
Abstract
Viekira Pak (ombitasvir/paritaprevir/ritonavir and dasabuvir) was one of the first interferon-free direct-acting antiviral (DAA) regimens to be approved for the treatment of genotype 1 HCV infection. The research and development team at Abbott/AbbVie based their approach to HCV cure on the use of three DAAs to avoid emergence of resistance, anchored by a potent protease inhibitor and NS5A inhibitor. Clinical trials were designed to answer multiple questions within a single study, in order to advance the regimen as quickly as possible in a highly competitive environment. The global phase 2 and 3 development program allowed for rapid identification of optimal treatment regimens and durations for populations defined by HCV subtype, prior treatment experience, and the presence of specific comorbidities such as cirrhosis and orthotopic liver transplant. The clinical trial program also clarified the limitations of this first-generation DAA regimen, including activity that was limited to genotypes 1 and 4 and the need for ribavirin for some patients, which defined a target product profile for a next-generation regimen. This continued research and development activity ultimately led to the approval of the pangenotypic regimen glecaprevir/pibrentasvir (Mavyret).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Clinical development
- Dasabuvir
- Direct-acting antivirals
- Glecaprevir
- Hepatitis C
- NS3 protease inhibitors
- NS5A inhibitors
- Ombitasvir
- Paritaprevir
- Pibrentasvir
1 Introduction
The advent of highly effective interferon-free curative regimens completely overturned the paradigm for treatment of hepatitis C virus (HCV) infection. Previous therapies had always included the inconvenience, toxicity, and poor tolerability of interferon injections. As a result, treatment was offered only to patients who met particular criteria of disease severity and overall health, who were willing to subject themselves to up to a year of difficult treatment for a chance of success significantly less than 100%. With the availability of in vitro systems like the replicon system that could assess the activity of compounds against HCV, medicinal chemistry rapidly began identifying compounds targeted against promising viral targets, including the NS3/4A protease enzyme, NS5A protein, and NS5B polymerase. However, it would require clinical trials of combinations of direct-acting antiviral agents (DAAs) to confirm that sustained virologic response (prolonged absence of detectable circulating HCV RNA months after the end of treatment) could be achieved without interferon. Once this fact had been demonstrated, innovators raced to identify the optimal combinations and shortest treatment durations that could offer the greatest number of patients a chance for cure. The field advanced rapidly, with breakthrough leapfrogging breakthrough resulting in incremental improvements in efficacy, safety, simplicity, and activity against a range of genotypes and resistant variants. The Abbott/AbbVie development of the protease inhibitor-based regimens ombitasvir/paritaprevir/ritonavir plus dasabuvir (Viekira, Viekirax plus Exviera) and glecaprevir/pibrentasvir (Mavyret, Maviret) epitomizes the rapid pace of science in this field.
2 Hepatitis C Therapy in 2003–2008
Abbott first initiated discovery activities aimed at identifying direct-acting inhibitors of HCV in the late 1990s, including programs directed at all three major targets (protease, NS5A protein, polymerase) [1, 2]. Although the tools to screen compounds for anti-HCV activity in vitro had recently become available, the strategy for developing and deploying these promising new tools had not yet been elucidated. Since the only therapies that had demonstrated SVR in the clinic included interferons, a logical first step was to combine these novel compounds with interferon, specifically pegylated interferon alfa 2a or 2b (peginterferon). This strategy had the advantage that a potential increase in efficacy could be demonstrated in a relatively simple placebo-controlled trial, with all subjects receiving the standard background regimen of peginterferon and ribavirin. A meaningful improvement over standard of care in a phase 2 trial would justify large global phase 3 trials. Furthermore, even if no improvement in SVR rate could be demonstrated, the new regimen might still be better than the standard of care if the total duration of the burdensome peginterferon could be reduced. Indeed, addition to standard of care was the path to approval for the first approved DAAs: boceprevir (Schering/Merck), telaprevir (Vertex), simeprevir (Janssen), and sofosbuvir (Gilead; for patients with genotype 1 or 4Footnote 1).
However, the add-on approach had drawbacks, the most obvious being that it did not eliminate interferon or ribavirin. Thus, although efficacy might be improved compared to the previous standard of care, safety and tolerability would be no better and might be significantly worse if the DAA caused additional side effects. Furthermore, an add-on approach was of little benefit to patients who were not candidates for interferon or ribavirin therapy (e.g., decompensated liver disease, autoimmune disease, severe cardiac or pulmonary disease) or those who wished to avoid the actual or perceived side effects of interferon. Finally, a path to regulatory approval that involved adding a new compound to peginterferon and ribavirin substantially limited the ability of a company like Abbott, a relatively late entrant to the HCV space, to differentiate its products from agents that were already in development. Early on, there was therefore a keen interest at Abbott in combining DAAs to obviate the need for interferon.
3 Sea Change: 2009–2010
3.1 The Perelson Paper
Early theoretical support for the feasibility of achieving SVR with a combination of DAAs alone was provided by a mathematical modeling exercise published by Alan S. Perelson and colleagues [3]. Perelson’s simulation suggested that, in order to completely suppress viral replication with a combination of DAAs, the regimen had to be able to inhibit growth of more than three mutations. This result suggested that SVR might be achieved just with inhibition of viral replication but that it would require a sufficient number of agents with nonoverlapping viral targets to overcome the ability of the virus to escape by selection of mutations that conferred resistance to each individual agent.
3.2 INFORM-1
In 2009 Ed Gane took the podium at the Annual Meeting of the American Association for the Study of Liver Diseases (AASLD) in Boston and presented “First-in-Man Demonstration of Potent Antiviral Activity with a Nucleoside Polymerase (R7128) and Protease (R7227/ITMN-191) Inhibitor Combination in HCV: Safety, Pharmacokinetics, and Virologic Results from INFORM-1.” This exploratory trial was sponsored by Roche/Pharmasset and demonstrated that two DAAs co-administered for 14 days could rapidly and profoundly suppress viral replication, as measured by serum HCV RNA, without interferon. Eight subjects who received the combination saw their HCV RNA levels reduced by a mean of 3.9 log10 IU/mL, including one subject whose HCV RNA level fell below the limit of quantification. Following the 14 days of DAA combination therapy in this trial, subjects were rolled onto standard therapy with peginterferon and ribavirin. Unfortunately, when the final results were ultimately published, there was no evidence that transiently lowering HCV RNA levels resulted in any improvement in the SVR rate compared to peginterferon and ribavirin alone [4]. Nevertheless, this presentation galvanized the room and caused people at Abbott to think more seriously about the possibility of an HCV cure without the need for interferon.
4 The Shift to Interferon-Free Therapy
4.1 Two Programs
Thus, the Abbott team was faced with a decision: whether to follow the same development path as others, i.e., adding a DAA to peginterferon and ribavirin to increase SVR rates and/or shorten treatment, or to pursue an interferon-sparing strategy, exploring DAA combinations intended to minimize or even dispense with interferon. The interferon-sparing DAA combination strategy was attractive. Abbott recognized that other pharmaceutical companies were already combining HCV protease inhibitors and nucleoside analog polymerase inhibitors with peginterferon in the clinic. Given their head start, it would be difficult for Abbott to bring a new entrant into this crowded competitive landscape, following the same path, and still be successful. Furthermore, Abbott had a robust internal discovery organization that could deliver assets in multiple classes, giving Abbott the ability to study combinations of internally owned compounds. Still, the all-oral DAA treatment strategy was completely unproven. There was as yet no clinical evidence that patients could be cured without interferon. Besides, even if an interferon-free DAA combination worked in some patients, it was possible that some population of more difficult-to-treat patients would always require the added potency of interferon. Abbott therefore decided to pursue a dual strategy: an interferon-free approach to explore DAA combinations and an interferon-containing strategy that would seek to combine one or more DAAs with peginterferon. It was felt that these two paths would provide treatment to the vast majority of HCV-infected patients at need, at least those infected with genotype 1. As would soon become apparent, combination DAA therapy would prove to be so efficacious as to make peginterferon obsolete for the treatment of HCV.
4.2 Pilot and Co-Pilot
Having taken the decision to pursue an interferon-free DAA combination regimen, the Abbott team was embarking on uncharted waters. There were no data demonstrating that a combination of small molecules could achieve SVR. While the Perelson modeling was provocative, and the INFORM-1 results were exciting, they left a lot of questions unanswered. How long could a 2-DAA combination maintain viral suppression? What proportion of patients would see their virus break through, and when? How many would experience viral relapse, and how long after the end of therapy? How many of these patients would fail with resistance to one or both DAA classes? What duration of therapy would be sufficient to cure a meaningful proportion of patients? Was it even possible to cure HCV infection without interferon? DAA clinical research was moving rapidly, and the Abbott team felt it was important to start generating internal data quickly. They decided to combine those investigational DAAs that were ready for use in HCV-infected subjects to start answering those questions, even if those DAAs might not constitute the intended final marketed product.
By late 2010, Abbott had produced sufficient drug supply and had generated sufficient toxiciology coverage, to dose three DAAs in humans for up to 12 weeks: the protease inhibitor ABT-450 (later paritaprevir; discovered in collaboration with Enanta Pharmaceuticals) and two non-nucleoside polymerase inhibitors, ABT-072 and ABT-333 (dasabuvir).Footnote 2 Abbott’s NS5A inhibitor, ABT-267 (ombitasvir), was still too early in development to be included in phase 2 trials, although the plan was still to initiate three-DAA combination trials as soon as possible, based on the Perelson modeling and in vitro data generated by Abbott’s virologists demonstrating superior suppression of virus in cell culture. Phase 1 and phase 2a studies had defined an efficacious dose range for all three DAAs given as monotherapy, and drug-drug interaction studies had already confirmed the absence of meaningful pharmacokinetic interactions between ABT-450 (with ritonavir) and either ABT-072 or ABT-333. Accordingly the team settled on a combination of ABT-450, a 100 mg boosting dose of ritonavir, ABT-072, and ribavirin dosed according to body weight, all given for 12 weeks. This trial was to be conducted by four experienced hepatologists and was referred to as the PILOT study [5].
The PILOT study would be a trial balloon intended to answer fundamental questions about rates of breakthrough and timing of relapse following a short course of DAA therapy. The Abbott team did not realistically expect to cure more than a minority of the patients. Anna Lok’s groundbreaking paper on the combination of daclatasvir and asunaprevir had not yet been published, but preliminary results presented at the 2010 AASLD meeting after the PILOT study had been started did not promise high SVR rates: 11 patients in that study received daclatasvir and asunaprevir alone for 24 weeks, and only 4 achieved SVR [6]. There was concern about the risk to participants in the PILOT study who might remain infected after study treatment, but in whom exposure to the DAA regimen might have selected viral variants with resistance to the protease inhibitor and non-nucleoside polymerase inhibitor classes. Indeed, an ongoing concern in studying DAAs was the risk of selecting long-lasting resistance that could eliminate an entire class of potential therapies in the future. With combination DAA regimens, there was the chance that multidrug resistance could render a patient incapable of being cured with DAAs at all. Accordingly, the PILOT study incorporated several safeguards to preserve additional chances for cure. Ribavirin was included in the regimen to maximize treatment response and hopefully forestall emergence of resistant variants. Enrollment in the trial would be small (N = 11) and limited to treatment-naïve patients without cirrhosis, for whom there was less urgency to treat and possibly a better chance of success. Finally, only patients with the favorable IL28B CC genotypeFootnote 3 were to be enrolled, so that patients who failed would have a high likelihood of achieving SVR with a subsequent course of peginterferon and ribavirin should that prove necessary.
It turned out to be less necessary than expected. Not only were rapid declines in HCV RNA seen in all 11 subjects, but all subjects also had unquantifiable levels by the end of treatment, and over the next 24 weeks of follow-up only 1 subject relapsed. This SVR rate of 91% was unexpectedFootnote 4 and constituted the first evidence that high cure rates would be achievable with a treatment duration less than 24 weeks. This result recalibrated the Abbott team’s thinking about what was achievable with a short course of all-oral therapy.
Having concluded that high SVR rates were possible with DAA combination therapy, the team launched a phase 2 program to confirm these findings and investigate factors like prior treatment history and ABT-450 dosage, leading up to a planned phase 2b duration-ranging trial. Like the PILOT study, the CO-PILOT study (Fig. 1) also evaluated a two-DAA regimen with ribavirin for 12 weeks, with some differences. Due primarily to greater ease of formulation, a strategic decision had been made to advance ABT-333 instead of ABT-072, so ABT-333 replaced ABT-072 in this study. CO-PILOT included three sequentially enrolled treatment groups and evaluated a higher dose (250 mg daily) of ABT-450, the more potent DAA, as well as the performance of the regimen in subjects with the less favorable CT or TT IL28B alleles or with prior treatment experience. Among treatment-naïve subjects, there was no difference in efficacy between 150 mg of ABT-450 and 250 mg (no virologic failures occurred at either dose); however, one patient who received the higher dose experienced an episode of asymptomatic alanine aminotransferase elevation. IL28B genotype had no impact in the treatment-naïve population, but 9 of 14 treatment-experienced subjects failed either during or after treatment [8].

Study design of CO-PILOT
5 The AVIATOR Study
The PILOT and CO-PILOT studies established 12 weeks as the baseline duration for treatment-naïve patients. In the meantime, the ABT-267 team had dramatically accelerated the development program, and ABT-267 was now available for combination trials. A number of questions remained to be answered before a regimen(s) could be advanced into phase 3 trials:
-
What is the optimal drug combination? Are all three DAAs better than any two-DAA regimen?
-
Is ribavirin necessary?
-
What is the optimal treatment duration? Can treatment be shortened to less than 12 weeks if the regimen includes three DAAs? Or conversely, will 24 weeks show better efficacy than 12 weeks?
-
What is the optimal ABT-450 dose? The transaminase elevation seen at the 250 mg dose made that dose unacceptable, but would doses of 100 mg or 200 mg have advantages over 150 mg?
-
Finally, since CO-PILOT showed that prior peginterferon treatment can affect the response to an interferon-free regimen, would the optimal treatment be different for patients with prior treatment failure?
The team finally decided to address as many questions as possible in a single multiple-arm study. The phase 2b trial, to be known as the AVIATOR study, eventually included a treatment-naïve cohort and a null responder cohort (prior null responders were considered to be the most interferon nonresponsive, so it was assumed that results in this population could be extrapolated to patients with prior partial response or relapse) and multiple arms within each cohort [9]. The base case was the maximal regimen, i.e., three DAAs with ribavirin, which was assumed to be maximally efficacious for all patients. Changes to that regimen would be compared with the base case for safety and efficacy (Fig. 2).

Study design of AVIATOR
In the naïve cohort, 12 weeks of treatment would be compared to durations of 8 and 24 weeks to establish an optimal duration. Regimens with fewer active components (two DAAs with ribavirin or three DAAs without ribavirin) would be compared to the three-DAA base case. A lower dose of ABT-450 (100 mg daily) would be compared with the 150 mg CO-PILOT dose in the 12- and 24-week treatment arms.
The null responder cohort would explore durations of 12 or 24 weeks, as well as ABT-450 doses of 100 and 150 mg daily. Since CO-PILOT already demonstrated that the efficacy of ABT-450, ABT-333, and ribavirin was inferior among treatment-experienced patients, this regimen was not assessed in null responders in AVIATOR; however, since ABT-267 had substantially greater potency than ABT-333, the study evaluated ABT-450 with ABT-267 and ribavirin in null responders.
Patients with cirrhosis were excluded from AVIATOR. Neither the safety nor the efficacy of these regimens had been established in cirrhotic patients. This population would be key to the success of any DAA regimen, since these patients were at the most urgent need of treatment, being at the highest risk of adverse outcomes. In addition, patients with cirrhosis responded most poorly to interferon-based therapy, so more efficacious treatment options were clearly needed. However, it was felt necessary to demonstrate safety and to define a dose-exposure relationship in patients with less advanced liver disease prior to administering the regimen to cirrhotic patients. Plasma levels of some components of the regimen were increased in individuals with cirrhosis compared to healthy volunteers, and accumulation of ABT-450 in the liver might pose a risk in these patients. In the interest of efficiency, the team therefore deferred assessment of the regimen in cirrhotic patients until phase 3, when the optimal regimen would be established for patients without cirrhosis.
The primary efficacy analysis for AVIATOR was comparison of the SVR rate among naïve patients treated for 8 versus 12 weeks. However, all the relevant comparisons were important in achieving the ultimate study objective, identifying optimal treatment regimens and durations. It would clearly not be possible to power such an ambitious trial to make statistically significant inferences about all the comparisons. Indeed, with only 20–40 subjects per arm, the trial would enroll a whopping 560 subjects. The team determined to make decisions based on directionality of differences in safety or efficacy among the various treatment arms, irrespective of statistical significance.
Beyond the study questions the trial was intended to answer, execution of a study of this scope and complexity would also answer numerous operational questions that would prove crucial in designing and executing a huge international phase 3 program. AVIATOR was a proving ground for Abbott’s global regulatory, clinical operations, and site management and monitoring organizations. It provided the first opportunity to gain experience with a large number of hepatology clinical research sites in North America, Europe, Australia, and New Zealand. Finally, AVIATOR helped to cement the crucial working relationships between Abbott’s clinical development and virology teams and the experienced hepatologists and infectious disease experts who would provide insight and advice to inform the Abbott HCV program, through phase 3 and into the next generation.
AVIATOR opened in the fall of 2011 and included patients from the United States, Canada, France, Germany, Spain, the United Kingdom, Australia, and New Zealand. The trial enrolled with gratifying speed, reflecting the desire of patients and their treaters for alternatives to the standard of care and excitement at the prospect of short-course, highly effective therapy. A total of 571 subjects were enrolled between October 2011 and April 2012. Because of the short treatment durations and the propensity of treatment failures to occur early during treatment or follow-up (the vast majority of relapses occurred within 4 weeks after the end of treatment), some differences between treatment groups became obvious quite soon. The 8-week treatment group quickly began to demonstrate a higher relapse rate than those treated for 12 weeks. Likewise, it was quickly obvious that across the genotype 1 population as a whole, the regimen was more efficacious with ribavirin than without. Lastly, although final data would not be available until the last subjects in the 24-week treatment groups completed follow-up, it was obvious early on that treatment failures were extremely uncommon among patients treated with the three-DAA regimen with ribavirin for 12 weeks and that extending treatment duration to 24 weeks would provide no incremental benefit in this population.
The final topline efficacy and safety findings from AVIATOR are summarized in Table 1. The team concluded the following:
-
For patients without cirrhosis, regardless of prior interferon treatment experience, the optimal treatment regimen comprised ABT-450/r, ABT-267, ABT-333, and ribavirin.
-
The optimal treatment duration in this population was 12 weeks.
-
There was no clear safety benefit to ABT-450 100 mg compared to 150 mg. Transaminase elevations were uncommon at both doses. While the two doses performed similarly in the treatment-naïve arms, 3 out of 46 null responders (6.5%) receiving the 100 mg dose experienced on-treatment virologic failure, compared to 1 out of 42 (2.4%) at the 150 mg dose.
-
The regimen appeared to have greater activity against genotype 1b virus: three DAAs appeared to be equally efficacious without ribavirin in these patients, and the results suggested that 8 weeks of treatment might be sufficient.
-
Among the few patients who failed, mutations conferring resistance to all three drug classes were frequently seen. However, emergence of resistance was less frequent in patients who failed after 8 weeks compared to 12 weeks.
6 The Phase 3 Program and Regulatory Approval
The results from AVIATOR and the remaining unanswered questions determined the configuration of the phase 3 program. It would confirm the efficacy and safety of the three-DAA regimen in treatment-naïve and treatment-experienced patients without cirrhosis, and the intriguing finding that ribavirin might not be needed in genotype 1b infection. It also included populations that were not studied in AVIATOR and in whom the activity, safety, and optimal regimen and duration were still unknown: patients with cirrhosis, HIV-1 coinfection, or prior liver transplantation. The planned phase 3 program would be the largest to date for an interferon-free regimen for hepatitis C. The pivotal phase 3 trials are summarized in Table 2.
Two double-blind placebo-controlled trials, the SAPPHIRE studies, confirmed the efficacy and safety of the three-DAA regimen with ribavirin in genotype 1-infected patients without cirrhosis. SAPPHIRE-I was conducted in treatment-naïve patients and SAPPHIRE-II in patients with prior peginterferon treatment. These trials demonstrated superiority to a historic control regimen of telaprevir with peginterferon and ribavirin [10,11,12].
The three PEARL studies elucidated the role of ribavirin, extending on the initial AVIATOR findings. PEARL-III was a large double-blind trial comparing the three-DAA regimen with ribavirin or with placebo in treatment-naïve patients with genotype 1b infection. A smaller phase 3 trial, PEARL-II, compared the regimen with or without ribavirin in open-label fashion in treatment-experienced patients with genotype 1b infection. While AVIATOR suggested that the regimen was less efficacious against genotype 1a without ribavirin, there was great interest in numerous quarters, including the US FDA, in understanding the magnitude of the loss, to guide risk-benefit decisions in patients who might be unable to take ribavirin, and in identifying possible subgroups that might have a better response without ribavirin. For this reason AbbVie conducted PEARL-IV, a double-blind comparison of the three-DAA regimen with ribavirin or placebo in treatment-naïve patients with genotype 1a infection [13, 14].
Two duration-ranging trials, the TURQUOISE studies, assessed the optimal treatment duration (12 or 24 weeks) of the three-DAA regimen in the so-called special populations, i.e., patients with characteristics that might impact the safety or activity of a DAA regimen. TURQUOISE-I was a phase 2/3 trial in patients coinfected with HIV-1, and TURQUOISE-II was a phase 3 trial in patients with compensated cirrhosis. TURQUOISE-I was complicated by the fact that the three-DAA regimen could potentially interact with numerous antiretroviral medications, principally because it contained ritonavir, a potent inhibitor of cytochrome P450 3A. As a result, several drug-drug interaction studies preceded its implementation. At the end of the day, the safety and efficacy of the three-DAA regimen in this population were consistent with the results of the pivotal studies, confirming the growing consensus that HIV-coinfected patients were no longer a “special” population in the interferon-free DAA era [15,16,17,18,19].
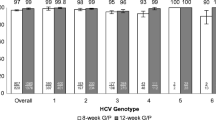
TURQUOISE-II evaluated the regimen in patients with compensated cirrhosis, with treatment durations of 12 and 24 weeks. At 380 subjects enrolled, this was at the time the largest dedicated cirrhotic trial of HCV therapy. In contrast to the experience in patients without cirrhosis, the results of this trial suggested that some patients did benefit from extending treatment duration to 24 weeks: prior null responders to peginterferon with genotype 1a infection had an SVR rate of 80% following 12 weeks of treatment and 93% after 24 weeks (Table 3). There was little difference between 12 and 24 weeks among patients without prior null response, and the regimen was again highly efficacious in patients with genotype 1b infection [20].
The three-DAA regimen was investigated in patients with a prior orthotopic liver (or kidney) transplant. This trial was amended several times to include different patient populations and different treatment durations. As with the HIV-coinfected population, drug-drug interactions were a major concern in transplant recipients, because the most important immunosuppressants in this population (cyclosporine A, tacrolimus, sirolimus, everolimus) all had important interactions with the three-DAA regimen that either necessitated substantial dose reductions of the immunosuppressant and close monitoring of immunosuppressant blood levels or prevented their co-administration [21, 22].
The results of all these trials have been reported elsewhere. Generally, they confirmed the efficacy and safety seen in AVIATOR and established the optimal regimens for genotype 1a infection (three DAAs with ribavirin) and genotype 1b infection (three DAAs alone). Comparable safety and efficacy were seen in the HIV-1-coinfected patients, for whom the indicated treatment regimens would be the same as in the HCV-monoinfected patients.
Ombitasvir/paritaprevir/ritonavir and dasabuvir were submitted for marketing approval as a single regimen (Viekira Pak) in the United States and as two products (Viekirax and Exviera) in the EU, and by January 2015 they were approved in both regions. Ongoing research continued to further refine the optimal use of these regimens. Some of these studies and their key findings are summarized in Table 4.
6.1 2 DAAs
Of note, the 2-DAA fixed-dose combination of ombitasvir/paritaprevir/ritonavir was also developed on its own for two distinct indications. Since both ombitasvir and paritaprevir (but not dasabuvir) had in vitro activity against genotype 4 HCV, a separate 2-DAA development program was undertaken both globally and in Egypt specifically, which demonstrated the safety and efficacy of this regimen in combination with ribavirin in genotype 4 infection [26,27,28,29]. This product was approved globally for this indication, marketed in the United States as Technivie and in most of the rest of the world as Viekirax. Finally, because of somewhat higher drug exposures (especially for paritaprevir) in Japanese subjects and the predominance of genotype 1b infection in Japan, the 2-DAA regimen was approved in Japan for treatment of genotypes 1 and 2.
7 The Next-Generation Development Program: From ABT-493/ABT-530 to Mavyret
7.1 The Case for a Next Generation
The approval of Viekira Pak and Harvoni, which occurred within weeks of each other in the United States and Europe, marked the definitive end of the previous era of interferon-containing therapies for genotypes 1 and 4 and made highly effective curative therapy simple and convenient for the majority of these patients. These regimens were so effective and saw such rapid uptake that it was by no means clear that there was a major unmet need remaining to justify developing an improved next generation of DAA therapy. Nevertheless, by the time Viekira Pak was approved, a discovery effort was well underway to identify new protease inhibitors and NS5A inhibitors that could address the two major needs not met by the first-generation assets: activity against resistant variants and across all six major genotypes.
7.2 Closing the Gaps
The discovery efforts were driven by the assumption that there would be little value in a next generation unless the compounds could fill the gaps in the Viekira profile. Besides limited genotypic coverage and susceptibility to resistance, those gaps included the need for ritonavir to enable once-daily administration and the need for ribavirin in a significant proportion of patients. Accordingly, it was essential for the next-generation protease inhibitor to both have robust pangenotypic activity and activity against the typical genotype 1-resistant variants with mutations at positions 155, 156, and 168, to have at least nanomolar potency against genotypes 1–6, and to have metabolic stability enabling once-daily dosing. The medicinal chemistry effort that led to the identification of ABT-493 (glecaprevir) is described in [30].
The first-generation NS5A inhibitor ombitasvir already had broad genotypic activity and a half-life allowing for once daily dosing. However, it shared the liability of all members of its class in that a number of single mutations would confer clinically significant resistance. A high priority in the NS5A discovery effort therefore centered around engineering a molecule that would retain activity against mutants selected by first-generation NS5A inhibitors, particularly mutations at position 93, which confer high-level resistance across the NS5A inhibitor class.
The clinical development program for glecaprevir/pibrentasvir was in some ways simpler than the first-generation effort. The universe of the possible had been outlined by the demonstration of short-course curative therapy with earlier DAA combination regimens, and the populations of interest with their respective challenges were well described. However, the bar for success was also considerably higher. It was no longer adequate to show improved efficacy compared to a historical interferon-containing control; instead, approval would require demonstrating efficacy comparable to the expected SVR rates of greater than 90% achieved with first-generation regimens in similar populations. In order to be competitive in the marketplace, the regimen needed to be simple and as short as possible in duration. The ultimate goal was a regimen that was uniformly efficacious and safe in the majority of patients, regardless of genotype, which could simplify treatment and enable patients to be successfully treated by healthcare providers who were not specialists. What follows is a high-level survey of a program that compressed hundreds of person-hours into a timeline of unprecedented speed.
7.3 Learning from the First Generation
The Mavyret clinical development program was conducted in a strikingly abbreviated time frame, with the first NDA submission occurring less than 3 years after the first patients were dosed in phase 2. The AbbVie team evaluated the process for submission of Viekira to identify areas for increased efficiency with the next-generation program. Under the breakthrough therapy designation, the team was able to interact frequently with regulatory agencies to understand agency expectations and to determine which study strategies would be acceptable as a basis for approval. This allowed the team to use innovative trial designs, with multiple treatment arms activated based on pre-specified safety and efficacy results from previous treatment groups.
SURVEYOR-II, a complex, staged phase 2–3 trial, was one of the most informative studies in the Mavyret program. This ambitious trial spanned 3 years and comprised four parts: a supportive/exploratory (phase 2) portion of the trial (parts 1 and 2), and a confirmatory/registrational (phase 3) portion (parts 3 and 4). In all the study included 22 separate dosing groups and was amended five times. This rolling study thus allowed numerous study questions to be answered and results from one set of analyses to inform the design and conduct of subsequent treatment groups, with the efficiency of a single protocol. It will be informative to examine the design of the four parts of this trial and to contrast it with the corresponding phase 2 AVIATOR trial from the Viekira program (Fig. 3).

Study design of SURVEYOR-II, parts 1 and 2
As with the development plan for Viekira, the primary objective was to determine how best to utilize active individual agents together in a regimen that would be simple to use across a range of patient types. In contrast to the Viekira program, however, the next-generation regimen needed excellent activity against multiple genotypes. The initial questions to be answered were fundamental and included dose ranging both agents and assessing the role of RBV. Treatment duration in the first arms (confined to patients without cirrhosis) was 12 weeks, but pharmacokinetic modeling suggested an 8-week duration might be sufficient for some genotypes, if not all.
By the time part 2 was finished, it was clear that, for genotypes 2 and 3, an 8-week course of treatment appeared to be as efficacious as 12 weeks, at least among treatment-naïve patients. Safety and efficacy in cirrhotic patients remained to be studied, as did activity in the less common genotypes 4–6. Since a single dosage of glecaprevir and of pibrentasvir had been identified across genotypes 1–3, the confirmatory, phase 3 portion of the study was able to utilize the final coformulated product, comprising three tablets containing a total of 300 mg of glecaprevir and 120 mg of pibrentasvir. Given the evident challenge posed by genotype 3-infected patients who had previous treatment failure, additional duration ranging was required to determine if an additional 4 weeks of treatment would improve efficacy. Finally, part 4 of the trial was dedicated to assessing efficacy in patients with the less common genotypes. In vitro systems allowed determination of EC50 values, which led the AbbVie team to hypothesize that the regimen would show activity against genotypes 4–6 comparable to that seen with genotypes 1 and 2; therefore, a single treatment arm with a duration of 8 weeks was expected to be sufficient for patients without cirrhosis (Fig. 4).

Study design of SURVEYOR-II, parts 3 and 4
The AbbVie team was thus able to identify optimal study drug dosages, treatment duration, and regimen across multiple genotypes and in patients with and without cirrhosis, in the setting of a single ongoing trial. This strategy even allowed the newly developed final commercial formulation to be incorporated into the trial “on the fly” [22, 31, 32].
The rest of the phase 2–3 registrational program was both straightforward and comprehensive. The ENDURANCE studies 1–4 confirmed safety and efficacy in patients without cirrhosis infected with genotypes 1, 2, 3, and 4–6, respectively. The MAGELLAN-1 study explored the glecaprevir/pibrentasvir regimen in patients who had failed prior DAA regimens [33]. The highest priority subgroups of HCV-infected patients were again investigated in dedicated trials: patients with cirrhosis were assessed in EXPEDITION-1 (in addition to part 3 of SURVEYOR-2), patients with HIV-1 coinfection in EXPEDITION-2, patients with renal insufficiency in EXPEDITION-4, and patients with prior liver or kidney transplant in MAGELLAN-2 [17, 31, 34, 35]. The efficacy of the regimen was confirmed in all populations, except that the data from MAGELLAN-1 was not sufficient to confirm efficacy in patients with genotypic resistance to both NS5A and protease inhibitors. The approved Mavyret indications from the US Prescribing Information are summarized in the text box.
Treatment-naïve
GTs 1, 2, 3, 4, 5, or 6 subjects without cirrhosis for 8 weeks
GTs 1, 2, 3, 4, 5, or 6 with compensated cirrhosis (Child-Pugh A) for 12 weeks
Treatment-experience-PRS (defined as prior treatment experience with regimens containing interferon, pegylated interferon, ribavirin, and/or sofosbuvir but no prior treatment experience with an NS5A inhibitor or NS3/4A protease inhibitor)
GTs 1, 2, 4, 5, or 6 without cirrhosis for 8 weeks
GTs 1, 2, 4, 5, or 6 with compensated cirrhosis for 12 weeks
GT3 with or without cirrhosis for 16 weeks
Treatment-experienced with a regimen containing an HCV NS5A inhibitor or NS3/4A inhibitor (not both)
GT1 with or without compensated cirrhosis and any prior treatment regimen containing an NS5A inhibitor (no prior NS3/4A protease inhibitor) for 16 weeks
GT1 with or without compensated cirrhosis and any prior treatment regimen containing an NS3/4A protease inhibitor (no prior NS5A inhibitor) for 12 weeks
8 The Future of HCV Therapy
Further investigations are underway in an ongoing effort to characterize the optimal use of Mavyret in as many populations as possible, including pediatric patients and those in diverse geographies. While the standard of care for treatment of HCV infection may continue to evolve, currently available therapies like Mavyret appear to address a substantial portion of the infected population. Public health efforts are already turning from the technical questions of efficacy and safety, to issues of diagnosis and access to treatment. Safe and simple therapeutic options can expand the pool of treating healthcare providers beyond specialists, at least for a subset of the population. Ultimately, the long-term goal of elimination of chronic hepatitis C may no longer be simply a dream.
Notes
- 1.
In addition to its approval in combination with peginterferon and ribavirin for 12 weeks for genotype 1 or 4, sofosbuvir was approved in combination with ribavirin alone for genotypes 2 and 3 and for interferon-intolerant patients with genotype 1, the first approved interferon-free all-oral therapy for chronic hepatitis C.
- 2.
ABT-072 and ABT-333 were closely related members of the same chemical series, with identical binding sites and similar antiviral activity. There was never any intention to combine these two drugs; rather the plan was to advance whichever one proved to have better properties. Although ABT-072 had a half-life that permitted once-daily dosing, due to formulation challenges, ABT-333 was ultimately selected.
- 3.
A number of polymorphisms near the IL28B gene (also known as the interferon lambda gene) were found to be associated with the probability of achieving SVR following treatment with interferon. One of the most frequently studied was the IL28B single-nucleotide polymorphism rs12979860. For this polymorphism, patients homozygous for the C allele (CC) had the most favorable prognosis; those with a TT genotype had the worst prognosis, and heterozygotes (CT) had an intermediate prognosis [7]. Highly effective DAA regimens ultimately obviated the need for IL28B genotype testing.
- 4.
To everyone’s greater surprise, a second subject relapsed at a follow-up visit 36 weeks after the end of treatment. This finding raised considerable discussion about the mechanism behind a delayed relapse, whether patients with the IL28B CC genotype might be uniquely prone to manifesting relapses at a later time point due to more robust immunologic control and whether SVR resulting from an interferon-free regimen might be less durable than that resulting from interferon. The latter question appears to have been conclusively answered in the negative.
References
McDaniel K (2018) The discovery and development of HCV NS3 protease inhibitor paritaprevir. Top Med Chem. https://doi.org/10.1007/7355_2018_42
Wagner R (2018) HCV NS5A as an antiviral therapeutic target. From validation to the discovery and development of ombitasvir and paritaprevir as components of IFN-sparing HCV curative treatments. Top Med Chem. https://doi.org/10.1007/7355_2018_50
Rong L, Dahari H, Ribeiro RM, Perelson AS (2010) Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci Transl Med 2:30ra32
Gane EJ, Roberts SK, Stedman CAM et al (2010) Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 376:1467–1475
Lawitz E, Poordad F, Kowdley KV et al (2013) A phase 2a trial of 12-week interferon-free therapy with two direct-acting antivirals (ABT-450/r, ABT-072) and ribavirin in IL28B C/C patients with chronic hepatitis C genotype 1. J Hepatol 59:18–23
Lok AS, Gardiner DF, Lawitz E et al (2012) Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 366:216–224
Ge D, Fellay J, Thompson AJ et al (2009) Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461:399–401
Poordad F, Lawitz E, Kowdley KV et al (2013) Exploratory study of oral combination antiviral therapy for hepatitis C. N Engl J Med 368:45–53
Kowdley K, Lawitz E, Poordad F et al (2014) Phase 2b trial of interferon-free therapy for hepatitis C virus genotype 1. N Engl J Med 370:222–232
Feld JJ, Kowdley KV, Coakley E et al (2014) Treatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med 370:1594
Zeuzem S, Jacobson IM, Baykal T et al (2014) Retreatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med 370:1604
Zeuzem S, Foster GR, Wang S et al (2018) Glecaprevir–pibrentasvir for 8 or 12 weeks in HCV genotype 1 or 3 infection. N Engl J Med 378:354–369
Andreone P, Colombo MG, Enejosa JV et al (2014) ABT-450, ritonavir, ombitasvir, and dasabuvir achieves 97% and 100% sustained virologic response with or without ribavirin in treatment-experienced patients with HCV genotype 1b infection. Gastroenterology 147:359
Ferenci P, Bernstein D, Lalezari J et al (2014) ABT-450/r-ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med 370:1983
Rockstroh J, Lacombe K, Viani RM et al (2017a) Efficacy and safety of glecaprevir/pibrentasvir in patients co-infected with hepatitis C virus and human immunodeficiency virus-1: the EXPEDITION-2 study. J Hepatol 66(1):S102–S103
Rockstroh JK, Orkin C, Viani RM et al (2017b) Safety and efficacy of ombitasvir, paritaprevir with ritonavir ± dasabuvir with or without ribavirin in patients with human immunodeficiency virus-1 and hepatitis C virus genotype 1 or genotype 4 coinfection: TURQUOISE-I part 2. Open Forum Infect Dis 4(3):ofx154. https://doi.org/10.1093/ofid/ofx154
Rockstroh JK, Lacombe K, Viani RM et al (2018) Efficacy and safety of glecaprevir/pibrentasvir in patients co-infected with hepatitis C virus and human immunodeficiency virus-1: the EXPEDITION-2 study. Clin Infect Dis 67:1010–1017. https://doi.org/10.1093/cid/ciy220
Sulkowski MS, Eron JJ, Wyles D et al (2015) Ombitasvir, paritaprevir co-dosed with ritonavir, dasabuvir, and ribavirin for hepatitis C in patients co-infected with HIV-1: a randomized trial. JAMA 313(12):1223–1231
Wyles D, Saag M, Viani RM et al (2017) TURQUOISE-I part 1b: ombitasvir/paritaprevir/ritonavir and dasabuvir with ribavirin for hepatitis C Virus infection in HIV-1 coinfected patients on darunavir. J Infect Dis 215:599–605
Poordad F, Hezode C, Trinh R et al (2014) ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med 370:1973–1982
Kwo PY, Mantry PS, Coakley E et al (2014) An interferon-free antiviral regimen for HCV after liver transplantation. N Engl J Med 371:2375–2382
Kwo PY, Poordad F, Asatryan A et al (2017) Glecaprevir and pibrentasvir yield high response rates in patients with HCV genotype 1-6 without cirrhosis. J Hepatol 67:263–271
Feld JJ, Moreno C, Trinh R et al (2016) Sustained virologic response of 100% in HCV genotype 1b patients with cirrhosis receiving ombitasvir/paritaprevir/r and dasabuvir for 12 weeks. J Hepatol 64:301–307
Mantry P, Reddy R, Cohen E et al (2017) Efficacy and safety of ombitasvir/paritaprevir/ritonavir ± dasabuvir with ribavirin in adults with genotype 1 or genotype 4 chronic hepatitis C virus infection and child-pugh B decompensated cirrhosis. J Hepatol 66(1):S728–S729
Welzel TM, Asselah T, Dumas EO et al (2017) Ombitasvir, paritaprevir, and ritonavir plus dasabuvir for 8 weeks in previously untreated patients with hepatitis C virus genotype 1b infection without cirrhosis (GARNET): a single-arm, open-label, phase 3b trial. Lancet Gastroenterol Hepatol 2:494–500
Asselah T, Hézode C, Qaqish RB et al (2016) Ombitasvir, paritaprevir, and ritonavir plus ribavirin in adults with hepatitis C virus genotype 4 infection and cirrhosis (AGATE-I): a multicentre, phase 3, randomised open-label trial. Lancet Gastroenterol Hepatol 1(1):25–35
Asselah T, Kowdley KV, Zadeikis N et al (2018) Efficacy of glecaprevir/pibrentasvir for 8 or 12 weeks in patients with hepatitis C virus genotype 2, 4, 5, or 6 infection without cirrhosis. Clin Gastroenterol Hepatol 16(3):417–426
Hézode C, Asselah T, Reddy KR et al (2015) Ombitasvir plus paritaprevir plus ritonavir with or without ribavirin in treatment-naive and treatment-experienced patients with genotype 4 chronic hepatitis C virus infection (PEARL-I): a randomised, open-label trial. Lancet 385:2502–2509
Waked I, Shiha G, Qaqish RB et al (2016) Ombitasvir, paritaprevir, and ritonavir plus ribavirin for chronic hepatitis C virus genotype 4 infection in Egyptian patients with or without compensated cirrhosis (AGATE-II): a multicentre, phase 3, partly randomised open-label trial. Lancet Gastroenterol Hepatol 1:36–44
Or YS, Wang G (2018) Discovery and development of the next generation HCV NS3 protease inhibitor glecaprevir. Top Med Chem. https://doi.org/10.1007/7355_2018_55
Gane E, Lawitz E, Pugatch D et al (2017) Glecaprevir and pibrentasvir in patients with HCV and severe renal impairment. N Engl J Med 377:1448–1455
Wyles D, Poordad F, Wang S et al (2018) Glecaprevir/pibrentasvir for hepatitis C virus genotype 3 patients with cirrhosis and/or prior treatment experience: a partially randomized phase 3 clinical trial. Hepatology 67:514–523
Poordad F, Pol S, Asatryan A et al (2018) Glecaprevir/pibrentasvir in patients with hepatitis C virus genotype 1 or 4 and past direct-acting antiviral treatment failure. Hepatology 67:1253–1260
Forns X, Lee SS, Valdes J et al (2017) Glecaprevir plus pibrentasvir for chronic hepatitis C virus genotype 1, 2, 4, 5, or 6 infection in adults with compensated cirrhosis (EXPEDITION-1): a single-arm, open-label, multicentre phase 3 trial. Lancet Infect Dis 17:1062–1068
Reau N, Kwo PY, Rhee S et al (2018) Glecaprevir/pibrentasvir treatment in liver or kidney transplant patients with hepatitis C virus infection. Hepatology 68:1298–1307
Compliance with Ethical Standards
Funding Daniel E Cohen did not receive any compensation for this chapter.
Conflict of Interest
Daniel E Cohen is an employee of AbbVie.
Ethical Approval
All clinical trials were approved by the independent ethics committee or institutional review board for each trial center and were conducted in accordance with the Good Clinical Practice Guidelines and the ethical principles of the Declaration of Helsinki.
Informed Consent
All study participants (or their legal guardians) provided written informed consent prior to any study procedures being performed.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Cohen, D.E. (2019). Clinical Development of Viekira Pak to Mavyret. In: Sofia, M. (eds) HCV: The Journey from Discovery to a Cure. Topics in Medicinal Chemistry, vol 32. Springer, Cham. https://doi.org/10.1007/7355_2018_60
Download citation
DOI: https://doi.org/10.1007/7355_2018_60
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28399-5
Online ISBN: 978-3-030-28400-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)