
Abstract
The development of well-tolerated treatments that attain nearly universal cure of hepatitis C virus (HCV) infection, less than 30 years after the long-sought discovery of the causative agent, ranks as a landmark achievement of modern medicine. In the broadest sense, the international effort to address this global public health problem can be divided into an era of nonspecifically targeted therapy centering on interferon, a relatively brief “hybrid period” combining interferon and ribavirin with direct-acting antiviral agents (DAAs), and the latest era of DAA combination regimens. One of the most notable features of this story is the quantum leap in efficacy for DAA therapy to extraordinarily high levels instead of the years-long incremental steps that might have been anticipated. Similarly gratifying is the foundation on which the concept of curability, unique to HCV thus far in human virology, has been solidified based on the combination of our understanding of the molecular biology of the virus and the rarity, dating back to the interferon era, of virologic relapse after attainment of sustained virologic response. Although, at least until recently, the number of therapeutic agents was very limited, the combination of viral and host diversity ensured the development of a rich literature reflecting hundreds of treatment studies which dominated the scientific programs of the international liver meetings for many years. Viewed panoramically through a retrospective lens, the field developed in a logical sequence by first making the most out of the limited tools which were available and later by building on the remarkable elucidation of HCV biology by the scientific community and the paradigm of combination therapy for viral infection established in the HIV field to get us where we are today.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The ability to cure hepatitis C virus (HCV) infection in nearly all recipients of currently available direct-acting antiviral (DAA) treatments, attained less than 30 years after the landmark publications heralding the discovery of this elusive virus in 1989, ranks as a major triumph of modern medicine [1,2,3]. The ingenuity of the scientific community, the resources dedicated by the pharmaceutical industry, the energetic involvement of the medical community, and the motivation of untold thousands of patients to participate in clinical trials were instrumental elements in the effort to address this enormous international public health problem. The consistent observation that the virus seldom reappears after it has been undetectable by molecular assays such as polymerase chain reaction (PCR) for a few months after completion of treatment, combined with our understanding of the life cycle of a virus that does not have a phase involving genomic archiving, has vindicated the distinctive status of HCV as a virus about which we can uniquely use the word “cure” to describe the outcome of successful therapy.
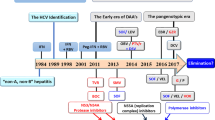
A retrospective assessment of the evolution of clinical trials for hepatitis C results in a division of the process into two major phases. The first was the interferon era, which had its onset over 30 years ago, while an intense search for the mysterious causative agent of what had become known as “non-A, non-B hepatitis” was still ongoing. Including the latter 1980s, when interferon was undergoing clinical trials, the “interferon phase” lasted over a quarter century and featured the addition of ribavirin as an adjunct to interferon as well as the development of pegylated interferon. Numerous clinical trials evaluated critical aspects of interferon-based therapy such as different interferon formulations, doses of both interferon and ribavirin, duration of therapy, response-guided therapy, and many specific populations. The new era of interferon-free DAA therapy was preceded by a “hybrid model” in which pegylated interferon and ribavirin were combined with either of the two HCV protease inhibitors, telaprevir or boceprevir, for the first time in 2011 in the United States. Hailed as a great advance at the time, it is a reflection of the accelerated pace of the field that the use of these two drugs, along with any role for interferon, vanished in many countries within 2–3 years.
The goal of this chapter is to correlate drug development with scientific advances in understanding the biology of HCV, highlight the processes that led to the selection of the various agents used to treat hepatitis C patients over the years, influenced trial design, and culminated in the current highly effective regimens, resting on the fundamental principle of combinations of DAAs with great antiviral specificity and potency. In doing so, one cannot escape the parallel with earlier developments in antiretroviral therapy from which so much was learned. The evolution of treatment with interferon prior to DAA therapy is covered here to provide a comprehensive overview, but greater focus is on the direct-acting antivirals, initially with then without interferon. Further information about interferon therapy is available in [4]. The reader should bear in mind that while of necessity this review focuses on the clinical trials of the drugs that “made it to the finish line,” many other agents, whether other formulations of interferon or ribavirin or members of the DAA classes (protease inhibitors, NS5A inhibitors, nucleotide polymerase inhibitors, non-nucleotide inhibitors, and other agents with alternative mechanisms of action, such as cyclophilin inhibitors and miR-122 inhibitors) failed because of efficacy or safety limitations, lack of partner drugs, or arrival on the scene too late to make the costs of further development worthwhile.
2 Early Days: Interferon
Approximately four decades ago, the scientific and medical communities began to focus on the potential therapeutic role of interferon in a variety of contexts because of its recognized combination of antiviral, immune modulatory, and antiproliferative properties [5,6,7]. Naturally derived interferon from sources such as fibroblasts and leukocytes excited such interest as a potential cancer treatment that readers of a feature article in The New York Times in 1981 could have been forgiven for taking away an impression that a miracle drug for cancer was on the horizon (http://www.nytimes.com/1981/04/26/magazine/putting-interferon-to-the-test.html). Early reports suggested potential benefit for hepatitis B [8,9,10,11,12,13]. The eventual role of nucleotides for viral hepatitis was also presaged by studies of adenine arabinoside for hepatitis B, alone or in combination with interferon [14]. The interest in interferon as an antiviral therapy made it logical to initiate studies in patients with a liver disease of viral etiology for which a causative agent had not yet been determined: non-A, non-B hepatitis. The major limitation of the early studies of what proved to be hepatitis C, which persisted through the initial approval of interferon in the early 1990s, was the need to rely upon serum aminotransferases as the endpoint of therapy because virologic testing was not yet available to serve as the far more appropriate endpoint of therapy which it soon became with the advent of polymerase chain reaction (PCR) technology.
The international effort to study the therapeutic applications of interferon in human medicine was greatly facilitated by the development of recombinant interferon, which allowed for the availability of large quantities of purified preparations of interferon. The Liver Diseases Section at the National Institutes of Health, led by Dr. Jay Hoofnagle, pioneered the effort to study the effectiveness of recombinant human alpha interferon in patients with non-A, non-B hepatitis. In a case series of ten patients published in 1986, interferon given at an initial dose of 1 MU or 5 MU, at first once daily and then three times weekly for up to 12 months, resulted in rapid decreases of serum aminotransferase levels, often with normalization, in most patients for as long as treatment was continued, along with histologic improvement [15].
In the 2 years following the initial NIH publication, several similar studies were published using various interferons, including beta interferon, recapitulating the theme of interferon’s ability to effect normalization of transaminase levels. Posttransfusion patients and others with classic risk factors quickly came to dominate most of the early study populations, still in the absence of available virologic markers [16, 17]. The capacity to achieve sustained biochemical response, a harbinger of the later concept of sustained virologic response (SVR), was demonstrated.
The field catapulted forward in 1989 with the publication of two landmark US studies. The first, representing an extension of the initial work at the NIH, was a double-blind, placebo-controlled trial of 41 patients who received 2 MU of recombinant human interferon alfa-2b three times weekly or placebo for 6 months. Nearly half the patients treated with interferon had normalization of aminotransferases on therapy, but only 10% had sustained biochemical response [18]. A simultaneously published multicenter US study randomized patients to 3 or 1 MU of recombinant interferon alfa-2b three times weekly for 6 months or to placebo. Response rates were higher in the 3 MU group, with 46% achieving normalization or near normalization of ALT by 6 months. Again, however, relapse was common [19].
A memorable feature of both trials establishing the efficacy of recombinant interferon alfa in non-A, non-B hepatitis is that the remarkable discovery of the hepatitis C virus was reported toward the completion phase of both trials by Michael Houghton and colleagues at the Chiron Corporation [1, 2]. Serologic testing of patient samples from both studies revealed that most patients had antibody to the newly discovered hepatitis C virus [3]. It is for this reason that the titles of both papers reporting the NIH and multicenter interferon were published in the New England Journal of Medicine indicated that they were studies on the treatment of chronic hepatitis C rather than the originally intended non-A, non-B hepatitis [18, 19]. These papers were among the first that featured the name for recombinant alpha interferon adopted in the INN (International Nonproprietary Name) classification, interferon alfa.
The studies on recombinant interferon alfa-2b led to its approval at a dose of 3 MU three times weekly for hepatitis C by the US Food and Drug Administration early in 1991 on the basis of the improvement in liver test parameters noted in the clinical trials. Now obsolete for hepatitis C, interferon in pegylated form remains a frontline recommended therapy for hepatitis B, although the better tolerated oral nucleosides or nucleotides are currently far more commonly used for this disease.
A critical development at this time was the development of testing for HCV RNA by polymerase chain reaction assays. Early studies combining assessment of biochemical and virologic response demonstrated a predictable, though not invariable, correlation between the two, including normalization of ALT with viral suppression both during and after completion of therapy, and increases in ALT levels concomitant with virologic relapse after therapy with interferon are discontinued [20,21,22,23,24,25,26]. However, it became clear that ALT normalization on treatment, as well as sustained ALT response, occurred more frequently than the responses at comparable time points for HCV RNA, thus indicating that from a virologic viewpoint the capacity to eradicate infection was lower than estimated from the early studies using ALT as the primary endpoint [27]. In addition to the obvious mandate to redefine primary outcome of treatment virologically, an important consequence of the development of virologic testing was the capacity to vastly expand the identifiable population of infected patients, with a proliferation of studies now including “community acquired” hepatitis C [28].
The recognition that HCV consists of a population of viruses with substantial genomic variation followed the advent of virologic testing, and by 1991 the phrase HCV “genotypes” was appearing in the literature [29,30,31,32,33,34]. Simmons et al. laid the foundation for what became the standard classification of six major HCV genotypes based upon phylogenetic analysis of nucleotide sequences derived from part of the gene encoding a nonstructural protein (NS5, [35]). Subsequent studies showed that similar classifications could be derived by analysis of one of the envelope proteins as well as the highly conserved 5’ untranslated region [36, 37]. Different genotypes were found to have up to 40% variability in genomic sequence with lesser degrees of heterogeneity characterizing different subtypes subsumed under individual genotypes, the most prevalent of which have been genotype 1a and 1b in the United States, Europe, Japan, and other areas [38, 39]. This classification was subsequently incorporated into the design of virtually all clinical trials of antiviral therapy for HCV and has persisted to the present era of direct-acting antiviral agents. It was not long before considerable variability in response to interferon therapy corresponding to HCV genotype was recognized, with genotype 1 being the least responsive and genotypes 2 and 3 considerably more so [40,41,42]. Genotype 4, which proved to be highly prevalent in the Middle East, especially Egypt, had an intermediate rate of response [43].
As clinical trials and observational studies on duration of therapy, variable doses, pretreatment viral load, predictors of response, rates of response in different populations, and side effects quickly proliferated [44,45,46,47,48,49,50], another alpha interferon, recombinant interferon alfa-2a, was developed. This molecule varies from alfa-2b by 1 amino acid in the 166 amino acid sequence of the protein, with efficacy and tolerability equivalent to that of interferon alfa-2b [51,52,53,54,55,56]. Interferon alfa-2a was approved for the treatment of hepatitis C in 1996, 5 years after the approval of interferon alfa-2b.
Yet another interferon alpha called consensus interferon marked the third and final commercially approved interferon to become available. Approved in 1997, consensus interferon was derived by placing the most common amino residue at each position of the alpha interferon molecule into a synthetic interferon molecule [57]. A phase 3 trial in treatment-naïve patients showed that 9 mcg three times weekly was superior to 3 mcg three times weekly for 24 weeks, leading to approval of the 9 mcg dose for treatment-naïve HCV-infected patients. Comparable rates of SVR were obtained in the same trial with interferon alfa-2b 3 MU three times weekly [58]. A second phase 3 trial in patients who had failed previous interferon therapy and received 15 mcg three times weekly yielded SVR over five times more frequently in relapsers than nonresponders treated for 24 weeks, and 48 weeks was superior to 24 weeks [59]. The longer duration of therapy became the approved dose for prior interferon failures. Consensus interferon received considerable attention and uptake in clinical practice for several years, but its use diminished, and eventually disappeared, with the advent of ribavirin in combination with interferons alfa-2a and alfa-2b and subsequently with the development of pegylated interferon alfa-2a and alfa-2b.
Concomitant with the advent of these alpha interferon molecules, the 1990s featured many advances in the understanding of virus-, host-, and treatment-related factors determining response beyond the differential rates of SVR across various HCV genotypes. The demonstrated capacity of a longer duration of therapy to attain higher rates of SVR, not by increasing rates of on-treatment response but by decreasing relapse, led to expansion of the approval of interferon alfa-2b to 18 to 24 months of treatment, although these prolonged durations of therapy were infrequently adopted in practice as opposed to 12 months [60,61,62]. It was also during this era that lower response rates were noted in African-American persons, even when corrected for the higher prevalence of genotype 1 in this population, as well as patients with hepatic cirrhosis, HIV coinfection, and other populations [63,64,65].
3 Interferon and Ribavirin Combination Therapy
The next leap forward in the evolution of HCV therapy was the introduction of ribavirin, a guanosine nucleoside analogue in search of a “therapeutic home” after it failed to fulfill its initial promise for HIV infection in the 1980s. One of the earliest clinical studies of ribavirin suggested efficacy in reducing ALT levels at a time when HCV RNA testing was still not available [66], with the observation on ALT normalization confirmed in a US study from the National Institutes of Health [67]. A subsequent multicenter study indicated that ribavirin monotherapy indeed resulted in normalization of ALT in up to half of HCV-infected patients but had very little antiviral efficacy [68].
Despite the lack of significant antiviral activity as monotherapy, ribavirin was combined with interferon alfa-2b in landmark phase 3 trials and significantly augmented the rates of SVR compared to those obtained with interferon alone. In a US phase 3 trial, 912 treatment-naïve patients received interferon alfa-2b alone or in combination with ribavirin in a weight-based dose of 1,000–1,200 mg/day for 24 or 48 weeks. SVR was assessed at follow-up period of 24 weeks and was higher in patients who received combination therapy for 24 or 48 weeks (31–38%) than in those receiving monotherapy (6–13%). Patients with genotype 1 drove the difference between 24 and 48 weeks, with lower relapse rates in the 48-week group [69]. In an international phase 3 trial, interferon alfa-2b combined with ribavirin for 48 weeks resulted in SVR in 43% as compared with 35% treated with the combination regimen for 24 weeks and only 19% treated with interferon alfa-2b for 48 weeks. Again, patients with genotypes 2 and 3 fared better, as did patients with viral levels less than 2 million copies/ml, age 40 or less, minimal fibrosis, and female gender [70]. A third phase 3 trial in interferon monotherapy relapsers yielded SVR nearly ten times more frequently in patients given combination therapy rather than monotherapy for 6 months [71]. Other studies showed that nonresponders to interferon monotherapy had lower rates of SVR after combination therapy than prior relapsers [72]. It was with the advent of interferon and ribavirin that the already recognized difference in responsiveness to interferon-based therapy between genotypes 1 versus 2 and 3 was accentuated, and a difference in recommended treatment duration (48 versus 24 weeks) emerged.
The emergence of ribavirin as a useful adjunct to interferon generated much discussion, but no final resolution, of the question of what mechanism was responsible for the augmentation of response rates when a relatively ineffective antiviral drug in its own right was added to interferon. Potential explanations included IMPDH inhibition, immunomodulatory effects, direct inhibition of viral replication as a guanosine analogue, and “error catastrophe,” based on the concept of incorporation of ribavirin into growing HCV RNA chains and the generation of an expanding population of defective virions [73,74,75,76,77,78,79,80,81].
4 Pegylated Interferon and Ribavirin
As interferon and ribavirin became established as the new standard of care, modifications of interferon in the form of pegylation were being studied. The addition of polyethylene glycol polymers of varying sizes to protein pharmaceutical agents had become established as a way to prolong the half-life of such products, minimize the peaks and valleys characterizing the pK profiles of standard interferon, decrease the dosing frequency to once weekly, and potentially improve the efficacy of therapy. Programs to pegylate interferon centered on the use of 12 kDa polyethylene residues for interferon alfa-2b and 40 kDa for interferon alfa-2a [82, 83]. In dose-ranging studies of peginterferon alfa-2b monotherapy, at three different doses, higher doses administered once weekly were more effective than a lower dose and also more effective than standard interferon 3 MU three times weekly [84].
Following phase 2 dose-ranging studies of peginterferon alfa-2b and ribavirin [85], the major phase 3 trial of peginterferon-2b in combination with ribavirin centered on a dose of 1.5 μg/kg weekly as the starting dose. In 1,530 patients assigned to 1 of 3 arms, patients received interferon alfa-2b 3 MU three times weekly plus ribavirin 1,000–1,200 mg/day for 48 weeks, PEG IFN alfa-2b 1.5 μg/kg/week plus ribavirin 800 mg/day for 48 weeks, or PEG IFN alfa-2b 1.5 μg/kg/week for the first 4 weeks and then 0.5 μg/kg/week plus ribavirin 1,000–1,200 mg/day for 48 weeks. SVR occurred in 54%, 47%, and 47% of patients, respectively. In GT1 patients, the SVR rates were 42%, 34%, and 33%, while they were in the range of 80% patients with GT2 or GT3 [86].
Studies of peginterferon alfa-2a appeared contemporaneous with those on peginterferon alfa-2b. In one study, PEG IFN alfa-2a 180 μg was compared with interferon-2a 6 MU three times weekly for 12 weeks followed by 3 MU three times weekly for 36 weeks, with SVR rates of 39% and 19%, respectively [87]. In a second study of patients with bridging fibrosis or cirrhosis, interferon-2a at a dose of 3 MU for 48 weeks was compared with 90 μg or 180 μg of PEG IFN alfa-2a SVR24 rates were 8%, 15%, and 30%, respectively [88].
With the dose of pegylated alfa-2a 180 μg weekly now established, the major pivotal trial of combination therapy plus ribavirin compared 48 weeks of peginterferon alfa-2a 180 μg once weekly plus ribavirin 1,000–1,200 mg, peginterferon alfa-2a alone, or interferon alfa-2b 3 million units three times weekly plus daily ribavirin. SVR occurred in 56%, 29%, and 44%, respectively, with rates of 46%, 21%, and 36%, respectively, in genotype 1 [89]. A second phase 3 trial with four arms compared peginterferon alfa-2a 180 μg weekly for 24 or 48 weeks plus ribavirin at a low dose (800 mg/day) versus weight-based dose 1,000–1,200 mg/day. For patients with genotype 1, SVR rates were higher with 48 weeks, while neither duration of therapy nor ribavirin dose led to statistically different SVR rates for genotypes 2 or 3 [90].
The two pegylated interferons had similar adverse effect profiles and were approved in combination with ribavirin for 48 weeks for genotype 1 and 24 weeks for genotypes 2 and 3. Nearly all trials from this era combined genotypes 2 and 3, obscuring what later emerged as higher SVR rates for genotype 2 than genotype 3, but with genotype 3 still easier to eradicate than genotype 1, a situation that was to reverse itself early in the era of DAA therapy when the first DAA drugs were designed primarily to target genotype 1.
Successive FDA approvals of peginterferon alfa-2b and alfa-2a as monotherapies and of each in combination with ribavirin occurred between 2001 and 2003. There followed a period of intense competition in the marketplace, with proponents of one side or the other referring to such features as the simplicity of fixed- (PEG IFN alfa-2a) versus weight-based dosing (PEG IFN alfa-2b) of the two peginterferons, considerations of volume of distribution putatively favoring weight-based dosing, and purported variability in rates of sustained response with fixed dosing across different body weights.
Debate persisted for years and generated several comparative studies, culminating in the massive IDEAL study, a 3,000+ patient study in genotype 1 HCV infection comparing PEG IFN alfa-2b 1.0 μg/kg/week or PEG IFN alfa-2b 1.5 μg/kg/week, each with ribavirin 800–1,400 mg/day, versus PEG IFN alfa-2a 180 μg/week plus ribavirin 1,000–1,200 mg/day [91]. The trial yielded statistically equivalent rates of SVR of 40%, 39%, and 38%, respectively, with PEG IFN alfa-2a attaining higher rates of on-treatment response but also higher rates of posttreatment relapse, resulting in the similar SVR rates. By the time this study was published, PEG IFN alfa-2a had for some time become the market leader, though both remained in widespread use and both were combined with the first two protease inhibitors, telaprevir (PEG IFN alfa-2a) and boceprevir (PEG IFN alfa-2b), along with ribavirin. However, most of the DAA inhibitors were subsequently studied in combination with peginterferon alfa-2a.
The years that followed the approval of each of the first two pegylated interferons in combination with ribavirin early in the new millennium can be characterized as an “era of refinement,” during which their efficacy and safety were evaluated in diverse patient populations, including patients with normal ALT, HIV-/HCV-coinfected persons, African–Americans, liver transplant recipients, and patients with kidney failure, among others. Viral kinetic studies improved our ability to predict therapeutic outcomes, with the recognition that failure to attain at least a 2 log drop after 12 weeks of treatment predicted ultimate failure with such a high level of confidence that treatment could be discontinued at that point. Similarly, failure to clear HCV RNA by 24 weeks was highly predictive of failure, and a strategy of stopping therapy under those conditions at that time point was adopted, as was the 12-week “stopping rule” (ref). Trials suggested potential efficacy for prolonged duration of therapy to as long as 72 weeks in patients with genotype 1 with “slow response patterns” such as persistent viremia at week 4 or, more commonly, by a >2 log reduction at week 12 with attainment of HCV RNA undetectability at week 24 [92,93,94,95,96]. Conversely, other studies suggested that viral clearance by week 4 in patients with genotype 1 was conducive to shortened duration of therapy to 24 weeks in patients with low baseline viral levels [97,98,99]. Still other studies examined the possibility of shortening duration of therapy in patients with HCV genotype 2 or 3 to 12–16 weeks, with mixed results [100,101,102,103].
In a recapitulation of what happened when ribavirin was introduced, the development of peginterferon and ribavirin spawned many studies on retreatment of patients who had failed previous regimens, including both relapsers and nonresponders to standard interferon with or without ribavirin. The results were modest, with success in only a minority of patients who had failed IFN and RBV and were retreated with PEG IFN and RBV but, in the absence of other prevailing options, led to considerable real-world use. It became clear that prior relapsers had a higher chance of SVR than prior nonresponders to IFN and RBV, but even in prior relapsers, SVR was attained in only a minority of patients who had failed a combination of standard interferon and ribavirin [104,105,106,107,108,109].
In patients failing to attain SVR on interferon-based therapy, long-term maintenance therapy with interferon monotherapy was studied, building upon the histologic improvement noted on liver biopsy, extending even to virologic nonresponders, after courses of interferon in biopsy-containing studies [110]. The most important of these studies was the HALT-C trial, an NIH-funded study conducted in the United States, which compared 3.5 years of PEG IFN alfa-2a 90 μg/week (n = 517) to the same duration of no therapy (n = 533) in nonresponders to previous nonresponders to PEG IFN and ribavirin. Although serum aminotransferases, the level of serum hepatitis C virus RNA, and histologic necroinflammatory scores all decreased significantly with PEG IFN alfa-2a, there was no difference in any of the primary clinical outcomes in death, liver decompensation, or hepatocellular carcinoma [111]. As a result of this and other trials, maintenance therapy never became a standard approach in clinical practice.
Trials were also designed to evaluate the optimal dosing of ribavirin, including what was at the time the largest HCV treatment study yet conducted, which showed that weight-based dosing in a range of 800–1,400 mg/day was superior to flat dosing in patients with genotype 1 receiving peginterferon alfa-2b and ribavirin [112]. The incremental efficacy of weight-based dosing of ribavirin was greatest in African–Americans, signaling that ribavirin’s greatest impact may have been in patients with intrinsically poor response to interferon, with a doubling of SVR with weight-based dosing in this population from 10% to 21% [113]. Even with this increment in response, however, absolute response rates remained much lower in this population. With pegylated interferon alfa-2a, the dosing range of ribavirin 1,000–1,200 mg/day was applied from the time this regimen was introduced.
As an antiproliferative agent, interferon suppressed bone marrow production of all blood cell lines, but the capacity of ribavirin to cause hemolysis resulted in anemia being the most common hematologic problem associated with interferon and ribavirin combination therapy. Studies demonstrated that erythropoietin allowed for maintenance of higher ribavirin doses by reducing the need for, or degree of, ribavirin dose reduction engendered by anemia [114]. However, there were no randomized trials showing convincingly that such adjuvant therapy led to higher SVR rates. The use of erythropoietin remained common through the introduction of telaprevir and boceprevir in combination with interferon and ribavirin because of the incremental anemia induced by these protease inhibitors. However, significant concerns arose about thrombotic events with these agents, and the need for their use abated with the advent of DAA therapy [115].
African–Americans represented perhaps the quintessential population in which interferon-based therapy did not present a “level playing field” in terms of the opportunity for response. In one of the most notable trials evaluating this issue, Muir et al. found that PEG IFN and ribavirin therapy yielded markedly disparate SVR rates of 52% for non-African–Americans and 19% for African–Americans [116]. The explanation for the disparate response rates to interferon in HCV-infected African–American persons was in large part, though not wholly, elucidated in a brief landmark paper in 2009. In a genome-wide association study (GWAS), a single nucleotide polymorphism in the region of the IL-28B locus was pinpointed as a key differentiator of response to interferon, with the CC genotype associated with markedly superior response to CT or, even more so, TT. Persons of African descent, for undetermined reasons, had a higher prevalence of the T allele, accounting in large part for the reduced efficacy of interferon-based therapy [117,118,119]. In the last phase of the interferon era, IL28B (subsequently called interferon lambda 4 (IFNL4)) testing became commonplace among clinicians who used the predictive value of the test to help determine whether patients with relatively mild disease should undergo treatment or have it withheld in favor of the hoped-for interferon-free era that had appeared on the horizon. Despite minor signals of a potential role of IL28B variants in influencing SVR rates with DAA therapy in a few studies [120], most studies showed no such signals, and few if any clinicians perform the test any longer.
HIV coinfection with HCV was consistently associated with a greater likelihood of progressive liver fibrosis and adverse liver-related outcomes [121]. As in monoinfected patients, studies in focusing on coinfected patients suggested higher response rates in HIV-/HCV-coinfected persons with PEG IFN plus ribavirin compared to standard IFN plus ribavirin [122,123,124,125,126]. Accordingly, peginterferon and ribavirin therapy was adopted as the standard approach to HCV in HIV-coinfected persons. However, only peginterferon alfa-2a and ribavirin were approved for this population by the US Food and Drug Administration.
One of the most challenging populations throughout the interferon era consisted of patients with renal failure. Patients on hemodialysis have a high prevalence of HCV infection, estimated at 9.3% in the United States [127]. For years, many kidney centers placed a high priority on curative HCV therapy before renal transplantation was offered, especially in patients with more advanced fibrosis, because of the perception that (a) interferon posed too high a risk of precipitating graft rejection after transplantation and (b) HCV-associated liver disease could progress more rapidly after transplantation [128]. PEG IFN monotherapy had reported success rates of up to 40%, with even higher rates reported when ribavirin was added, but many clinicians did not encounter such rates of success, and the severity of ribavirin-induced anemia in these patients was a major obstacle [129].
It was during the “era of refinement” with pegylated interferon and ribavirin as the centerpiece that the concept of SVR as tantamount to virologic cure firmly took hold, based upon the relative rarity, in the range of 1%, of virologic relapse after the standard SVR time point at that time of 24 weeks after discontinuation of therapy [130, 131]. This time point was subsequently modified to SVR12 with DAA therapy. In addition to the overwhelming weight of these empirical observations, collective confidence in the concept of curability of HCV infection arose from the maturation of our understanding of the HCV life cycle, which appears to involve no form of genomic archiving analogous to that which occurs with hepatitis B and HIV.
5 The Era of Direct-Acting Antiviral (DAA) Therapy
The limited efficacy of interferon-based therapy, especially in genotype 1 infection, and its poor tolerability profile further exacerbated by ribavirin led to a massive effort to develop specifically targeted antiviral agents. The deep-rooted conviction that the paradigm would eventually change was fueled by the successful development of antiretroviral therapy for HIV infection in the 1990s and by remarkable advances in HCV biology.
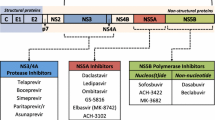
The elucidation of the organization of the HCV genome led to an understanding of the viral proteins – the NS3/4A serine protease, NS5A, and NSB HCV polymerase – that are critical for HCV replication and came to serve as the therapeutic targets against which their corresponding inhibitors have revolutionized the field. A critical juncture in the evolution of HCV therapy was the development and refinement of replicon systems which made it possible to subject putative antiviral agents to in vitro testing – an advance that was all the more historic because of the lack of animal models for HCV infection other than chimpanzees, at least until chimeric mouse models were developed much later [132]. The initial subgenomic in vitro replicon systems developed in the late 1990s [133], with subsequent refinements including adaptive mutations that increased their replicative efficiency [134,135,136,137], were of profound importance in later providing the opportunity to screen many putative antiviral agents for potency. They also became critical in the development of our understanding of the role of resistant variants in altering the sensitivity of the virus to the suppressive effects of these classes of agents.
The HCV NS3 protein contains the viral serine protease activity responsible for much of the polyprotein processing as well as an RNA helicase activity that is likely involved in genome replication. The NS4A protein serves as a cofactor for the activities of NS3 and is important in attaching NS3 to cellular membranes [138,139,140]. Critical to HCV RNA replication within the lipid-rich membranous web formed within the hepatocyte cytoplasm, the NS5A protein has also been suggested to be important for viral assembly [141, 142]. The NS5B protein serves as the viral RNA-dependent RNA polymerase. The NS5B RNA-dependent RNA polymerase can be inhibited by nucleos(t)ide or non-nucleoside inhibitors, the former by binding with the active site, which leads to chain termination of RNA synthesis, and the latter by allosteric effects.
The NS3/NS4A serine protease mediates proteolysis at the NS3/NS4A, NS4A/NS4B, NS4B/NS5A, and NS5A/NS5B junctions, suggesting a key role in HCV polyprotein processing and, therefore, viral replication [143,144,145]. The structure of the NS3/NS4A serine protease of HCV was determined by two different groups in the mid-1990s [144,145,146]. Given that the protease is critical to viral replication, and the profound importance that the development of HIV protease inhibitors played in advancing the field of HIV therapy, the identification and development of clinically useful HCV inhibitors became a goal of urgent priority.
The first HCV protease inhibitor studied in humans was BILN 2061 [147,148,149]. Studies of this agent in patients with HCV genotype 1 infection given 2 days of dosing demonstrated potent viral suppression with 2–3 log reductions of HCV RNA levels during exposure [150]. Viral rebound occurred soon after therapy was stopped. The results of these studies, representing a groundbreaking proof of concept, garnered enormous attention in an oral presentation at the 2002 meeting of the American Association for the Study of Liver Diseases [151]. Unfortunately, development of the drug was halted because of cardiotoxicity in monkeys, and it would be several years before further clinical data were reported with other protease inhibitors [152]. For the remainder of the first decade of the twenty-first century, while the “era of refinement” of peginterferon therapy moved steadily forward, the development of protease inhibitors proceeded at an accelerating pace and ultimately became the first class of DAAs approved for clinical use in patients with hepatitis C.
The development of nucleotide polymerase inhibitors was an inevitable development in light of the success of this class of agents for human immunodeficiency virus (HIV) and hepatitis B virus (HBV) infections. The active site of the HCV polymerase is relatively highly conserved [153] compared to the sequences of the other viral proteins that have been therapeutically targeted, accounting for the better pangenotypic coverage, and the higher barrier to resistance, of even the early polymerase inhibitors than was the case for the first generation of protease and NS5A inhibitors. An early agent studied clinically in this class was NM283 (valopicitabine), which conferred <2 log reduction in HCV RNA and had gastrointestinal effects, never progressing to phase 3 [154]. Subsequent agents in this class had superior potency (≥2 log early reduction in HCV RNA), including IDX-184, R1479, R1626, and mericitabine (RG-7128), but there were significant adverse effects in certain cases. For a time, mericitabine, which was well tolerated, appeared poised for advanced development when it became the first polymerase inhibitor to be combined with a protease inhibitor (danoprevir, see below) in the landmark INFORM study, demonstrating profound if transient inhibition of viral replication over a dosing period of 28 days [155, 156]. However, mericitabine was supplanted by PSI-7977, which eventually became known as sofosbuvir (SOF), a central drug in the HCV therapeutic revolution owing to its 4 log potency, excellent safety, and very high barrier to resistance attributable to the low replicative fitness of the signature resistance-associated substitution (S282T) demonstrable in vitro [157]. A comprehensive early review of the development of nucleotides, featuring a rich discussion of the medicinal chemistry as well as the early clinical studies, is available from Dr. Michael Sofia, who played a key role in the development of sofosbuvir [158], earning a 2016 Lasker Award for his work.
Before the early 2000s, only limited characterization of the NS5A protein was available. Examination of NS5A using bioinformatics tools suggested the protein consisted of three domains and contained a zinc-binding motif within the N-terminal domain. Four essential cysteine residues within domain 1 collectively bind to a single structural zinc ion, and mutation of these residues results in the complete inhibition of RNA replication [159]. NS5A proved to be a nonenzymatic protein which plays a critical role in the viral life cycle, essential not only in facilitating HCV replication in the replicase complex but appearing also to play a role in viral assembly [160,161,162]. The initial report of clinical testing in HCV patients of the first-in-class NS5A inhibitor, daclatasvir (DCV), was greeted with fascination by a large audience congregating for hours around the relevant poster at the AASLD meeting in 2009. It was shown that a single 100 mg dose resulted in viral suppression for an entire week before the appearance of virologic rebound [163]. Years later, NS5A inhibitors have come to comprise a critical component of nearly all DAA regimens currently administered to hepatitis C patients because of their potency, tolerability, and relative lack of drug–drug interactions.
The final category of DAAs that has reached clinical practice are non-nucleotide polymerase inhibitors, which bind to sites on the NS5B polymerase away from the active site and confer allosteric inhibition rather than chain termination as do nucleotide polymerase inhibitors. The former proved to be less potent than the more potent nucleotide polymerase inhibitors and have a lower barrier to resistance [164, 165]. A number of such drugs underwent trials, but only one, dasabuvir, entered the clinic in combination with paritaprevir and ombitasvir and is seldom used any longer (see below).
6 Interferon-Based DAA Regimens
Up to 60% of patients with hepatitis C virus (HCV) genotype 1 infection failed to have a sustained virologic response to therapy with peginterferon alfa plus ribavirin. The direct-acting antiviral (DAA) era of HCV therapy arrived in 2011 with the introduction of the NS3/4A protease inhibitors (PIs) telaprevir (TVR) and boceprevir (BOC) for HCV genotype 1 patients. The development program for these drugs lasted for several years and captivated a global audience as it became progressively more apparent that approval would be forthcoming based upon the incremental efficacy when either PI was added to peginterferon and ribavirin.
Early results with both PIs made it clear that in genotype 1 patients higher response rates resulted from combining either agent with peginterferon and ribavirin [166,167,168,169,170,171,172]. Both programs also moved the field forward by highlighting the role of resistance in virologic failure; delineating the resistant variants, largely common to both agent, which were the basis of this clinical problem; underscoring the variability in replicative fitness of resistant variants, a concept that later carried over into the other classes of antiviral agents; and determining the longevity of the resistant variants often found in patients who had suffered virologic failure [173,174,175,176,177,178]. An early understanding emerged of the variability in time to spontaneous clearance of resistant variants after conclusion of an unsuccessful course of treatment. It became apparent, for example, that with either TVR or BOC the resistant variants emerging after a failed course of therapy cleared more quickly in patients with genotype 1b than genotype 1a.
The phase 3 development programs for the two initial PIs were similar in important respects, but there were also significant differences. Both sets of phase 3 trial programs evaluated treatment-naïve patients and interferon-experienced patients in separate studies. Patients with cirrhosis were admixed with noncirrhotic patients, and subanalyses were performed that showed SVR rates to be significantly lower in cirrhotics, just as had been the case with peginterferon and ribavirin alone, but clearly superior to the results obtained with peginterferon and ribavirin alone. Both programs evaluated on-treatment viral kinetics carefully to establish “stopping rules” for futility, and both programs incorporated truncation of therapy to 24–28 weeks for treatment naïve patients with rapid virologic response. Throughout most of the TVR development program, all three drugs were started simultaneously. In contrast, the phase 3 BOC regimen was founded upon utilization of a 4-week “lead-in” of peginterferon and ribavirin followed by triple therapy. For both regimens, the PI was given with peginterferon and ribavirin for 12 weeks followed by completion of therapy with peginterferon and ribavirin alone. Both development programs explored the utility of response-guided therapy, in which treatment duration was governed by attainment of virologic response at predefined time points.
ADVANCE was a phase 3 double-blind placebo-controlled trial in which 1,088 patients with HCV treatment-naïve GT1 patients were randomized to one of three groups: a group receiving TVR combined with peginterferon alfa-2a and ribavirin for 12 weeks (T12PR group), followed by peginterferon–ribavirin alone for 12 weeks if HCV RNA was undetectable at weeks 4 and 12 (termed an extended rapid virologic response, or eRVR) or for 36 weeks if HCV RNA was detectable at either time point; a group receiving telaprevir with peginterferon–ribavirin for 8 weeks and placebo with peginterferon–ribavirin for 4 weeks (T8PR group), followed by 12 or 36 weeks of peginterferon–ribavirin on the basis of the same HCV RNA response criteria; or a group receiving placebo with peginterferon–ribavirin for 12 weeks, followed by 36 weeks of peginterferon–ribavirin (PR group). Significantly more patients in the T12PR or T8PR group than in the PR group had a sustained virologic response (75% and 69%, respectively, versus 44%) [179]. Although 8 weeks of TVR came close to 12 weeks, this trial established that the optimal duration of TVR in combination with PR was 12 weeks, which became the standard when the regimen was approved. The ADVANCE trial also established a strong foundation for response-guided duration of therapy with peginterferon, ribavirin, and TVR.
The ILLUMINATE trial enrolled patients with chronic HCV GT 1 infection who had not previously received treatment. All patients received telaprevir, peginterferon alfa-2a weekly, and ribavirin for 12 weeks (T12PR12), followed by peginterferon–ribavirin. Patients who had an eRVR were randomly assigned after week 20 to receive the dual therapy for 4 more weeks (T12PR24) or 28 more weeks (T12PR48). Patients without an eRVR were assigned to T12PR48. Of 540 patients, 65% had an extended rapid virologic response. The overall rate of sustained virologic response was 72%. Among the 322 patients with an eRVR, 92% in the T12PR24 group and 88% in the T12PR48 group had a sustained virologic response [180]. This trial was instrumental in establishing a 24-week duration of total therapy as sufficient in patients meeting the criteria for rapid virologic response.
In the REALIZE study, 663 treatment (interferon)-experienced GT1 patients received 12 weeks of PR plus TVR followed by 36 weeks of PR alone, or a 4-week lead-in of PR followed by 12 weeks of triple therapy and 32 weeks of PR, or 48 weeks of PR therapy alone. SVR rates were 83% in prior relapsers, 59% in prior partial responders, and 29% in “null” responders, with no significant difference in overall rates of response from the patients treated with a lead-in phase but significantly superior to PR alone. The results of this trial indicated that a lead-in PR phase did not add significant efficacy to this regimen and that the addition of a potent protease inhibitor to PR could not overcome the disadvantage inherent in intrinsic nonresponsiveness to interferon, as defined by decremental gradients of response to earlier unsuccessful therapy [181].
Based on these pivotal trials, both treatment-naïve patients and relapsers, but not nonresponders, were considered eligible for response-guided therapy in practice. HCV RNA was determined at week 4 of therapy, and if it remained >1,000 IU/mL, the entire treatment regimen was discontinued. At week 12, TVR was discontinued, and an HCV RNA assay was performed, with continuation of PEG IFN and RBV alone. However, if the HCV RNA was >1,000 IU/mL at week 12 and/or the HCV RNA declined <2 log10, then the entire regimen was to be discontinued. The stopping rules were identical for treatment-naïve and treatment-experienced patients [182, 183].
The BOC phase 3 program consisted of two trials, one in treatment-naïve and one in treatment-experienced patients. SPRINT-2 evaluated BOC in combination with PR (peginterferon alfa-2b 1.5 mcg/kg/week with weight-based ribavirin 600–1,400 mg/day) in treatment-naïve patients with HCV GT1. Group 1 received PR for 48 weeks (PR48). Group 2 received PR for 4 weeks followed by PR with BOC 800 mg three times daily × 24 weeks. If the treatment week (TW) 8 HCV RNA was undetectable (early responder or EVR) and TW24 HCV RNA was undetectable, treatment was discontinued at TW28. If the TW8 or any subsequent treatment week HCV RNA was detectable but not detectable at TW24 (late responder), PR was continued for another 20 weeks for a total treatment duration of 48 weeks (BOC-response-guided therapy or RGT). Group 3 received PR for 4 weeks followed by BOC 800 mg three times daily plus PR for 44 weeks. Subjects with detectable virus at TW24 were discontinued. The overall SVR 24 rates for the 3 groups were 40%, 67%, and 68%, respectively. Subjects with an EVR had SVR rates of 86%, 89%, and 91%, respectively versus 31%, 37%, and 43%, respectively, if the subject did not have an EVR [184]. Other than the lead-in phase, these results were thematically similar to those in the treatment-naïve telaprevir studies with regard to the capacity to stop therapy earlier in the face of a rapid response and the higher SVR rates in patients with rapid responses than in those with slower responses even when the latter group received a longer duration of total therapy.
RESPOND-2 was the pivotal BOC trial in patients with genotype 1 who had previously failed PR. It compared PR for 48 weeks versus a 4 week lead-in of PR, followed by PR plus BOC for an additional 32 weeks or an additional 12 weeks of PR if HCV RNA was detectable at week 8 of treatment, versus a 4 week lead-in of PR plus 44 weeks of PR plus BOC. The overall SVR 24 rates were 21%, 59%, and 66%, respectively. Prior relapsers to PR had SVR24 rates of 29%, 69%, and 75%, respectively, while prior nonresponders to PR had SVR 24 rates of 7%, 40%, and 52%, respectively [185]. As a result of the way the phase 3 trials of BOC had been conducted, the approval for BOC included a 4-week lead-in with PR followed by BOC-RGT to determine the duration of therapy.
Post hoc analyses using data from the phase 3 trials were undertaken to determine whether protocol-specified stopping rules (detectable HCV RNA at week 24 for SPRINT-2 and at week 12 for RESPOND-2) could be refined and harmonized. They concluded that week 12 HCV RNA levels ≥100 IU/mL almost universally predicted a failure to achieve SVR in both treatment-naïve and treatment-experienced patients. In boceprevir recipients, the combination of two stopping rules – an HCV RNA level ≥100 IU/mL at week 12 and detectable HCV RNA at week 24 – maximized the early discontinuation of futile therapy and minimized premature treatment discontinuation [186].
The introduction of TVR and BOC was hailed as a major advance in the treatment of genotype 1 HCV infection in 2011. Unfortunately, the enthusiasm for these medications was tempered by the added burden of adverse effects, including exacerbation of the anemia already engendered by peginterferon and ribavirin, and the adverse cutaneous effects of TVR, including the development of grade 3 rashes that could even include Stevens–Johnson syndrome. Neither PI was incorporated into pivotal trials in combination with other DAAs, and within 3 years the two initially approved protease inhibitors that had made medical history were obsolete.
While the trials of BOC and TVR were moving into the advanced phases of testing and then approval, another protease inhibitor, simeprevir (SIM), was also being developed and showed early promise of better tolerability and at least equivalent efficacy in genotype 1 HCV infection. In the phase 3 QUEST-1 trial, treatment-naïve HCV genotype 1 infection patients were randomly assigned in a 2:1 ratio to receive SIM or placebo plus peginterferon alfa-2a plus ribavirin for 12 weeks, followed by peginterferon alfa-2a plus ribavirin. Total treatment was 24 weeks if HCV RNA <25 IU/mL (undetectable or detectable) at week 4 and <25 IU/mL undetectable at week 12, otherwise 48 weeks, and 48 weeks in the placebo group. Treatment with SIM, peginterferon alfa-2a, and ribavirin was superior to placebo, peginterferon alfa-2a, and ribavirin, with SVR12 in 80% versus 50%, respectively [187].
In the phase 3 QUEST-2 trial, treatment-naïve patients with HCV genotype 1 infection were randomly assigned to receive SIM, peginterferon alfa-2a or alfa-2b, and ribavirin (SIM group) for 12 weeks, followed by peginterferon alfa-2a or alfa-2b plus ribavirin, versus placebo plus peginterferon alfa-2a or alfa-2b, plus ribavirin (placebo group) for 12 weeks, followed by just peginterferon alfa-2a or alfa-2b plus ribavirin. Total treatment duration was 24 weeks or 48 weeks (SIM group) based on criteria for response-guided therapy or 48 weeks (placebo). SVR was seen in 81% of the patients in the SIM group and 50% in the placebo group, clearly establishing that the addition of SIM improved SVR 12 in HCV GT1 treatment-naïve patients [188]. In a phase 2b study of treatment-experienced GT 1 patients in whom the two regimens were compared, with 12, 24, or 48 weeks of SIM versus placebo plus peginterferon and ribavirin, with all patients receiving 48 weeks of total therapy, the SIM recipients had higher SVR12 rates, and there were increasingly high rates of SVR12 in null responders, partial responders, and relapsers, respectively [189].
A distinctive feature of the SIM development program emerging from the studies on simeprevir was the finding that the Q80K polymorphism in the protease domain, present in up to 50% of US GT1a patients but a smaller percentage of European patients, impaired the chance of SVR with the triple regimen of PEG IFN, ribavirin, and SIM, but only in GT1a patients (the polymorphism is much less common in GT1b). An inkling of this had emerged in the phase 2 program but became quantitatively better established in phase 3. This polymorphism results in a modest loss of antiviral activity in in vitro assays. The clinical findings led to the first approval of a regimen for HCV infection bearing the stipulation that baseline resistance testing was required for a subgroup of patients, i.e., those with GT1a, to identify patients in whom a suboptimal response could be expected.
Had SIM been the first protease inhibitor developed for HCV infection, it would likely have dominated the landscape for treatment of GT1 patients during the interlude between PR therapy and interferon-free DAA therapy. It had efficacy that easily matched that of its two forerunners, and its tolerability was superior, with the major adverse effects including photosensitivity and a benign effect on bilirubin transporters that caused occasional hyperbilirubinemia which seldom required discontinuation of therapy. As it happened, its major contribution to patient care was in combination with SOF without interferon in the interval lasting through most of 2014, before NS5A inhibitor-containing therapy was approved (see below).
The culmination of the interferon era, albeit too late in that era to enjoy more than a brief period of use, was the combination of pegylated interferon, ribavirin, and SOF. In the phase 3 NEUTRINO clinical trial, subjects previously untreated with chronic HCV infection with genotypes 1, 4, 5, or 6 were enrolled in an open-label single-treatment group with pegylated interferon alfa-2a and weight-based ribavirin and SOF for 12 weeks. The overall SVR rate was 90%, the highest SVR rate, with the shortest duration of treatment, for any interferon-based regimen [190]. In the simultaneously published FISSION study, 24 weeks of peginterferon alfa-2a and ribavirin 800 mg was compared to 12 weeks of SOF and ribavirin in treatment-naive patients with genotypes 2 and 3, with SVR rates of 67% in each group [190].
As these important refinements of PEG IFN-based therapy for HCV were being made, the development of DAA therapy had been moving forward rapidly. Ironically, the most attractive interferon-based regimen in the history of the field in terms of efficacy, tolerability, and shortened duration of therapy, the combination of peginterferon, ribavirin, and SOF, quickly lost its relevance as the development of interferon-free DAA-based therapy bore fruit less than 5 years after the earliest glimmerings of what such therapy could achieve.
7 A Historic Proof of Concept: Curability of HCV Without Interferon
In 2010, the first demonstration of potent viral suppression with a non-interferon containing DAA combination regimen was published from the INFORM-1 trial [155]. Treatment of 73 patients for 13 days with a combination of 2 oral DAAs, the nucleoside polymerase inhibitor (RG7128, meracitabine) and an NS3/4A PI (danoprevir), without peginterferon or ribavirin profoundly suppressed HCV RNA levels in patients with genotype 1 infection. The median change in HCV RNA concentration from baseline to day 14 ranged from −3.7 to −5.2 log(10) IU/mL in the cohorts that received 13 days of combination treatment. At the highest combination doses, the median change in HCV RNA concentration from baseline to day 14 was −5.1 log(10) IU/mL in treatment-naïve patients and −4.9 log(10) IU/mL in previous standard of care null responders to interferon-based therapy versus an increase of 0.1 log(10) IU/mL in the placebo group. Minority PI-resistant variants present at baseline were suppressed by mericitabine [156]. However, the later INFORM-SVR study of this regimen with or without ribavirin for 24 weeks yielded low rates of SVR with 24 weeks of therapy [191].
Subsequently, Gane and colleagues evaluated SOF-based interferon-free regimens for untreated patient with HCV genotype 2 and 3 in the ELECTRON study, which spawned a number of arms before its ultimate completion. At the first presentation by Dr. Gane of the findings, many who were present recall to this day the several overflow rooms required to accommodate an audience correctly sensing it was witnessing a milestone in the history of medicine [192]. Forty patients were randomly assigned to four groups; all four groups received SOF plus ribavirin for 12 weeks. Three of these groups also received peginterferon alfa-2a for 4, 8, or 12 weeks. Two additional groups of previously untreated patients with HCV genotype 2 or 3 infection received SOF monotherapy for 12 weeks or SOF plus peginterferon alfa-2a and ribavirin for 8 weeks. Two groups of patients with HCV genotype 1 infection received SOF and ribavirin for 12 weeks: 10 patients with no response to prior treatment and 25 with no previous treatment. Of the 40 patients who underwent randomization, 100% who received SOF plus ribavirin without interferon and 100% who received SOF plus ribavirin for 12 weeks and interferon for 4, 8, or 12 weeks had a sustained virologic response at 24 weeks. For the other patients with HCV genotype 2 or 3 infection, 100% of the patients who received SOF plus peginterferon alfa-2a and ribavirin for 8 weeks had a sustained virologic response at 24 weeks, as did 60% who received SOF monotherapy. Among patients with HCV genotype 1 infection, 84% previously untreated patients had a sustained virologic response at 24 weeks. However, only 10% of HCV GT1 prior null responders to interferon and ribavirin had SVR [193], one of several early studies with DAA therapy that showed a deleterious impact of prior interferon nonresponse on response to non-interferon-based DAA therapy, a gap that was ultimately overcome with combination regimens.
Another landmark proof of concept study, performed by Lok and colleagues, was an open-label, phase 2a study in patients with HCV genotype 1a or 1b without cirrhosis who had not had a response to therapy with peginterferon and ribavirin. Patients were assigned in a 1:1 ratio to receive DCV (NS5A inhibitor) and asunaprevir (NS3 PI) for 24 weeks (11 patients) or DCV, asunaprevir, peginterferon alfa-2a, and ribavirin (10 patients). Coadministration of DCV and asunaprevir alone to 11 patients led to a rapid reduction in HCV RNA. Of these 11 patients, five had undetectable HCV RNA at the end of the treatment period and four had sustained virologic response at weeks 12 and 24 after treatment. SVR24 occurred in 9 of the 10 quadruple regimen patients, but it was the four patients with SVR to interferon-free DAA therapy who provided proof-of-concept for interferon-free cure [194]. These data a glimmering of what became a major theme in subsequent years, namely, the difference in resistance barrier of first-generation protease and especially NS5A inhibitors to genotype subtypes 1a versus 1b. Patients with the latter subtype were less prone to develop resistance during exposure to the first-generation members of these two classes because of differences in the number of nucleotide substitutions at critical loci needed to generate resistance – fewer for 1a than for 1b [195].
8 Further Early Studies of DAA Combination Therapy
With the proof of concept for curability of HCV infection without interferon now established, intense activity in the field fueled a number of development programs that not only affirmed the concept of curability but soon resulted in the stunning realization that extraordinarily high rates of cure could be attained with first-generation antiviral regimens. Many had anticipated that progress in the field would be incremental and that it would take years for cure rates in most patients could occur, but within a 2-year period, it became clear that history would record a quantum leap forward.
An early trial of combination therapy with daclatasvir, the first-in-class NS5A inhibitor to be tested in patients, and SOF was one of the first studies to establish that very high rates of SVR could be attained in most patients. In a trial by Sulkowski et al., 44 previously untreated patients with HCV genotype 1 infection and 44 patients with HCV genotype 2 or 3 were randomly assigned to DCV plus SOF daily, with or without ribavirin, for 24 weeks. The study was expanded to include 123 additional patients with genotype 1 infection who were randomly assigned to daclatasvir plus sofosbuvir, with or without ribavirin, for 12 weeks (82 previously untreated patients) or 24 weeks (41 patients who had previous virologic failure with TVR or BOC plus peginterferon alfa and ribavirin). Among patients with genotype 1 infection, 98% of 126 previously untreated patients had SVR, as did 98% of 41 patients who had not attained SVR to HCV protease inhibitors in combination with peginterferon and ribavirin. A total of 92% of 26 patients with genotype 2 infection and 89% of 18 patients with genotype 3 infection had a sustained virologic response at week 12 [196].
The theme of an incipient quantum leap in HCV curability was not limited to nucleotide-containing regimens. The AVIATOR trial evaluated a combination of paritaprevir (protease inhibitor) with low-dose ritonavir boosting, ombitasvir (NS5A inhibitor), and dasabuvir (a non-nucleotide polymerase inhibitor) in several hundred noncirrhotic treatment-naïve patients who received a variety of two or three drug combinations with or without ribavirin. Of the nine arms, SVR rates varied between 85% and 99%. Two 8-week regimens fell just short of 90% SVR, and the highest rates of SVR were attained in treatment-naïve patients who received 12 weeks of the three-drug regimen plus ribavirin (99%) and 24 weeks of the same regimen in prior interferon null responders (98%) [197].
The FISSION (treatment-naïve) and POSITRON (treatment-experienced) trials were instrumental in providing a portent, contrary to the expectations arising from the ELECTRON study, that genotype 3 would emerge as the “problem child” in the early era of DAA therapy, with lower SVR rates for GT3 patients, particularly those with cirrhosis, when treated with SOF and RBV for 12 weeks. In cirrhotic patients, SVR12 was achieved at a rate of 34% for treatment-naïve and 21% for treatment-experienced patients. By extending this regimen to 16 weeks, SVR12 rates could be increased to 61% in treatment-experienced patients with HCV GT3 infection [198]. The subsequent VALENCE trial confirmed that high SVR12 rates could be achieved in HCV GT2 patients with cirrhosis after 12 weeks of therapy (100% for treatment-naïve, 88% for treatment-experienced). Extending treatment to 24 weeks for HCV GT3 patients allowed for an improvement to 92% in treatment-naïve patients, but those who were treatment-experienced remained at 62% [199]. As a result of the cumulative studies up to that time, the initial approval of SOF and ribavirin for GT3 entailed a recommended treatment duration of 24 weeks rather than the 12-week approval garnered for GT2.
9 The Era of Approved Interferon-Free Therapy Begins
The first approval of oral, interferon-free treatment occurred in late 2013 for sofosbuvir and ribavirin in HCV genotypes 2 and 3 infection. Contemporaneously, sofosbuvir and ribavirin combined with peginterferon was approved for all genotypes based on data showing SVR rates of about 90%, with a treatment duration of for 12 weeks for all patients. Also around the same time, the protease inhibitor simeprevir (SIM) was approved with peginterferon and ribavirin in combination for genotype 1. These three seemingly disparate developments proved fateful because, as the year 2014 dawned, it was apparent to HCV treaters in countries where SIM and SOF had each been approved with interferon that it would be more effective to combine these agents with each other and leave interferon and even ribavirin aside.
By the time peginterferon, ribavirin, and simeprevir were approved in combination, the phase 2 COSMOS trial had shown the combination of SIM and SOF to confer very high rates of SVR with excellent tolerability. This was a phase 2, four-arm trial evaluating SMV+SOF without or with ribavirin and for 12 versus 24 weeks in genotype 1 patients across the fibrosis range of F0–F4. The trial demonstrated SVR rates over 90% in all arms [200]. Based on the COSMOS data, many clinicians prescribed the regimen for their patients with excellent results that generally emulated the trial, despite initial concerns about whether payers would cover the combination regimen in the absence of FDA approval for the two drugs together. By the time the combination of SIM and SOF was approved in late 2014 in the United States, thousands of patients had benefitted from the “head start” they had been given on the opportunity to cure their HCV infections with SIM and SOF in combination.
Atypically, the publication of the phase 3 trials of the combination of SIM and SOF was released after the US FDA had already approved it based on the results of COSMOS in the context of the pressing unmet need for interferon-free therapy and higher rates of SVR. Subsequently, in the phase 3 OPTIMIST-1 trial, a randomized open-label study assessed the efficacy and safety of 12 and 8 weeks of simeprevir and sofosbuvir in HCV GT1-infected treatment-naïve and treatment-experienced patients without cirrhosis. Patients were randomly assigned to simeprevir 150 mg once daily and sofosbuvir 400 mg once daily for 12 or 8 weeks with primary endpoint of SVR12. Superiority in SVR12 was assessed for SIM and SOF at 12 and 8 weeks versus a composite historical control SVR rate. SVR12 with SIM and SOF for 12 weeks was 97% versus 83% in the 8-week arm. Patients in the 8-week arm with GT1a and the Q80K polymorphism had lower SVR rates [201]. OPTIMIST-2 evaluated the combination of SIM and SOF in GT1 treatment-naïve or treatment-experienced cirrhotic patients for 12 weeks, with SVR in 83% overall (88% and 79% in naïve and experienced patients, respectively). Patients with GT1a infection and the Q80K polymorphism had lower rates of SVR than those without Q80K [202].
Nearly contemporaneous with the approval of simeprevir and sofosbuvir in combination was the approval of ledipasvir/sofosbuvir (LDV/SOF) in late 2014 based upon a very large phase 3 development program. The phase 3 ION-1 and ION-2 studies evaluated the fixed-dose combination (FDC) of SOF and the first-generation NS5A inhibitor LDV in GT1 treatment-naïve (ION-1) and treatment-naïve and treatment-experienced (ION-2) patients without or with cirrhosis. Each trial contained four arms, featuring LDV/SOF without or with ribavirin for 12 or 24 weeks. SVR12 rates in ION-1 were 99%, 97%, 98%, and 99% with 12 weeks of LDV/SOF without ribavirin and with ribavirin and 24 weeks without and with ribavirin, respectively [203]. ION-2 included treatment-experienced patients who achieved SVR12 rates after 12 weeks of treatment of 82–86% (with or without ribavirin, respectively) and 100% in each of the 24-week arms, respectively. In both studies the inclusion of RBV appeared to make no difference to the overall SVR rates in cirrhosis, nor was there a difference in results between genotype 1a and 1b patients. Results in ION-2 were similar in patients with or without exposure to a protease inhibitor combined with PEG IFN and RBV [204].
The pivotal phase 3 ION-3 LDV/SOF study reflected the widespread interest in shortening duration of DAA therapy without significantly compromising the chance of SVR. With three arms containing LDV/SOF for 8 weeks with or without ribavirin or for 12 weeks without ribavirin, all in treatment-naïve noncirrhotic patients with GT1, SVR rates varied between 93% and 95% with no significant differences among them. Retrospective analysis indicated that relapse rates were higher in the 8-week ribavirin-free arm when patients had baseline viral load of >6,000,000 IU/mL, accounting for about 30% of GT1 patients [205]. The AASLD/IDSA guidelines subsequently recommended against the adoption of the 8-week regimen in African–Americans and HIV-/HCV-coinfected patients based on data extrapolated from other studies [206]. In one of the clearest examples of the impact of real-world post-marketing studies with DAA regimens, a high proportion of such studies vindicated the hypothesis that treatment in GT1 patients with “low” baseline viral level was equally effective for 8 as for 12 weeks [207,208,209,210].
Subsequently, the SIRIUS trial randomized 155 HCV-1 patients with compensated cirrhosis who had failed PI therapy to either LDV/SOF FDC plus RBV for 12 weeks, or 24 weeks of the FDC alone, and found similar SVR12 rates between the two regimens (96% versus 97%) [211]. Concomitantly, a pooled analysis of all phase 2b and phase 3 trials that included cirrhotic patients with HCV-1 treated with this DAA combination (n = 513), including the SIRIUS population, demonstrated that RBV may improve SVR12 rates in treatment-experienced patients receiving 12 weeks of therapy (96% versus 90%). There was no difference seen in SVR12 rates between those receiving the LDV/SOF with RBV for 12 weeks and those receiving 24 weeks of FDC without RBV (96% versus 98%) [212], an unexpected finding after the earlier and smaller ION-2 study [205].
Nearly simultaneous with approval of LDV/SOF came the approval of the first nucleotide-free regimen: paritaprevir/r (ritonavir boosting)/ombitasvir and dasabuvir. Paritaprevir, a protease inhibitor, was the first drug, and remains the only drug to date, in the HCV armamentarium to be co-administered with ritonavir for pharmacologic boosting of the PI, a concept borrowed from the HIV field. It was formulated in a single-tablet regimen with the NS5A inhibitor, ombitasvir, and a non-nucleotide inhibitor, dasabuvir, was administered as a separate tablet. This regimen has been replaced by the pangenotypic combination of glecaprevir and pibrentasvir in many countries (see below) but retains an important place in the history of the first generation of DAA regimens.
In the SAPPHIRE-1 phase 3 trial, the three-drug regimen was evaluated in previously untreated patients with HCV genotype 1 infection and no cirrhosis. Treatment with this regimen included: single-tablet coformulation of ABT-450(paritaprevir)/r–ombitasvir and dasabuvir (250 mg twice daily) with ribavirin. The overall rate of sustained virologic response in this group was 96.2%. The response rates in this group were 95.3% among patients with HCV genotype 1a infection and 98.0% among those with HCV genotype 1b infection [213].
In the SAPPHIRE-2 trial, patients with HCV genotype 1 infection and no cirrhosis, who had been previously treated with peginterferon–ribavirin, were randomly assigned to receive co-formulated paritaprevir/ritonavir/ombitasvir and dasabuvir with ribavirin or to matching placebos during the 12-week double-blind period. In the active treatment group, an overall rate of 96.3% virologic response at posttreatment week 12 was seen. This rate was superior to the historical control rate. Rates were 95.3% among patients with a prior relapse, 100% among patients with a prior partial response, and 95.2% among patients with a prior null response [214].
The role of ribavirin with this triple regimen was investigated extensively in two phase 3 trials known as PEARL-III and PEARL-IV. Patients with HCV genotype 1b infection (PEARL-III) and HCV genotype 1a infection (PEARL-IV) were randomized to 12 weeks of paritaprevir/r–ombitasvir, dasabuvir, and ribavirin or to matching placebo for ribavirin. The rate of SVR among patients with HCV genotype 1b infection was 99.5% with ribavirin and 99.0% without ribavirin, and among those with genotype 1a infection was 97.0% and 90.2%, respectively. Response rates in all treatment groups were superior to the historical response rate with a peginterferon-containing TVR-based regimen [215].
The phase 3 TURQUOISE-II trial evaluated the above regimen with ribavirin in treatment-naïve or treatment-experienced patients with compensated HCV GT1 cirrhosis and compared 12 to 24 weeks of treatment. In this study, SVR12 rates were 92% and 96% following 12 and 24 weeks therapy, respectively. Results varied according to HCV GT1 subtype, higher in HCV GT1b with SVR12 of 98.5% and 100%, compared with 89% and 94% in HCV GT1a subtype following 12 and 24 weeks, respectively [216]. With these results in HCV GT1b cirrhosis, the phase 3b TURQUOISE-III study evaluated the three DAA regimens without RBV in HCV GT1b compensated cirrhosis. One hundred percent of the enrolled patients achieved SVR12 including 33 patients with prior PegIFN/RBV treatment experience. Based on these results, for all HCV GT1 cirrhosis patients, except prior HCV GT1a null responders who needed 24 weeks, 12 weeks of ritonavir-boosted paritaprevir, dasabuvir, and ombitasvir was sufficient, with RBV still needed in those with HCV GT1a [217].
In late 2014 the regimen of paritaprevir/ritonavir, ombitasvir, and dasabuvir with and without RBV were approved to treat HCV GT1 patients in the United States. Following approval of this regimen, post-marketing surveillance identified several patients with cirrhosis who developed hepatic decompensation and/or liver failure while receiving this therapy. This led to the US FDA issuing a warning that treatment with ritonavir-boosted paritaprevir, dasabuvir, and ombitasvir can cause serious liver injury in patients with advanced liver disease (www.fda.gov/drugs/drugsafety/ulm468634.htm).
The first DAA regimen approved for treatment of genotype 3 without ribavirin in the United States was daclatasvir and sofosbuvir (2015), followed in early 2016 by expanded approval for use with or without ribavirin in genotype 1 patients, including patients with cirrhosis, post-liver transplant HCV, and HIV coinfection [218, 219]. The ALLY-3 study evaluated 12 weeks of DCV plus SOF in treatment-naïve and treatment-experienced GT3 patients without or with cirrhosis. SVR occurred in 96% of the noncirrhotic patients but in only 63% of those with cirrhosis [220]. Other studies demonstrated substantial improvement in SVR rates in GT3 cirrhotic patients with 24 weeks of treatment, with no augmentation with RBV [221]. Daclatasvir was an important drug in the evolution of HCV therapy but suffered from the lack of a companion drug.
In 2016 another DAA regimens were approved by the FDA: elbasvir (EBR), an NS5A inhibitor, and grazoprevir (GZR), a NS3/4A protease inhibitor, co-formulated in a single tablet. The phase 3 C-EDGE treatment-naïve (TN) trial evaluated chronic HCV genotype 1, 4, and 6 treatment-naïve with and without cirrhosis given EBR/GZR 50/100 mg tab daily for 12 weeks. The overall SVR rate was 95%. The SVR rate for GT1a was 92% and 99% for GT1b. Lower SVR12 rates occurred in patients with baseline NS5A resistance-associated substitutions (RASs) associated with >fivefold loss of EBR susceptibility [222]. These included substitutions at the 28, 30, 31, and 93 positions of the NS5A molecule. The phase 3 open-label trial C-EDGE treatment-experienced (TE) for HCV GT1 peginterferon plus RBV failures with and without cirrhosis evaluated fixed-dose elbasvir–grazoprevir daily for 12 or 16 weeks with or without ribavirin. There were four treatment arms, EBR/GZR × 12 weeks, EBR/GZR + RBV × 12 weeks, EBR/GZR × 16 weeks, and EBR/GZR + RBV × 16 weeks. SVR rates were 92.4%, 94.2%, 92.4%, and 98.1%, respectively. Virologic failure occurred only in prior nonresponders, not relapsers. No virologic failures occurred in patients treated for 16 weeks with ribavirin [223].
An analysis of six clinical trials assessed the safety and efficacy of EBR/GZR in patients with compensated cirrhosis and compared 12 versus 16–18 weeks of treatment without or with ribavirin. Ribavirin did not add significantly to the efficacy of 12 weeks of treatment. Among treatment-experienced patients, only those treated for 16–18 weeks with ribavirin had no virologic failures. In genotype 1a patients, baseline RASs were the major driver of virologic failure [224].
The cumulative data on this regimen led to GZR/EBR for 12 weeks in treatment-naïve or treatment-experienced genotype 1a patients with and without compensated cirrhosis without NS5A RAVs and GZR/EBR + RBV for 16 weeks in GT1a patients with NS5A RASs. For genotype 1b patients with and without compensated cirrhosis, treatment-naïve or treatment-experienced, GZR/EBR for 12 weeks without RAS testing was recommended based on data across a broad spectrum of patient populations, except for decompensated cirrhotics in whom no protease inhibitor is recommended [225].
10 The Issue of NS5A Inhibitor Resistance
Resistance to NS5A inhibitors emerged as a major theme during the era of the first-generation DAA regimens. Most patients who failed to have SVR on such regimens had NS5A resistance-associated substitutions (RASs) in their viral populations at the time of virologic failure, which usually took the form of posttreatment relapse rather than on-treatment breakthrough or failure to suppress HCV RNA to undetectable levels. Most of the relevant RASs were in the 28, 30, 31, and 93 positions. Approximately 15% of patients had such variants at baseline as detected by population sequencing, which required a threshold of roughly 15–20% of the viral population within an individual patient to be detected; deep or “next-generation” sequencing had a lower threshold in the range of 1% but proved to have lower predictive value for virologic failure [226].
Most of what was learned about the impact of baseline RASs, and the need for adjustment of the regimen prior to treatment initiation, was gleaned from retrospective analyses of data from studies in which patients were not stratified by the presence or absence of baseline RASs. This proved to be most impactful for the regimen of elbasvir/grazoprevir, the phase 3 trials of which had arms with or without ribavirin for treatment durations of 12 or 16 weeks. It emerged that in genotype 1a patients the chance of SVR was significantly impacted by RASs in the four positions cited above and that this adverse impact was overcome by the addition of ribavirin and extension to 16 weeks in patients with genotype 1 (the regimen was approved only for genotypes 1 and 4). This resulted in the regimen being the only one with a stipulation in its package insert in the United States that baseline RAS testing was advised before treatment of genotype 1a patients, with adjustment of the regimen accordingly if it was to be used at all in such patients with baseline RASs. Although a signal of an impact of baseline RASs could be shown with other genotype 1 regimens, e.g., LDV/SOF in some populations [227], the impact was not such as to lead to advice to obtain RAS testing in the package insert nor in the AASLD or EASL guidelines [228]. In genotype 3 patients, however, the regimen of SOF/VEL generated recommendations for baseline RAS testing to assess for the presence of the Y93H variant in interferon-experienced or cirrhotic patients with genotype 3 and the addition of ribavirin should this variant, which confers substantial resistance to VEL, be present (see below) [228].
11 The Advent of Pangenotypic DAA Regimens
The era of pangenotypic HCV DAA therapy was ushered in with publication of the double-blind, placebo-controlled ASTRAL-1 study involving untreated and previously interferon-treated patients with chronic HCV (n = 624) with genotypes 1a, 1b, 2, 4, 5, or 6 infection, including those with compensated cirrhosis (19%) and treatment-experienced (32%), who received the nucleotide polymerase inhibitor sofosbuvir and the NS5A inhibitor velpatasvir in a once-daily, fixed-dose combination tablet or matching placebo for 12 weeks. The rate of SVR 12 among patients receiving sofosbuvir–velpatasvir (SOF/VEL) was 99% with only 2 virologic failures, both in genotype 1, and a small number of nonvirologic failures [229].
The ASTRAL-2 study was a randomized, phase 3 studies for patients HCV genotype 2 treatment-naïve and treatment-experienced, including patients with compensated cirrhosis. In one of the trials, patients with HCV GT2 were randomly assigned to sofosbuvir–velpatasvir or sofosbuvir plus weight-based ribavirin for 12 weeks. The SVR rate was 99% in the sofosbuvir–velpatasvir group versus 94% in the sofosbuvir–ribavirin group, with no virologic failures in the SOF/VEL group [230].
The same regimen for HCV genotype 3 was evaluated separately in the ASTRAL-3 study. This phase 3 study evaluated 12 weeks of SOF/VEL without RBV versus 24 weeks of sofosbuvir plus RBV, including patients with compensated cirrhosis. In patients without cirrhosis, treatment-naïve patients had SVR in 98% versus 91% of interferon-experienced patients. Among the patients with cirrhosis receiving SOF/VEL, SVR12 rates were 93% in treatment-naïve patients and 89% in those with prior treatment failure. Overall, the rate of sustained virologic response in the SOF/VEL group was 95% and 80% in the sofosbuvir–ribavirin group [230]. Cumulatively, ASTRAL-1, ASTRAL-2, and ASTRAL-3 and the subsequent approval of SOF/VEL in 2016 signaled the end of the era of combination therapy with sofosbuvir and ribavirin alone for any patients with hepatitis C.
Voxilaprevir (VOX) is a second-generation HCV protease inhibitor with coverage across genotypes and against most PI-resistant variants. A triple regimen of SOF/VEL/VOX appeared highly promising in phase 2 trials when given for 8 or 12 weeks and was subjected to a series of four trials called the POLARIS studies in phase 3. POLARIS-2 and POLARIS-3 evaluated DAA-naïve patients, including both noncirrhotic and cirrhotic patients except genotype 3 patients with cirrhosis. POLARIS-2, the larger of the two trials, was designed to assess the efficacy of 8 weeks of SOF/VEL/VOX versus 12 weeks of SOF/VEL single in DAA treatment-naïve subjects. SVR was 95% versus 98% of subjects, respectively, with genotype 1a driving the SVR rate in the 8-week regimen to below the noninferiority endpoint established in the protocol [231]. POLARIS-3, the trial in genotype 3 patients with cirrhosis, yielded identical SVR rates by intent-to-treat analysis of 96% in each group [231]. Since SOF/VEL performed well in these trials, SOF/VEL/VOX did not garner FDA approval in an 8-week regimen in DAA-naïve patients, although it did succeed in doing so in Europe.
Glecaprevir (GLE) and pibrentasvir (PIB) are a second-generation NSA 3/4A protease inhibitor and NS5A inhibitor, respectively. These are pangenotypic drugs that cover a broad range of RASs associated with the first-generation protease inhibitors and NS5A inhibitors. In a study in which the resistance profiles of the HCV NS5A inhibitors were evaluated in an independent laboratory, PIB had the broadest range of coverage within the NS5A class but was still susceptible to resistance in the setting of certain dual variants [232].
Zeuzem and colleagues conducted a randomized trial in over 600 patients (ENDURANCE-1) with genotype 1 infection randomly assigned in a 1:1 ratio to receive once-daily GLE/PIB for either 8 or 12 weeks. The rate of sustained virologic response at 12 weeks among genotype 1-infected patients was 99.1% in the 8-week group and 99.7% in the 12-week group, with only one virologic failure in the 8-week group and none in the 12-week group [233]. To establish the clinical pangenotypic efficacy expected from the in vitro properties of these drugs, the GLE/PIB combination was evaluated in three open-label studies (SURVEYOR-II, Part 4, ENDURANCE-4, ENDURANCE-5,6) and a randomized, double-blind, placebo-controlled study (ENDURANCE-2). In the ENDURANCE-2 study, adult patients with untreated or previously treated HCV genotype 2 infection without cirrhosis were randomly assigned (2:1) to groups given once-daily oral glecaprevir/pibrentasvir or placebo for 12 weeks. In the SURVEYOR-II, Part 4, and ENDURANCE-4 studies, adult patients with untreated or previously treated patients with HCV genotype 2, 4, 5, or 6 infection, without cirrhosis, were given once-daily oral GLE/PIB for 12 or 8 weeks, respectively. Among patients receiving GLE/PIB for 8 weeks, rates of SVR12 were 98% in those infected with HCV genotype 2 and 93% in those infected with HCV genotypes 4, 5, or 6. Among patients receiving GLE/PIB for 12 weeks, rates of SVR12 were 99.5% (95% CI, 98.5–100) in those infected with HCV genotype 2 and 99% (95% CI, 97.6–100) in those infected with HCV genotype 4, 5, or 6. In the 8 week treated patients, no virologic failures occurred in the patients with genotypes 4, 5, or 6 [234]. Similarly high rates of success, with rare virologic failure, were observed in ENDURANCE-5,6 with 8 weeks of treatment for HCV GT5 and 6 without cirrhosis and 12 weeks with cirrhosis [235].
For HCV genotype 3 patients, the ENDURANCE-3 study enrolled 505 treatment-naïve patients without cirrhosis and randomized 2:1 to receive 12 weeks of once-daily therapy to three arms, consisting of GLE/PIB for 12 weeks, sofosbuvir + daclatasvir (SOF + DCV) for 12 weeks, or GLE/PIB for 8 weeks. SVR 12 was achieved in 95%, 97%, and 95% in each arm, respectively, with SVR 12 from GLE/PIB for 8 weeks meeting noninferiority compared to the other two arms. However, there were arithmetically greater numbers of patients with virologic failure in the 8- and 12-week GLE/PIB arms, particularly the former [233]. The clinical significance of this is unclear, and the 8-week regimen was approved for GT3 noncirrhotic patients, along with all other genotypes in noncirrhotics, in the United States in 2017. There were a small number of patients in ENDURANCE-3 with a baseline A30 RAS, and these patients had a lower rate of SVR, but the significance of this, too, is unclear, and there has been no recommendation for baseline RAS testing with this regimen [228, 236].
The EXPEDITION-1 study evaluated 12 weeks of GLE/PIB in patients with compensated cirrhosis across genotypes 1–6; no attempt was made in the phase 3 program to compare 8 versus 12 weeks in cirrhotic patients. Uniformly high rates of SVR12 (≥98%) were seen in this study, establishing 12 weeks as the approved treatment duration in this population when the GLE/PIB regimen was approved [237]. As with other protease inhibitor-containing regimens, GLE/PIB is not recommended for use in patients with decompensated cirrhosis.
12 Special Populations
12.1 Decompensated Cirrhosis and Pre-liver Transplant (LT)
In patients with HCV infection awaiting LT, the primary aim of antiviral therapy is to prevent recurrent HCV infection of the new liver, which is associated with reduced graft and patient survival [238]. A key study that set the tone for what has followed in transplant candidates with HCV infection was conducted in patients with HCV genotypes 1–4 awaiting LT for HCC who were treated with sofosbuvir and RBV. Seventy percent of those with an undetectable HCV RNA at the time of transplantation achieved a posttransplant virologic response, defined as a negative HCV RNA 12 weeks after LT. Those with an undetectable HCV RNA on treatment for >30 days prior to LT had a low risk of viral relapse and recurrent HCV infection in the graft [239]. A contemporaneously reported retrospective database study showed improved posttransplant survival in recipients with a listing diagnosis of hepatitis C who were HCV RNA negative at the time of transplantation [240]. Another study showed that there has been improvement in posttransplant survival in the DAA era compared to the pre-DAA era attributable to DAA-associated SVR, whether attained on the wait list or after transplantation [241]. Even in the absence of liver transplantation, the attainment of SVR in patients with decompensated cirrhosis may improve liver function and, in some cases, reduce portal hypertension [242,243,244,245,246,247].
The SOLAR program evaluated LDV/SOF and RBV for 12–24 weeks in patients with HCV genotypes 1 and 4 infection (mostly genotype 1) and decompensated cirrhosis. In the US SOLAR-1 trial, SVR rates of 87% were achieved after 12 weeks of treatment and 89% after 24 weeks in patients who had not undergone transplantation, with similar response rates in patients with Child–Pugh B or C [242]. There was improvement in synthetic liver function in the majority of patients and subsequent increases in both MELD and CTP scores. The international SOLAR-2 trial investigated the same regimens in similar cohorts. In GT1 non-transplanted patients with decompensated cirrhosis, SVR was achieved in 87% and 96% of the Child–Pugh B patients and 85% and 78% of the CP B patients treated for 12 and 24 weeks, respectively [243].
Neither SOLAR-1 nor SOLAR-2 evaluated ribavirin-free therapy in patients with decompensated cirrhosis. The ASTRAL-4 phase 3 study filled this gap by evaluating SOF/VEL with and without RBV for 12 weeks or without ribavirin for 24 weeks, in previously treated and untreated patients with HCV genotypes 1–6 and decompensated cirrhosis. Overall rates of SVR12 were 83% in those receiving 12 weeks of the FDC, 94% in those receiving 12 weeks of FDC plus RBV, and 86% in those receiving 24 weeks of FDC without RBV. The difference between 12 weeks of SOF/VEL and ribavirin and 24 weeks of SOF/VEL was relatively small in HCV GT1 but much larger in HCV GT3, with SVR rates of 86% and 50%, respectively [244].
12.2 Post-liver Transplant
Although SVR was sometimes attainable with interferon-based therapy in post-liver patients, with greater frequency after the protease inhibitors were introduced, toxicity was a major problem. The introduction of interferon-free DAA therapy radically transformed the therapeutic landscape for posttransplant patients. Dramatic evidence for this came from a study early in the DAA era demonstrating sometimes striking clinical improvement with sofosbuvir and ribavirin in a group of posttransplant patients with decompensated cirrhosis [248]. In posttransplant patients without cirrhosis in SOLAR-1, SVR was attained in 96% and 98% with 12 or 24 weeks of treatment. Child–Pugh A patients had similar rates of SVR, but there were lower response rates in Child–Pugh B and C patients: 85–88% and 60–75% in those with CTP B and C cirrhosis, respectively [242]. In the cohort of GT1 transplanted patients in SOLAR-2, 93–100% achieved SVR whether noncirrhotic or cirrhotic with Child–Pugh scores of A or B, regardless of duration of treatment. All five patients with fibrosing cholestatic hepatitis had SVR [243]. The use of ritonavir-boosted paritaprevir/r, ombitasvir, and dasabuvir achieved an SVR rate of 97% in noncirrhotic patients with recurrent HCV GT1 infection [249]. Daclatasvir and sofosbuvir also showed high levels of efficacy in both decompensated hepatitis C cirrhosis and patients with post-liver transplantation HCV infection recurrence [218, 250]. The MAGELLAN-2 trial evaluated the safety and efficacy of GLE/PIB in liver or renal transplant adults with chronic hepatitis C genotype 1–6 infection. GLE/PIB in liver- or kidney-transplanted patients for 12 weeks achieved 99% SVR, and the treatment was tolerated well [251], thereby earning this newest regimen a firm place in the therapeutic armamentarium for post-liver transplant HC-infected patients.
Other published “real-world” studies have similarly shown high rates of SVR in patients post-liver transplantation [252]. As a result, many centers have adopted a policy of withholding antiviral therapy until after transplantation, lest viral eradication in an advanced decompensated cirrhotic delay transplantation by blunting the progression of the MELD score and/or precluding access to an HCV-positive organ [253]. This approach is most often adopted in patients with MELD scores of over 20 or those with CTP C [254]. For all regimens used after liver transplantation or, increasingly, after transplantation of other HCV-positive organs to facilitate access to organ transplant (see below), attention must be paid to potential drug–drug interactions, which have been extensively studied and for which specific information is available in the package inserts.
12.3 Renal Failure
Patients with HCV and chronic kidney disease have historically not had good treatment options. Ribavirin is associated with a high incidence of hemolytic anemia because of drug accumulation in these patients. Interferon-based antiviral therapy was highly problematic in patients after renal transplantation because of the risk of graft rejection with interferon. In the era of DAA therapy, the potential use of SOF in the renal failure population has been considered potentially problematic because of the up to 20-fold accumulation of the major metabolite of SOF, which undergoes renal excretion. Although such toxicity has not been recognized in several case series, the use of SOF in this population has not been recommended.
As a result of the restrictions on SOF use in this population, two major trials were performed with nucleotide-free therapy that changed the paradigm for these patients. The C-SURFER trial was a phase 3 randomized study of safety and observational study of efficacy; patients with HCV genotype 1 infection and chronic kidney disease (stage 4–5 with or without hemodialysis dependence) were randomly assigned to receive GZR and EBR or placebo once daily for 12 weeks. SVR12 in the combined immediate treatment group by per protocol analysis, leaving out a small number of nonvirologic failures, was 99% [255]. The subsequent EXPEDITION-4 study of GLE/PIB, including over 100 treated patients, demonstrated a similarly high SVR rate of 98%, with the only two failures representing nonvirologic failure [256].
The “other side of the coin” in patients with renal failure historically has been the difficulty in treating these patients after kidney transplantation because of the high risk of interferon-induced rejection of the graft. As a result, patients had to be treated pretransplant, but this led to patients being deprived of the opportunity to receive an HCV-positive kidney, waiting times for which have been significantly shorter in many geographic areas than waiting times for HCV-negative organs. This changed dramatically with a phase 2, open-label clinical trial that evaluated the safety and efficacy of the daily fixed-dose combination of LDV/SOF in 114 kidney transplant recipients who were more than 6 months posttransplant enrolled patients that had genotype 1 (91%) or 4 infection; 69% were treatment-naïve and 15% had compensated cirrhosis. Patients were randomized to 12 weeks or 24 weeks of LDV/SOF. Median eGFR prior to treatment was 50 mL/min for patients in the 12-week study arm and 60 mL/min for those in the 24-week arm. Overall SVR12 was 100% excluding nonvirologic failures. Adverse events were common (64%), and serious adverse events occurred in 11% of the patients. Four patients with an eGFR >40 mL/min at baseline experienced a decrease to 30 mL/min at the last visit recorded; one patient who had interrupted study treatment had a final value of 14.4 mL/min. All but one of the six patients with compensated cirrhosis whose eGFR decreased to <40 mL/min continued study treatment without interruption [257].
Given the simplification of HCV treatment in the last few years and the efficacy of the new regimens, a major paradigm shift has occurred in end-stage renal disease patients as a result of the prolonged kidney transplant wait times for HCV-negative organs in some parts of the United States. In 2017, Goldberg et al. reported the THINKER pilot trial evaluating the safety and efficacy of transplantation of the kidneys from HCV genotype 1-viremic donors into HCV-negative patients, followed by 12 weeks of elbasvir–grazoprevir upon the appearance of viremia soon after transplantation. All ten recipients achieved SVR 12 [258]. An additional ten GT1 patients were subsequently treated successfully by the same group. Nineteen of the 20 patients in total had detectable HCV RNA at days 2–4 postoperatively and the remaining patient on day 5. Seventeen of the patients received 12 weeks of treatment, while 3 received 16 weeks including ribavirin because of baseline NS5A RASs [259]. Another group treated ten patients with one dose of elbasvir–grazoprevir pretransplant and 12 weeks of follow-up therapy (GT1) with sofosbuvir added for patients with GT2 and 3, again with 100% SVR [260]. Based on these and other emerging studies, transplant centers around the United States are offering HCV-infected kidney organs to HCV-negative recipients in hopes to decrease the waiting times for transplantation and time on dialysis in most cases. Recently, this concept has been extended to other transplants, including liver, cardiac, and lung transplantation [261,262,263].
12.4 HIV Coinfection
Coinfection with HIV-1 and hepatitis C virus (HCV) appears to accelerate the course of HCV-associated liver disease [264]. Historically, as discussed earlier HIV-/HCV-coinfected patients did not respond as well to interferon-based therapy compared to HCV infection alone. This discordance in ability to respond faded in the interferon-free DAA era. Nearly all the development programs for the current DAA regimens included separate trials dedicated to HIV-/HCV-coinfected patients, although occasional trials included HIV-infected subjects within the larger study population [233].
The ION-4 open-label study involved patients coinfected with HIV-1 and genotype 1 or 4 HCV receiving an antiretroviral regimen of tenofovir and emtricitabine with efavirenz, rilpivirine, or raltegravir. All patients received LDV/SOF as a single fixed-dose combination for 12 weeks. Overall, 96% had a sustained virologic response at 12 weeks after the end of therapy, including rates of 96% in patients with HCV genotype 1a, 96% in those with HCV genotype 1b, and 100% in those with HCV GT4. Rates of sustained virologic response were similar regardless of previous treatment or the presence of cirrhosis [265]. However, black race and the TT allele at the IL28B locus were associated with virologic relapse, one of the few DAA studies with a signal of such an impact, and, with only 12 weeks having been studied, likely contributing to the stipulation in the AASLD Guidance that black patients and those with HIV coinfection should not receive 8 weeks of LDV/SOF [206].
The C-EDGE CO-INFECTION study assessed the efficacy, safety, and tolerability of GZR/EBR in patients with HCV and HIV coinfection. In this phase 3, open-label, single-arm study, treatment-naïve patients with chronic HCV genotype 1, 4, or 6 infection and HIV coinfection, with or without cirrhosis, were enrolled from 37 centers in nine countries across Europe, the United States, and Australia. Patients were either naïve to treatment with any antiretroviral therapy (ART) or stable on ART for at least 8 weeks. All patients received EBR/GZR in a fixed-dose combination tablet once daily for 12 weeks. SVR12 was achieved in 96% of patients. All patients with cirrhosis achieved SVR12 [266].
The ASTRAL-5 study evaluated SOF/VEL for 12 weeks in a cohort of 106 patients with HIV-HCV coinfection across genotypes 1–4. SVR12 was attained in 101/106 (95%), including 19 of 19 patients with cirrhosis. Three of the five subjects who failed to attain SVR were nonvirologic failures [267].
The EXPEDITION-2 trial, evaluated an 8-week regimen of GLE/PIB for people with both HIV and hepatitis C. About two-thirds of the patients had HCV genotype 1 (mostly with harder-to-treat subtype 1a), followed by genotypes 3 (17%) and 4 (11%); only a small number had genotypes 2 or 6. Sixteen patients (10%) had cirrhosis. Study participants had well-controlled HIV infection with a median CD4 count of nearly 600 cells/mm3. Participants without cirrhosis received GLE/PIB for 8 weeks, while those with cirrhosis were treated for 12 weeks. Ninety-eight percent of participants achieved SVR 12 and 99%, with no virologic failures, for those without cirrhosis who were treated for 8 weeks [268].
The rates of SVR after treatment have been in line with HCV-monoinfected patients, thus resulting in harmonization of treatment recommendations of regimens for HCV-monoinfected and HIV-/HCV-coinfected patients [269]. However, one consideration in treating these patients with DAAs is potential drug–drug interaction with HIV antiretrovirals. The clinical trial development programs involved investigation of the potential interactions between HCV DAAs and HIV antiretrovirals. Careful consideration to avoid such drug–drug interactions in this population has to be given when choosing regimens, and modification of the antiretroviral regimen may be required.
12.5 DAA Failures
Combination regimens of direct-acting antiviral agents (DAAs) provide rates of sustained virologic response exceeding 90%, regardless of HCV genotype, disease stage, or treatment history. Treatment options for patients who failed previous DAA-containing regimens, particularly those with nonstructural protein 5A inhibitors, had been limited, with no FDA-approved regimens for this populations until mid-2017. This changed with the advent of the two pangenotypic regimens SOF/VEL/VOX and GLE/PIB.
Two phase 3 trials evaluated patients who had been previously treated with a DAA-containing regimen. In POLARIS-1, patients with HCV genotype 1 infection who had previously received a regimen containing an NS5A inhibitor were randomly assigned in a 1:1 ratio to receive SOF/VEL/VOX (n = 150) or matching placebo (n = 150) once daily for 12 weeks. Patients who were infected with HCV of other genotypes (114 patients) were enrolled in the SOF/VEL/VOX group. In POLARIS-4, patients with HCV genotype 1, 2, or 3 infection who had previously received a DAA regimen without an NS5A inhibitor were randomly assigned in a 1:1 ratio to receive SOF/VEL/VOX (n = 163) or SOF/VEL (n = 151) for 12 weeks. An additional 19 patients with HCV genotype 4 infection were enrolled in the SOF/VEL/VOX group. In POLARIS-1, the rate of sustained virologic response was 96% with SOF/VEL/VOX, as compared with 0% with placebo. Baseline RASs did not appear to affect response. In POLARIS-4, the rate of response was 98% with SOF/VEL/VOX and 90% with SOF/VEL. The overall rate of SVR in the SOF/VEL group was driven down by patients with GT1a and GT3, where there was no clear advantage over SOF/VEL alone [270]. Accordingly, SOF/VEL/VOX became approved in the United States for patients with GT1-6 who have failed a regimen with an NS5A inhibitor and for GT1b, 2, 4, 5, and 6 if the patient failed sofosbuvir without an NS5A inhibitor.
A phase 2, open-label study (MAGELLAN-1) evaluated the efficacy and safety lower dose GLE/PIB without RBV (n = 6), higher dose GLE/PIB plus RBV (n = 22), or higher-dose GLE/PIB without RBV (n = 22). By intent-to-treat analysis, sustained virologic response at posttreatment week 12 was achieved in 100% (6/6, 95% confidence interval 61–100), 95% (21/22, 95% confidence interval 78–99), and 86% (19/22, 95% confidence interval 67–95) of patients in arms A, B, and C, respectively [271]. There were 0, 1 and 1 virologic failures, respectively.
In the MAGELLAN-1 part 2 study, GLE/PIB was given to patients with HCV genotype 1 or 4 and prior DAA treatment failure for 12 or 16 weeks. In this study patients with prior failure to PI-containing regimens (NS5A inhibitor naïve) had an SVR of 100% with both 12 and 16 weeks of GLE/PIB. In patients with prior failure to NS5A inhibitors but NS3/4A PI-naïve there was a 94% SVR 12 rate with 16 weeks of GLE/PIB and slightly lower with 12 weeks. SVR rates were lower in patients with prior exposure to both PI’s and NS5A inhibitors, leading to FDA approval of the G/P regimen only for genotype 1 patients with prior exposure to NS5A inhibitors (16 weeks) or PI inhibitors (12 weeks) alone, but not both [272].
13 Conclusion
The development of HCV therapy ranks among the great achievements of medicine in the era spanning the close of the twentieth century and the opening of the twenty-first century. The conceptual framework for the development of direct-acting antiviral therapy was provided by the advances in treatment of HIV that occurred in the last decade of the millennium, with vital contributions from the fields of virology and medicinal chemistry. The lack of genomic archiving for HCV has made it possible to cure, rather than suppress, a human viral infection for the first time. We now have treatment that is almost universally capable of effecting virologic cure across viral genotypes, and we have salvage therapy that can cure most of the few who fail an initial course of treatment. It is even possible that our salvage regimens can be used, for a longer duration or with ribavirin, to cure the approximately 0.1% of patients who fail repeated courses of therapy, including one of the currently approved salvage regimens, despite being compliant with treatment, or that elements from different regimens can be combined to accomplish the same goal.
The extraordinary success in treating HCV infection has been richly complemented by a large and growing body of literature, dating back to the interferon era and being amplified in the DAA era, demonstrating improved clinical outcomes following virologic cure. Not only does cure prevent the progression of hepatic fibrosis and decompensation [273,274,275,276,277,278] but, as in other liver diseases in which the offending agent or pathologic process has been suppressed or treated, regression of fibrosis or even cirrhosis can ensue, as can reduction in portal hypertension [274, 279, 280]. Overwhelming evidence indicates that the risk of hepatocellular carcinoma in patients with advanced fibrosis or cirrhosis is markedly reduced, although not to the point of obviating the need for ongoing screening [276, 281,282,283,284]. Patients who have been cured virologically have higher rates of overall survival [241, 275, 276], as well as improved outcomes after transplantation [285,286,287]. Extrahepatic conditions associated with HCV infection can also be ameliorated or prevented, such as de novo diabetes [288,289,290], cryoglobulinemia [291], non-Hodgkin’s lymphoma [292,293,294], and renal and cardiovascular or cerebrovascular disease [295]. Improvement in patient-reported outcomes and health-related quality of life has been well documented [296, 297].
With the advent of the recent pangenotypic regimens, a high bar has been set for further development of antiviral regimens. It remains possible that we will see the development of novel regimens that will be capable of curing patients with a shorter duration of therapy requiring only one prescription, or even, perhaps, with the parenteral administration of a drug with established or novel mechanisms of action, with or without a short oral course of agents in the existing classes. For the most part, however, the focus on hepatitis C has shifted toward the realm of social science and public health policy, with identification of infected people and affordable access to treatment dominating the landscape on an international scale.
References
Choo Q-L, Kuo G, Weiner AJ et al (1989) Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362
Kuo G, Choo H, Alter G et al (1989) An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science 244:362–364
Alter HJ, Purcell RH, Shih JW et al (1989) Detection of antibody to hepatitis C virus in prospectively followed transfusion recipients with acute and chronic non-A, non-B hepatitis. N Engl J Med 321:1494–1500
Alqahtani SA, Sulkowski MS (2019) The role of interferon for the treatment of chronic hepatitis C virus infection. Top Med Chem. https://doi.org/10.1007/7355_2018_59
Finter NB (1986) The classification and biological functions of interferons. J Hepatol 3(Suppl 2):S157–S160
Dianzani F (1993) Biological basis for the clinical use of interferon. Gut 34(2 Suppl):S74–S76. Review
Vilcek J (2006) Fifty years of interferon research: aiming at a moving target. Immunity 25:343–348
Greenberg HB, Pollard RB, Lutwick LI et al (1976) Effect of human leukocyte interferon on hepatitis B virus infection in patients with chronic active hepatitis. N Engl J Med 295:517–522
Kingham JG, Ganguly NK, Shaari ZD et al (1978) Treatment of HBsAg-positive chronic active hepatitis with human fibroblast interferon. Gut 19:91–94
Scullard GH, Alberti A, Wansbrough-Jones MH et al (1979) Effects of human leucocyte interferon on hepatitis B virus replication and immune responses in patients with chronic hepatitis B infection. J Clin Lab Immunol 1(4):277–282
Ponzetto A, Zucca M, Marucci F et al (1979) Normal lymphocyte interferon production in adult HBsAg-positive chronic active liver disease. J Med Virol 4:43–50
Merigan TC, Robinson WS, Gregory PB (1980) Interferon in chronic hepatitis infection. Lancet 1(8165):422–423
Weimar W, Heijtink RA, ten Kate FJ et al (1980) Double-blind study of leucocyte interferon administration in chronic HBsAg-positive hepatitis. Lancet 1(8164):336–338
Sacks SL, Scullard GH, Pollard RB, Gregory PB, Robinson WS, Merigan TC (1982) Antiviral treatment of chronic hepatitis B virus infection: pharmacokinetics and side effects of interferon and adenine arabinoside alone and in combination. Antimicrob Agents Chemother 21:93–100
Hoofnagle J, Mullen K, Jones B, Rustoli V, Di Bisceglie A, Peters M, Wagonner J, Park Y, Jones A (1986) Treatment of chronic non-A non-B hepatitis with recombinant human alpha interferon. N Engl J Med 315:1575–1578
Ohnishi K, Nomura F, Linda S (1989) Treatment of posttransfusion on-A,non-B acute and chronic hepatitis with human fibroblast beta-interferon: a preliminary report. Am J Gastroenterol 84(6):596–600
Hoofnagle JH, Di Bisceglie AM (1989) Treatment of chronic type C hepatitis with alpha interferon. Semin Liver Dis 9:259–263
Di Bisceglie A, Martin P, Kassianides C (1989) Recombinant interferon alfa therapy for chronic hepatitis C: a randomized, double-blind, placebo-controlled trial. N Engl J Med 321:1506–1510
Davis GL, Balart LA, Schiff ER et al (1989) Treatment of chronic hepatitis C with recombinant interferon alfa. A multicenter randomized, controlled trial. N Engl J Med 321:1501–1506
Kanai K, Iwata K, Nakao K et al (1990) Suppression of hepatitis C virus RNA by interferon-alpha. Lancet 336(8709):245
Chayama K, Saitoh S, Arase Y et al (1991) Effect of interferon administration on serum hepatitis C virus RNA in patients with chronic hepatitis C. Hepatology 13:1040–1043
Shindo M, Di Bisceglie AM, Cheung L et al (1991) Decrease in serum hepatitis C viral RNA during alpha interferon therapy for chronic hepatitis C. Ann Intern Med 115:700–794
Brillanti S, Garson J, Tuke P et al (1991) Effect of α-Interferon therapy on hepatitis C viraemia in community-acquired chronic non-A, non-B hepatitis: a quantitative polymerase chain reaction study. J Med Virol 34:136–141
Garson JA, Brillanti S, Ring C et al (1992) Hepatitis C viraemia rebound after “successful” interferon therapy in patients with chronic non-A, non-B hepatitis. J Med Virol 37:210–214
Haqiwara H, Hayashi N, Mita E et al (1992) Detection of hepatitis C virus RNA in serum of patients with chronic hepatitis C treated with interferon-alpha. Hepatology 15:37–41
Bresters D, Mauser-Bunschoten EP et al (1993) Long term treatment of chronic hepatitis C with interferon alfa-2b: disappearance of HCV RNA in a pilot study of eight hemophilia patients. Gut 34(2 Suppl):S124–S125
Alyama T, Yoshioka K, Hirofuji H, Cuypers HT et al (1994) Changes in serum hepatitis C virus RNA titer and response to interferon therapy in patients with chronic hepatitis C. Dig Dis Sci 39:2244–2249
Alberti A, Chemello L, Bonetti P et al (1993) Treatment with interferon(s) of community-acquired chronic hepatitis and cirrhosis type C. J Hepatol 17(suppl 3):S123–S126
Nakao T, Enomoto N, Takada N et al (1991) Typing hepatitis C virus genomes by restriction fragment length polymorphism. J Gen Virol 72:2105–2112
Li JS, Tong SP, Vitvitski L et al (1991) Evidence of two major genotypes of hepatitis C virus in France and close relatedness of the predominant one with the prototype virus. J Hepatol 13(Suppl 4):S33–S37
Kanai K, Kako M, Okamoto H (1992) HCV genotypes in chronic hepatitis C and response to interferon. Lancet 339(8808):1543
Takada N, Takase S, Takada A (1993) Effects of genotypes of hepatitis C virus on interferon treatment for chronic type C hepatitis. Gastroenterol J 28(2):268–275
Takada N, Matsuda Y, Takase S, Takada A, Date T (1993) New genotypes of hepatitis C virus. Gastroenterol J 28(2):323
Okamoto H, Mishiro S (1994) Genetic heterogeneity of hepatitis C virus. Intervirology 37:68–76
Simmonds P, Holmes EC, Cha TA et al (1993) Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS-5 region. J Gen Virol 74:2391–2399
Simmonds T, Smith DB, McOmish F et al (1994) Identification of genotypes of hepatitis C virus by sequence comparisons in the core, E1 and NS-5 regions. J Gen Virol 75(Pt 5):1053–1061
Lau JY, Mizokami M, Kelberg JA et al (1995) Application of six hepatitis C virus genotyping systems to sera from chronic hepatitis C patients in the United States. J Infect Dis 171:281–289
Dusheiko G, Schmilovitz-Weiss H et al (1994) Hepatitis C virus genotypes: an investigation of type specific differences in geographic origin and disease. Hepatology 19:13–18
Mahaney K, Tedeschi V, Maertens G et al (1994) Genotypic analysis of hepatitis C virus in American patients. Hepatology 44:410–414
Chemello L, Alberti A, Rose K, Simmonds P (1994) Hepatitis C serotype and response to interferon therapy. N Engl J Med 330(2):143
Kanai K, Kako M, Aikawa T et al (1995) Clearance of serum hepatitis C virus RNA after interferon therapy in relation to virus genotype. Liver 15:185–188
Pozatto G, Moretti M, Croce LS et al (1995) Interferon therapy in chronic hepatitis C virus: evidence of different outcome with respect to different viral strains. J Med Virol 45:445–450
Kamal SM, El Kamary SS, Shardell MD et al (2007) Pegylated interferon alpha-2b plus ribavirin in patients with genotype 4 chronic hepatitis C: the role of rapid and early virologic response. Hepatology 46:1732–1740
Garson JA, Brillanti S, Whitby K et al (1995) Analysis of clinical and virological factors associated with response to alpha interferon therapy in chronic hepatitis C. J Med Virol 45:348–353
Chemello L, Cavalletto L, Noventa F et al (1995) Predictors of sustained response, relapse and no response in patients with chronic hepatitis C treated with interferon-alpha. J Viral Hepat 2(2):91–96
Lindsay K, Davis G, Schiff E et al (1996) Response to higher doses of interferon alfa-2b in patients with chronic hepatitis C: a randomized multicenter trial. Hepatology 24(5):1034–1040
Davis GL, Lau JY (1997) Factors predictive of a beneficial response to therapy of hepatitis C. Hepatology 26(Suppl 1):122S–127S
Martinot-Peignoux M, Boyer N et al (1998) Predictors of sustained response to alpha interferon therapy in chronic hepatitis C. J Hepatol 29:214–223
Wada M, Kang KB, Nishigami T, Shimoyama T (1997) Importance of pretreatment viral load and monitoring of serum hepatitis C virus RNA in predicting responses to interferon alpha2a treatment of chronic hepatitis C. Hanshin Chronic Hepatitis C Study Group. J Interferon Cytokine Res 17:707–712
Izopet J, Payen JL, Alric L et al (1998) Baseline level and early suppression of serum HCV RNA for predicting sustained complete response to alpha-interferon therapy. J Med Virol 54:86–91
Diodati C, Bonetti P, Noventa F et al (1994) Treatment of chronic hepatitis C with recombinant human interferonalfa-2a: results of a randomized controlled clinical trial. Hepatology 19:1–5
Negro F, Baldi M, Mondardini A et al (1994) Continuous versus intermittent therapy for chronic hepatitis C with recombinant interferon alfa-2a. Gastroenterology 107:479–485
Chemello L, Bonetti P, Cavallett L et al (1995) Randomized trial comparing three different regimens of alpha-2a-interferon in chronic hepatitis C. Hepatology 22(4):700–606
Rumi M, del Ninno E, Parravicini MLK et al (1996) A prospective, randomized trial comparing lymphoblastoid to recombinant interferon alfa-2a as therapy for chronic hepatitis C. Hepatology 24:1366
Imai Y, Kawata S, Tamura S et al (1997) recombinant interferon-alpha-2a for treatment of chronic hepatitis C: results of a multicenter randomized controlled dose study. Liver 17:88–92
Lee W (1997) Therapy of hepatitis C: interferon alfa-2a trials. Hepatology 26(3 Suppl 1):89S–95S
Keeffe EB, Hollinger FB (1997) Therapy of hepatitis C: consensus interferon trials. Consensus Interferon Study Group. Hepatology 26(3 Suppl 1):101S–107S
Tong MJ, Reddy KR, Lee WM et al (1997) Treatment of chronic hepatitis C with consensus interferon: a multicenter, randomized, controlled trial. Consensus Interferon Study Group. Hepatology 26:747–754
Heathcote EJ, Keeffe EB, Lee SS et al (1998) Re-treatment of chronic hepatitis C with consensus interferon. Hepatology 28:599
Poynard T, Bedossa P, Chevallier M et al (1995) A comparison of three interferon alfa-2b regimens for the long-term treatment of chronic non-A, non-B hepatitis. Multicenter Study Group. N Engl J Med 332:1457–1462
Farrell GC (1996) Two years versus 6 months of interferon therapy for chronic hepatitis C. Dig Dis Sci 41(12 Suppl):93S–98S
Payen JL, Izopt J, Galindo-Migot V et al (1998) Better efficacy of a 12 month interferon alfa-2b retreatment in patients with chronic hepatitis C relapsing after a 6 month treatment: a multicenter, controlled, randomized trial. LeGroupe D’etude et DeTraitement du Virus De L’hepatite C (Get.VHC). Hepatology 28:1680–1686
Sieck JO, Ellis ME, Alfurayh O et al (1993) Histologically advanced chronic hepatitis C treated with recombinant alpha-interferon: a randomized placebo-controlled double-blind cross-over study. J Hepatol 19:418–423
Soriano V, García-Samaniego J, Bravo R et al (1996) Interferon alpha for the treatment of chronic hepatitis C in patients infected with human immunodeficiency virus. Hepatitis-HIV Spanish Study Group. Clin Infect Dis 23:585–591
Howell C, Jeffers L, Hoofnagle JH (2000) Hepatitis C in African-Americans: summary of a workshop. Gastroenterology 119:1385–1396
Reichard O, Andersson J, Schvarcz R, Weiland O (1991) Ribavirin treatment for chronic hepatitis C. Lancet 337:1058–1061
Di Bisceglie AM, Shindo M, Fong TL et al (1992) A pilot study of ribavirin therapy for chronic hepatitis C. Hepatology 16:649–654
Bodenheimer H, Lindsay K, Davis G et al (1997) Tolerance and efficacy of oral ribavirin treatment of chronic hepatitis C: a multicenter trial. Hepatology 26:473–477
McHutchison JG, Gordon SC, Schiff ER et al (1998) Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. N Engl J Med 338:1485–1492
Poynard T, Marcellin P, Lee SS et al (1998) Randomised trial of interferon alpha2b plus ribavirin for 48 weeks or for 24 weeks versus interferon alpha2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. International Hepatitis Interventional Study Group (IHIT). Lancet 352:1426–1432
Davis GL, Esteban-Mur R, Rustgi V et al (1998) Interferon alfa-2b alone or in combination with ribavirin for the treatment of relapse of chronic hepatitis C. International Hepatitis Interventional Therapy Group. N Engl J Med 339:1493–1499
Cummings KJ, Lee SM, West ES et al (2001) Interferon and ribavirin vs interferon alone in the re-treatment of chronic hepatitis C previously nonresponsive to interferon: a meta-analysis of randomized trials. JAMA 285:193–199
Crotty S, Maag D, Arnold JJ et al (2000) The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat Med 6:1375–1379
Vo NV, Young KC, Lai MM (2003) Mutagenic and inhibitory effects of ribavirin on hepatitis C virus RNA polymerase. Biochemistry 42:10462–10471
Zhou S, Liu R, Baroudy BM et al (2003) The effect of ribavirin and IMPDH inhibitors on hepatitis C virus subgenomic replicon RNA. Virology 310:333–342
Crotty S, Cameron CE, Andino R (2001) RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci U S A 98:6895–6900
Contreras AM, Hiasa Y, He W et al (2002) Viral RNA mutations are region specific and increased by ribavirin in a full-length hepatitis C virus replication system. J Virol 76:8505–8517
Asahina Y, Izumi N, Enomoto N et al (2005) Mutagenic effects of ribavirin and response to interferon/ribavirin combination therapy in chronic hepatitis C. J Hepatol 43:623–629
Fang SH, Hwang LH, Chen DS et al (2000) Ribavirin enhancement of hepatitis C virus core antigen-specific type 1 T helper cell response correlates with the increased IL-12 level. J Hepatol 33:791–798
Dixit NM, Layden-Almer JE, Layden TJ et al (2004) Modeling how ribavirin improves interferon response rates in hepatitis C virus infection. Nature 432:922–924
Feld JJ, Hoofnagle JH (2005) Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436:967–972
Glue P, Fang JW, Rouzier-Panis R et al (2000) Pegylated interferon-alfa2b: pharmacokinetics, pharmacodynamics, safety, and preliminary efficacy data. Hepatitis C Intervention Therapy Group. Pharmacol Ther 68:556–567
Bailon P, Palleroni A, Schaffer CA et al (2001) Rational design of a potent, long- lasting form of interferon: a 40 kDa branched polyethylene glycol-conjugated interferon alfa-2a for the treatment of hepatitis C. Bioconjug Chem 12:195–202
Lindsay KL, Trepo C, Heintges T et al. Hepatitis Interventional Therapy Group (2001) A randomized, double-blind trial comparing pegylated interferon alfa-2b to interferon alfa-2b as initial treatment for chronic hepatitis C. Hepatology 34:395–403
Glue P, Rouzier-Panis R, Raffanel C et al (2000) A dose-ranging study of pegylated interferon alfa-2b and ribavirin in chronic hepatitis C. The Hepatitis C Intervention Therapy Group. Hepatology 32(2):647–653
Manns MP, McHutchison JG, Gordon SC et al (2001) Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomized trial. Lancet 358:958–965
Zeuzem S, Feinman SV, Rasenack J et al (2000) Peginterferon alfa-2a in patients with chronic hepatitis C. N Engl J Med 343:1666–1672
Heathcote EJ, Shiffman ML, Cooksley WG (2000) Peginterferon alfa-2a in patients with chronic hepatitis C and cirrhosis. N Engl J Med 343:1673–1680
Fried MW, Shiffman ML, Reddy KR et al (2002) Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 347:975–982
Hadziyannis SJ, Sette Jr H, Morgan TR et al (2004) Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med 140:346–355
McHutchison JG, Lawitz EJ, Shiffman ML et al. IDEAL Study Team (2009) Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med 361:580–593
Berg T, von Wagner M, Nasser S et al (2006) Extended treatment duration for hepatitis C virus type 1: comparing 48 versus 72 weeks of peginterferon-alfa-2a plus ribavirin. Gastroenterology 130:1–86-97
Sánchez-Tapias JM, Diago M et al (2006) Peginterferon-alfa2a plus ribavirin for 48 versus 72 weeks in patients with detectable hepatitis C virus RNA at week 4 of treatment. Gastroenterology 131:451–460
Pearlman BL, Ehleben C, Saifee S (2007) Treatment extension to 72 weeks of peginterferon and ribavirin in hepatitis C genotype 1-infected slow responders. Hepatology 46(6):1688–1694
Ferenci P, Laferl H, Scherzer TM et al (2010) Peginterferon alfa-2a/ribavirin for 48 or 72 weeks in hepatitis C genotypes 1 and 4 patients with slow virologic response. Gastroenterology 138:503–512
Zeuzem S, Poordad F (2010) Pegylated-interferon plus ribavirin therapy in the treatment of CHC: individualization of treatment duration according to on-treatment virologic response. Curr Med Res Opin 26:1733–1743
Ferenci P, Laferl H, Scherzer TM et al (2008) Peginterferon alfa-2a and ribavirin for 24 weeks in hepatitis C type 1 and 4 patients with rapid virological response. Gastroenterology 135:451–458
Zeuzem S, Buti M, Ferenci P et al (2006) Efficacy of 24 weeks treatment with peginterferon alfa-2b plus ribavirin in patients with chronic hepatitis C infected with genotype 1 and low pretreatment viremia. J Hepatol 44(1):97–103
Jensen DM, Morgan TR, Marcellin P et al (2006) Early identification of HCV genotype 1 patients responding to 24 weeks peginterferon alpha-2a (40 kd)/ribavirin therapy. Hepatology 43(5):954–960
Mangia A, Santoro R, Minerva N et al (2005) Peginterferon alfa-2b and ribavirin for 12 vs. 24 weeks in HCV genotype 2 or 3. N Engl J Med 352:2609–2617
Shiffman ML, Suter F, Bacon BR et al (2007) Peginterferon alfa-2a and ribavirin for 16 or 24 weeks in HCV genotype 2 or 3. N Engl J Med 357:124–134
Dalgard O, Bjøro K, Ring-Larsen H et al (2008) Pegylated interferon alfa and ribavirin for 14 versus 24 weeks in patients with hepatitis C virus genotype 2 or 3 and rapid virological response. Hepatology 47:35–42
Lagging M, Langeland N, Pedersen C et al (2008) Randomized comparison of 12 or 24 weeks of peginterferon alpha-2a and ribavirin in chronic hepatitis C virus genotype 2/3 infection. Hepatology 47:1837–1845
Shiffman ML, Di Bisceglie AM, Lindsay KL et al (2004) Peginterferon alfa-2a and ribavirin in patients with chronic hepatitis C who have failed prior treatment. Gastroenterology 126:1015–1023
Jacobson IM, Gonzalez SA, Ahmed F et al (2005) A randomized trial of pegylated interferon alpha-2b plus ribavirin in the retreatment of chronic hepatitis C. Am J Gastroenterol 100:2453–2462
Mathew A, Peiffer LP, Rhoades K, McGarrity T (2006) Sustained viral response to pegylated interferon alpha-2b and ribavirin in chronic hepatitis C refractory to prior treatment. Dig Dis Sci 51:1956–1961
Taliani G, Gemignani G, Ferrari C et al (2006) Pegylated interferon alfa-2b plus ribavirin in the retreatment of interferon-ribavirin nonresponder patients. Gastroenterology 130:1098–1106
Parise E, Cheinquer H, Crespo D et al (2006) Peginterferon alfa-2a (40KD) (PEGASYS) plus ribavirin (COPEGUS) in retreatment of chronic hepatitis C patients, nonresponders and relapsers to previous conventional interferon plus ribavirin therapy. Braz J Infect Dis 10:11–16
Poynard T, Colombo M, Bruix J et al (2009) Peginterferon alfa-2b and ribavirin: effective in patients with hepatitis C who failed interferon alfa/ribavirin therapy. Gastroenterology 136:1618–1628
Poynard T, Moussali J, Ratziu V et al (1999) Effects of interferon therapy in “nonresponder” patients with chronic hepatitis C. J Hepatol 31(Suppl 1):178–183
Di Bisceglie AM, Shiffman ML, Everson GT et al (2008) Prolonged therapy of advanced chronic hepatitis C with low-dose peginterferon. N Engl J Med 359:2429–2441
Jacobson IM, Brown Jr RS, Freilich B et al (2007) Peginterferon alfa-2b and weight-based versus flat dosing of ribavirin in patients with chronic hepatitis C. Hepatology 46:971–981
Jacobson IM, Brown Jr RS, McCone J et al (2007) Impact of weight based ribavirin with pegylated alfa-2b in African Americans with HCV genotype 1. Hepatology 46:982–990
Afdhal N, Sherman M, Cohen L et al (2006) Clinical recommendations emerged for the use of recombinant human erythropoietin in patients with hepatitis C virus being treated with ribavirin. Can J Gastroenterol 20:479–485
Kouloridis I, Alfayez M, Trikalinos TA et al (2013) Dose of erythropoiesis-stimulating agents and adverse outcomes in CKD: a metaregression analysis. Am J Kidney Dis 61:44–56
Muir AJ, Bornstein JD, Killenberg PG, Atlantic Coast Hepatitis Treatment Group (2004) Peginterferon alfa-2b and ribavirin for the treatment of chronic hepatitis C in blacks and non-Hispanic whites. N Engl J Med 350:2265–2271
Ge D, Fellay J, Thompson AJ et al (2009) Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461:399–401
Thomas DL, Thio CL, Martin MP et al (2009) Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 461:798–801
Thompson AJ, Muir AJ, Sulkowski MS et al (2010) Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 139:120–129
Naggie S, Cooper C, Saag M et al (2015) Ledipasvir and sofosbuvir for hepatitis virus in patients coinfected with HIV-1. N Engl J Med 373:705–713
Hernandez MD, Sherman KE (2011) HIV/HCV coinfection natural history and disease progression, a review of the most recent literature. Curr Opinion HIV AIDS 6:478–482
Carrat F, Bani-Sadr F, Pol S et al (2004) Pegylated interferon alfa-2b vs standard interferon alfa-2b, plus ribavirin, for chronic hepatitis C in HIV-infected patients: a randomized controlled trial. JAMA 292:2839–2848
Chung RT, Andersen J, Volberding P et al (2004) Peginterferon Alfa-2a plus ribavirin versus interferon alfa-2a plus ribavirin for chronic hepatitis C in HIV-coinfected persons. AIDS Clinical Trials Group A5071 Study Team. N Engl J Med 351:451–459
Laguno M, Murillas J, Blanco JL et al (2004) Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for treatment of HIV/HCV co-infected patients. AIDS 18:F27–F36
Torriani FJ, Rodriguez-Torres M, Rockstroh JK et al (2004) Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection in HIV-infected patients. APRICOT Study Group. N Engl J Med 351:438–450
Kim AI, Dorn A, Bouajram R et al (2007) The treatment of chronic hepatitis C in HIV-infected patients: a meta-analysis. HIV Med 8:312–321
Mbaeyi C, Thompson ND (2013) Hepatitis C virus screening and management of seroconversions in hemodialysis. Semin Dial 26:438–446
Kidney Disease: Improving Global Outcomes (KDIGO) (2008) KDIGO clinical practice guidelines for the prevention, diagnosis, evaluation, and treatment of hepatitis C in chronic kidney disease. Kidney Int Suppl 73(Suppl 109):S1–S99
Tseng PL, Chen TC, Chien YS et al (2013) Efficacy and safety of pegylated interferon alfa-2b and ribavirin combination therapy versus pegylated interferon monotherapy in hemodialysis patients: a comparison of 2 sequentially treated cohorts. Am J Kidney Dis 62:789–795
Maylin S, Martinot-Peignoux M, Moucari R et al (2008) Eradication of hepatitis C virus in patients successfully treated for chronic hepatitis C. Gastroenterology 135:821–829
Giannini EG, Basso M, Savarino V, Picciotto A (2010) Sustained virological response to pegylated interferon and ribavirin is maintained during long-term follow-up of chronic hepatitis C patients. Aliment Pharmacol Ther 31:502–508
Mercer DF, Schiller DE, Elliott JF et al (2001) Hepatitis C virus replication in mice with chimeric human livers. Nat Med 7:927–933
Lohmann V, Korner F, Koch J et al (1999) Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113
Blight KJ, Kolykhalov AA, Rice CM (2000) Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974
Krieger N, Lohmann V, Bartenschlager R (2001) Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J Virol 75:4614–4624
Blight KJ, McKeating JA, Rice CM (2002) Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76(24):13001–13014
Blight KJ, McKeating JA, Marcotrigiano J, Rice CM (2003) Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J Virol 77:3181–3190
Failla C, Tomei L, DeFrancesco R (1994) Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J Virol 68:3753–3760
Lin C, Thomson JA, Rice CM (1995) A central region in the hepatitis C virus NS4A protein allows formation of an active NS3-NS4A serine proteinase complex in vivo and in vitro. J Virol 69:4373–4380
Pang PS, Jankowsky E, Planet PJ, Pyle AM (2002) The hepatitis C viral NS3 protein is a processive DNA helicase with cofactor enhanced RNA unwinding. EMBO J 21:1168–1176
Egger D, Wolk B, Gosert R et al (2002) Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76:5974–5984
Evans MJ, Rice CM, Goff SP (2004) Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc Natl Acad Sci U S A 101:13038–13043
Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobsen H (1993) Non-structural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J Virol 67:3835–3844
Grakoui A, Wychowski C, Lin C et al (1993) Expression and identification of hepatitis C virus polyprotein cleavage products. J Virol 67:1385–1395
Kim JL, Morgenstern KA, Lin C et al (1996) Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 87:343–355
Love RA, Parge HE, Wickersham JA et al (1996) The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell 87:331–342
Lamarre D, Anderson PC, Bailey M et al (2003) An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426:186–189
Thibeault D, Bousquet C, Gingras R et al (2004) Sensitivity of NS3 serine proteases from hepatitis C virus genotypes 2 and 3 to the inhibitor BILN 2061. J Virol 78:7352–7359
Llinàs-Brunet M, Bailey MD, Bolger G et al (2004) Structure-activity study on a novel series of macrocyclic inhibitors of the hepatitis C virus NS3 protease leading to the discovery of BILN 2061. J Med Chem 47:1605–1608
Hinrichsen H, Benhamou Y, Wedemeyer H et al (2004) Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology 127:1347–1355
Hinrichsen H, Benhamou Y, Reiser M et al (2002) The first report of the antiviral efficacy of BILN-2061, a novel oral HCV serine protease inhibitor, in patients with chronic hepatitis C genotype 1. Hepatology 36:379A
Vanwolleghen T, Meuleman P, Libbrecht L et al (2007) Ultra-rapid cardiotoxicity of the hepatitis C virus protease inhibitor BILN 2061 in the urokinase-type plasminogen activator mouse. Gastroenterology 133:1144–1155
Haqshenas G (2012) The conserved lysine 151 of HCV NS5B modulates viral genome replication and infectious virus production. J Viral Hepat 19:862–866
Afdhal N et al (2007) Valopicitabine (NM 283), alone or with peg-interferon, compared to peg-interferon/ribavirin (PEGIFN/RBV) retreatment in patients with HCV-1 infection and prior non-response to PEGIFN/RBV: one year results. J Hepatol 46(Suppl. 1):S5
Gane EJ, Roberts SK, Stedman CA et al (2010) Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 376:1467–1475
Le Pogam S, Yan JM, Chhabra M et al (2012) Characterization of hepatitis C Virus (HCV) quasispecies dynamics upon short-term dual therapy with the HCV NS5B nucleoside polymerase inhibitor mericitabine and the NS3/4 protease inhibitor danoprevir. Antimicrob Agents Chemother 56:S494–S502
Svarovskaia ES, Dvory-Sobol H, Parkin N et al (2014) Infrequent development of resistance in genotype 1-6 hepatitis C virus-infected subjects treated with sofosbuvir in phase 2 and 3 clinical trials. Clin Infect Dis 59:1666–1674
Sofia MJ (2011) Nucleotide prodrugs for HCV therapy. Antiviral Chem Chemother 22:23–49
Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM (2004) The NS5A protein of hepatitis C virus is a zinc metalloprotein. J Biol Chem 279:48576–48587
Tellinghuisen TL, Foss KL, Treadaway JC, Rice CM (2008) Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J Virol 82:1073–1083
Tellinghuisen TL, Foss KL, Treadaway J (2008) Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog 4(3):e1000032. https://doi.org/10.1371/journal.ppat.1000032
Guedj J, Dahari H, Uprichard SL, Perelson AS (2013) The hepatitis C virus NS5A inhibitor daclatasvir has a dual mode of action and leads to a new virus half-life estimate. Expert Rev Gastroenterol Hepatol 7:397–399
Nettles RE, Chien C, Chung E et al (2008) BMS-790052 us a first-in-class potent hepatitis C virus (HCV) NS5A inhibitor for patients with chronic HCV infection: results from a proof-of-concept study. 59th annual meeting of the American Association for the Study of Liver Diseases. LB12
McCown MF, Rajyaguru S, Le Pogam S et al (2008) The hepatitis C virus replicon presents a higher barrier to resistance to nucleoside analogs than to nonnucleoside polymerase or protease inhibitors. Antimicrob Agents Chemother 52:1604–1612
Le Pogam S, Seshaadri A, Kosaka A et al (2008) Existence of hepatitis C virus NS5B variants naturally resistant to non-nucleoside, but not to nucleoside, polymerase inhibitors among untreated patients. J Antimicrob Chemother 61:1205–1216
Reesink HW, Zeuzem S, Weegink CJ et al (2006) Rapid decline of viral RNA in hepatitis C patients treated with VX-950: a phase Ib, placebo-controlled, randomized study. Gastroenterology 131:997–1002
Forestier N, Reesink HW, Weegink CJ et al (2007) Antiviral activity of telaprevir (VX-950) and peginterferon alfa-2a in patients with hepatitis C. Hepatology 46:640–648
Lawitz E, Rodriguez-Torres M, Muir AJ et al (2008) Antiviral effects and safety of telaprevir, peginterferon alfa-2a, and ribavirin for 28 days in hepatitis C patients. J Hepatol 49:163–169
McHutchison JG, Everson GT, Gordon SC et al (2009) Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 360:1827–1838
McHutchison JG, Manns MP, Muir AJ et al (2010) Telaprevir for previously treated chronic HCV infection. N Engl J Med 362:1292–1303
Sarrazin C, Kieffer TL, Bartels D et al (2007) Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767–1777
Kwo PY, Lawitz EJ, McCone J et al (2010) Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial. Lancet 376:705–716
Sullivan JC, De Meyer S, Bartels DJ et al (2013) Evolution of treatment-emergent resistant variants in telaprevir phase 3 clinical trials. Clin Infect Dis 57(2):221–229
Bartels DJ, Sullivan JC, Zhang EZ et al (2013) Hepatitis C virus variants with decreased sensitivity to direct-acting antivirals (DAAs) were rarely observed in DAA-naive patients prior to treatment. J Virol 87:1544–1553
Kieffer TL, Sarrazin C, Miller JS et al (2007) Telaprevir and pegylated interferon-alpha-2a inhibit wild-type and resistant genotype 1 hepatitis C virus replication in patients. Hepatology 46:631–639
Susser S, Flinders M, Reesink HW et al (2015) Evolution of hepatitis C virus quasispecies during repeated treatment with the NS3/4A protease inhibitor telaprevir. Antimicrob Agents Chemother 59(5):2746–2755
Sarrazin C (2016) The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J Hepatol 64:486–504
Pawlotsky JM (2016) Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 151:70–86
Jacobson I, McHutchison J, Dusheiko G et al (2011) Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 364:2405–2416
Sherman K, Flamm S, Afdhal N et al (2011) Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 365(16):1551
Zeuzem S, Andreone P, Pol S et al (2011) Telaprevir for retreatment of HCV infection. N Engl J Med 362:2417–2428
Liapakis AM, Jacobson I (2012) Telaprevir user’s guide. Liver Int 32(Suppl 1):17–25
Tura C, Planas R (2013) Clinical use of telaprevir: stopping rules, predicting response, treatment length and management of adverse effects. Enferm Infecc Microbiol Clin 31:19–25
Poordad F, McCone J, Bacon B et al (2011) Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 364:1195–2006
Bacon B, Gordon S, Lawitz E et al (2011) Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 364:1207–1217
Jacobson I, Marcellin P, Zeuzem S et al (2012) Refinement of stopping rules during treatment of hepatitis C genotype 1 infections with boceprevir and peginterferon/ribavirin. Hepatology 56:567–575
Jacobson I, Dore G, Foster G et al (2014) Simeprevir with pegylated interferon alfa 2a plus ribavirin in treatment-naïve patients with chronic hepatitis C virus genotype 1 infection (QUEST-1) a phase 3 randomised, double-blind, placebo-controlled trial. Lancet 384:403–413
Manns M, Marcellin P, Poordad F et al (2014) Simeprevir with pegylated interferon alfa 2a or 2b plus ribavirin in treatment-naïve patients with chronic hepatitis C virus genotype 1 infection (QUEST-2): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 384:414–426
Zeuzem S, Berg T, Gane E et al (2014) Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology 146:430–441
Lawitz E, Mangia A, Wyles D et al (2013) Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 368:1878–1887
Gane EJ, Pockros PJ, Zeuzem S et al (2015) Meracitabine and ritonavir-boosted danoprevir with or without ribavirin in treatment-naïve hepatitis C virus genotype 1 patients: INFORM-SVR study. Liver Int 35:79–89
Gane EJ, Stedman CA, Hyland RH et al (2011) Pegylated interferon alfa-2a not required for complete rapid viral response in treatment-naïve patients with HCV GT2 or GT3. 62nd annual meeting of the American Association for the Study of Liver Diseases, abstract 34
Gane E, Stedman C, Hyland R et al (2013) Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 368:34–44
Lok A, Gardiner DF, Lawitz E et al (2012) Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 366:216–224
Wyles D, Gutierrez J (2014) Importance of HCV genotype 1 subtypes for drug resistance and response to therapy. J Viral Hepat 21(4):229–240
Sulkowski MS, Gardiner DF, Rodriguez-Torres M et al (2014) Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med 370:211–221
Kowdley KV, Lawitz E, Poordad F et al (2014) Phase 2b trial of interferon-free therapy for hepatitis C virus genotype 1. N Engl J Med 370:222–232
Jacobson IM, Gordon SC, Kowdley KV et al (2013) Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 368:1867–1877
Zeuzem S, Dusheiko G, Salupere R et al (2014) Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N Engl J Med 370:1993–2001
Lawitz E, Sulkowski MS, Ghalib R et al (2014) Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naïve patients: the COSMOS randomised study. Lancet 384:1756–1765
Kwo P, Gitlin N, Nahass R et al (2016) Simeprevir plus sofosbuvir (12 and 8 weeks) in hepatitis C virus genotype 1-infected patients without cirrhosis: OPTIMIST-1, a phase 3, randomized study. Hepatology 64:370–380
Lawitz E, Matusow G, De Jesus E et al (2016) Simeprevir plus sofosbuvir in patients with chronic hepatitis C virus genotype 1 infection and cirrhosis: a phase 3 study (OPTIMIST-2). Hepatology 64:360–369
Afdhal N, Zeuzem S, Kwo P et al (2014) Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 370:1889–1898
Afdhal N, Reddy KR, Nelson DR et al (2014) Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med 370:1483–1493
Kowdley KV, Gordon SC, Reddy KR et al (2014) Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med 370:1879–1888
American Association for the Study of Liver Diseases and Infectious Diseases Society of America. Recommendations for testing, managing, and treating hepatitis C. http://www.hcvguidelines.org. 16 Sept 2016
Terrault NA, Zeuzem S, Di Bisceglie AM et al (2016) Effectiveness of ledipasvir-sofosbuvir combination in patients with hepatitis C virus infection and factors associated with sustained virologic response. Gastroenterology 151:1131–1140.e5. https://doi.org/10.1053/j.gastro.2016.08.004. Epub 2016 Aug 24
Younossi ZM, Park H, Gordon SC et al (2016) Real-world outcomes of ledipasvir/sofosbuvir in treatment-naïve patients with hepatitis C. Am J Manag Care 22:SP205–SP211
Ingiliz P, Christensen S, Kimhofer T et al (2016) Sofosbuvir and ledipasvir for 8 weeks for the treatment of chronic hepatitis C virus (HCV) infection in HCV-monoinfected and HIV-HCV-coinfected individuals: results from the German Hepatitis C Cohort (GECCO-01). Clin Infect Dis 63:1320–1324
Marcus JL, Hurley LB, Chamberland S et al (2018) No difference in effectiveness of 8 vs 12 weeks of ledipasvir and sofosbuvir for treatment of hepatitis C in black patients. Clin Gastroenterol Hepatol 16:927–935
Bourliere M, Bronowicki J, de Ledinghen V et al (2015) Ledipasvir and sofosbuvir with or without ribavirin to treat patients with hepatitis C virus genotype 1 infection and cirrhosis non-resoponsive to previous protease inhibitor therapy: a randomized, double-blind phase 2 trial (SIRIUS). Lancet Infect Dis 15:397–404
Reddy KR, Bourliere M, Sulkowski M et al (2015) Ledipasvir and sofosbuvir in patients with genotype 1 hepatitis C virus infection and compensated cirrhosis: an integrated safety and efficacy analysis. Hepatology 62:79–86
Feld JJ, Kowdley KV, Coakley E et al (2014) Treatment of HCV with ABT-450/r–ombitasvir and dasabuvir with ribavirin. N Engl J Med 370:1594–1603
Zeuzem S, Jacobson IM, Baykal T et al (2014) Retreatment of HCV with ABT-450/r–ombitasvir and dasabuvir with ribavirin. N Engl J Med 370:1604–1614
Ferenci P, Bernstein D, Lalezari J et al (2014) ABT-450/r-ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med 370:1983–1992
Poordad F, Hezode C, Trinh R et al (2014) ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med 370:1973–1982
Feld JJ, Moreno C, Trinh R et al (2016) Sustained virologic response of 100% in HCV genotype 1b patients with cirrhosis receiving ombitasvir/paritaprevir/r and dasabuvir for 12 weeks. J Hepatol 64:301–307
Poordad F, Schiff ER, Vierling J et al (2016) Daclatasvir with sofosbuvir and ribavirin for hepatitis C virus infection with advanced cirrhosis or post-liver transplantation recurrence. Hepatology 63:1493–1505
Wyles DL, Ruane PJ, Sulkowski MS et al (2015) Daclatasvir plus sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med 373:714–725
Nelson DR et al (2015) All-oral 12-week treatment with daclatasvir plus sofosbuvir in patients with hepatitis C virus genotype 3 infection: ALLY-3 phase III study. Hepatology 61:1127–1135
Hezode C, Lebray P, De Ledinghen V et al (2017) Daclatasvir plus sofosbuvir, with or without ribavirin, for hepatitis C virus genotype 3 in a French early access programme. Liver Int 37:1314–1324
Zeuzem S, Ghalib R, Reddy KR et al (2015) Grazoprevir-elbasvir combination therapy for treatment-naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection: a randomized trial. Ann Intern Med 163:1–13
Kwo P, Gane E, Penguin CY (2017) Effectiveness of elbasvir grazoprevir combination, with or without ribavirin, treatment-experienced patients with chronic hepatitis C infection. Gastroenterology 152:164–175
Jacobson I, Lawitz E, Kwo P et al (2017) Safety and efficacy of elbasvir and grazoprevir in patients with hepatitis C virus infection and compensated cirrhosis: an integrated analysis. Gastroenterology 152:1372–1382
Zeuzem S, Serfaty L, Vierling J et al (2018) The safety and efficacy of elbasvir and grazoprevir in participants with hepatitis C virus genotype 1b infection. J Gastroenterol 53:679–688
Jacobson IM, Asante-Appiah E, Wong P et al (2016) Prevalence and impact of baseline NS5A resistance associated variants (RAVs) on the efficacy of elbasvir/grazoprevir (EBR/GZR) against GT1a infection. 66th annual meeting of the American Association for the Study of Liver Diseases. LB-22
Sarrazin C, Dvory-Sobol H, Svarovskaia ES et al (2016) Prevalence of resistance-associated substitutions in HCV NS5A, NS5B, or NS3 and outcomes of treatment with ledipasvir and sofosbuvir. Gastroenterology 151:501–512
American Association for the Study of Liver Diseases and Infectious Diseases Society of America. HCV guidance: recommendations for testing, managing, and treating hepatitis C. www.hcvguidelines.org. 24 May 2018
Feld JJ, Jacobson IM, Hezode C et al (2015) Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N Engl J Med 373:2599–2607
Foster GR, Afdhal N, Roberts SK et al (2015) Sofosbuvir and velpatasvir for HCV genotype 2 and 3 infection. N Engl J Med 373:2608–2617
Jacobson IM, Lawitz E, Gane EJ et al (2017) Efficacy of 8 weeks of sofosbuvir, velpatasvir, and voxilaprevir in patients with chronic HCV infection: 2 phase 3 randomized trials. Gastroenterology 153:113–122
Gottwein JM, Pham LV, Mikkelsen LS et al (2018) Efficacy of NS5A inhibitors against hepatitis C virus genotypes 1-7 and escape variants. Gastroenterology 154:1435–1448
Zeuzem S, Foster GR, Wang S et al (2018) Glecaprevir–pibrentasvir for 8 or 12 weeks in HCV genotype 1 or 3 Infection. N Engl J Med 378:354–369
Asselah T, Kowdley KV, Zadeikis N et al (2018) Efficacy of glecaprevir/pibrentasvir for 8 or 12 weeks in patients with hepatitis C virus genotype 2, 4, 5, or 6 infection without cirrhosis. Clin Gastroenterol Hepatol 16:417–426
Asselah T, Lee SS, Yao BB et al (2019) Efficacy and safety of glecaprevir/pibrentasvir in patients with chronic hepatitis C virus genotype 5 or 6 infection (ENDURANCE-5,6): an open-label, multicenter, phase 3b trial. Lancet Gastroenterol Hepatol 4:45–51
Zeuzem S, Mizokami M, Pianko S et al (2017) NS5A resistance-associated substitutions in patients with genotype 1 hepatitis C virus: prevalence and effect on treatment outcome. J Hepatol 66:910–918
Forns X, Lee SS, Valdes J et al (2017) Glecaprevir plus pibrentasvir for chronic hepatitis C virus genotype 1, 2, 4, 5, or 6 infection in adults with compensated cirrhosis (EXPEDITION-1): a single-arm, open-label, multicentre phase 3 trial. Lancet Infect Dis 17:1062–1068
Gane EJ (2008) The natural history of recurrent hepatitis C and what influences this. Liver Transpl 14(Suppl 2):S36–S44
Curry MP, Forns X, Chung RT et al (2015) Sofosbuvir and ribavirin prevent recurrence of HCV infection after liver transplantation: an open-label study. Gastroenterology 148:100–107
Fortune BE, Martinez-Camacho A, Kreidler S et al (2015) Post-transplant survival is improved for hepatitis C recipients who are RNA negative at time of liver transplantation. Transpl Int 28:980–989
Crespo G, Trota N, Londoño MC et al (2018) The efficacy of direct anti-HCV drugs improves early post-liver transplant survival and induces significant changes in waiting list composition. J Hepatol 69:11–17
Charlton M, Everson GT, Flamm SL et al (2015) Ledipasvir and sofosbuvir plus ribavirin for treatment o.f HCV infection in patients with advanced liver disease. Gastroenterology 149:649–659
Manns M, Samuel D, Gane EJ et al (2016) Ledipasvir and sofosbuvir plus ribavirin in patients with genotype 1 or 4 hepatitis C virus infection and advanced liver disease: a multicentre, open-label, randomised, phase 2 trial. Lancet Infect Dis 16:685–697
Curry MP, O’Leary JG, Bzowej N et al (2015) Sofosbuvir and velpatasvir for HCV in patients with decompensated cirrhosis. N Engl J Med 373:2618–2628
Welzel TM, Petersen J, Herzer K et al (2016) Daclatasvir plus sofosbuvir, with or without ribavirin, achieved high sustained virological response rates in patients with HCV infection and advanced liver disease in a real-world cohort. Gut 65:1861–1870
Afdhal N, Asselah T, Everson GT et al (2016) HCV eradication results in reduction of hepatic venous pressure gradient 48 weeks after end of treatment; final results of the study of sofosbuvir plus ribavirin in patients with cirrhosis and portal hypertension. J Hepatol 64:S221–S222
Mandorfer M, Kosbial K, Schwabl P et al (2016) Sustained virologic response to interferon-free therapies ameliorates HCV-induced portal hypertension. J Hepatol 65:692–699
Forns X, Charlton M, Denning J et al (2015) Sofosbuvir compassionate use program for patients with severe recurrent hepatitis C after liver transplantation. Hepatology 61:1485–1494
Kwo PY, Mantry PS, Coakley E et al (2014) An interferon-free antiviral regimen for HCV after liver transplantation. N Engl J Med 18(371):2375–2382
Kwo P, Fried MW, Reddy KR et al (2018) Daclatasvir and sofosbuvir treatment of decompensated liver disease or post-liver transplant hepatitis C virus recurrence in patients with advanced liver disease/cirrhosis in a real-world cohort. Hepatol Commun 27(2):354–363
Reau N, Kwo PY, Rhee S et al (2018) Glecaprevir/Pibrentasvir treatment in liver or kidney transplant patients with hepatitis C virus infection. Hepatology 68:1298–1307
Saxena V, Khungar V, Verna E et al (2017) Safety and efficacy of current direct-acting antiviral regimens in kidney and liver transplant recipients with hepatitis C: results from the HCV-TARGET Study. Hepatology 66:1090–1101
El-Sherif O, Jiang ZG, Tapper E et al (2018) Baseline factors associated with improvements in decompensated cirrhosis after direct-acting antiviral therapy for hepatitis C virus infection. Gastroenterology 154:2111–2121
Terrault N, McCaughan G, Curry M et al (2017) International Liver Transplantation Society Consensus Statement on hepatitis C management in liver transplant candidates. Transplantation 101:945–955
Roth D, Nelson DR, Bruchfeld A et al (2015) Grazoprevir plus elbasvir in treatment-naive and treatment-experienced patients with hepatitis C virus genotype 1 infection and stage 4-5 chronic kidney disease (the C-SURFER study): a combination phase 3 study. Lancet 386:1537–1545
Gane E, Lawitz E, Pugatch D et al (2017) Glecaprevir and pibrentasvir in patients with HCV and severe renal impairment. N Engl J Med 377:1448–1455
Colombo M, Aghemo A, Liu H et al (2017) Treatment with ledipasvir-sofosbuvir for 12 or 24 weeks in kidney transplant recipients with chronic hepatitis C virus genotype 1 or 4 infection: a randomized trial. Ann Intern Med 166:109–117
Goldberg D, Abt PL, Reese PP, THINKER Trial Investigators (2017) Transplanting HCV-infected kidneys into uninfected recipients. N Engl J Med 377:1103–1105
Reese PP, Abt PL, Blumberg EA et al (2018) Twelve-month outcomes after transplant of hepatitis C-infected kidneys into uninfected patients: a single-group trial. Ann Intern Med 169:273–281
Durand CM, Bowring MG, Brown DM et al (2018) Direct-acting antiviral prophylaxis in kidney transplantation from hepatitis C virus-infected donors to noninfected recipients: an open-label nonrandomized trial. Ann Intern Med 168:533–540
Selzner N, Berenguer M (2018) Should organs from hepatitis C-positive donors be used in hepatitis C-negative recipients for liver transplantation? Liver Transpl 24:831–840
Liapakis A, Formica RN, Levitsky J (2018) Solid organ transplantation of viral hepatitis C positive donor organs into viral hepatitis C negative recipients. Curr Opin Organ Transplant 23:257–263
Bethea E, Gaj K, Gustafson J et al (2018) Preemptive DAA therapy in donor HCV-positive to recipient HCV-negative cardiac transplantation. Hepatology 68(1 Suppl):4A. Abstract 7
Reiberger T, Ferlitsch A, Sieghart W et al (2010) HIV-HCV co-infected patients with low CD4+ cell nadirs are at risk for faster fibrosis progression and portal hypertension. J Viral Hepat 17:400–409
Naggie S, Cooper C, Saag M et al (2015) Ledipasvir and sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med 373:705–713
Rockstroh JK, Nelson M, Katlama C et al (2015) Efficacy and safety of grazoprevir (MK-5172) and elbasvir (MK-8742) in patients with hepatitis C virus and HIV co-infection (C-EDGE CO-INFECTION): a non-randomised, open-label trial. Lancet HIV 2(8):e319–e327
Wyles D, Brau N, Kottilil S et al (2017) Sofosbuvir and velpatasvir for the treatment of hepatitis C virus in patients coinfected with human immunodeficiency virus type 1: an open-label, phase 3 study. Clin Infect Dis 65:6–12
Rockstroh J, Lacombe K, Viani R et al (2018) Efficacy and safety of glecaprevir/pibrentasvir in patients co-infected with hepatitis C virus and human immunodeficiency virus-1: the EXPEDITION-2 study. Clin Infect Dis 67:1010–1017
European Association for the Study of the Liver (2018) EASL recommendations on treatment of hepatitis C 2018. J Hepatol. https://doi.org/10.1016/j.jhep.2018.03.026
Bourliere M, Gordon SC, Flamm SL et al (2017) Sofosbuvir, velpatasvir, and voxilaprevir for previously treated HCV infection. N Engl J Med 376:2134–2146
Poordad F, Felizarta F, Asatryan A et al (2017) Glecaprevir and pibrentasvir for 12 weeks for hepatitis C virus genotype 1 infection and prior direct-acting antiviral treatment. Hepatology 66:389–397
Poordad F, Pol S, Asatryan A et al (2018) MAGELLAN-1, part 2: Glecaprevir/Pibrentasvir in patients with hepatitis C virus genotype 1 or 4 and past direct-acting antiviral treatment failure. Hepatology 67:1253–1260
Veldt BJ, Heathcote EJ, Wedemeyer H et al (2007) Sustained virologic response and clinical outcomes in patients with chronic hepatitis C and advanced fibrosis. Ann Intern Med 147:677–684
George SL, Bacon BR, Brunt EM et al (2009) Clinical, virologic, histologic, and biochemical outcomes after successful HCV therapy: a 5-year follow-up of 150 patients. Hepatology 49:729–738
Backus LI, Boothroyd DB, Phillips BR et al (2011) A sustained virologic response reduces risk of all-cause mortality in patients with hepatitis C. Clin Gastroenterol Hepatol 9:509–516
Van der Meer AJ, Veldt BJ et al (2012) Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA 308:2584–2593
Perricone G, Duvoux C, Berenguer M et al (2018) Delisting HCV-infected liver transplant candidates who improved after viral eradication: outcome 2 years after delisting. Liver Int 38:2170–2177
Young K, Liu B, Bhuket T et al. Improved liver transplant waitlist mortality and lower risk of disease progression among chronic hepatitis C patients awaiting liver transplantation after the introduction of direct-acting antiviral therapies in the United States. J Viral Hepat. 9 Nov 2018. Doi:https://doi.org/10.1111/jvh.13039. Epub ahead of print
Lee YA, Friedman SL (2014) Reversal, maintenance or progression: what happens to the liver after a virologic cure of hepatitis C? Antiviral Res 107:23–30
Lens S, Alvarado-Tapias E, Mariño Z et al (2017) Effects of all-oral anti-viral therapy on HVPG and systemic hemodynamics in with hepatitis C virus-associated cirrhosis. Gastroenterology 153:1273–1283
Cardoso AC, Figueredo-Mendes C, Ripault MP et al (2010) Impact of peginterferon and ribavirin therapy on hepatocellular carcinoma: incidence and survival in hepatitis C patients with advanced fibrosis. J Hepatol 52:652–657
Morgan RL, Baack B, Smith BD et al (2013) Eradication of hepatitis C virus infection and the development of hepatocellular carcinoma: a meta-analysis of observational studies. Ann Intern Med 158:329–337
Calvaruso V, Cabibbo G, Cacciola I et al (2018) Incidence of hepatocellular carcinoma in patients with HCV-associated cirrhosis treated with d-acting antiviral agents. Gastroenterology 155:411–421
Van der Meer AJ, Feld JJ, Hofer H et al (2017) Risk of cirrhosis-related complications in patients with advanced fibrosis following hepatitis C virus eradication. J Hepatol 66:485–493
Tanaka T, Setzner N, Therapondos G et al (2015) Virological response for recurrent hepatitis C improves long-term survival in liver transplant recipients. Transpl Int 26:42–49
Saab S, Challita Y, Chen PH et al (2018) Elimination of hepatitis C in liver transplant recipients. J Clin Transl Hepatol 6:347–250
Martini S, Sacco M, Strona S et al (2017) Impact of viral eradication with sofosbuvir-based therapy on the outcome of post-transplant hepatitis C with severe fibrosis. Liver Int 37:62–70
Arase Y, Suzuki F, Suzuki Y et al (2009) Sustained virological response reduces incidence of onset of type 2 diabetes in chronic hepatitis C. Hepatology 49:739–744
Romero-Gómez M, Fernández-Rodríguez CM et al (2008) Effect of sustained virological response to treatment on the incidence of abnormal glucose values in chronic hepatitis C. J Hepatol 48:721–727
Li J, Zhang T, Gordon SC et al (2018) Impact of sustained virologic response on risk of type 2 diabetes among hepatitis C patients in the United States. J Viral Hepat 25:952–958
Bonacci M, Lens S, Londoño MC (2017) Virologic, clinical, and immune response outcomes of patients with hepatitis C virus-associated cryoglobulinemia treated with direct-acting antivirals. Clin Gastroenterol Hepatol 15:575–583
Rossotti R, Travi G, Pazzi A et al (2015) Rapid clearance of HCV-related splenic marginal zone lymphoma under an interferon-free, NS3/NS4A inhibitor-based treatment. A case report. J Hepatol 62:234–237
Merli M, Frigeni M, Alric L et al (2018) Direct-acting antivirals in hepatitis C virus-associated diffuse large B-cell lymphomas. Oncologist. pii: 2018-0331. Epub ahead of print
Su TH, Liu CJ, Tseng TC et al (2019) Early antiviral therapy reduces the risk of lymphoma in patients with chronic hepatitis C infection. Aliment Pharmacol Ther 49:331–339
Hsu YC, Ho HJ, Huang YT et al (2015) Association between antiviral treatment and extrahepatic outcomes in patients with hepatitis C virus infection. Gut 64:495–503
Younossi ZM, Stepanova M, Esteban R et al (2017) Superiority of interferon-free regimens for chronic hepatitis C: the effect on health-related quality of life and work productivity. Medicine (Baltimore) 96:e5914. https://doi.org/10.1097/MD.0000000000005914
Cacoub P, Bourliere M, Asselah T et al (2018) French patients with hepatitis C treated with direct-acting antiviral combinations: the effect on patient-reported outcomes. Value Health 21:1218–1225
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
Conflict of Interest: Ira M. Jacobson has received research grants from Assembly, BMS, Gilead, Janssen, Enanta, Merck, and Genfit. He has received a honorarium from Novo Nordisk, Siemens, Gilead, Springbank, Janssen, AbbVie, and Intercept for consulting.
Mary Olson has received research grants from Merck.
Viviana Figueroa-Diaz has received research grants from Eiger, Conatus, and TARGET.
Ethical Approval: Not applicable.
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Diaz, V.F., Olson, M., Jacobson, I.M. (2019). The Evolution of Clinical Trials for Hepatitis C. In: Sofia, M. (eds) HCV: The Journey from Discovery to a Cure. Topics in Medicinal Chemistry, vol 32. Springer, Cham. https://doi.org/10.1007/7355_2019_64
Download citation
DOI: https://doi.org/10.1007/7355_2019_64
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28399-5
Online ISBN: 978-3-030-28400-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)