ZI, Hughes SE, Muelbaier M, Dai H, Smith IED, Kerr WJ, Burley GA, Bantscheff M, Harling JD (2019) PROTAC-mediated degradation of Bruton’s tyrosine kinase is inhibited by covalent binding. ACS Chem Biol 14:342–347. https://doi.org/10.1021/acschembio.8b01094
Cohen MS, Zhang C, Shokat KM, Taunton J (2005) Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 308:1318–1321. https://doi.org/10.1126/science1108367
Cohen MS, Hadjivassiliou H, Taunton J (2007) A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nat Chem Biol 3:156–160. https://doi.org/10.1038/nchembio859
Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J (2013) Electrophilic fragment-based design of reversible covalent kinase inhibitors. J Am Chem Soc 135:5298–5301. https://doi.org/10.1021/ja401221b
Lebraud H, Coxon CR, Archard SV, Bawn MC, Carbain B, Matheson JC, Turner MD, Cano C, Griffin JR, Hardcastle RI, Baisch U, Harrington WR, Golding TB (2014) Model system for irreversible inhibition of Nek2: thiol addition to ethynylpurines and related substituted heterocycles. Org Biomol Chem 12:141–148. https://doi.org/10.1039/C3OB41806E
Mitcheson DF, Bottrill AR, Carr K, Coxon CR, Cano C, Golding BT, Griffin RJ, Fry AM, Doerig C, Bayliss R, Tobin AB (2016) A new tool for the chemical genetic investigation of the Plasmodium falciparum Pfnek-2 NIMA-related kinase. Malar J 15:535. https://doi.org/10.1186/s12936-016-1580-3
Henise JC, Taunton J (2011) Irreversible Nek2 kinase inhibitors with cellular activity. J Med Chem 54:4133–4146. https://doi.org/10.1021/jm200222m
Pearson RJ, Blake DG, Mezna M, Fischer PM, Westwood NJ, McInnes C (2018) The Meisenheimer complex as a paradigm in drug discovery: reversible covalent inhibition through C67 of the ATP binding site of PLK1. Cell Chem Biol 25:1107–1116.e4. https://doi.org/10.1016/j.chembiol.2018.06.001
Dittus L, Werner T, Muelbaier M, Bantscheff M (2017) Differential kinobeads profiling for target identification of irreversible kinase inhibitors. ACS Chem Biol 12:2515–2521. https://doi.org/10.1021/acschembio.7b00617
Zhou W, Hur W, McDermott U, Dutt A, Xian W, Ficarro SB, Zhang J, Sharma SV, Brugge J, Meyerson M, Settleman J, Gray NS (2010) A structure-guided approach to creating covalent FGFR inhibitors. Chem Biol 17:285–295. https://doi.org/10.1016/j.chembiol.2010.02.007
Tan L, Wang J, Tanizaki J, Huang Z, Aref AR, Rusan M, Zhu S-J, Zhang Y, Ercan D, Liao RG, Capelletti M, Zhou W, Hur W, Kim N, Sim T, Gaudet S, Barbie DA, Yeh J-RJ, Yun C-H, Hammerman PS, Mohammadi M, Jänne PA, Gray NS (2014) Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. PNAS 111:E4869–E4877. https://doi.org/10.1073/pnas.1403438111
Brameld KA, Owens TD, Verner E, Venetsanakos E, Bradshaw JM, Phan VT, Tam D, Leung K, Shu J, LaStant J, Loughhead DG, Ton T, Karr DE, Gerritsen ME, Goldstein DM, Funk JO (2017) Discovery of the irreversible covalent FGFR inhibitor 8-(3-(4-acryloylpiperazin-1-yl)propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)pyrido[2,3-d]pyrimidin-7(8H)-one (PRN1371) for the treatment of solid tumors. J Med Chem 60:6516–6527. https://doi.org/10.1021/acs.jmedchem.7b00360
Kalyukina M, Yosaatmadja Y, Middleditch MJ, Patterson AV, Smaill JB, Squire CJ (2019) TAS-120 cancer target binding: defining reactivity and revealing the first fibroblast growth factor receptor 1 (FGFR1) irreversible structure. ChemMedChem 14:494–500. https://doi.org/10.1002/cmdc.201800719
Kwarcinski FE, Fox CC, Steffey ME, Soellner MB (2012) Irreversible inhibitors of c-Src kinase that target a nonconserved cysteine. ACS Chem Biol 7:1910–1917. https://doi.org/10.1021/cb300337u
Hagel M, Miduturu C, Sheets M, Rubin N, Weng W, Stransky N, Bifulco N, Kim JL, Hodous B, Brooijmans N, Shutes A, Winter C, Lengauer C, Kohl NE, Guzi T (2015) First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov 5:424–437. https://doi.org/10.1158/2159-8290.CD-14-1029
Joshi JJ, Coffey H, Corcoran E, Tsai J, Huang C-L, Ichikawa K, Prajapati S, Hao M-H, Bailey S, Wu J, Rimkunas V, Karr C, Subramanian V, Kumar P, MacKenzie C, Hurley R, Satoh T, Yu K, Park E, Rioux N, Kim A, Lai WG, Yu L, Zhu P, Buonamici S, Larsen N, Fekkes P, Wang J, Warmuth M, Reynolds DJ, Smith PG, Selvaraj A (2017) H3B-6527 is a potent and selective inhibitor of FGFR4 in FGF19-driven hepatocellular carcinoma. Cancer Res 77:6999–7013. https://doi.org/10.1158/0008-5472.CAN-17-1865
Fairhurst AR, Knoepfel T, Leblanc C, Buschmann N, Gaul C, Blank J, Galuba I, Trappe J, Zou C, Voshol J, Genick C, Brunet-Lefeuvre P, Bitsch F, Graus-Porta D, Furet P (2017) Approaches to selective fibroblast growth factor receptor 4 inhibition through targeting the ATP-pocket middle-hinge region. MedChemComm 8:1604–1613. https://doi.org/10.1039/C7MD00213K
Yamaura T, Nakatani T, Uda K, Ogura H, Shin W, Kurokawa N, Saito K, Fujikawa N, Date T, Takasaki M, Terada D, Hirai A, Akashi A, Chen F, Adachi Y, Ishikawa Y, Hayakawa F, Hagiwara S, Naoe T, Kiyoi H (2018) A novel irreversible FLT3 inhibitor, FF-10101, shows excellent efficacy against AML cells with FLT3 mutations. Blood 131:426–438. https://doi.org/10.1182/blood-2017-05-786657
Ward RA, Colclough N, Challinor M, Debreczeni JE, Eckersley K, Fairley G, Feron L, Flemington V, Graham MA, Greenwood R, Hopcroft P, Howard TD, James M, Jones CD, Jones CR, Renshaw J, Roberts K, Snow L, Tonge M, Yeung K (2015) Structure-guided design of highly selective and potent covalent inhibitors of ERK1/2. J Med Chem 58:4790–4801. https://doi.org/10.1021/acs.jmedchem.5b00466
Aronchik I, Dai Y, Labenski M, Barnes C, Jones T, QIao L, Beebe L, Malek M, Elis W, Shi T, Mavrommatis K, Bray GL, Filvaroff EH (2018) Efficacy of a covalent ERK1/2 inhibitor, CC-90003, in KRAS mutant cancer models reveals novel mechanisms of response and resistance. Mol Cancer Res Mol 17:642–654. https://doi.org/10.1158/1541-7786.MCR-17-0554
Mita MM, LoRusso P, McArthur GA, Kim ES, Bray GL, Hock NH, Laille EJ, Aronchik I, Filvaroff E, Wu X, Bendell JC (2017) A phase Ia study of CC-90003, a selective extracellular signal-regulated kinase (ERK) inhibitor, in patients with relapsed or refractory BRAF or RAS-mutant tumors. JCO 35:2577–2577. https://doi.org/10.1200/JCO.2017.35.15_suppl.2577
Sipthorp J, Lebraud H, Gilley R, Kidger AM, Okkenhaug H, Saba-El-Leil M, Meloche S, Caunt CJ, Cook SJ, Heightman TD (2017) Visualization of endogenous ERK1/2 in cells with a bioorthogonal covalent probe. Bioconjug Chem 28:1677–1683. https://doi.org/10.1021/acs.bioconjchem.7b00152
Yang Z, Liu H, Pan B, He F, Pan Z (2018) Design and synthesis of (aza)indolyl maleimide-based covalent inhibitors of glycogen synthase kinase 3β. Org Biomol Chem. https://doi.org/10.1039/C8OB00642C
Wissner A, Fraser HL, Ingalls CL, Dushin RG, Floyd MB, Cheung K, Nittoli T, Ravi MR, Tan X, Loganzo F (2007) Dual irreversible kinase inhibitors: Quinazoline-based inhibitors incorporating two independent reactive centers with each targeting different cysteine residues in the kinase domains of EGFR and VEGFR-2. Bioorg Med Chem 15:3635–3648. https://doi.org/10.1016/j.bmc.2007.03.055
Redenti S, Marcovich I, De Vita T, Pérez C, De Zorzi R, Demitri N, Perez DI, Bottegoni G, Bisignano P, Bissaro M, Moro S, Martinez A, Storici P, Spalluto G, Cavalli A, Federico S (2019) A Triazolotriazine-based dual GSK-3β/CK-1δ ligand as a potential Neuroprotective agent presenting two different mechanisms of enzymatic inhibition. ChemMedChem 14:310–314. https://doi.org/10.1002/cmdc.201800778
Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, Jenkins CE, Hannett NM, McMillin D, Sanda T, Sim T, Kim ND, Look T, Mitsiades CS, Weng AP, Brown JR, Benes CH, Marto JA, Young RA, Gray NS (2014) Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511:616. https://doi.org/10.1038/nature13393
Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, Ficarro SB, Elkins JM, Liang Y, Hannett NM, Manz T, Hao M, Bartkowiak B, Greenleaf AL, Marto JA, Geyer M, Bullock AN, Young RA, Gray NS (2016) Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat Chem Biol 12:876–884. https://doi.org/10.1038/nchembio.2166
Gao Y, Zhang T, Terai H, Ficarro SB, Kwiatkowski N, Hao M-F, Sharma B, Christensen CL, Chipumuro E, Wong K, Marto JA, Hammerman PS, Gray NS, George RE (2018) Overcoming resistance to the THZ series of covalent transcriptional CDK inhibitors. Cell Chem Biol 25:135–142.e5. https://doi.org/10.1016/j.chembiol.2017.11.007
Andersen JL, Gesser B, Funder ED, Nielsen CJF, Gotfred-Rasmussen H, Rasmussen MK, Toth R, Gothelf KV, Arthur JSC, Iversen L, Nissen P (2018) Dimethyl fumarate is an allosteric covalent inhibitor of the p90 ribosomal S6 kinases. Nat Commun 9:4344. https://doi.org/10.1038/s41467-018-06787-w
Dong T, Li C, Wang X, Dian L, Zhang X, Li L, Chen S, Cao R, Li L, Huang N, He S, Lei X (2015) Ainsliadimer A selectively inhibits IKKα/β by covalently binding a conserved cysteine. Nat Commun 6:ncomms7522. https://doi.org/10.1038/ncomms7522
Uhlenbrock N, Smith S, Weisner J, Landel I, Lindemann M, Le TA, Hardick J, Gontla R, Scheinpflug R, Czodrowski P, Janning P, Depta L, Quambusch L, Müller MP, Engels B, Rauh D (2019) Structural and chemical insights into the covalent-allosteric inhibition of the protein kinase Akt. Chem Sci 10:3573–3585. https://doi.org/10.1039/C8SC05212C
Craven GB, Affron DP, Allen CE, Matthies S, Greener JG, Morgan RML, Tate EW, Armstrong A, Mann DJ High-throughput kinetic analysis for target-directed covalent ligand discovery. Angew Chem Int Ed 57:5257–5261. https://doi.org/10.1002/anie.201711825
Bührmann M, Hardick J, Weisner J, Quambusch L, Rauh D (2017) Covalent lipid pocket ligands targeting p38α MAPK mutants. Angew Chem Int Ed 56:13232–13236. https://doi.org/10.1002/anie.201706345
Dalton SE, Dittus L, Thomas DA, Convery MA, Nunes J, Bush JT, Evans JP, Werner T, Bantscheff M, Murphy JA, Campos S (2018) Selectively targeting the Kinome-conserved lysine of PI3Kδ as a general approach to covalent kinase inhibition. J Am Chem Soc 140:932–939. https://doi.org/10.1021/jacs.7b08979
Anscombe E, Meschini E, Mora-Vidal R, Martin MP, Staunton D, Geitmann M, Danielson UH, Stanley WA, Wang LZ, Reuillon T, Golding BT, Cano C, Newell DR, Noble MEM, Wedge SR, Endicott JA, Griffin RJ (2015) Identification and characterization of an irreversible inhibitor of CDK2. Cell Chem Biol 22:1159–1164. https://doi.org/10.1016/j.chembiol.2015.07.018
Dahal UP, Gilbert AM, Obach RS, Flanagan ME, Chen JM, Garcia-Irizarry C, Starr JT, Schuff B, Uccello DP, Young JA (2016) Intrinsic reactivity profile of electrophilic moieties to guide covalent drug design: N-α-acetyl-L-lysine as an amine nucleophile. Med Chem Commun 7:864–872. https://doi.org/10.1039/C6MD00017G
Zhao Q, Ouyang X, Wan X, Gajiwala KS, Kath JC, Jones LH, Burlingame AL, Taunton J (2017) Broad-Spectrum kinase profiling in live cells with lysine-targeted Sulfonyl fluoride probes. J Am Chem Soc 139:680–685. https://doi.org/10.1021/jacs.6b08536
Hatcher JM, Wu G, Zeng C, Zhu J, Meng F, Patel S, Wang W, Ficarro SB, Leggett AL, Powell CE, Marto JA, Zhang K, Ngo JCK, Fu X-D, Zhang T, Gray NS (2018) SRPKIN-1: a covalent SRPK1/2 inhibitor that potently converts VEGF from pro-angiogenic to anti-angiogenic isoform. Cell Chem Biol 25:460–470.e6. https://doi.org/10.1016/j.chembiol.2018.01.013
Acknowledgments
The author thanks Kristine Schmidt for language proofreading and editing. Dr. Michael Forster, Dr. Marcel Günther, and Dr. Apirat Chaikuad are gratefully acknowledged for fruitful discussion and suggestions. Dr. Apirat Chaikuad and Prof. Dr. Stefan Knapp are further acknowledged for providing the data of their analysis of the protein kinases’ cysteinome. The author appreciates financial support by the Postdoctoral Fellowship Program of the Baden-Württemberg Stiftung, the Institutional Strategy of the University of Tübingen (ZUK 63, German Research Foundation), the RiSC Program of the State Ministry of Baden-Württemberg for Sciences, Research and Arts, and the Max Buchner Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
Conflict of Interest
The author declares that he has no conflict of interest.
Funding
While preparing the manuscript, the author received funding from the Postdoctoral Fellowship Program of the Baden-Württemberg Stiftung, the Institutional Strategy of the University of Tübingen (ZUK 63, German Research Foundation), the RiSC Program of the State Ministry of Baden-Württemberg for Sciences, Research and Arts, and the Max Buchner Research Foundation.
Ethical Approval
This article does not contain any studies with human participants or animals performed by the author.
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gehringer, M. (2020). Covalent Kinase Inhibitors: An Overview. In: Laufer, S. (eds) Proteinkinase Inhibitors. Topics in Medicinal Chemistry, vol 36. Springer, Cham. https://doi.org/10.1007/7355_2020_103
Download citation
DOI: https://doi.org/10.1007/7355_2020_103
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-68179-1
Online ISBN: 978-3-030-68180-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)