
Abstract
Mutated or dysregulated protein kinases represent major oncogenic drivers in cancer. Due to the general druggability of these potential oncoproteins, protein kinases have been regarded the most significant drug targets in cancer cells for the past three decades. Starting with the approval of imatinib for targeting BCR-ABL in leukemia positive for Philadelphia chromosome, a multitude of different kinase inhibitors have been developed and approved for the market so far. Additionally, many new compounds with increased efficacy and target specificity are under development and clinical testing. While several of these compounds allow for an efficient temporary treatment success in different tumor entities, long-term cancer control is often limited due to the development of therapy resistance. Thus, overcoming drug resistance in tumors represents a major challenge for successful cancer therapies in the future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Over the past 20 years, research revealed that many diseases emerge from impairments in signal transduction. This insight has been used by scientists to unravel molecular mechanisms that drive complex diseases such as solid tumors, leukemias, systemic autoimmune diseases, and inflammatory diseases. Hence, biologists, chemists, physicians, and pharmacologists have focused their clinical research toward development of specific molecules targeting key signaling cascades of these diseases. To this point, most molecules aiming to this direction of treatment represented protein kinase inhibitors. Kinases are proteins that play a critical role in cellular signal transduction by phosphorylating downstream targets. Because dysregulation and mutations of protein kinases play major roles in human diseases, this family of enzymes has become one of the most significant drug targets over the past three decades. It all started in 1978, when the protein kinase c-SRC was found to share high similarity to a protein from sarcoma virus and act as an oncogene [1]. In addition, studies in the early 1980s pointed out that hyperactivation of a protein kinase (protein kinase C) represents a key mechanism for tumor promotion [2]. The idea to target this group of enzymes therapeutically was also fueled by findings showing that naphthalene-sulphonamides were able to block kinases [3]. These molecules were used as a starting point to further synthetize drugs that inhibit protein kinases.
One of the key experiments for the development of kinase inhibitors was the crystallization of protein kinase A in 1991. Susan Taylor and colleagues revealed the structure of the kinase core for the very first time, giving insight into a key element of all kinases in the genome. This study demonstrated that residues involved in the binding of ATP were conserved among kinases [4, 5]. The crystal structure of PKA gave valuable information for the structural function of these enzymes. However, since core domains of kinases are highly conserved, the idea of selective inhibition of a protein kinase was also considered to be a major challenge.
Starting from the late 1980s, molecules targeting more than one kinase with different efficacies were developed. Some years before that, the only purely isolated tyrosine protein kinases were epidermal growth factor receptor (EGFR) and insulin receptor. The new molecules were 1,000-fold more potent against EGFR than against insulin receptor kinase. Interestingly, these drugs were found to be inactive against serine/threonine kinases [6]. Based on this evidence, scientists could then develop more inhibitors against these kinases that show structure/activity relationships.
Later on, new findings strengthened the idea of targeted kinase drug development. In particular, ATP mimics were found to selectively inhibit platelet-derived growth factor receptor (PDGFR), while they were not potent against other protein kinases. In addition, a study in the mid-1990s showed that quinoxalines are potent inhibitors of PDGFR though not able to interact with EGFR. Accordingly, quinazolines showed the opposite effect [7, 8]. Based on this finding, years later, Zeneca developed the inhibitor gefitinib that targets EGFR. Since 1988, when the first study showing targeted inhibition of the catalytic activity of EGFR was published, the number of protein kinase inhibitor agents developed climbed steadily. It is interesting to note that although EGFR and receptor tyrosine-protein kinase erbB-2 (HER2) share high homology, scientists were able to develop selective molecules against these targets with low cross-reaction already since 1993 [9].
A breakthrough was the first approval of a protein kinase inhibitor by the FDA (2001). This molecule was imatinib, firstly developed by Zeneca as a PDGFR inhibitor. Interestingly, it was later shown that the drug had also high efficacy against BCR-ABL, making it suitable for treatment of chronic myelogenous leukemia (CML) and acute lymphocytic leukemia (ALL) patients positive for Philadelphia chromosome [10, 11]. Since 2001, 48 kinase inhibitors have been approved to the market (Table 1) [12]. The vast majority are drugs against tyrosine protein kinases and receptors for the treatment of cancer. Only a few, ten of them, target serine/threonine kinases. The main difficulty of developing selective agents against serine/threonine kinases is the high similarity of the ATP-binding domain of these enzymes. In addition, more serine/threonine kinases (420) than tyrosine kinases (90) were found in the human kinome. In the future, the use of different development strategies along with alternative targeting domains might extend the numbers of FDA-approved inhibitors [13, 14].
2 EGFR Inhibitors
The epidermal growth factor receptor (EGFR) is a transmembrane protein that acts as a receptor for ligands of the epidermal growth factor (EGF) family [15]. The EGFR gene is located at chromosome 7, and the encoding protein product encompasses 1,210 amino acids. EGFR is a member of the ErbB family, which is a family of four similar receptors with tyrosine kinase activity. The family consists of EGFR or HER1, HER2, HER3, and HER4. EGFR is a cell surface receptor and represents the starting point of signal transduction mechanisms controlling diverse cellular responses such as cell proliferation, migration, survival, and apoptosis [16]. Mutations and amplification of the EGFR gene can lead to overexpression of the receptor. This results in constant kinase activity and uncontrolled activation of downstream pathways. In breast cancer patients, the incidence of overexpressed EGFR is approximately 10–30% [17]. Apart from breast cancer, upregulated EGFR can also be found in several other epithelial tumor entities such as lung cancer, prostate cancer, and squamous carcinomas of head and neck [17,18,19] (Fig. 1).

Chemical structures of EGFR inhibitors
In addition, deletion of EGFR can also be found in several malignancies. One of the most common deletions in the EGFR locus is EGFRvIII, where exons 2–7 of EGFR are deleted giving rise to a receptor lacking ligand-binding domain but remaining constantly active [20, 21]. Amplification of this mutant is present in gliomas such as glioblastomas with a frequency of 64% (grade IV) but also in head and neck squamous carcinomas and medulloblastomas [21, 22]. High expression of EGFR has been also correlated with short survival time of cancer patients [23].
Lapatinib is an inhibitor of both EGFR and Her2 receptor tyrosine kinases. It was approved in 2007 for treatment of breast cancer, non-small cell lung cancer (NSCLC), head and neck cancer, as well as gastric cancer. The use of lapatinib can inhibit the signaling of MAPK and PI3K pathways in patients with overexpressing EGFR and HER2. In particular, the response to lapatinib is linked to HER2 overexpression. The dual specificity of this drug results in inhibition of phosphorylation of AKT, RAF, and ERK. Interestingly, breast cancer patients positive for HER2 amplification with brain metastases are treated with lapatinib in combination with capecitabine for improvement of survival rates [24,25,26,27]. Gefitinib is an inhibitor targeting selectively EGFR (Fig. 1). Patients with locally advanced or metastatic NSCLC experienced beneficial outcome when treated with gefitinib [28]. In addition, treatment of EGFR mutation-positive NSCLC patients with gefitinib improved progression-free survival in comparison with chemotherapy. This was the first study that showed longer progression-free survival of patients treated with selective therapy compared to classic chemotherapy [29, 30]. Erlotinib (another kinase inhibitor targeting EGFR) is used for treatment of locally advanced or metastatic NSCLC (Fig. 1). A clinical study published in 2011 revealed that use of erlotinib prolongs survival of NSCLC patients, previously treated with first-line chemotherapy, leading to its approval for this use [31]. In addition, erlotinib combined with gemcitabine increases overall survival of patients with unresectable pancreatic cancer positive for mutant EGFR [32]. Despite providing therapeutic benefit, use of erlotinib has severe side effects such as breathing abnormalities, skin rush, diarrhea, and cough, and the recommended dosage is close to the maximum tolerated dose [33].
Afatinib is an irreversible inhibitor of ErbB family of kinase receptors (Fig. 1). As a first-line treatment of patients with lung adenocarcinoma carrying activating mutations in EGFR, afatinib increased progression-free survival but not overall survival, when compared to gefitinib [34]. In addition, the LUX-Lung 6 trial revealed that patients with advanced lung adenocarcinoma treated with afatinib had prolonged progression-free survival and time to treatment failure in comparison with those treated with gemcitabine in combination with cisplatin [35].
3 ALK Inhibitors
Anaplastic lymphoma kinase (ALK) is a tyrosine kinase receptor. In 1994, ALK was described for the first time as a component of a fusion protein derived from translocation t2;5 in anaplastic large cell lymphoma [36]. Several years later, the full length of ALK receptor tyrosine kinase was characterized. It consists of an extracellular ligand-binding domain, a transmembrane domain, and an intracellular kinase domain that shares high similarity with the insulin receptor (ER) [37, 38]. Although the physiological function of ALK is not completely revealed, it has been described to play a critical role in early embryo development and neural system development [38,39,40,41]. Furthermore, activation of ALK is involved in activation of PI3K-AKT, CRKL-C3G, MEKK2/3-MEK5-ERK5, JAK-STAT, and MAPK signaling pathways [38, 42,43,44,45]. Around 3–7% of NSCLC patients (usually non-smokers) have a particular mutation, where echinoderm microtubule-associated protein-like 4 (EML4) gene is fused to ALK gene. This inversion of chromosome 2 results in the expression of the fusion protein EML4-ALK consisting of the N-terminal region of EML4 and the intracellular/kinase region of ALK [46, 47] (Fig. 2).

Chemical structures of ALK inhibitors
The first inhibitor of this category approved by FDA was crizotinib (2011) Fig. 2. Initially, it was developed as a c-Met inhibitor but is also able to target ALK, proto-oncogene tyrosine-protein kinase (ROS1), and hepatocyte growth factor receptor (HGFR) [48]. Profile 10,019 phase-I clinical trial and profile 100,513 showed significant objective response rates, prolonged progression-free survival, and median progression-free survival in pretreated ALK-positive (EML4-ALK) NSCLC patients [49, 50]. Based on this evidence, crizotinib received conditional approval for use also in Canada, in 2012. Subsequently, phase-III trials revealed superior median progression-free survival and greater reduction in symptoms related with lung cancer when treated with crizotinib compared to chemotherapy [51, 52]. The results from these studies led to full approval of crizotinib marking it as the “gold standard” of ALK-positive NSCLC.
Ceritinib represents a next-generation ALK inhibitor and showed a higher potency than first-generation inhibitors such as crizotinib (Fig. 2). Ceritinib showed a potency in inhibiting ALK-positive NSCLC that were previously treated and resistant to crizotinib [53]. This finding suggested potency in treatment of mutated and therapy-resistant ALK tumors. Indeed, phase-I ASCEND-1 trial resulted in significant overall response rates in patients pre-acquired with both identified and non-identified resistance mechanisms to crizotinib [54]. An ASCEND-2 phase-II trial showed beneficial response of ceritinib in patients pretreated with crizotinib or chemotherapy and with or without brain metastases [55]. These data resulted in the approval of ceritinib as the first-choice treatment for crizotinib-resistant, ALK-positive NSCLC patients in 2014. In addition, an ASCEND-5 phase-III trial confirmed superior response of ceritinib. In this trial, NSCLC patients with brain metastases that were previously treated either with crizotinib or platinum-based chemotherapy were treated with ceritinib or chemotherapy [56]. Finally, ceritinib demonstrated potency against naive ALK-inhibitor NSCLC patients. The ASCEND-4 study revealed a median progression-free survival of 16.6 months of patients with advanced ALK-positive NSCLC treated with ceritinib versus 8.1 months of the chemotherapy-treated group [57]. The results from this study led to approval of ceritinib in 2017 by FDA as first-line treatment for patients with ALK-positive NSCLC [58]. Alectinib is another selective ALK inhibitor (Fig. 2). It was approved by FDA in 2015 for the treatment of NSCLC patients with acquired resistance to crizotinib (NP28673 and NP28761 phase-II clinical trials) [59, 60]. Later, the randomized phase-III clinical trial ALEX showed extended beneficial activity of alectinib in ALK-positive NSCLC patients. In particular, results demonstrated a superior progression-free survival rate of alectinib compared to crizotinib in naive ALK-inhibitor patients. In addition, alectinib was found to be less toxic and more active toward CNS. Only 12% of patients in the alectinib group showed a CNS progression event compared to 45% of the crizotinib group. Taking into account the previous results, FDA approved alectinib as first-line treatment of ALK-positive, metastatic NSCLC in 2017 [58, 61]. Latest additions to the ALK inhibitors’ list include brigatinib and lorlatinib. Brigatinib is an ALK inhibitor used for NSCLC resistant to crizotinib (Fig. 2). It has received accelerated approval by the FDA in 2017 after phase-II clinical trial ALTA demonstrated significant results for the treatment of patients with progressed NSCLC [62, 63]. Lorlatinib was accepted for the same treatment in 2018 (Fig. 2). Both drugs demonstrate significant intracranial activity making them very potent in decreasing the formation of brain metastasis [63, 64]. Phase-III clinical trial CROWN is currently ongoing for comparison of lorlatinib with crizotinib as first-line treatments [58, 65].
4 VEGFR Inhibitors
More than 40 years ago, the hypothesis of targeting angiogenesis as a tumor therapy was established [66]. Although many factors are involved in mechanisms leading to blood vessel formation, activation of vascular endothelial growth factor (VEGF) pathways was described to be critical in pro-angiogenic signaling. Several types of solid cancers overexpress VEGF-A, making initially this protein to a highly relevant target for selective antiangiogenic therapeutic strategy [67, 68]. Another strategy for inhibiting angiogenesis is the blockage of tyrosine kinase activity of the corresponding receptors VEGFR1, VEGFR2, and VEGFR3. Many receptor tyrosine kinase inhibitors targeting VEGFR have been approved so far. Sorafenib, sunitinib, axitinib, regorafenib, pazopanib, vandetanib, cabozantinib, and lenvatinib are used for treatment of different solid carcinomas such as renal cell carcinoma (RCC), hepatocellular carcinoma (HCC), thyroid cancer, pancreatic neuroendocrine tumor, gastrointestinal stromal tumor (GIST), and metastatic colorectal cancer (CRC) (Fig. 3). Sorafenib and sunitinib represent pioneer kinase inhibitors for the inhibition of angiogenic signaling in cancer. These multikinase inhibitors were initially approved for the treatment of advanced renal cell carcinoma. The pivotal phase-III study TARGET resulted in significantly prolonged progression-free survival time of patients with resistant, advanced renal cell carcinoma when treated with sorafenib. Sunitinib showed similar results in the randomized phase-III trial, where it was compared as first-line treatment to subcutaneous injection of interferon-α for treatment of metastatic RCC. Patients of sunitinib group showed improvement in median progression-free survival and objective response rate [69,70,71,72]. Sorafenib and sunitinib have been also accepted by FDA for treatment of HCC and advanced pancreatic neuroendocrine tumors, respectively [73, 74] (Fig. 3).

Chemical structures of VEGFR inhibitors
Regorafenib was the first therapeutic agent to show improvement in the overall survival of patients with metastatic CRC, previously progressed on classic therapies [75]. Based on these results, regorafenib was approved by FDA in 2012 for treatment of metastatic CRC. Half a year later, regorafenib was also accepted for the treatment of advanced GIST [76]. Regorafenib also shows beneficial outcome when used as treatment of HCC, significantly longer overall survival in second-line HCC patients, leading to its approval by the FDA for this use in 2017 [77].
Second-generation VEGFR/multikinase inhibitors include pazopanib, cabozantinib, lenvatinib, axitinib, and vandetanib (Fig. 3). All of them have been approved by the FDA for the treatment of one or several cancer types including thyroid cancer, RCC, soft tissue sarcoma, and medullary thyroid cancer [78].
5 BCR-ABL Inhibitors
The ABL protein family consists of two members: c-ABL and ARG. Physiologically, c-ABL is involved in actin remodeling, cell adhesion, motility, DNA damage response, and microbial pathogen response. In several types of cancer, deregulation and uncontrolled expression of c-ABL kinase has been described [79, 80]. When phosphorylated, c-ABL induces activity of downstream targets, activating ERK5, RAC/JNK, and STAT 1/3 pathways. C-ABL is also a molecular component driving CML. Translocation of part of chromosome 9 to chromosome 22 (Philadelphia chromosome) leads to the expression of oncogenic fusion protein BCR-ABL [81] highlighting ABL is an important target for the development of selective inhibitors. Imatinib was the first kinase inhibitor to be approved by FDA (2001) (Fig. 4). It is an inhibitor of three different targets: ABL, mast/stem cell growth factor receptor (tyrosine kinase KIT or CD117), and PDGFR [82]. After phase-III clinical trial showed improved cytogenetic response rates of CML patients treated with imatinib, the drug was accepted for treatment of CML in blast, accelerated, and chronic phases [83]. Later, in 2002 and 2008, imatinib was approved also for treatment of GIST both for advanced, metastatic tumors and previously resected tumors [84, 85]. Unfortunately, imatinib treatment is not successful in around 30% of patients [86]. The reason is acquired resistance, based on either a reduced cellular uptake of the drug, an increased activity of efflux transporters, or point mutations leading to conformational changes of BCR-ABL and therefore to a reduced binding to imatinib. In addition, resistance is acquired by amplification and overexpression of BCR-ABL gene [87]. Second-generation ABL inhibitors such as nilotinib, dasatinib, and bosutinib were developed, in order to overcome mutation-related resistance (Fig. 4). They were all approved for the treatment of CML: nilotinib and dasatinib as first- or second-line treatment and bosutinib as second-line therapy [88]. Nilotinib showed highly promising results because it was potent against almost all mutations resulting in BCR-ABL-dependent resistance [89]. Dasatinib showed high potency in patients with chronic phase CML and a faster treatment response when it was compared to imatinib [90]. Due to its unique structure, dasatinib is also potent against some conformation-altering mutations of BCR-ABL [91, 92]. Bosutinib has a much different structure. It was initially designed as a SRC inhibitor but found to have activity against ABL [93]. Although bosutinib is not potent against major resistant mutants and does not have high selectivity for BCR-ABL, it has the benefit to be not sensitive to resistance efflux transporters and remains in the cells [94, 95]. Therefore, bosutinib is approved for second-line treatment of CML, while trials that test it as first-line treatment are ongoing [88, 96] (Fig. 4).

Chemical structures of ABL inhibitors
The only approved third-generation inhibitor against ABL is ponatinib (Fig. 4). It is a dual SRC/ABL inhibitor that is accepted for the treatment of CML and Ph+ ALL. The structure of ponatinib was modified accordingly so that is highly potent against resistant mutants [97]. Clinically, it shows potency in the treatment of progressed and pretreated Ph+ leukemias. In addition, patients with resistant mutations also benefit from ponatinib treatment. In the corresponding study with 43 patients harboring the abovementioned characteristics, 98% showed a complete hematologic response and 72% a major cytogenetic response [98]. Finally, ponatinib has proven to be a valuable alternative to stem cell transplantation in patients with mutant, advance CML and Ph+ ALL [88, 99].
6 RAF Inhibitors
The RAS/MAPK pathway controls cell growth, proliferation, and survival in a broad range of different tumor entities. Activation of membrane-associated RAS proteins (KRAS, NRAS, HRAS) results in a recruitment of RAF proteins (ARAF, BRAF, and RAF1) leading to a phosphorylation of MEK1 and MEK2 which in turn phosphorylate and activate extracellular signal-regulated kinase (ERK1 and ERK2) (Fig. 5).

Chemical structures of RAF inhibitors
RAS represents an important proto-oncogene and a major oncogenic driver. It was found mutated in around 30% of all human cancer entities [100]. Due to the fact that RAS proteins do not harbor any cavities for small molecule interaction, approaches to directly inhibit the function of RAS have not been successful so far. Therefore, the inhibition of RAS downstream factors such as RAF, MEK, and ERK has gained interest for the treatment of cancer [100].
RAF monomers are usually inactive, since the N-terminal domain of BRAF triggers autoinhibition [101, 102]. Upon activation, RAF forms homo- and heterodimers which induces downstream signaling to MEK. While physiological RAS activation induces MEK activation mainly via the formation of BRAF dimers [103], oncogenic RAS often triggers the formation of BRAF-RAF1 heterodimers [101, 104, 105].
BRAF mutations are present in 8% of all human tumors [106]. They were found in more than 50% of melanoma patients and were also identified in CRC (5–10%), hairy cell leukemia (∼100%), thyroid carcinomas (25–45%), and, as a rare event, ovarian and lung cancer [106, 107]. Ninety percent of all BRAF mutations account for a substitution of valine with glutamic acid at position 600 (V600E) [100]. This mutation results in a constitutive kinase activity of BRAF monomers and protects BRAF from ERK-mediated negative feedback signaling [102].
The identification of BRAF mutations as oncogenic drivers led to intensified efforts in order to develop more selective and potent BRAF inhibitors. This work yielded in the development of vemurafenib (Zelboraf) and dabrafenib (Tafinlar) as FDA-approved drugs for the treatment of BRAFV600E-mutated advanced melanoma [108,109,110] (Fig. 5).
Vemurafenib is a BRAFV600E inhibitor with an IC50 of 31 nM, which inhibits also BRAF proteins with other mutations (V600D, V600K, and V600R) as well as RAF1 (IC50 = 48 nM) [109]. It shows only a low affinity to wild-type BRAF (IC50 = 100 nM) [111]. In preclinical treatment studies, vemurafenib was effective in xenograft models of BRAFV600E-mutated melanoma [108] and showed efficacy against BRAF-mutated melanoma cell lines [112].
Upon successful phase-I and phase-II clinical trials, a phase-III clinical trial of vemurafenib was initiated on 675 patients suffering from metastatic, BRAFV600E-mutated melanomas (who did not receive any treatment before). This trial showed a median overall survival of 13.2 months for patients under vemurafenib treatment, while patients under dacarbazine (as a control) showed only a survival of 9.9 months. Furthermore, vemurafenib resulted in a significant response in 48.4% of patients (in comparison with 5.5% in the dacarbazine-treated group) [113, 114].
Although vemurafenib was in general well tolerated by patients, several adverse symptoms were found upon treatment such as fatigue, nausea, alopecia, lymphopenia, neutropenia, headache, and diarrhea [113, 115,116,117,118].
Dabrafenib was shown to be in general a more potent Raf inhibitor than vemurafenib. It inhibits BRAFV600E with an IC50 of 0.8 nM, wild-type BRAF with an IC50 of 3.2 nM, and RAF1 with an IC50 of 5 nM [109, 119]. In preclinical assays, dabrafenib also showed efficacy against BRAF-mutated cell lines and reduced tumor development in xenograft melanoma mouse models [119]. A phase-III clinical trial of dabrafenib was performed in a total of 250 patients suffering from BRAFV600E-mutated metastatic melanoma. While 187 patients received 150 mg dabrafenib twice per day, 63 patients received dacarbazine treatment [120]. In the dabrafenib-treated group, 6 patients (3%) showed a complete and 87 patients (47%) partial response, while 78 patients (42%) displayed a stable disease. In the dacarbazine group, one patient (2%) showed a complete and three patients (5%) partial response. A stable disease was seen in 30 patients (48%). The median progression-free survival in the dabrafenib group was 5.1 months, while dacarbazine-treated patients showed a progression-free survival rate of 2.7 months.
Side effects found often associated with dabrafenib treatment were pyrexia, headache, neutropenia, fatigue, thrombocytopenia, leukopenia, asthenia, hyponatremia, arthralgia, nausea, chills, myalgia, vomiting, diarrhea, and hair loss [108, 109, 117, 118, 120].
Overall, vemurafenib and dabrafenib induced initial therapeutic effects against BRAFV600 mutant melanomas. However, the long-term treatment success is limited due to the development of secondary resistance. Thus, most patients relapse after 1 year of treatment [121]. In addition, other tumor entities with BRAFV600E mutation, such as colorectal, pancreatic, and thyroid cancer, mostly show a primary resistance to these drugs [122, 123]. It was hypothesized that long-term control of tumor development by these inhibitors is limited by the fact that they do not efficiently inhibit the dimerization of RAF and are partly unsuccessful in targeting BRAF and RAF1 dimers. Thus, BRAF homodimeric or BRAF-RAF1 heterodimeric signaling can trigger therapy resistance [100,101,102]. In addition, it was shown that therapy resistance of RAF inhibition can be also induced by the formation of different BRAFV600E splice variants which can form resistant dimers [124].
Of note, RAF inhibitors were also applied in tumors without BRAF mutation. For example, the previously mentioned multikinase inhibitor sorafenib represents a RAF inhibitor and showed certain efficacy in the treatment of patients with HCC and RCC. Sorafenib was approved by the FDA for the treatment of these tumor types [125,126,127,128,129]. However, development of resistance against sorafenib is a frequent incident in treated patients.
Sorafenib inhibits RAF1 with an IC50 of 6 nM, wild-type BRAF with an IC50 of 25 nM, and BRAFV600E with an IC50 of 38 nM. Of note, it is hypothesized that the effect of sorafenib is based on a combined inhibition of RAF and other kinases such as VEGFR. Sorafenib inhibits VEGFR1 with an IC50 of 26 nM and VEGFR2 with an IC50 = 90 nM. Other kinases that were found to be influenced by sorafenib are FLT-3 (IC50 = 33 nM), p38 (IC50 = 38 nM), RET (IC50 = 47 nM), c-KIT (IC50 = 68 nM), and FGFR1 (IC50 = 580 nM) [109, 126, 130]. A phase-III clinical trial testing sorafenib in advanced HCC (SHARP trial) was performed with 602 patients that were treated either with 400 mg sorafenib twice daily (299 patients) or with placebo (303 patients) [74, 129]. In that trial, 7 patients (2%) showed partial response, and 211 patients (71%) had a stable disease in the sorafenib group, whereas 2 patients (1%) showed partial response and 204 patients (67%) had a stable disease in placebo group. The median overall survival was 10.7 months upon sorafenib and 7.9 months upon placebo treatment. Median time to symptomatic progression was 4.1 months in the sorafenib group and 4.9 months in the placebo group. In addition, median time to radiological progression was 5.5 months in the sorafenib group and 2.8 months in the placebo group [109].
As mentioned before, phase-III clinical trials of sorafenib were also conducted against other tumor entities such as RCC and thyroid carcinoma [74, 128, 131]. In a trial against radioactive iodine refractory thyroid cancer (DECISION), 417 patients were treated either with 400 mg sorafenib twice per day (207 patients) or with placebo (210 patients) [132]. Sorafenib triggered a partial response in 12.2% of patients which was seen in only 0.5% of patients treated with the placebo. Furthermore, the sorafenib-treated group showed a median overall survival of 10.8 months compared to 5.8 months in the placebo-treated group.
Adverse effects associated with sorafenib were fatigue, anorexia, hypertension, nausea, vomiting, alopecia, flushing, constipation, voice change, diarrhea, headache, joint pain, pruritus, weight loss, hemorrhage (upper GI), neuropathy, stomatitis, hypophosphatemia, musculoskeletal pain, and abdominal pain [74, 109, 118, 125, 129, 132].
The molecular mechanisms for resistance against sorafenib are complex. It was shown that activation of p38alpha signaling during sorafenib therapy circumvents sorafenib-mediated inhibition of Raf in HCC. In line with this, inhibition of p38alpha improved the outcome of sorafenib in HCC mouse models [133].
For patients that show a progressive disease under sorafenib, recently the RAF inhibitor regorafenib (which targets also several other kinases, such as VEGFR, PDGFR) was approved by the FDA [77] (Fig. 5). In the RESORCE trial, 573 patients that progressed under sorafenib received either 160 mg regorafenib or placebo. The median survival under regorafenib was 10.6 months compared to 7.8 months under placebo treatment [77]. Furthermore, regorafenib was approved by the FDA for the treatment of metastatic colorectal cancer and advanced gastrointestinal stromal tumor (GIST) [75, 76].
7 MEK Inhibitors
The observation of therapy resistance and paradoxical ERK activation upon RAF inhibition resulted in increased effort to develop inhibitors targeting MEK. Several MEK inhibitors, such as refametinib, selumetinib, cobimetinib, and trametinib, have been tested in clinical trials for different tumor entities, e.g., NSCLC and melanoma [134, 135] (Fig. 6).

Chemical structures of MEK inhibitors
Also combination therapies of RAF and MEK inhibitors were tested and FDA approved and were able to further increase the treatment responses in melanoma patients [136]. In patients with metastatic melanoma, two phase-III trials were performed in order to test the combination of BRAF inhibitors and MEK inhibitors. In the COMBI-d trial, 423 patients with BRAFV600 mutations, which were not treated before, received either dabrafenib in combination with trametinib or dabrafenib alone [137]. The application of the combination therapy resulted in a 3-year overall survival of 44% compared to 32% in the group that received a dabrafenib monotherapy. Adverse effects of the trametinib and dabrafenib combination were pyrexia, fatigue, nausea, headache, diarrhea, rash, and arthralgia [137, 138].
In addition, also a combination of cobimetinib and vemurafenib was tested in 495 patients with untreated advanced BRAFV600-mutated melanoma (coBRIM trial) [139]. In this trial, the 3-year rate of relapse-free survival was 58% in the group that received the combination therapy compared to 39% in the placebo group. The 3-year overall survival rate was 86% in the combination-therapy group compared to 77% in the placebo group [139].
In addition, different studies tested also the combination of RAF/MEK inhibitors with immunotherapies [140].
8 Next Clinical Developments
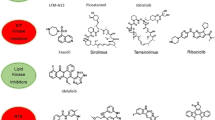
The human genome encodes for over 500 kinases which gives ample scope for novel target finding and drug development in cancer therapy [141]. In addition to the 48 FDA-approved kinase inhibitors, a huge range of potential inhibitors are currently in clinical or preclinical trials. For example, clinical phase-I trials of compounds targeting nerve growth factor receptors [142], polo-like kinase 1 [143], phosphatidylinositol 4,5-bisphosphate 3-kinase delta and gamma [144], protein kinase B [145], focal adhesion kinase [146], casein kinase II [147], and Aurora kinases [148] were performed within the last years (Fig. 7).

Chemical structures of next clinical development
Notably, the Aurora A kinase inhibitor alisertib could finish two clinical phase-II trials with promising responses in patients with neuroendocrine prostate cancer [NCT01799278] as well as advanced breast cancer and small cell carcinoma of the lung [NCT01045421] (Fig. 7). However, a phase-III trial in patients with relapsed/refractory peripheral T-cell lymphoma [NCT01482962] was announced to be discontinued based on a pre-specified interim analysis by Takeda.
In parallel, the research on PI3K and mTOR inhibiting compounds was heavily impelled in recent years [13]. In addition to idelalisib which was the first FDA-approved compound to inhibit a lipid kinase (PI3Kδ isoform) [149, 150], in total seven dual PI3K/mTOR small molecule inhibitors are tested in advanced clinical trials [13] (Fig. 7). These comprise PKI587 (advanced solid malignancies) [151], quinacrine (various leukemias) [152, 153], GSK2126458 (colorectal, breast, NSCLC, and pancreatic cancer) [154], PF04691502 (breast cancer) [155], GDC0980 (mRC) [156], XL765 (breast cancer) [157], and NVP-BEZ235 (glioblastomas) [158].
The oral pan-PI3K inhibitor buparlisib which targets all four isoforms of class I PI3K was registered for three phase-III clinical trials against breast cancer. Buparlisib was tested in combination with fulvestrant in an advanced breast cancer study (Fig. 7). Due to the safety profile, the results do not endorse an expansion in this clinical setting [159, 160].
Interestingly, patients with a PIK3CA mutation have shown a median progression-free survival of 4.2 months (95% CI 2.8–6.7) after buparlisib treatment compared to 1.6 months (95% CI 1.4–2.8) in the placebo group. These results support the application of PI3K inhibitors combined with endocrine therapy in this genetic background [160].
Nearly all hallmarks of cancer can be targeted by approved protein kinase inhibitors. However, there are currently no FDA-accepted kinase inhibitors influencing genome instability and DNA damage response. The success story of olaparib, a Poly [ADP-ribose] polymerase 1 (PARP-1) inhibitor, underlines the high potential of small molecules in this field. In general, the incidence of DNA single- or double-strand breaks is mainly controlled by the network of ATR/ATM and CHK1/CHK2 signaling pathways leading to active DNA repair mechanisms and cell cycle checkpoint regulation [161]. At the moment, two different ATR inhibitors, AZD6738 and M6620, are tested in clinical phase-II trials with four and six studies. They are administered in prostate cancer, CLL, recurrent ovarian cancer, progressive metastatic gastric or gastroesophageal junction cancer, small cell lung carcinoma, as well as metastatic tumors including RCC, urothelial carcinoma, ovarian cancer, and PDAC [NCT03787680, NCT03328273, NCT03641313, NCT03517969, NCT03682289, NCT02595892, NCT02567409, NCT03462342, NCT02627443, NCT02487095].
Furthermore, checkpoint kinases are valid targets in DNA damage response. The CHK1/CHK2 inhibitor prexasertib is tested in small cell lung cancer, ovarian cancer, breast cancer, and prostate cancer in phase-II clinical trials [NCT02735980, NCT03414047, NCT02873975, NCT02203513] (Fig. 7).
In BRCA wild-type recurrent high-grade serous ovarian cancer, prexasertib could exhibit clinical activity and was in general well tolerated by treated patients [162]. Particularly patients with platinum-resistant or platinum-refractory cancer could profit here from further drug development [162].
9 Challenges
Beside numerous advances of kinase inhibitors, profound understanding of mechanisms in vivo is needed to overcome actual limitations in clinical oncology [13].
Secondary therapy resistance based on kinase mutations is an abundant phenomenon arising after kinase inhibition [163]. The diversity of such mutations among different kinases hampers the overall treatment success in cancer patients [164]. Acquired resistance is the most common resistance type caused by kinase inhibitors and relates to tumors that respond to therapy initially but show posterior resistance to permanent delivered therapy [13]. Secondary resistance can be induced by changes in the kinase gatekeeper residue since hydrophobic interactions in the sub-pocket are decisive for the inhibitor binding affinity [165, 166]. The gatekeeper residue interacts with Type I and Type II kinase inhibitors and sterically influences inhibitor binding to the hydrophobic region in the binding pocket [167]. In addition to gatekeeper mutations in BCR-ABL kinases inducing imatinib resistance, numerous other targets are affected by gatekeeper mutations [168,169,170,171,172,173]. A prominent example is the T790M mutation in EGFR kinase leading to boosted affinity toward ATP that triggers resistance to quinazoline inhibitors as well as gefitinib and erlotinib [174,175,176]. To circumvent drug resistance in the clinic, structural optimization of small molecule inhibitors is required [13]. In case of mutated EGFR-induced resistance to gefitinib and erlotinib, newly developed EGFR inhibitors can covalently bind to the ATP-binding site of EGFR [177, 178]. That represents an example for highly selective inhibitors against mutated targets [13].
To further counter kinase inhibitor resistance, scientists break fresh ground with innovative strategies. In the context of gatekeeper mutations, currently developed inhibitors are going to accept varying amino acids at the gatekeeper mutation site [179, 180]. In a second approach, kinases will be targeted at alternative binding sites to avoid the ubiquitous ATP-binding pocket by a presumable unique cavity [181, 182]. Apart from that, also indirect kinase targeting via inhibition of kinase transformers would be a valid option to overcome resistance [183].
An additional clinical challenge represents the reduction or elimination of critical toxicities associated with kinase inhibitors, such as proteinuria, skin reactions, hypertension, or cardiotoxicity [184, 185].
Well-known examples are associated side effects of BCR-ABL inhibitors, including cytopenia, cardiotoxicity, and cardiac sequela. HER2 and ALK inhibition cause gastric problems and dermatological irregularities. EGFR inhibition is linked to dermatological issues, and VEGFR inhibition can trigger cardiotoxicity [186, 187].
To exclude toxicities triggered by off-target binding of the inhibitor, more specific therapeutic strategies are required. RNA interference is not only a powerful tool for specific gene knockdown in basic research, but it also raises expectations as a therapeutic approach to inhibit crucial players in cancer such as kinases [13]. However, since important drug targets cannot be efficiently eradicated by RNA interference so far, clinical resistance to kinase inhibitors will continue to be an important challenge to kinase-associated therapies [13, 188].
Altogether, the development of clinical relevant kinase inhibition has just started, but the rapid progress in the development of molecular technologies and engineering raises confidence for further success stories.
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- CEL:
-
Chronic eosinophilic leukemia
- CLL:
-
Chronic lymphoblastic leukemia
- CML:
-
Chronic myeloid leukemia
- CNS:
-
Central nervous system
- CRC:
-
Colorectal cancer
- DFSP:
-
Dermatofibrosarcoma protuberans
- ER:
-
Estrogen receptor
- FDA:
-
Food and Drug Administration
- GI:
-
Gastrointestinal
- GIST:
-
Gastrointestinal stromal tumor
- HCC:
-
Hepatocellular carcinoma
- HES:
-
Hypereosinophilic syndrome
- MDS/MDP:
-
Myelodysplastic/myeloproliferative diseases
- NRY:
-
Non-receptor protein-tyrosine kinase
- NSCLC:
-
Non-small cell lung carcinoma
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PH:
-
Philadelphia chromosome
- PNET:
-
Primitive neuroectodermal tumor
- RCC:
-
Renal cell carcinoma
- RY:
-
Receptor protein-tyrosine kinase
- S/T:
-
Protein-serine/threonine protein kinase
- SEGA:
-
Subependymal giant cell astrocytoma
- shRNA:
-
Short hairpin RNA
- T/Y:
-
Threonine/tyrosine dual specificity protein kinase
References
Collett MS, Erikson RL (1978) Protein kinase activity associated with the avian sarcoma virus SRC gene product. Proc Natl Acad Sci 75(4):2021–2024
Castagna M et al (1982) Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem 257(13):7847–7851
Hidaka H et al (1984) Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide-dependent protein kinase and protein kinase C. Biochemistry 23(21):5036–5041
Knighton D et al (1991) Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253(5018):407–414
Zheng J et al (1993) Crystal structures of the myristylated catalytic subunit of cAMP-dependent protein kinase reveal open and closed conformations. Protein Sci 2(10):1559–1573
Yaish P et al (1988) Blocking of EGF-dependent cell proliferation by EGF receptor kinase inhibitors. Science 242(4880):933–935
Kovalenko M et al (1994) Selective platelet-derived growth factor receptor kinase blockers reverse sis-transformation. Cancer Res 54(23):6106–6114
Gazit A et al (1996) Tyrphostins. 5. Potent inhibitors of platelet-derived growth factor receptor tyrosine kinase: structure−activity relationships in quinoxalines, quinolines, and indole tyrphostins. J Med Chem 39(11):2170–2177
Osherov N et al (1993) Selective inhibition of the epidermal growth factor and HER2/neu receptors by tyrphostins. J Biol Chem 268(15):11134–11142
Levitzki A, Mishani E (2006) Tyrphostins and other tyrosine kinase inhibitors. Annu Rev Biochem 75(1):93–109
Hunter T (2007) Treatment for chronic myelogenous leukemia: the long road to imatinib. J Clin Invest 117(8):2036–2043
Roskoski R Jr (2020) FDA-approved protein kinase inhibitors. Blue Ridge Institute for Medical Research, Horse Shoe
Bhullar KS et al (2018) Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 17(1):48
Manning G et al (2002) The protein kinase complement of the human genome. Science 298(5600):1912–1934
Herbst RS (2004) Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys 59(2, Supplement):S21–S26
Oda K et al (2005) A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol 1:2005.0010
Lee HJ et al (2015) Prognostic and predictive values of EGFR overexpression and EGFR copy number alteration in HER2-positive breast cancer. Br J Cancer 112(1):103–111
Yang C-H et al (2015) EGFR over-expression in non-small cell lung cancers harboring EGFR mutations is associated with marked down-regulation of CD82. Biochim Biophys Acta 1852(7):1540–1549
Bossi P et al (2016) Prognostic and predictive value of EGFR in head and neck squamous cell carcinoma. Oncotarget 7(45):74362–74379
Gan HK, Cvrljevic AN, Johns TG (2013) The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J 280(21):5350–5370
An Z et al (2018) Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene 37(12):1561–1575
Han J et al (2015) CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep 5:11483
Sacher AG et al (2016) Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol 2(8):1014–1022
De Silva N et al (2015) Molecular effects of Lapatinib in the treatment of HER2 overexpressing oesophago-gastric adenocarcinoma. Br J Cancer 113:1305
D’Amato V et al (2015) Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat Rev 41(10):877–883
Bachelot T et al (2013) Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): a single-group phase 2 study. Lancet Oncol 14(1):64–71
Long X-H et al (2014) Lapatinib alters the malignant phenotype of osteosarcoma cells via downregulation of the activity of the HER2-PI3K/AKT-FASN axis in vitro. Oncol Rep 31(1):328–334
Chen J-C et al (2014) Suppression of Dicer increases sensitivity to gefitinib in human lung cancer cells. Ann Surg Oncol 21(4):555–563
Mok TS et al (2009) Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361(10):947–957
Sim EHA et al (2018) Gefitinib for advanced non-small cell lung cancer. Cochrane Database Syst Rev 1:CD006847
Neal JW (2010) The SATURN trial: the value of maintenance erlotinib in patients with non-small-cell lung cancer. Future Oncol 6(12):1827–1832
Wang JP et al (2015) Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: a randomized, open-label, prospective trial. Oncotarget 6(20):18162–18173
Cappuzzo F (2013) Erlotinib in the first-line treatment of non-small-cell lung cancer AU – D’Arcangelo, Manolo. Expert Rev Anticancer Ther 13(5):523–533
Paz-Ares L et al (2017) Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: overall survival data from the phase IIb LUX-Lung 7 trial. Ann Oncol 28(2):270–277
Wu YL et al (2013) LUX-Lung 6: a randomized, open-label, phase III study of afatinib (A) versus gemcitabine/cisplatin (GC) as first-line treatment for Asian patients (PTS) with EGFR mutation-positive (EGFR M+) advanced adenocarcinoma of the lung. J Clin Oncol 31(15_suppl):8016–8016
Morris S et al (1994) Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 263(5151):1281–1284
Morris SW et al (1997) ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 14:2175
Hallberg B, Palmer RH (2016) The role of the ALK receptor in cancer biology. Ann Oncol 27(suppl_3):iii4–iii15
Stute C et al (2004) Myoblast determination in the somatic and visceral mesoderm depends on Notch signalling as well as on milliways(miliAlk) as receptor for Jeb signalling. Development 131(4):743–754
Janoueix-Lerosey I et al (2018) The ALK receptor in sympathetic neuron development and neuroblastoma. Cell Tissue Res 372(2):325–337
Yao S et al (2013) Anaplastic lymphoma kinase is required for neurogenesis in the developing central nervous system of zebrafish. PLoS One 8(5):e63757
Yip PY (2015) Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Transl Lung Cancer Res 4(2):165–176
An R et al (2016) CRKL mediates EML4-ALK signaling and is a potential therapeutic target for ALK-rearranged lung adenocarcinoma. Oncotarget 7(20):29199–29210
Umapathy G et al (2014) The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci Signal 7(349):ra102
Hallberg B, Palmer RH (2013) Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 13:685
Soda M et al (2007) Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 448:561
Hofman P (2017) ALK in non-small cell lung cancer (NSCLC) pathobiology, epidemiology, detection from tumor tissue and algorithm diagnosis in a daily practice. Cancers 9(8):107
Nwizu T et al (2011) Crizotinib (PF02341066) as a ALK/MET inhibitor – special emphasis as a therapeutic drug against lung cancer. Drugs Future 36(2):91–99
Kwak EL et al (2010) Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N Engl J Med 363(18):1693–1703
Crinò L et al (2011) Initial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): PROFILE 1005. J Clin Oncol 29(15_suppl):7514
Shaw AT et al (2013) Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 368(25):2385–2394
Solomon BJ et al (2014) First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 371(23):2167–2177
Friboulet L et al (2014) The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov 4(6):662–673
Kim D-W et al (2016) Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol 17(4):452–463
Crinò L et al (2016) Multicenter phase II study of whole-body and intracranial activity with ceritinib in patients with ALK-rearranged non–small-cell lung cancer previously treated with chemotherapy and crizotinib: results from ASCEND-2. J Clin Oncol 34(24):2866–2873
Bearz A et al (2016) Ceritinib vs chemotherapy (CT) in patients (pts) with advanced anaplastic lymphoma kinase (ALK)-rearranged (ALK+) non-small cell lung cancer (NSCLC) previously treated with CT and crizotinib (CRZ): results from the confirmatory phase 3 ASCEND-5 study. Ann Oncol 27(suppl_6):1–36
Soria J-C et al (2017) First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 389(10072):917–929
Rothenstein JM, Chooback N (2018) ALK inhibitors, resistance development, clinical trials. Curr Oncol 25(Suppl 1):S59–S67
Zeaiter A et al (2016) Updated efficacy and safety from the global phase II NP28673 study of alectinib in patients (pts) with previously treated ALK+ non-small-cell lung cancer (NSCLC). Ann Oncol 27(suppl_6):1263P
Yang JC-H et al (2017) Pooled systemic efficacy and safety data from the pivotal phase II studies (NP28673 and NP28761) of alectinib in ALK-positive non-small cell lung cancer. J Thorac Oncol 12(10):1552–1560
Peters S et al (2017) Alectinib versus crizotinib in untreated ALK-positive non–small-cell lung cancer. N Engl J Med 377(9):829–838
Huang W-S et al (2016) Discovery of brigatinib (AP26113), a phosphine oxide-containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J Med Chem 59(10):4948–4964
Kim D-W et al (2017) Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase–positive non–small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 35(22):2490–2498
Solomon B et al (2017) OA 05.06 phase 2 study of lorlatinib in patients with advanced ALK+/ROS1+ non-small-cell lung cancer. J Thorac Oncol 12(11):S1756
Solomon BJ et al (2018) Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol 19(12):1654–1667
Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285(21):1182–1186
Ferrara N, Gerber H-P, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9:669
Ferrara N (2002) VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer 2:795
Di Lorenzo G et al (2010) Third-line sorafenib after sequential therapy with sunitinib and mtor inhibitors in metastatic renal cell carcinoma. Eur Urol 58(6):906–911
Motzer RJ et al (2007) Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 356(2):115–124
Carducci MA et al (2015) Atrasentan in patients with advanced renal cell carcinoma: a phase 2 trial of the ECOG-ACRIN Cancer Research Group (E6800). Clin Genitourin Cancer 13(6):531–539.e1
Bhargava P, Robinson MO (2011) Development of second-generation VEGFR tyrosine kinase inhibitors: current status. Curr Oncol Rep 13(2):103–111
Raymond E et al (2011) Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 364(6):501–513
Llovet JM et al (2008) Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359(4):378–390
Grothey A et al (2013) Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381(9863):303–312
Demetri GD et al (2013) Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381(9863):295–302
Bruix J et al (2017) Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 389(10064):56–66
Zirlik K, Duyster J (2018) Anti-angiogenics: current situation and future perspectives. Oncol Res Treat 41(4):166–171
Srinivasan D, Plattner R (2006) Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res 66(11):5648–5655
Sirvent A et al (2007) The tyrosine kinase Abl is required for Src-transforming activity in mouse fibroblasts and human breast cancer cells. Oncogene 26:7313
Mgbemena VE (2014) Re-evaluating the role of BCR/ABL in chronic myelogenous leukemia AU – Ross, Theodora S. Mol Cell Oncol 1(3):e963450
Schenone S et al (2015) Analogs, formulations and derivatives of imatinib: a patent review AU – Musumeci, Francesca. Expert Opin Ther Pat 25(12):1411–1421
Cohen MH et al (2002) Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res 8(5):935–942
Dematteo RP et al (2009) Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 373(9669):1097–1104
Dagher R et al (2002) Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res 8(10):3034–3038
Hasford J et al (2011) Predicting complete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score. Blood 118(3):686–692
Hochhaus A et al (2002) Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 16:2190
Rossari F, Minutolo F, Orciuolo E (2018) Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. J Hematol Oncol 11(1):84–84
Kantarjian HM et al (2011) Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol 12(9):841–851
Kantarjian HM et al (2012) Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood 119(5):1123–1129
Skora L et al (2013) NMR reveals the allosteric opening and closing of Abelson tyrosine kinase by ATP-site and myristoyl pocket inhibitors. Proc Natl Acad Sci 110(47):E4437–E4445
Shah NP et al (2004) Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 305(5682):399–401
Boschelli DH et al (2001) Optimization of 4-Phenylamino-3-quinolinecarbonitriles as potent inhibitors of Src kinase activity. J Med Chem 44(23):3965–3977
Boschelli F, Arndt K, Gambacorti-Passerini C (2010) Bosutinib: a review of preclinical studies in chronic myelogenous leukaemia. Eur J Cancer 46(10):1781–1789
Redaelli S et al (2009) Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J Clin Oncol 27(3):469–471
Cortes JE et al (2017) Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: results from the randomized BFORE trial. J Clin Oncol 36(3):231–237
Huang W-S et al (2010) Discovery of 3-[2-(Imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. J Med Chem 53(12):4701–4719
Cortes JE et al (2012) Ponatinib in refractory Philadelphia chromosome–positive leukemias. N Engl J Med 367(22):2075–2088
Nicolini FE et al (2015) The impact of ponatinib versus allogeneic stem cell transplant (SCT) on outcomes in patients with chronic myeloid leukemia (CML) or Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) with the T315I mutation. Blood 126(23):480–480
Samatar AA, Poulikakos PI (2014) Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 13(12):928–942
Agianian B, Gavathiotis E (2018) Current insights of BRAF inhibitors in cancer. J Med Chem 61(14):5775–5793
Lavoie H, Therrien M (2015) Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 16(5):281–298
Roring M et al (2012) Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J 31(11):2629–2647
Rajakulendran T et al (2009) A dimerization-dependent mechanism drives RAF catalytic activation. Nature 461(7263):542–545
Heidorn SJ et al (2010) Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140(2):209–221
Davies H et al (2002) Mutations of the BRAF gene in human cancer. Nature 417(6892):949–954
Schubbert S, Shannon K, Bollag G (2007) Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 7(4):295–308
Zambon A et al (2012) Small molecule inhibitors of BRAF in clinical trials. Bioorg Med Chem Lett 22(2):789–792
Rahman MA et al (2014) BRAF inhibitors: from the laboratory to clinical trials. Crit Rev Oncol Hematol 90(3):220–232
Alcala AM, Flaherty KT (2012) BRAF inhibitors for the treatment of metastatic melanoma: clinical trials and mechanisms of resistance. Clin Cancer Res 18(1):33–39
Bollag G et al (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467(7315):596–599
Tap WD et al (2010) Pharmacodynamic characterization of the efficacy signals due to selective BRAF inhibition with PLX4032 in malignant melanoma. Neoplasia 12(8):637–649
Chapman PB et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364(26):2507–2516
da Rocha Dias S et al (2013) The European Medicines Agency review of vemurafenib (Zelboraf(R)) for the treatment of adult patients with BRAF V600 mutation-positive unresectable or metastatic melanoma: summary of the scientific assessment of the Committee for Medicinal Products for Human Use. Eur J Cancer 49(7):1654–1661
Flaherty KT et al (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 363(9):809–819
Ravnan MC, Matalka MS (2012) Vemurafenib in patients with BRAF V600E mutation-positive advanced melanoma. Clin Ther 34(7):1474–1486
Mattei PL et al (2013) Cutaneous effects of BRAF inhibitor therapy: a case series. Ann Oncol 24(2):530–537
Anforth R, Fernandez-Penas P, Long GV (2013) Cutaneous toxicities of RAF inhibitors. Lancet Oncol 14(1):e11–e18
Gibney GT, Zager JS (2013) Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies. Expert Opin Drug Metab Toxicol 9(7):893–899
Hauschild A et al (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380(9839):358–365
Zhang W (2015) BRAF inhibitors: the current and the future. Curr Opin Pharmacol 23:68–73
Barras D (2015) BRAF mutation in colorectal cancer: an update. Biomark Cancer 7(Suppl 1):9–12
Witkiewicz AK et al (2015) Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6:6744
Poulikakos PI et al (2011) RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480(7377):387–390
Eisen T et al (2006) Sorafenib in advanced melanoma: a phase II randomised discontinuation trial analysis. Br J Cancer 95(5):581–586
Wilhelm SM et al (2004) BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64(19):7099–7109
Rimassa L et al (2013) A phase II randomized dose escalation trial of sorafenib in patients with advanced hepatocellular carcinoma. Oncologist 18(4):379–380
Escudier B et al (2009) Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol 27(20):3312–3318
Rimassa L, Santoro A (2009) Sorafenib therapy in advanced hepatocellular carcinoma: the SHARP trial. Expert Rev Anticancer Ther 9(6):739–745
Wilhelm S et al (2006) Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 5(10):835–844
Brose MS et al (2011) Rationale and design of decision: a double-blind, randomized, placebo-controlled phase III trial evaluating the efficacy and safety of sorafenib in patients with locally advanced or metastatic radioactive iodine (RAI)-refractory, differentiated thyroid cancer. BMC Cancer 11:349
Brose MS et al (2014) Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet 384(9940):319–328
Rudalska R et al (2014) In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat Med 20(10):1138–1146
Janne PA et al (2017) Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: the SELECT-1 randomized clinical trial. JAMA 317(18):1844–1853
Kim C, Giaccone G (2018) MEK inhibitors under development for treatment of non-small-cell lung cancer. Expert Opin Investig Drugs 27(1):17–30
Moriceau G et al (2015) Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 27(2):240–256
Long GV et al (2017) Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol 28(7):1631–1639
Long GV et al (2017) Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med 377(19):1813–1823
Ascierto PA et al (2016) Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol 17(9):1248–1260
Griffin M et al (2017) BRAF inhibitors: resistance and the promise of combination treatments for melanoma. Oncotarget 8(44):78174–78192
Faivre S, Kroemer G, Raymond E (2006) Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov 5:671
Anagnostopoulou V et al (2013) Differential effects of dehydroepiandrosterone and testosterone in prostate and colon cancer cell apoptosis: the role of nerve growth factor (NGF) receptors. Endocrinology 154:2446
WeiSz L, Efferth T (2012) Polo-like kinase 1 as target for cancer therapy. Exp Hematol Oncol 1:38
Akinleye A et al (2013) Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol 6:88
Fayard E et al (2011) Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. In: Phosphoinositide 3-kinase in health and disease. Springer, New York, pp 31–56
Cance WG et al (2013) Disrupting the scaffold to improve focal adhesion kinase–targeted cancer therapeutics. Sci Signal 6:pe10
Ahmed K et al (2015) Targeting CK2 for cancer therapy using a nanomedicine approach. In: Protein kinase CK2 cellular function in normal and disease states. Springer, New York, pp 299–315
Hilton JF, Shapiro GI (2014) Aurora kinase inhibition as an anticancer strategy. J Clin Oncol 32:57
Jones JA et al (2017) Efficacy and safety of idelalisib in combination with ofatumumab for previously treated chronic lymphocytic leukaemia: an open-label, randomised phase 3 trial. Lancet Haematol 4(3):e114–e126
Pandey R, Kapur R (2018) Kinase inhibitors in clinical practice: an expanding world. J Allergy Clin Immunol 141(2):522–524
Shapiro GI et al (2015) First-in-human study of PF-05212384 (PKI-587), a small-molecule, intravenous, dual inhibitor of PI3K and mTOR in patients with advanced cancer. Clin Cancer Res 21(8):1888–1895
Eriksson A et al (2015) Drug screen in patient cells suggests quinacrine to be repositioned for treatment of acute myeloid leukemia. Blood Cancer J 5:e307
Haïk S et al (2014) Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 13:150
Bedard PL et al (2014) Abstract CT205: a phase I dose-escalation study of trametinib (T) in combination with continuous or intermittent GSK2126458 (GSK458) in patients (pts) with advanced solid tumors. Cancer Res 74:CT205
Dowsett M et al (2012) Phase II randomized study of pre-operative pf-04691502 plus letrozole compared with letrozole (L) in patients with estrogen receptor-positive, HER2-negative early breast cancer (BC). Ann Oncol 23:44
Powles T et al (2014) A randomized phase II study of GDC-0980 versus everolimus in metastatic renal cell carcinoma (mRCC) patients (pts) after VEGF-targeted therapy (VEGF-TT). J Clin Oncol 32(15_suppl):4525
Yu P et al (2014) Characterization of the activity of the PI3K/mTOR inhibitor XL765 (SAR245409) in tumor models with diverse genetic alterations affecting the PI3K pathway. Mol Cancer Ther 13:1078
Mukherjee B et al (2012) The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM-and DNA-PKCs-mediated DNA damage responses. Neoplasia 14:34
Bauer TM, Patel MR, Infante JR (2015) Targeting PI3 kinase in cancer. Pharmacol Ther 146:53–60
Di Leo A et al (2018) Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 19(1):87–100
Manic G et al (2015) Trial watch: targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol Cell Oncol 2(4):e1012976
Lee J-M et al (2018) Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol 19(2):207–215
Wilson TR et al (2012) Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487:505
Lito P, Rosen N, Solit DB (2013) Tumor adaptation and resistance to RAF inhibitors. Nat Med 19:1401
Chell V et al (2013) Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene 32:3059
Foguel D et al (2014) The importance of a gatekeeper residue on the aggregation of transthyretin implications for transthyretin-related amyloidoses. J Biol Chem 289(41):28324–28337
Liu Y, Gray NS (2006) Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2:358–364
Gibbons DL et al (2012) The rise and fall of gatekeeper mutations? The BCR-ABL1 T315I paradigm. Cancer 118:293
Heinrich MC et al (2006) Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 24:4764
Metzgeroth G et al (2012) Limited clinical activity of nilotinib and sorafenib in FIP1L1-PDGFRA positive chronic eosinophilic leukemia with imatinib-resistant T674I mutation. Leukemia 26:162
Nishikawa S et al (2018) Selective gene amplification to detect the T790M mutation in plasma from patients with advanced non-small cell lung cancer (NSCLC) who have developed epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) resistance. J Thorac Dis 10(3):1431–1439
Pauwels D, Sweron B, Cools J (2012) The N676D and G697R mutations in the kinase domain of FLT3 confer resistance to the inhibitor AC220. Haematologica 97:1773
Xavier CP et al (2009) Luteolin, quercetin and ursolic acid are potent inhibitors of proliferation and inducers of apoptosis in both KRAS and BRAF mutated human colorectal cancer cells. Cancer Lett 281:162
Kobayashi S et al (2005) EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N Engl J Med 352:786
Pao W et al (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2:e73
Yun CH et al (2008) The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci 105:2070
Heymach JV et al (2006) Epidermal growth factor receptor inhibitors in development for the treatment of non–small cell lung cancer. Clin Cancer Res 12:4441s
Kwak EL et al (2005) Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A 102:7665
Modugno M et al (2007) Crystal structure of the T315I Abl mutant in complex with the aurora kinases inhibitor PHA-739358. Cancer Res 67:7987
Weisberg E et al (2005) Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 7:129
Gumireddy K et al (2005) A non-ATP-competitive inhibitor of BCR-ABL overrides imatinib resistance. Proc Natl Acad Sci U S A 102:1992
Gumireddy K et al (2005) ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell 7:275
Copland M et al (2008) BMS-214662 potently induces apoptosis of chronic myeloid leukemia stem and progenitor cells and synergizes with tyrosine kinase inhibitors. Blood 111:2843
Orphanos GS, Ioannidis GN, Ardavanis AG (2009) Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol 48:964
Shah DR, Shah RR, Morganroth J (2013) Tyrosine kinase inhibitors: their on-target toxicities as potential indicators of efficacy. Drug Saf 36:413
Mayor S (2006) Targeting cardiovascular complications. Lancet Oncol 7:282
Moslehi JJ, Deininger M (2015) Tyrosine kinase inhibitor–associated cardiovascular toxicity in chronic myeloid leukemia. J Clin Oncol 33:4210
Bernards R, Brummelkamp TR, Beijersbergen RL (2006) shRNA libraries and their use in cancer genetics. Nat Methods 3:701
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Ethics declarations
Funding: L.Z. and D.D. are supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) [FOR2314 (D.D., L.Z.), SFB-TR209 (D.D., L.Z.,), SFB-TR240 (L.Z.), Gottfried Wilhelm Leibniz Program (L.Z.)], the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s excellence strategy – EXC 2180 – 390900677 [Image Guided and Functionally Instructed Tumour Therapies (iFIT)], the Landesstiftung Baden-Wuerttemberg [‘Improve CRC’ (D.D., L.Z.)], the European Research Council [‘CholangioConcept’ (L.Z.)] and the German Center for Translational Cancer Research (DKTK).
Ethical Approval: This chapter does not contain any studies with human participants or animals performed by of the authors.
Informed Consent: All authors have given their consent to this book chapter.
Conflict of Interest: The authors declare no competing financial interests.
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Moschopoulou, A., Zwirner, S., Zender, L., Dauch, D. (2020). Exploiting Kinase Inhibitors for Cancer Treatment: An Overview of Clinical Results and Outlook. In: Laufer, S. (eds) Proteinkinase Inhibitors. Topics in Medicinal Chemistry, vol 36. Springer, Cham. https://doi.org/10.1007/7355_2020_100
Download citation
DOI: https://doi.org/10.1007/7355_2020_100
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-68179-1
Online ISBN: 978-3-030-68180-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)