
Abstract
The advent of direct-acting antiviral agents (DAAs) has revolutionized the treatment and cure of chronic hepatitis C virus (HCV) infection. Herein is described the discovery of ledipasvir (LDV), an orally available HCV nonstructural protein 5A inhibitor with picomolar antiviral potency and a long pharmacokinetic half-life. The combination of LDV with the nonstructural protein 5B inhibitor sofosbuvir (SOF) is Harvoni® and represents the first approved single-tablet regimen for the treatment of HCV infection. This safe simple and efficacious regimen affords clinical trial cure rates over 95% and comparable effectiveness in real-world studies and has treatment durations as short as 8 weeks. The approval of Harvoni® heralded a new era for the treatment of HCV infection.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Direct-acting antiviral
- GS-5885
- Harvoni®
- HCV
- LDV/SOF
- Ledipasvir
- NS5A inhibitor
- NS5B nucleotide inhibitor
- Single-tablet regimen
1 Introduction
Prior to the direct-acting antiviral (DAA) era, the standard of care for HCV-infected individuals included toxic, poorly tolerated regimens based on ribavirin (RBV) and some form of interferon (IFN) and resulted in low SVR (cure) rates. Among the six major HCV genotypes, IFN-based regimens unfortunately resulted in the lowest cure rates for genotype 1 (GT1) patients, the most prevalent genotype worldwide (Fig. 1, US Veterans Administration) [1].

SVR rates are the lowest for GT1 patients with IFN-based regimens
The complexity, poor response rates, and toxicity of IFN-based regimens are underrepresented by the SVR rates reported from clinical studies and the common description that these regimens engender “flu-like” symptoms. Real-world results present a more accurate picture of the patient’s experience. The real-world efficacy of interferon regimens (cure rates outside of clinical trials) is vastly lower than even the ~30–50% SVR results reported from controlled clinical trials. Real-world efficacy late in the IFN era produced SVR as low as 3% in a report of 13,000 patients in 25 real-world studies across the United States (Fig. 2) [2]. This exceedingly low cure rate is a consequence of many factors, including a large number of exclusion criteria and significant toxicity and complexity of the regimen that precipitates patient discontinuation, hesitance to start therapy, and viral breakthrough and relapse [2].

Real-world IFN regimen results. 25 studies (2002–2012) show much lower SVR rates than those reported from clinical trials
Based on the poor results observed from the IFN-based standard of care, many patients deferred therapy in hope of newer treatment approaches while their liver disease progressed to advanced stages of fibrosis or cirrhosis (Fig. 3) [1]. In 2013 as a consequence of the aging HCV-infected patient population progressing to liver cirrhosis, fibrosis, and hepatocellular carcinoma, the Center for Disease Control disclosed that deaths arising from HCV infection in the United States had surpassed those of all other notifiable infections combined in the United States (including human immunodeficiency virus [HIV], tuberculosis, and influenza) [3]. There was a critical need for improved therapies.

Treatment rates increase sharply with the introduction of DAAs, particularly the STR LDV/SOF. Interferon beta-interferon, PEG pegylated interferon, PrOD paritaprevir, ritonavir, ombitasvir, dasabuvir
The advent of DAA therapies marked a revolution in the treatment and cure of HCV infection. The rapid uptake of DAA therapy underscores the high unmet need that existed in HCV therapy, with telaprevir (2011) and then sofosbuvir (2013)Footnote 1 , Footnote 2 [4] and finally the combination drug ledipasvir/sofosbuvir (2014)Footnote 3 [5] each becoming the largest drug launches in history [6,7,8,9]. The benefit to patients in this progression of DAAs can be seen in Figs. 3 and 4 which show treatment rates and cure rates in the US Veterans Administration progressing from the IFN era to the approval and uptake of LDV/SOF [1].

The low SVR rates from the IFN era (compare to Fig. 3) are transformed during the DAA era. Interferon beta-interferon, PEG pegylated interferon, PrOD paritaprevir, ritonavir, ombitasvir, dasabuvir
With the high level of unmet need for HCV patients in the IFN era as a backdrop, we pursued multiple viral and host targets for the treatment of HCV infection. At the time of initiating our efforts to discover an HCV nonstructural protein 5a (NS5A) inhibitor, we had over 20 HCV research programs ongoing, and several compounds undergoing, or selected to enter clinical trials. The more advanced agents, GS-9190 (NS5B non-nucleotide polymerase inhibitor), GS-9256, and GS-9451 (NS3/4a protease inhibitors), were directed at inhibiting GT1 HCV [10,11,12]. As a result of both the lower response rates to IFN therapy and high prevalence (with GT1 infection estimated to be as high as 60% of HCV-infected individuals worldwide [13]), there was a dominant unmet need for improved treatment of GT1 HCV infection. Based on this epidemiological and therapeutic landscape, we initiated our NS5A inhibitor program with primary potency assays directed toward GT1 HCV antiviral activity. This chapter details the discovery and early development of the potent HCV NS5A inhibitor ledipasvir (1, GS-5885, Table 1) and its clinical combination with sofosbuvir [14]. The discovery program toward the pan-genotypic NS5A inhibitor velpatasvir initiated immediately following our ledipasvir discovery work. The discovery of our pan-genotypic NS5A inhibitor velpatasvir (VEL, GS-5816) is described in Volume II, HCV: The Journey from Discovery to a Cure and the following references: [15,16,17]. The discovery of our pan-genotypic NS3/4a protease inhibitor voxilaprevir (VOX, GS-9857) and its combination with SOF and VEL as Vosevi® is described in Volume I, HCV: The Journey from Discovery to a Cure and the following references: [18]. (https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209195s000lbl.pdf. Accessed 10 June 2018).

We targeted our NS5A inhibitor to possess properties appropriate for incorporation with one or more other HCV antiviral agents in a single-tablet regimen (STR). Single-tablet regimens have proven beneficial for patient compliance and efficacy in the chronic treatment of HIV infection [19, 20]. We saw a similar utility for an STR in the treatment of HCV infection. Thus the attributes of our NS5A inhibitor required sufficient potency and metabolic stability to achieve a low dose, a long pharmacokinetic half-life compatible with once-daily dosing, and a drug interaction profile suitable for combination with other HCV antivirals of complementary mechanism. The research program focused on these principles to guide the optimization of potency and pharmacokinetic (PK) parameters in the discovery of LDV. LDV is combined with SOF as Harvoni®, and LDV was the first US Food and Drug Administration (FDA) approved NS5A inhibitor (October 10, 2014). Harvoni® was approved for the treatment and cure of GT1 HCV-infected individuals in as short as 8 weeks of therapy and was the first approved HCV STR. Based on the potent antiviral activity of LDV against GT4, GT5, and GT6 HCV (Table 1), and the concomitant high SVR rates for LDV/SOF in GT4–6 patients, Harvoni® was additionally approved for treatment of GT4–6 patients (https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/205834s001lbl.pdf. Accessed 10 June 2018) [5].
2 Discovery Work Leading to Ledipasvir
We focused on discovering an NS5A inhibitor that could be utilized in a single-tablet regimen in combination with other HCV agents. Further, we sought high antiviral potency and a long PK half-life to reduce the potential for emergence of viral breakthrough or resistance during treatment [14]. A large number of diverse cores were designed and synthesized in the discovery of LDV [21]. In early studies we investigated symmetric core inhibitors (where the core is the portion of the inhibitor structure that spans from between the C2 positions of each [modified] pyrrolidine – as colored in blue in Table 2). Potency proved challenging to attain against the genotype 1a subtype (GT1a) replicon but was more readily attained against the genotype 1b subtype (GT1b). Thus potency discussions herein most typically refer to GT1a potency, with GT1b potency provided in tables for reference. Throughout this manuscript, potency values represent effective concentration to reduce replication by 50% (EC50) in cell lines with engineered replicons.

Table 2 outlines potency optimization for structural variation within the core between two benzimidazoles. With directly linked benzimidazoles, compound 2 does not achieve an EC50 against GT1a at the top concentration of 44,000 pM. Alkyne 3 is 138 pM against GT1b, but again is not active against GT1a. Bis-alkyne 4 improves GT1a potency to 11,000 pM. Incorporation of ring systems further improves potency. Thiophene- and phenyl-based cores improve potency from 1,700 to 500 pM, respectively. Biphenyl (6) loses activity relative to phenyl (5), while fused-ring systems provide highly potent inhibitors with naphthyl (7) and benzodithiophene (8) achieving 110 pM and 200 pM GT1a inhibition, respectively.
The inhibitors in Table 3 represent a shift in our thinking and provide the initiation of a fruitful path that we investigated throughout our NS5A inhibitor program. The inhibitors in Table 3 have unsymmetric cores, where one end of the core is a benzimidazole and the other end an imidazole. The use of unsymmetric cores has implications that will be discussed further (vide infra). Our unsymmetric approach afforded intriguing structural variation and properties to our inhibitors and afforded striking divergences from the results for the bis-benzimidazole cores in Table 2. In the imidazole/benzimidazole series, interestingly the phenyl-based core inhibitor 9 does not achieve 50% inhibition at the highest assay concentration and is >88-fold weaker in activity than in the bis-benzimidazole example (5). Most striking is that replacement of phenyl (9) by naphthyl (10) in this series affords an increase in potency by 620-fold. Despite significant divergence in potency for the phenyl inhibitors between these series, in both series the naphthyl-based core provides the most potent inhibitor against GT1a observed within each table of inhibitors (compounds 7 and 10). It is also notable the unsymmetric core inhibitor 10 at 71 pM in GT1a is more potent than the symmetric inhibitor 7. Phenyl-alkynyl inhibitors 11 and 12 reveal another important facet of these unsymmetric cores. With the core possessing unsymmetric ends (imidazole/benzimidazole), and with the presence of unsymmetric central “-X-” groups, there is a matched and mismatched combination within the core. The core with the phenyl attached to the imidazole (inhibitor 12, 380 pM versus GT1a) affords 6.6-fold more potency than the core with the alkynyl attachment to the imidazole. Finally, replacement of the alkyne with an aryl group provides potent biaryl inhibitors. The biphenyl inhibitor 14 is 22-fold more potent at 170 pM in GT1a than the corresponding inhibitor in the bis-benzimidazole series.

Fused-ring cores in Tables 2 and 3 afforded three out of the top four most potent inhibitors (7, 8, and 10, ranging from 70 to 110 pM versus GT1a). The biphenyl-based inhibitor 14 was the only non-fused-ring core that attained high potency (170 pM) comparable to the fused-ring cores. Importantly inhibitor 14 was stable at the lower measured limit of our routine human liver microsomes (HLM) stability assay (predicted clearance <0.16 L/h/kg), whereas the naphthyl inhibitor 10 demonstrated less metabolic stability in microsomes. The favorable potency and metabolic stability of the biaryl inhibitor 14, along with our observation that fused-ring systems provide high potency, prompted us to combine these concepts through constraint of the biphenyl to afford a tricyclic fused-ring inhibitor series.
These tricycle-based core inhibitors (Fig. 5) were synthesized in a symmetric bis-imidazole core series allowing for an easier synthetic path than the unsymmetric series and a more rapid assessment of the tricyclic systems. Fluorene 15 showed good potency but was unstable even to air oxidation. The oxidation product, fluorenone 16, was less potent, and dimethyl substitution to block the system from oxidation lost over 13-fold in potency. The exo-dimethylmethylene was synthesized to avoid the out-of-plane bulk of 17 and the dipole of 16 but suffered a 78-fold potency loss relative to fluorene 15. (It is intriguing that GT1b activity tightly ranges from 13 to 18 pM over this series.)

Fused tricyclic core-based inhibitor potency
We sought to synthesize the difluorofluorene to block the fluorene methylene oxidation with a relatively small group – installation of two fluorines on the methylene proved challenging, and we incorporated them in the high value unsymmetric series (Fig. 6). The unsymmetric difluorofluorene-based core afforded inhibitor 19 with the highest potency in this series at 40 pM in GT1a and 3 pM in GT1b and was stable under the conditions of the HLM assay.

Difluorofluorene 14 affords improved potency and oxidative stability and has good PK half-lives
Unsymmetric biphenyl (14) and difluorofluorene (19) inhibitors were progressed into rat and dog PK (Table 4). Both inhibitors displayed good pharmacokinetic properties with moderate steady-state volumes of distribution (V ss) higher than total body water. Difluorofluorene 19 had superior and low clearance in both rat and dog, longer half-lives, and greater bioavailability in rat than biphenyl inhibitor 14 (36.7% versus 11.5%). Thus difluorofluorene 19 showed improvements in potency and pharmacokinetic properties over biphenyl 14.
Next a number of terminal heterocycle modifications were undertaken, again in a symmetric series for simplification of chemical synthesis. The azabicyclo[2.2.1] inhibitor 21 was more potent than piperidine 20 (Table 5). Although not as potent as the more advanced inhibitors such as 19, azabicyclic inhibitor 21 displayed favorable PK properties in dog as well as a very low clearance, a 5.29 h half-life and 35% oral bioavailability in rat (Table 4).

We sought to incorporate this pharmacokinetic enhancing azabicyclo[2.2.1] ring system into our unsymmetric imidazole/benzimidazole series. In this process we discovered a further important facet of structure-activity relationships in the unsymmetric core series. Earlier we found that the directionality of the central portion of the core (phenyl-alkyne) relative to the unsymmetric imidazole/benzimidazole groups produced a matched and mismatched pairing (compounds 11 and 12). This core directionality again provided a matched and mismatched pairing but this time with positioning of the terminal azabicyclo[2.2.1] ring system relative to the unsymmetric core (i.e., positioning of the azabicyclo[2.2.1] system on either end of the core). When the bicyclic ring system is proximal to the benzimidazole (22), the inhibitor is more potent than when the bicyclic ring system is proximal to the imidazole (23) (Fig. 7). Further, the matched case inhibitor is more potent (22, GT1a = 160 pM) than the azabicyclic symmetric inhibitor (21, GT1a = 210 pM, Table 5), while the mismatched case inhibitor 23 is less potent (GT1a = 660 pM) than the symmetric case 21. These results highlight a critical discovery; unsymmetric cores allow for diversification of structure that can provide beneficial properties (matched cases, with beneficial properties such as enhanced potency and/or pharmacokinetics) over the more limited symmetric cases. Finally, we had sought to determine if the beneficial PK properties imparted by the azabicyclo[2.2.1] ring in symmetric inhibitor 21 might translate in the unsymmetric series. Indeed, the half-lives in rat and dog are improved in azabicyclo[2.2.1] inhibitor 22 (unsymmetric “matched case”) over the corresponding pyrrolidine 14 (Table 4).

Differential end-substitution with an unsymmetric core leads to divergence in potency. Bridged azabicyclic ring system is more potent on benzimidazole side of the core
Having improved the PK half-life with azabicyclic inhibitor 22, we sought to enhance its potency. As demonstrated earlier, fused-ring systems within the inhibitor’s core can provide potency enhancements. Accordingly, incorporation of the difluorofluorene ring system in inhibitor 24 (Fig. 8) afforded a GT1a potency of 56 pM, which is an ~3fold improvement over the biaryl core inhibitor 22.

Difluorofluorene core improves potency in combination with azabicyclic pyrrolidine
The inhibitors described herein are highly protein bound, even in the replicon cellular assay which includes 10% fetal bovine serum (FBS). We utilize a dialysis methodology to assess relative inhibitor free fraction based on plasma protein binding and then generate a value for the protein-binding-adjusted potency. In this method, the inhibitor of interest is dialyzed between 100% human plasma in one well and replicon cell culture medium (including 10% FBS) in a second well [22]. The measured concentration ratio between wells can be multiplied by the replicon potency to “cancel” the cell culture medium binding and provide a human plasma protein-binding-adjusted potency (PA EC50). This methodology affords a GT1a PA EC50 for inhibitor 24 of 784 pM. We sought to improve upon this protein-binding-adjusted potency.
3 Ledipasvir (1, LDV, GS-5885)
During our final phase of discovery, one of the directions we undertook was modification of the pyrrolidine of inhibitor 24. Incorporation of a spirocyclopropyl ring provided the most potent inhibitor (compound 1) in the series with improvements in both the replicon potency and the plasma protein-binding-adjusted potency (GT1a EC50 = 31 pM, PA EC50 = 208 pM, Fig. 9) over inhibitor 24; interestingly, the PA EC50 of 1 versus 24 was differentially improved (3.6-fold, 208 versus 740 pM) relative to the EC50 (1.8-fold, 31 versus 56 pM). The protein-binding ratio of human plasma versus cell culture medium of 6.7-fold for inhibitor 1 as measured by dialysis was the lowest for any advanced inhibitor we had assessed. Compound 1 is highly protein bound, with only 1% free drug even in the replicon cell culture medium (containing 10% fetal bovine serum). Accounting for free drug, the intrinsic replicon GT1a and GT1b EC50 values for compound 1 are 310 femtomolar (fM) and 40 fM, respectively (Fig. 9).

Ledipasvir structure, replicon cellular antiviral potency, intrinsic replicon potency, and human PK half-life
Pharmacokinetic curves and PK parameters for inhibitor 1 are found in Fig. 10 and Table 6. Compound 1 has the longest half-lives (from 4.7 to 10.3 h in rat, dog, and monkey) among the compounds described herein and has high metabolic stability across preclinical species. Based on its exceptional replicon and PA potency, excellent pharmacokinetic half-lives, bioavailability, and low predicted clearance, compound 1 was selected for development and is now known as ledipasvir (LDV, GS-5885).

Pharmacokinetic curves for ledipasvir (1) in rat, dog, and cynomolgus monkey. LDV has long half-lives in preclinical species
A synthesis of LDV is depicted in Scheme 1. The azabicyclo[2.2.1] ester 1a [23] is debenzylated with palladium hydroxide and hydrogen, Boc protected, and the ester hydrolyzed to form bicyclic acid 1b. Coupling of 1b with 4-bromo-1,2-diaminobenzene and heating in ethanol affords benzimidazole 1c, which is borylated to form pinacol boronate ester 1d. Bromo-iodofluorene 1e is difluorinated in a mild and novel “one-pot” procedure by treatment with N-fluorobenzenesulfonimide followed by KHDMS in THF (1f). The Grignard of 1f is selectively formed and reacted with 2-chloro-N-methoxy-N-methylacetamide to generate the chloroketone 1g which is alkylated with the potassium salt of spirocyclic acid 1h. Heating of the resulting keto-ester with ammonium acetate in toluene affords cyclization to difluorofluorene-imidazole 1i. Coupling of pinacol boronate 1d with bromo-fluorene 1i completes the core of ledipasvir 1j. Double Boc deprotection of 1j and HATU-mediated coupling with Moc-valine (1k) affords ledipasvir, 1 [14].

ledipasvir synthesis. (a) Pd(OH)2/C, H2, EtOH; (b) Boc2O, i-Pr2NEt, CH2Cl2; (c) LiOH, THF/MeOH/H2O; (d) 4-bromo-1,2-diaminobenzene, HATU, 4-methylmorpholine, DMF; (e) EtOH; (f) bis(pinacolato)diboron, PdCl2(dppf)2, KOAc, 1,4-dioxane; (g) N-fluorobenzenesulfonimide, KHMDS, THF; (h) i-PrMgCl, 2-chloro-N-methoxy-N-methylacetamide, THF; (i) 1h, K2CO3, KI, acetone; (j) NH4OAc, PhMe; (k) 1d, Pd(OAc)2, PPh3, NaHCO3, DME/H2O; (l) HCl/dioxane/DCM; (m) 1k, HATU, i-Pr2NEt, DMF
Further data supporting the decision to undertake the clinical development of ledipasvir follows. LDV showed no measurable instability at the lower limit of detection in our in vitro liver microsome assays in preclinical species and human (human predicted CL <0.16 L/h/kg). Therefore 3H-LDV was incubated in hepatocytes to measure low-level metabolites for calculation of the predicted CL. This approach afforded an exceptionally low predicted human metabolic clearance of 0.012 L/h/kg (Table 6). In bile duct-cannulated dogs, LDV was slowly excreted, and over 24 h 65% of the dose was recovered as parent drug in bile consistent with the low hepatic oxidative metabolism measured across species in microsomes and human hepatocytes. (Less than 1% of LDV was recovered in urine.)
Despite its high protein binding, LDV displays moderate volumes of distribution (V ss). It is interesting to note that although high serum protein binding often leads to a low volume of distribution [24], the V ss of LDV in preclinical species (1.2–2.7 L/h/kg) is significantly higher than the plasma volume. The moderate V ss of LDV in concert with its high metabolic stability contributes to its long PK half-life. As a consequence of its low oxidative metabolism in human hepatocytes, moderate V ss in rat, dog, and monkey, and slow biliary excretion in dog, LDV was predicted to have a long human half-life. Based on its PK and potency, LDV was predicted to have a sufficiently low once-daily dose that would be compatible with dosing in a single-tablet regimen. As a result, LDV was progressed into clinical development. In our discovery of LDV, we targeted a long human half-life to enable once-daily dosing in an STR and to ensure that drug trough concentrations would remain sufficiently high to suppress viral breakthrough and the emergence of resistance potentially even in the event of patient non-compliance.
As described herein, multiple unique motifs make up the complex structure of ledipasvir and contribute to its picomolar cellular and PA potency, high metabolic stability, and excellent pharmacokinetic properties. The structure of LDV has captured the interest of a number of authors and has been cited in a range of publications focusing on some of the intriguing structural elements now becoming utilized in medicinal chemistry, including benzimidazoles [25]; spirocyclic ring systems [26]; cyclopropanes [27], bridged heterocyclic ring systems [28]; incorporation of stereocenters (LDV has six) [29]; and the use of fluorine in drug discovery (whereas most fluorinated drugs include aryl or heteroaryl fluorides, few bear aliphatic fluorine substitution as in LDV) [30]. With these and other elements taken together, the unique structure of ledipasvir is highly complex within known drug space. In a recent publication detailing a computational algorithm that defines structural complexity in drugs, ledipasvir is the most structurally complex orally bioavailable drug among the examples discussed [31]. The chemical complexity of ledipasvir based on ring structure and calculated properties defines it as a “rulebreaker” drug by multiple metrics. LDV has been discussed in drugs “beyond the rule of 5” (bRo5) [32, 33], bRo5 “chameleonic” drugs [34] that display differential lipophilicity based on their conformational state, and has been provided as an example of the importance of organic synthesis in drug discovery [35].
Our discovery and implementation of the structural motifs incorporated in LDV followed a data-driven path that sought improvement of measured properties such as antiviral activity and in vitro and in vivo pharmacokinetic attributes and disregarded rule-based metrics. Indeed ledipasvir is a “rulebreaker” compound that defies much of the dogma dominating contemporary medicinal chemistry design that attempts to define “drug likeness” and potential for bioavailability. Rule limit values for molecular weight, calculated logP, the number of H-bond donors and acceptors [36], the number of rotatable bonds and polar surface area [37], the number of ring structures [38], and the number of aromatic rings [39] are provided in Table 7, along with the corresponding values for LDV. These rule-based metrics are provided here for reference and were not utilized in any way during the discovery of LDV. LDV lies outside most of these metric-based rules. Ledipasvir has proven to be a bioavailable, efficacious, safe, and well-tolerated drug.
4 Translation of Ledipasvir’s Preclinical Potency and PK Properties in Phase 1 Clinical Trials
The preclinical pharmacokinetic optimization efforts in the discovery of LDV proved fruitful. In human healthy volunteers the exposure of a single dose of LDV increases dose proportionally from 3 to 100 mg, and gratifyingly the half-lives are typically over 40 h (Fig. 11 and Table 8) [14]. The LDV 24 h trough drug concentration is well over the PA EC50 at all doses (depicted with the red dotted line, Fig. 11), ranging from 12-fold at the 3 mg dose to 470-fold at the 100 mg dose. For the 10–100 mg doses, the trough concentrations remain well over the PA EC50 even at 96 h post-dose.

LDV single oral dose pharmacokinetics in healthy volunteers
The PK curves and PK parameters post the third-daily dose of LDV administered to GT1 HCV-infected individuals are depicted in Fig. 12 and Table 9, respectively [40]. Here the exposures are approximately dose proportional over five dose levels from 1 to 90 mg, and the half-life for the 90 mg dose of LDV (the dose utilized in the LDV/SOF STR Harvoni®) is 49.7 h. Although not yet at steady state 24 h post the third dose, the drug concentration is 610-fold over the GT1a PA EC50 and 4,730-fold over the GT1b PA EC50. The 24 h drug concentration is above the GT1a PA EC50 even for the lowest total dose of 1 mg.

LDV PK in patients post the third and final dose. Depicted as mean with standard deviation. Day 3 dose administered at time zero
Accordingly, all monotherapy doses of LDV from 1 to 90 mgs displayed potent and rapid viral load reductions (VLR) in GT1-infected individuals (Fig. 13) [40]. Doses 3 mg and higher rapidly achieved VLR ~3 log10 within 24 h post the first dose and exceeded 3 log10 mean maximal viral load reductions during the dosing interval. Even the 1 mg total dose afforded a mean maximal VLR of 2.3 log10. The 30 and 90 mg doses maintained >2 log10 viral suppression at 144 h (4 days post the third and final dose), while the 10 mg dose in GT1b-infected individuals afforded a mean maximal VLR of 3.3 log10 and maintained ~2.5 log10 viral suppression 4 days post the final dose.

Viral load reduction, 3-day LDV dosing in monotherapy in GT1a (1–90 mg) or GT1b patients (10 mg). All cohorts included ten patients except the GT1a 10 mg dose (9) and the placebo (11)
The NS5B polymerase is highly error-prone. Based on the polymerase error rate, the HCV replication rate in vivo (1012 viral particles per day per patient), and the size of the HCV genome, it is estimated that every single, double, and some triple viral mutants are produced every day in a single patient [41]. In the NS5A gene sequence, variants present at gene positions 28, 30, 31, and 93 have shown reduced susceptibility to inhibitors [42]. Substitutions in the NS5A sequence are termed resistance-associated substitutions (RAS, plural RASs) and noted with the amino acid in the wild-type (WT) sequence first, the position next, and substituted amino acid last; e.g., Y93H denotes the RAS where a histidine is substituted for the WT tyrosine at position 93. Figure 14 and Table 10 depict the potency of LDV for a range of GT1a and GT1b RASs [43]. These RASs are present at low levels in most patients prior to treatment, but in some cases one or more RASs may be present at higher levels or may even represent dominant virus. The mean maximal viral load reduction for a given patient in monotherapy is defined by the titer of viruses with these RASs along with the inhibitor activity against these RASs.

Ledipasvir potency against clinically observed GT1 RAS (transient transfection in wild-type replicon)
It is interesting to note that there does not appear to be a dose-response for LDV in monotherapy for doses from 3 to 90 mg in the Phase 1b monotherapy study shown in Fig. 13 (VLR ranges from 3.1 to 3.3 log10 over this dose range) [40]. Assessment of baseline RASs helps to understand the apparent absence of dose-response. By chance, RASs were not present (at detectible levels) in any patient at baseline in the 1 and 3 mg GT1a cohorts (Fig. 15, baseline RAS assessments were determined post cohort randomization) [42]. Thus the patients in the 1 and 3 mg dosing cohorts had a disproportionately strong mean responses relative to the 30 and 90 mg cohorts which had relatively weaker responses in two patients showing high levels of the RAS Q30E/Q (30 mg cohort) or L31M (90 mg cohort). The weaker response of LDV against these RASs in vivo is consistent with the reduced susceptibility of the L31M and Q30E replicons to LDV in vitro (Table 10, Fig. 14). In our work to discover a pan-genotypic NS5A inhibitor, we improved activity across genotypes along with improved potency against NS5A RASs. The culmination of those efforts resulted in the discovery of velpatasvir [15,16,17], a potent pan-genotypic NS5A inhibitor with a high barrier to resistance that is combined with sofosbuvir in the first pan-genotypic STR Epclusa® and with sofosbuvir and voxilaprevir in the pan-genotypic STR Vosevi®. For both Harvoni® and Epclusa®, the presence of RASs detected in patients at baseline does not diminish the SVR relative to the SVR in patients with no detectible RASs [16, 44,45,46,47,48].

LDV clinical VLR by individual patient. Clinical RASs found at baseline are noted. RASs are measured by two methods, either by population sequencing (detectable as >25% of total viral population which are labeled and outlined in blue) or by deep sequencing (limit of detection ~1% of total population are labeled and outlined in black). The percentage of the resistant population is noted for RAS detected by deep sequencing. The horizontal lines represent the mean viral load reduction
5 LDV/SOF Approval and Real-World Data
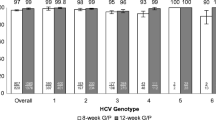
In October of 2014 LDV (90 mg) combined with sofosbuvir (400 mg) was approved under the name Harvoni®, for the treatment of GT1 HCV-infected non-cirrhotic and compensated cirrhotic patients based on the ION 1–4 Phase 3 clinical trials [44,45,46,47]. Administration of a single pill, once-daily for 8 or 12 weeks affords cure rates from 94 to 97%. In subsequent studies, Harvoni® was shown to afford high SVR rates in genotype 4, 5, and 6 patients (LDV displays potent GT4a–6a replicon potency, Table 1), and the prescribing label was accordingly expanded for treatment of these patients (https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/205834s001lbl.pdf. Accessed 10 June 2018) [5].
A measure of the practicality of a treatment regimen can be assessed by studies outside of the controlled environment of clinical trials in “real-world” “effectiveness” studies. There is no more extreme disparity between efficacy (risk-benefit in a clinical setting) in Phase 3 trials and real-world effectiveness (risk-benefit in real-world healthcare practice) than has been observed for SVR rates for IFN-based regimens. The poor tolerability, high complexity, and low efficacy of IFN-based therapy all conspire to afford real-world SVR rates that are dramatically lower than the ~60% [49, 50] achieved in later Phase 3 clinical studies. Strikingly, the real-world SVR in the US Veterans Administration (VA) is as low as 3.5%, with only 35.9% of the 99,156 HCV-infected veterans having no contraindications to this poorly tolerated regimen [51]. The attrition leading to this low SVR rate is outlined in Fig. 16 and includes patients unable or unwilling to undergo treatment, patients unable to complete treatment, and a high failure rate for those completing treatment. In contrast, a recent study of Harvoni® in this US VA HCV-infected patient population showed that 90% of patients had no contraindications, and of those patients initiating therapy the real-world SVR was 92–94% (4,365 patients, 8- and 12-week regimens, respectively). The high SVR of SOF/LDV in this population is even more notable since the authors of the study posited that advanced age, higher body mass index, ethnicity, and the prevalence of advanced liver disease (fibrosis and cirrhosis, including decompensated cirrhosis) are all factors defining this VA population as more difficult to treat than a typical cohort of HCV-infected individuals [51]. Accordingly, elimination of HCV within the 200,000 US VA HCV-infected individuals is projected for the end of the year 2018 [52].

Real-world SVR rates in the US Veterans Administration: IFN-based effectiveness contrasted with LDV/SOF. Tolerability and simplicity of LDV/SOF therapy present a striking real-world effectiveness advantage over IFN-based therapies. aVA calculated IFN-based SVR by including attrition from contraindications and those who did not receive treatment
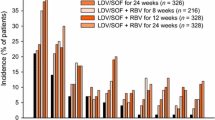
Additionally, two large real-world cohorts showed comparable results to those of the LDV/SOF Phase 3 ION-1 and ION-3 clinical trials as depicted in Fig. 17 ranging from 94 to 97% SVR. The Hepatitis C Therapeutic Registry and Research Network (HCV-TARGET) is a study comprised of North American and European academic and community medical centers. Trio Health Innervation Platform (TRIO) is a disease management cloud-based platform with data collection from specialty pharmacies [53]. It is probable that the simplicity, safety, and potency of the SOF/LDV single-tablet regimen are important attributes leading to this high level of translation from clinical trials to the real world [54].

Comparison of overall SVR rates from Phase 3 clinical studies relative to “real-world” studies (treatment-naïve patients). Bars colored as follows: red = Gilead Phase 3 clinical studies; dark blue = Trio Real-World Cohort; green = HCV Target Real-World Cohort
6 Conclusion
Ledipasvir is a highly potent NS5A (GT1a and GT1b EC50 values are 31 and 4 pM, respectively) inhibitor with a long pharmacokinetic half-life of 49.7 h in HCV-infected individuals. These attributes were critical aspects of the discovery LDV, making it a drug favorable for combination in a single-tablet regimen. Ledipasvir is the first FDA-approved NS5A inhibitor (October 10, 2014). LDV combined with sofosbuvir as Harvoni® is the first STR for the treatment and cure of HCV infection and the first HCV therapy to provide cure rates of 94–97% in as little as 8 weeks of treatment. Subsequent to approval for GT1 HCV infection, the label of Harvoni® was expanded to include treatment of GT4–6-infected individuals. The real-world effectiveness of Harvoni® is comparable to that achieved in controlled clinical trials, making it a valuable regimen for application in resource-limited settings and an important drug for HCV eradication programs [51, 52].
Notes
- 1.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/204671s002lbl.pdf. Accessed 4 Dec 2018.
- 2.
Volume I, HCV: The Journey from Discovery to a Cure.
- 3.
https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/205834s001lbl.pdf. Accessed 10 June 2018.
References
Moon AM, Green PK, Berry K, Ionnou GN (2017) Aliment Phamacol Ther 45:1201–1212
North CS, Hong BA, Adewuyi SA, Pollio DE, Jain MK, Devereaux R, Quartey NA, Ashitey S, Lee WM, Lisker-Melman M (2012) General Hosp Psychiatry 35:122
Ly KN, Hughes EM, Jiles RB, Holmberg SD (2016) Clin Infect Dis 62:1287
Sofia M (2015) J Med Chem Rev 50:397
Sofia MJ, Link JO (2017) In: Chackalamannil S, Rotella D, Ward S (eds) Comprehensive medicinal chemistry III. Elsevier, Amsterdam, p 558
Weisman R (2018) Boston Globe, Vertex to stop selling hepatitis C drug Incivek. https://www.bostonglobe.com/business/2014/08/12/vertex-stop-selling-hepatitis-drug-incivek/El0jtOpH9I1CaIgQpSUKWO/story.html. Accessed 19 June 2018
Palmer E FiercePharma, Vertex’s Incivek unseats Celebrex as fastest drug launch ever. https://www.fiercepharma.com/sales-and-marketing/vertex-s-incivek-unseats-celebrex-as-fastest-drug-launch-ever. Accessed 10 June 2018
Herper M, Forbes (2014) The top drug launches of all time. https://www.forbes.com/sites/matthewherper/2015/07/29/the-top-drug-launches-of-all-time/#6fe13a386512. Accessed 19 June 2018
EP Vantage (2018) The biggest drug launches – hep C dominates but Tecfidera stands out. http://www.epvantage.com/Universal/View.aspx?type=Story&id=766560&isEPVantage=yes. Accessed 21 June 2018
Hebner CM, Han B, Brendza KM, Nash M, Sulfab M, Tian Y, Hung M, Fung W, Vivian RW, Trenkle J, Taylor J, Bjornson K, Bondy S, Liu X, Link J, Neyts J, Sakowicz R, Zhong W, Tang H, Schmitz U (2012) PLoS One 7(6):e39163
Sheng XC, Casarez A, Cai R, Clarke MO, Chen X, Cho A, Delaney III WE, Doerffler E, Ji M, Mertzman M, Pakdaman R, Pyun HJ, Rowe T, Wu Q, Xu J, Kim CU (2012) Bioorg Med Chem Lett 22(3):1394–1396
Sheng X, Appleby T, Butler T, Cai R, Chen X, Cho A, Clarke MO, Cottell J, Delaney IV WE, Doerffler E, Link J, Ji M, Pakdaman R, Pyun HJ, Wu Q, Xu J, Kim CU (2011) Bioorg Med Chem Lett 22(7):2629–2634
Magiorkinis G, Sypsa V, Magiorkinis E, Paraskevis D, Katsoulidou A, Belshaw R, Fraser C, Pybus OG, Hatzakis A (2013) PLoS Comput Biol 9(1):e1002876
Link JO, Taylor JG, Xu L, Mitchell M, Guo H, Liu H, Kato D, Kirschberg T, Sun J, Squires N, Parrish J, Keller T, Yang ZY, Yang C, Matles M, Wang Y, Wang K, Cheng G, Tian Y, Mogalian E, Mondou E, Cornpropst M, Perry J, Desai MC (2014) J Med Chem 57:2033
Link JO, Taylor JG, Trejo-Martin TA, Kato D, Katana AA, Krygowski ES, Yang Z-Y, Zipfel S, Cottell JJ, Bacon EM, Tran CV, Yang CY, Wang Y, Wang K, Zhao G, Cheng G, Tian Y, Gong R, Lee J, Yu M, Gorman E, Mogalian E, Perry J. Bioorg Med Chem Lett. Submitted
Link JO (2018) Med Chem Rev 53:541–564
Bacon EM, Cottell JJ, Katana AA, Kato D, Krygowski ES, Link JO, Taylor J, Tran CV, Trejo-Martin TA, Yang Z-Y, Zipfel S (2012) Patent application WO 2012/068234 A2
Taylor JG, Zipfel S, Ramey K, Vivian R, Schrier A, Karki KK, Katana A, Kato D, Kobayashi T, Martinez R, Sangi M, Siegel D, Tran CV, Yang Z-Y, Zablocki J, Yang CY, Wang Y, Wang K, Chan K, Barauskas O, Cheng G, Jin D, Schultz B, Appleby T, Villasenor A, Link JO. Bioorg Med Chem Lett. Submitted
Porter DP, Guyer B (2013) In: Desai MC, Meanwell NA (eds) Successful strategies for the discovery of antiviral drugs. The Royal Society of Chemistry, Cambridge, p 482
Blanco JL, Montaner JS, Marconi VC, Santoro MM, Campos-Loza AE, Shafer RW, Miller MD, Paredes R, Harrigan R, Nguyen ML, Perno CF, Gonzalez-Hernandez LA, Gatell JM (2014) AIDS 28:2531–2539
Guo H, Kato D, Kirschberg TA, Liu H, Link JO, Mitchell ML, Parrish JP, Squires N, Sun J, Taylor J, Bacon EM, Canales E, Cho A, Cottel JJ, Desai M, Halcomb RL, Krygowski ES, Lazerwith SE, Mackman R, Pyun HJ, Saugier JH, Trenkle J, Tse W, Vivian RW, Schroeder SD, Watkins WJ, Xu L, Yang Z-Y, Kellar T, Sheng X, Clarke M, O’Neil H, Chou C-H, Graupe M, Jin H, McFadden R, Mish M, Metobo R, Phillips BW, Venkataramani C (2010) Patent application WO 2010/132601 A1
Mo H, Yang C, Wang K, Wang Y, Huang M, Murray B, Qi X, Sun SC, Deshpande M, Rhodes G, Miller MD (2011) J Viral Hepat 18:338
Stella L, Abraham H, Feneau-Dupont J, Tinant B, Declercq JP (1990) Tetrahedron Lett 31(18):2603–2606
Bruno LB, Agrawal VK (2014) Interdiscip Sci Comput Life Sci 6:71–83
Akhtar W, Khan MF, Verma G, Shaquiquzzaman M, Rizvi MA, Mehdi SH, Akhter M, Alam MM (2017) Eur J Med Chem 126:705–753
Zheng Y, Tice CM, Singh SB (2014) Bioorg Med Chem Lett 16:3673–3682
Talele TT (2016) J Med Chem 59:8712–8756
Degorce SL, Bodnarchuk MS, Cumming IA, Scott JS (2018) J Med Chem 61:8934–8943
Singh K, Shakya P, Kumar A, Alok S, Kamal M, Singh SP (2014) Int J Pharm Sci Res 5:4644–4659
Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Aceña JL, Soloshonok VA, Izawa K, Liu H (2016) Chem Rev 116:422–518
Proudfoot JR (2017) Bioorg Med Chem Lett 27:2014–2017
Doak BC, Over B, Giordanetto F, Kihlberg J (2014) Chem Biol 21:1115–1142
DeGoey DA, Chen HJ, Cox PB, Wendt MD (2018) J Med Chem 12:2636–2651
Rossi SM, Doak BC, Backlund M, Poongavanam V, Over B, Ermondi G, Caron G, Matsson P, Kihlberg J (2018) J Med Chem 61:4189–4202
Rotella DP (2016) ACS Chem Neurosci 7:1315–1316
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Adv Drug Deliv Rev 23:3
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD (2002) J Med Chem 45:2615
Taylor RD, MacCoss M, Lawson AD (2014) J Med Chem 57:5845
Ritchie TJ, Macdonald SJ (2009) Drug Discov Today 14:1011
Lawitz EJ, Gruener D, Hill JM, Marbury T, Moorehead L, Mathias A, Cheng G, Link JO, Wong KA, Mo H, McHutchison JG, Brainard DM (2012) J Hepatol 57:24–31
Nguyen T, Guedj J (2015) CPT Pharmacometrics Syst Pharmacol 4:231
Wong KA, Worth A, Martin R, Svarovskaia E, Brainard DM, Lawitz E, Miller MD, Mo H (2013) Antimicrob Agents Chemother 57(12):6333–6340
Cheng G, Tian Y, Doehle B, Peng B, Corsa A, Lee YJ, Gong R, Yu M, Han B, Xu S, Dvory-Sobol H, Perron M, Xu Y, Mo H, Pagratis N, Link JO, Delaney W (2016) Antimicrob Agents Chemother 60(3):1847–1853
Afdhal N, Zeuzem S, Kwo P, Chojkier M, Gitlin N, Puoti M, Romero-Gomez M, Zarski JP, Agarwal K, Buggisch P, Foster GR, Bräu N, Buti M, Jacobson IM, Subramanian GM, Ding X, Mo H, Yang JC, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Mangia A, Marcellin P (2014) N Engl J Med 370(20):1889–1898
Afdhal N, Reddy KR, Nelson DR, Lawitz E, Gordon SC, Schiff E, Nahass R, Ghalib R, Gitlin N, Herring R, Lalezari J, Younes ZH, Pockros PJ, di Bisceglie AM, Arora S, Subramanian GM, Zhu Y, Dvory-Sobol H, Yang JC, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Sulkowski M, Kwo P (2014) N Engl J Med 370(16):1483–1493
Kowdley KV, Gordon SC, Reddy KR, Rossaro L, Bernstein DE, Lawitz E, Shiffman ML, Schiff E, Ghalib R, Ryan M, Rustgi V, Chojkier M, Herring R, Di Bisceglie AM, Pockros PJ, Subramanian GM, An D, Svarovskaia E, Hyland RH, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Pound D, Fried MW (2014) N Engl J Med 370:1879–1888
Naggie S, Cooper C, Saag M, Workowski K, Ruane P, Towner WJ, Marks K, Luetkemeyer A, Baden RP, Sax PE, Gane E, Santana-Bagur J, Stamm LM, Yang JC, German P, Dvory-Sobol H, Ni L, Pang PS, McHutchison JG, Stedman CA, Morales-Ramirez JO, Bräu N, Jayaweera D, Colson AE, Tebas P, Wong DK, Dieterich D, Sulkowski M (2015) N Engl J Med 373:705–713
Feld JJ, Jacobson IM, Hezode C, Asselah T, Ruane PJ, Gruener N, Abergel A, Mangia A, Lai CL, Chan HL, Mazzotta F, Moreno C, Yoshida E, Shafran SD, Towner WJ, Tran TT, McNally J, Osinusi A, Svarovskaia E, Zhu Y, Brainard DM, McHutchison JG, Agarwal K, Zeuzem S (2015) N Engl J Med 373:2599
Strader DB, Seeff LB (2012) Clin Liver Dis 1:6
Hoofnagle JH, Seeff LB (2006) N Engl J Med 355:2444
Backus LI, Belperio PS, Shahoumian TA, Loomis TP, Mole LA (2016) Hepatology 64:405–414
US Medicine. http://www.usmedicine.com/agencies/department-of-veterans-affairs/va-could-soon-achieve-near-complete-eradication-of-hepatitis-c/. Accessed 16 June 2018
Younossi ZM, Park H, Gordon SC, Ferguson JR, Ahmed A, Dieterich D, Saab S (2016) Am J Manag Care 22. (6 Spec No)
Eichler HG, Abadie E, Breckenridge A, Flamion B, Gustafsson LL, Leufkens H, Rowland M, Schneider CK, Bloechl-Daum B (2011) Nat Rev Drug Discov 10:495–506
Acknowledgment
The author would like to thank the Gilead research and development colleagues that discovered and developed ledipasvir and the clinical collaborators and patients who made this work possible.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
Conflict of Interest: John O. Link is an employee of Gilead Sciences, Inc.
Ethical Approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent: Informed consent was obtained from all individual participants included in the study.
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Link, J.O. (2019). The Discovery of Ledipasvir (GS-5885): The Potent Once-Daily Oral HCV NS5A Inhibitor in the Single-Tablet Regimen Harvoni®. In: Sofia, M. (eds) HCV: The Journey from Discovery to a Cure. Topics in Medicinal Chemistry, vol 32. Springer, Cham. https://doi.org/10.1007/7355_2019_66
Download citation
DOI: https://doi.org/10.1007/7355_2019_66
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28399-5
Online ISBN: 978-3-030-28400-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)