
Abstract
Bacteria are becoming increasingly resistant to currently available antibiotics, and the development of new antibiotics is not keeping pace. Alternative approaches to combatting drug-resistant bacteria are sorely needed. One such approach is the development of small-molecule antibiotic adjuvants. Adjuvants that thwart resistance mechanisms and render bacteria susceptible to antibiotics have the potential to prolong the life span and also to extend the spectrum of our current armamentarium of drugs. Several approaches to the development of potential adjuvant therapeutics have been investigated, based upon combatting various resistance mechanisms, and have identified promising adjuvant classes. These classes include adjuvants that inhibit modification or degradation of the antibiotic by enzymes (such as β-lactamases or the aminoglycoside-modifying enzymes), adjuvants that increase the intracellular concentration of the antibiotic by inhibiting efflux or facilitating antibiotic uptake, adjuvants that interfere with bacterial signaling systems that drive or coordinate resistance mechanisms, and finally adjuvants that target nonessential steps in bacterial cell wall synthesis. The antibiotic adjuvant approach is a promising orthogonal strategy for the development of new antibiotics to combat drug-resistant bacteria.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The development of new antibiotics that are active against multidrug-resistant (MDR) bacteria is not keeping pace with the ever-increasing emergence of resistance, and there are few truly novel classes of antibiotics in clinical development [1]. While the traditional approach of developing new antibiotics remains a vital tool in the fight against MDR bacteria, one major issue with that approach is that bacteria inevitably evolve resistance to single-entity therapeutics that rely on a bacteriostatic or bactericidal mechanism. For example, resistance to the first-in-class antibiotics daptomycin and linezolid was observed after only 1 year of clinical use [2,3,4], and this resistance will likely continue to increase as their clinical use is prolonged.
There is, therefore, a pressing need to develop alternative new approaches to combat antibiotic resistance. One approach that is receiving increasing attention is the development of antibiotic adjuvants [5, 6]. This approach involves the combination of an antibiotic with a non-microbicidal compound that increases the activity of the antibiotic, for example by blocking the mechanism of resistance. Compounds that are not of themselves microbicidal are termed antibiotic adjuvants. This approach differs from the identification of synergistic antibiotic combinations involving two or more microbicidal agents that target essential gene products [7]. The development of antibiotic adjuvants has one potential advantage. Because they do not typically inhibit bacterial growth when administered alone, they may exert a lower evolutionary pressure on bacteria to evolve resistance. This outcome is in contrast to the evolution of resistance to synergistic antibiotic combinations, which has been demonstrated to be dependent on the evolutionary response to the constituent drugs. For example, if the mutational response to one drug results in increased resistance to the second drug, enhanced resistance evolution is likely. Conversely, a response to one drug that confers increased susceptibility to the other drug (known as collateral sensitivity) will likely result in reduced or slower evolution of resistance to the drug combination [8]. Though there are challenges associated with the use of combination therapies, such as optimizing dosing regimens, these drugs have the potential to allow the continued use of clinically approved antibiotics that may otherwise be rendered obsolete by increasing bacterial resistance.
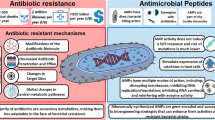
This chapter describes the identification of compounds that inhibit genotypic bacterial resistance mechanisms (as opposed to phenotypic drug tolerance such as that conferred by the formation of biofilms or persister cells). Genotypic antibiotic resistance occurs predominantly through one of three mechanisms [9]: (1) inactivation of the antibiotic by degradation or modification, (2) decreased accumulation of the antibiotic within the bacterial cell as a result of increased efflux or decreased uptake of the antibiotic, or (3) modification of the antibiotic target leading to reduced affinity for the antibiotic. Proteins involved in these resistance mechanisms are therefore attractive potential targets for the development of adjuvant drugs. Additionally, the signaling and regulatory pathways that control the activation of these resistance mechanisms are also potential adjuvant targets. A summary of potential adjuvant targets is shown in Fig. 1.


Targets of small-molecule adjuvants that suppress antibiotic resistance
2 Inhibition of Antibiotic-Modifying Enzymes
The production of enzymes that modify antibiotics such that they no longer have the required affinity for their target and thus render the antibiotic inactive is a common mechanism by which bacteria evade the action of these drugs. In chemical terms, one modification frequently used by bacteria is hydrolysis, for example, hydrolysis of the lactam bond of β-lactam antibiotics by β-lactamase enzymes, the hydrolysis of the lactone bond of macrolides by macrolide esterases, and the ring opening of the epoxide moiety of fosfomycin [10]. Another way in which bacteria modify antibiotics is to add a group to a key site of the molecule, the most well-known examples of which are the addition of an acetyl, adenyl, or phosphoryl group to aminoglycosides by the aminoglycoside-modifying enzymes (AMEs) [11]. Other examples of antibiotic-modifying enzymes include chloramphenicol acetyltransferases, macrolide kinases and macrolide glycosyltransferases [10]. Antibiotics can also be inactivated through redox reactions, as in the case of the oxidation of tigecycline by the monooxygenase TetX [12].
The classical examples of adjuvants that work by inhibiting modification of the antibiotic are β-lactamase inhibitors [13]. This class of adjuvants has been reviewed extensively [14,15,16,17], and as such only a brief overview is given in this chapter. Augmentin is a clinically approved combination therapy that consists of a β-lactam antibiotic (amoxicillin) and a β-lactamase inhibitor (clavulanic acid 1) (Fig. 2). The co-dosing of clavulanic acid with amoxicillin inhibits β-lactamase activity in vivo and allows amoxicillin to inhibit cell wall biosynthesis in strains that would be otherwise resistant. This combination ultimately has allowed the continued use of what may otherwise have become an obsolete antibiotic [18]. In 2001 Augmentin was the best-selling antibiotic, demonstrating the effectiveness of combining an antibiotic and an adjuvant in clinical settings [19]. Clavulanic acid, along with the other early β-lactam-containing β-lactamase inhibitors sulbactam 2 and tazobactam 3, is largely specific for class A β-lactamases and is not active against the class C β-lactamases or the Klebsiella pneumoniae carbapenemases (KPCs) or against the metallo-β-lactamases (MBLs) [20].


Adjuvants that inhibit β-lactamases
Recent focus has shifted to the development of non-β-lactam-derived β-lactam inhibitors. One such class is the diazabicyclooctanes (DBOs). This class exhibits a more potent and broader spectrum of activity than earlier inhibitors as it is active against the KPCs and the class C β-lactamases [20]. One member of this class is avibactam 4, which unlike β-lactam-derived inhibitors is not susceptible to hydrolysis upon binding to the β-lactamase, as the deacylation mechanism releases the intact inhibitor [21]. When examined against a collection of Enterobacteriaceae clinical isolates enriched for resistant strains possessing serine β-lactamases, minimum inhibitory concentrations (MICs) of the combination of ceftazidime-avibactam were significantly lower than those of piperacillin-tazobactam, cefotaxime, ceftriaxone, or cefepime and similar or superior to those of imipenem [22]. Avibactam was approved in 2015 in combination with ceftazidime as Avycaz® for the treatment of complicated intra-abdominal infections and complicated urinary tract infections [23]. Avibactam has also completed Phase I trials in combination with aztreonam. Another member of the DBO class of β-lactamase inhibitors is relebactam (MK-7665) 5, which is in Phase III clinical trials in combination with imipenem/cilastatin. Other β-lactamase inhibitors that have demonstrated promising activity against serine β-lactamases, including against several extended-spectrum β-lactamases (ESBLs), include the imidazole-substituted 6-methylidene-penem compound BLI-489 6 and the tricyclic carbapenem LK-157 7 (Fig. 2) [24,25,26].
The boronic acid class of β-lactamase inhibitors includes RPX7009 8 (also known as vaborbactam), which inhibits several class A and C β-lactamases including the KPCs. RPX7009 was initially developed for use in combination with biapenem [27]. RPX7009 has also demonstrated activity in combination with meropenem against KPC-producing Enterobacteriaceae [28]. The safety, tolerability, and pharmacokinetic profile of RPX7009 in a Phase I study was recently reported, and it is currently undergoing Phase III clinical investigation for the treatment of complicated urinary tract infections, acute pyelonephritis, and serious infections caused by carbapenem-resistant Enterobacteriaceae (CRE) [29].
Despite potent activity against serine β-lactamases, neither the early β-lactam-containing inhibitors, the DBO inhibitors such as avibactam, nor the boronic acid-based inhibitors are active against strains producing MBLs [30, 31]. In fact, there are as yet no clinically approved MBL inhibitors. This is partly due to the challenge of overcoming cross-reactivity with human metalloenzymes, and also due to the fact that until recently MBL-mediated β-lactam resistance was not considered a major clinical problem [32]. However, in recent years the emergence and dissemination of Gram-negative bacteria harboring plasmid-encoded MBLs such as the New Delhi metallo-β-lactamase (NDM-1) has increased the clinical importance of this class of β-lactamases [33, 34].
β-Lactamase inhibitors that are active against MBLs include the fumarate derivative ME1071 9 (Fig. 2), which inhibits the MBLs IMP-1 and VIM-2 and significantly enhances the activity of biapenem against Pseudomonas aeruginosa [25]. The triple combination cocktail BAL30367 combines the siderophore monobactam BAL19764 10, the bridged monobactam BAL29880 11 inhibitor of class C β-lactamases (Fig. 2), and clavulanic acid. BAL30367 has shown good in vitro activity against MBL-producing Enterobacteriaceae [35]. More recently, the bisthiazolidine (BTZ) class of compounds (including compound 12) has been reported to be micromolar inhibitors of several MBLs in vitro and to restore imipenem susceptibility to MBL-producing Escherichia coli [36]. In 2014 the Wright lab identified from a screen of natural products the fungal metabolite aspergillomarasmine A (AMA) 13 (Fig. 2) for its ability to inhibit NDM-1 [32]. AMA was previously investigated as an inhibitor of the mammalian metalloenzymes angiotensin-converting enzyme (ACE) and endothelin-converting enzyme and was well tolerated in mice making it a promising lead for the development of adjuvants active against MBL-producing bacteria. AMA selectively inhibited NDM-1 and the related MBL VIM-2 over rabbit lung ACE in vitro. This fact coupled with AMA as well tolerated by mice suggests that potential side effects caused by inhibition of mammalian metalloenzymes might be limited. AMA appears to act via a mechanism involving metal depletion. It was able to fully restore meropenem activity against a panel of clinical isolates of CRE, Acinetobacter spp., and Pseudomonas spp. harboring NDM-1 or VIM. AMA also demonstrated in vivo activity, restoring meropenem activity in mice infected with NDM-1-expressing K. pneumoniae, making AMA a promising potential adjuvant to address the significant clinical challenge of MBL-harboring Gram-negative pathogens [32].
Although the development of adjuvants that inhibit modification of other classes of antibiotics has not received the same degree of attention as the development of β-lactamase inhibitors, there are still several targets that have been investigated, in particular the inhibition of AMEs. AMEs are the major mechanisms of resistance to aminoglycoside antibiotics. Their catalysis of the addition of a functional group to a key site on the aminoglycoside achieves resistance as the added functional group disrupts the interaction of the antibiotic with the rRNA target. AMEs include nucleotidyltranferases, phosphotransferases, and acetyltransferases. AMEs effect modifications at both hydroxyl and amine groups, both on the 2-deoxystreptamine core of the aminoglycoside and on the appended saccharides to the core [11]. Given the prevalence of AMEs and the importance of the aminoglycoside class of antibiotics (particularly for the treatment of infections caused by Gram-negative bacteria), inhibitors of these enzymes are attractive prospective adjuvants. Several classes of AME inhibitors have been reported [11, 37]. We focus here on those that have demonstrated not only in vitro enzyme inhibition but also the ability to suppress aminoglycoside resistance in bacterial cells.
The development of aminoglycoside-coenzyme A conjugates as chemical probes to investigate the catalytic mechanism of aminoglycoside 6-N-acetyltransferases (AAC (6)-Ii), which transfer an acetyl group from acetyl-coenzyme A to the amino group at the 6 position of the aminoglycoside [11], led to the identification of the truncated conjugate 14 (Fig. 3). Compound 14 exhibited inhibitory activity against AAC (6)-Ii from Enterococcus faecium and was shown to act synergistically with kanamycin A against E. faecium harboring aac-(6)-Ii [38].


Adjuvants that inhibit antibiotic-modifying enzymes other than β-lactamases
A screen of metal cations for interference of acetylation of kanamycin and tobramycin by AAC (6)-Ib revealed that Zn(II) is an inhibitor of this enzyme, with ZnCl2 exhibiting an IC50 of 15 μM for inhibition of kanamycin acetylation. Suppression of amikacin resistance in Acinetobacter baumannii harboring aac-(6)-Ib by ZnCl2 was observed only at high ZnCl2 concentrations (800 μM). However, replacement of the chloride counter ions with the ionophore pyrithione to mediate internalization of the cation resulted in suppression of amikacin resistance in A. baumannii at concentrations as low as 4 μM, with the complex exhibiting no effect on bacterial growth alone. The zinc pyrithione complex 15 (Fig. 3) also suppressed resistance to amikacin in E. coli harboring aac-(6)-Ib [39] and was subsequently shown to suppress amikacin and tobramycin resistance in clinical isolates of several Gram-negative bacterial species including K. pneumoniae and Enterobacter cloacae [40]. Similarly, a copper pyrithione complex was reported to suppress amikacin resistance in K. pneumoniae [41].
The extensive attention that has been given to the development of human kinase inhibitors for the treatment of cancer, and the similarities in the 3-D fold structure shared among Ser/Thr/Tyr kinases were exploited by Shakya et al. in the screening of a diverse library of 80 known kinase inhibitors for inhibition of 14 bacterial kinases involved in antibiotic resistance [42]. This screen identified several active inhibitors possessing either broad- or narrow-spectrum inhibition profiles. The flavonol quercetin 16 (Fig. 3) inhibited 12 of the 14 kinases screened, including all of the aminoglycoside phosphotransferases (APHs). Quercetin significantly increased cell death caused by aminoglycoside antibiotics in E. coli expressing several APHs [42]. Resistance to arbekacin in methicillin-resistant Staphylococcus aureus (MRSA) is thought to be predominantly mediated by the bifunctional enzyme AAC(6′)/APH(2″), which catalyzes both phosphorylation and acetylation of aminoglycosides. The antitumor antibiotic aranorosin 17 has been reported to lower the MIC of arbekacin against resistant MRSA strains at sub-MIC concentrations by inhibiting AAC(6′)/APH(2″)-catalyzed phosphorylation of the aminoglycoside [43].
Another example of bacterial resistance mediated by modification of the antibiotic is the case of mycothiol (MSH). Mycobacterium species use the small-molecule MSH to maintain an intracellular reducing environment and for the detoxification of xenobiotics [44]. MSH has been reported to play a role in the resistance of Mycobacterium tuberculosis (Mtb) to several clinically important antibiotics including the first-line Mtb drug rifampin and the second-line drug streptomycin. Knocking out genes encoding for enzymes involved in MSH biosynthesis leads to increased sensitivity toward several antibiotics, making these enzymes promising targets for overcoming resistance in Mtb. It is important to note, however, that MSH also plays a role in the activation of some antibiotics used to combat Mtb such as isoniazid, and any inhibitor of MSH biosynthesis or activity would therefore be incompatible with any treatment regimen that uses MSH-dependent antibiotics. Dequalinium 18 (Fig. 3), which had been reported previously as an inhibitor of the MSH biosynthetic enzyme MshC [45], was identified from a high-throughput screen for compounds that enhance the activity of spectinomycin against Mycobacterium smegmatis. Dequalinium has potential for the sensitization of Mtb to antibiotics that are inactivated by MSH [46].
3 Inhibition of Target Alteration
Analogous to modification of the antibiotic, bacteria may also alter the target of the antibiotic to result in the antibiotic no longer having the required binding affinity to exert its effect. There are few examples of adjuvants successfully targeted at this resistance mechanism. One example is the ErmC inhibitor 19 (Fig. 4). The ErmC methyltransferase class of enzymes catalyzes methylation of an adenine residue of the bacterial 23S rRNA, disrupting binding of the macrolide-lincosamide-streptogramin-B (MLS) classes of antibiotics to the rRNA and thus rendering the bacteria resistant [47, 48]. A high-throughput screening by Clancy et al. identified inhibitors of ErmC including compound 19, which exhibited synergistic or additive activity with azithromycin against S. aureus and Enterococcus faecalis [49]. A separate virtual screen for inhibitors of ErmC identified several compounds (including compound 20, Fig. 4) that decreased erythromycin MICs against an E. coli strain constitutively expressing ErmC [50].


Compounds that inhibit the modification of antibiotic targets
4 Inhibition of Efflux
Another major resistance mechanism used by bacteria is efflux, in which membrane-bound efflux proteins pump toxic agents (including virtually all classes of antibiotics) out of the cell. This efflux results in a less-than-efficacious intracellular concentration of drug. Efflux pumps are ubiquitous in bacteria and present a significant challenge to the development of effective antibiotics. They may be specific for one substrate or one substrate class or may expel multiple unrelated classes of antibiotics, as is the case for the resistance-nodulation-division (RND) superfamily that includes the clinically relevant AcrAB-TolC and Mex pumps that contribute to multidrug resistance [51, 52]. Additionally, efflux pumps can act synergistically with other resistance mechanisms, such as the outer membrane permeability barrier in Gram-negative bacteria, exacerbating resistance [52]. Compounds that inhibit efflux pumps therefore have significant potential to sidestep antibiotic resistance and are attractive as potential adjuvants.
S. aureus possesses more than 15 different efflux pumps, both chromosomally and plasmid encoded, which contribute to resistance against various classes of antibiotics [53]. NorA is the best-studied S. aureus efflux pump. It is a multidrug efflux pump that plays a role in resistance to the fluoroquinolone antibiotics and to disinfectants. NorA is thought to be responsible for at least 10% of antibacterial resistance in MRSA strains [54]. The plant alkaloid reserpine 21 (Fig. 5) inhibits NorA-mediated drug efflux and decreases the MIC of the fluoroquinolone norfloxacin against S. aureus. Additionally, reserpine increases the bactericidal activity and post-antibiotic effect of ciprofloxacin on S. aureus and reduces the emergence of norfloxacin resistance. The activity of reserpine establishes NorA inhibitors as having potential as adjuvants. However, reserpine itself cannot be used in a clinical setting due to its neurotoxicity [55]. In an attempt to identify a more clinically useful potentiator of fluoroquinolone activity, a 9,600-member library was screened for NorA inhibition. Several structurally diverse compounds with increased potency compared to reserpine were identified. The most active compound (22) demonstrated synergy with ciprofloxacin against a resistant S. aureus strain and considerably reduced the emergence of ciprofloxacin resistance [56].


Inhibitors of S. aureus efflux pumps
Several other phytochemicals have also been reported to inhibit bacterial efflux pumps, as reviewed by Abreu et al. [54]. Examples include carnosic acid 23 and carnosol 24, which inhibit several efflux pumps of S. aureus and thus suppress resistance to tetracycline resulting from the TetA efflux pump and to erythromycin resulting from the MsrA efflux pump. Carnosic acid also demonstrated inhibition of ethidium bromide efflux in a NorA-expressing S. aureus strain [54]. Other reported inhibitors of NorA that potentiate antibiotic activity include the related abietanes ferruginol 25 and 5-epipisiferol 26, the polyphenol hydnocarpin D 26, the chlorophyll metabolite pheophorbide A 27, and the flavonoid baicalein 29 (Fig. 5). While baicalein potentiated ciprofloxacin against MRSA and gentamicin against vancomycin-resistant E. faecium (VRE) through a mechanism thought to involve inhibition of NorA, it also potentiated the effects of β-lactam antibiotics against MRSA and exhibited synergy with tetracycline against MRSA strains not possessing the TetK efflux pump. Baicalein may therefore act via multiple modes of action [54, 57].
Celecoxib 30 (Fig. 5) is a cyclooxygenase-2 (COX-2) inhibitor that also suppresses drug resistance in cancers by inhibiting the MDR1 efflux pump of the cancer cell. Celecoxib was reported additionally to suppress resistance to multiple antibiotic classes including ampicillin, kanamycin, chloramphenicol, and ciprofloxacin in S. aureus [58]. Celecoxib was later confirmed to inhibit NorA. Subsequent virtual screening of a library of 150 celecoxib analogs identified compound 31 as a more potent inhibitor than celecoxib, and as having synergistic activity with ciprofloxacin against a NorA-overexpressing strain of S. aureus [59]. The investigation of a series of indole derivatives as NorA inhibitors identified several active compounds including 32 and 33, which exhibited IC50 values of 12.5 μM and potentiated ciprofloxacin against a NorA-overexpressing strain of S. aureus, reducing the MIC by eightfold [60]. Phenothiazine antipsychotic drugs such as thioridazine 34 (Fig. 5) possess modest antimicrobial activity and have been reported to potentiate several classes of antibiotics against multiple bacterial species. This class of compounds inhibits both efflux mediated by NorA and by other efflux mechanisms in S. aureus and thus reduces norfloxacin MICs in strains overexpressing efflux pumps [61]. Another multidrug efflux pump in S. aureus is MdeA, which plays a role in resistance to several antibiotics including novobiocin and mupirocin [53]. The alkaloid piperine 35 (Fig. 5), which has also been reported as an inhibitor of NorA, markedly reduced the MIC of mupirocin against S. aureus, thought to occur by a mechanism likely involving inhibition of MdeA efflux. In an in vivo dermal infection model, mupirocin had increased efficacy at one-quarter the commercially available dose upon combination with piperine, compared to the full dose alone [62].
The inhibition of efflux pumps as a means of potentiating antibiotics in Gram-negative bacteria also has been extensively investigated. Several inhibitors of Gram-negative bacterial efflux pumps have been described. A screening program for inhibitors of P. aeruginosa efflux pumps identified the peptidomimetic Phe-Arg-β-naphthylamide (PAβN) 36 (Fig. 6) as an inhibitor of all four of the clinically relevant efflux pumps in this bacterium (MexAB-OprM, MexCD-OprJ, MexEF-OprN, and MexXY-OprM). PAβN decreased resistance to levofloxacin, reducing MICs by eightfold in wild-type strains of P. aeruginosa, while MICs were decreased by up to 64-fold in efflux pump-overexpressing strains [63,64,65]. Further illustrating the intricate role played by efflux pumps in multidrug resistance, the overexpression of MexAB-OprM in P. aeruginosa isolates from cystic fibrosis patients was reported to result in both derepression of the cephalosporinase AmpC and decreased membrane permeability. As a result, susceptibility to meropenem is decreased. Addition of PaβN abolishes this decrease [66]. PAβN also inhibits similar pumps in other MDR Gram-negative bacteria, including the clinically important RND family efflux pump system AcrAB-TolC [63]. AcrAB-TolC is the major multidrug resistance efflux pump in Enterobacteriaceae and is responsible for the efflux of a variety of structurally diverse compounds, including β-lactams, fluoroquinolones, and tetracyclines. Strains lacking this efflux system are hypersusceptible to several antimicrobials. AcrAB-TolC overexpression in response to antibiotic exposure contributes to multidrug resistance in Gram-negative bacteria [67, 68]. In the Enterobacteriaceae AcrAB–TolC production is regulated by the transcriptional activator RamA encoded by ramA. Inactivation of ramA results in decreased acrB expression, while high-level but not low-level overexpression of ramA leads to increased acrB expression [69]. Interestingly, inhibition of efflux by several inhibitors including PaβN results in upregulation of ramA, thought to be a response to increased cellular accumulation of internal metabolites [68]. PaβN binds to the bottom of the distal binding pocket of AcrB and interferes with the binding of drug substrates to the upper part of the binding pocket [70]. Counterintuitively, carbapenemase-producing Enterobacteriaceae mutants lacking AcrAB-TolC efflux pumps have been reported to exhibit elevated carbapenem resistance levels, a phenotype that was recapitulated by inhibitor-mediated loss of efflux pump function with 72% of clinical isolates tested being more resistant to ertapenem in the presence of PAβN. The increased resistance observed both for the mutants and in the presence of PAβN was attributed to a change in outer membrane porin production [71]. This result highlights the interdependent nature of bacterial resistance mechanisms and the need to evaluate the effect of any new adjuvant on other clinically important antibiotics.


Adjuvants that inhibit efflux in Gram-negative bacteria
Several other compounds have been reported to inhibit the AcrAB-TolC efflux system, including the previously mentioned NorA inhibitor thioridazine 34 and the related phenothiazine chlorpromazine 37, both of which effected an increase in ramA expression [68]. As mentioned earlier, high-level but not low-level overexpression of ramA leads to increased acrB expression, and chlorpromazine 37 induced modest overproduction of ramA, repressed acrB, and increased susceptibility to several antibiotics including nalidixic acid, norfloxacin, ciprofloxacin, chloramphenicol, and tetracycline. These results suggest phenothiazines are not direct efflux pump inhibitors, but suppress resistance by affecting the expression of efflux-related genes [69]. Trimethoprim 38 and epinephrine 39 (Fig. 6) have also been reported to exhibit synergy with ciprofloxacin, tetracycline, and chloramphenicol by inhibiting AcrAB-TolC [72]. The arylpiperazine and arylmorpholine scaffolds constitute two of the most well-studied classes of RND pump inhibitors, and the piperazine arylideneimidazolone 40 was shown to inhibit efflux in an acrAB-overexpressing strain of E. coli and to increase susceptibility to several antibiotics including levofloxacin, oxacillin, linezolid, and clarithromycin to levels close to those found in an acrAB-knockout strain [67]. A series of arylhydantoin derivatives were identified as potentiating the activity of nalidixic acid in strains of Enterobacter aerogenes overexpressing the AcrAB-TolC efflux pump [73]. Modulation of this scaffold led to the identification of 41 (Fig. 6) which exhibited synergy with both nalidixic acid and chloramphenicol against an acrAB-overexpressing strain of E. aerogenes [74].
A screen of 1,120 actinomycete fermentation extracts for rifampin potentiation against E. coli identified antibiotic 301A1 42 (Fig. 6). Antibiotic 301A1 does not possess antibiotic activity against Gram-negative bacteria itself, but displays high-level synergy with rifampin against E. coli. This compound also displayed synergy with rifampin against A. baumannii and with the Gram-positive-selective antibacterial linezolid against both E. coli and A. baumannii (both Gram-negatives). Inhibition of efflux was postulated to play a role in the adjuvant activity of 42 at least with respect to linezolid potentiation, with 42 competitively inhibiting extrusion of the AcrAB substrate Nile Red from E. coli and exhibiting a loss of activity in an A. baumannii strain overexpressing the AcrAB family efflux pump AdeIJK [75].
A screen for compounds that potentiate the Gram-positive-selective aminocoumarin antibiotic novobiocin against E. coli identified compound 43 (Fig. 6) that inhibited MreB, a component of the bacterial cytoskeleton with a role in cell division. Alterations in cell shape upon disruption of MreB correlated with decreased efflux and subsequent accumulation of normally extruded antibiotics, making this compound an example of synergy as a result of indirect inhibition of efflux [76].
5 Enhancement of Antibiotic Uptake
Antibiotics with targets that are located within the cytoplasm must cross the bacterial cell envelope in order to exert their effects. This crossing is achieved by several different mechanisms, depending on the antibiotic class and the bacterial species. Reduced permeability of the bacterial cell envelope confers increased antibiotic resistance, and various approaches to circumvent permeability-mediated resistance have been investigated. These studies include destabilization of the bacterial outer membrane and the hijacking of the transport mechanisms used by the bacteria for nutrient uptake [77].
The Gram-positive cell wall is relatively permeable to most antibiotics, and the Gram-positive cytoplasmic membrane is typically crossed by active transport. The additional outer membrane of Gram-negative bacteria poses a much greater barrier. While some hydrophilic antibiotics can traverse the outer membrane by passive diffusion through the pores of the porin proteins, these pores typically exclude larger antibiotics (Mw >800 Da). The cytoplasmic membrane must still be traversed for antibiotics with targets within the cytoplasm. The loss of outer membrane porins, or of active uptake pathways in the cytoplasmic membrane, contributes further to increased antibiotic resistance in Gram-negative bacteria [78, 79]. The development of antibiotics that are active against Gram-negative bacteria is therefore considerably more challenging than the development of Gram-positive antibiotics. Gram-negative bacteria are intrinsically resistant to several classes of antibiotics. Accordingly, breaching the Gram-negative cell envelope has the potential to render antibiotics that are currently only viable for use against Gram-positive bacteria clinically useful for a much broader spectrum of bacteria and is therefore another attractive adjuvant approach.
Several compounds that lack direct antimicrobial activity, but destabilize the Gram-negative outer membrane to an extent that allows antibiotics not normally able to cross the membrane to access the cell, have been investigated for their ability to enhance antibiotic activity [80]. One example is the truncated polymyxin, polymyxin B nonapeptide (PMBN) (44, Fig. 7). PMBN lacks the acyl tail and N-terminal aminoacyl residue of polymyxin B. As a result this compound lacks the antibacterial activity exhibited by native polymyxins against Gram-negative bacteria. PMBN increases the susceptibility of several species of Gram-negative bacteria, including K. pneumoniae and P. aeruginosa, to erythromycin, novobiocin, and fusidic acid. PMBN also exhibits in vivo activity in combination with erythromycin or novobiocin in mice infected with K. pneumoniae or P. aeruginosa [81, 82]. More recently, PMBN was reported to potentiate ceftazidime and ceftazidime-avibactam against clinical isolates of E. coli, K. pneumoniae, and E. aerogenes [83]. The renal toxicity associated with PMBN prevented its development as a clinically viable adjuvant and led to the creation of second-generation analogs with reduced positive charge (including SPR7061 45 and SPR741 46, Fig. 7) and with potentially reduced renal toxicity [80]. As with PMBN, these analogs lack significant antimicrobial activity against most Gram-negative bacteria, but potentiate the activity of several antibiotics against Gram-negative species including E. coli, K. pneumoniae, and A. baumannii. These second-generation analogs lack, however, activity against P. aeruginosa.


Adjuvants that enhance the uptake of antibiotics
A screen for potentiation of the tetracycline minocycline using a library of approved drugs identified the opioid receptor agonist loperamide 47 (Fig. 7). Loperamide potentiated the activity of minocycline (and other tetracycline antibiotics) against several species of Gram-negative bacteria including P. aeruginosa, E. coli, A. baumannii, K. pneumoniae, and Salmonella enterica. The use of previously approved drugs such as loperamide as potential antibiotic adjuvants has the advantage of identifying adjuvants with well-characterized toxicology and pharmacology. Loperamide 47 exhibited in vivo activity in a murine model of infectious colitis caused by S. enterica serovar Typhimurium. Loperamide increased membrane permeability in E. coli and P. aeruginosa and dissipated the membrane potential in E. coli enhancing the uptake of tetracycline antibiotics, which requires a pH gradient to traverse the Gram-negative outer membrane [84].
As mentioned earlier, the known efflux pump inhibitor PAβN 36 increases the susceptibility of P. aeruginosa to fluoroquinolone antibiotics. However, its ability to potentiate the β-lactam class of antibiotics was less studied. The Burrows group showed that while PAβN does indeed potentiate β-lactam antibiotics against P. aeruginosa, the mechanism of action is not due solely to efflux inhibition. PAβN additionally acts as a membrane-permeabilizing agent. PAβN enhanced the potency of β-lactam antibiotics against a P. aeruginosa strain deficient in all four major RND efflux pumps, effected increased uptake of the fluorescent probe 8-anilino-1-naphthylenesulfonic acid, caused a release of the AmpC β-lactamase from cells, and sensitized the bacterium to vancomycin, which under normal conditions is unable to cross the P. aeruginosa outer membrane. All of these effects indicate that membrane permeabilization could be a significant secondary mechanism through which PAβN acts to potentiate antibiotic activity. This property could even expand the scope of antibiotics for which it is effective [85].
Another strategy to increase antibiotic effectiveness by enhancing uptake is to take advantage of native transport systems that are used by bacteria for nutrient uptake. One such system is the iron transport system, which is vital to the ability of bacteria to cause infection, being required for both virulence and survival in the host. Bacteria secrete a variety of high affinity iron chelating small molecules known as siderophores that sequester and solubilize iron and facilitate iron entry into the bacterial cell through siderophore-specific receptors [86]. The ability of these molecules to gain entry into bacterial cells has been exploited to circumvent antibiotic resistance associated with limited uptake by way of a “trojan horse” approach, in which an antibiotic is linked to a siderophore and is transported into the cell via the iron transport system. One example of this approach is the synthesis of siderophore-aminopenicillin conjugates including compound 48 (Fig. 7), wherein the β-lactam ampicillin is conjugated to a biscatecholate moiety. Several conjugates displayed activity against multiple species of Gram-negative bacteria, effecting a >1,000-fold increase in activity compared to ampicillin against P. aeruginosa and Stenotrophomonas maltophilia laboratory strains and a >100-fold increase in activity against Enterobacteriaceae laboratory strains in vitro. The conjugates were active against carbapenem-resistant clinical isolates of P. aeruginosa and S. maltophilia and exhibited in vivo activity in a murine model of P. aeruginosa infection. Importantly 48 and several other conjugates tested were not substrates for the major P. aeruginosa efflux pumps MexAB-OprM, MexCD-OprJ, or MexEF-OprN. Although these β-lactam-siderophore conjugates are not typical adjuvants in that they are a single molecular entity, they represent a promising approach to circumventing permeability-mediated antibiotic resistance [77].
6 Interfering with Signaling Systems
An alternative to directly inhibiting the enzyme or protein responsible for imparting resistance to the bacteria, such as occurs with β-lactamase and efflux pump inhibitors, is to interfere with the ability of the bacteria to “switch on” their resistance machinery. Bacteria use various pathways to detect the presence of antibiotics and respond by either activating or upregulating the production of the proteins required for resistance. One such example is the detection of β-lactam antibiotics by the BlaR1 and MecR1 sensor systems of MRSA. Upon sensing a β-lactam antibiotic, BlaR1 and MecR1 are phosphorylated and subsequently initiate a series of events that culminate in the expression of genes encoding for a β-lactamase and penicillin binding protein 2a (PBP2a), respectively, both of which play a role in the resistance of MRSA to β-lactam antibiotics. A recent screen by the Mobashery group of a library of protein kinase inhibitors for their ability to lower the oxacillin MIC against MRSA identified the known mammalian serine/threonine kinase inhibitor 49 (Fig. 8). Compound 49 reduced the extent of phosphorylation of BlaR1 in the presence of a penicillin that otherwise was capable of inducing resistance, which correlated to a lack of induction of the bla system, accounting for the reduction in oxacillin resistance. Analog synthesis of compound 49 led to compound 50, which lowered oxacillin MICs by up to 64-fold at a concentration of just 7 μg/mL [87].


Inhibitors of bacterial signaling systems involved in antibiotic resistance
One of the most prominent signaling and regulatory systems used by bacteria to control behaviors in response to external stimuli and stresses are two-component systems (TCS). TCS regulate the expression of genes in response to external stimuli and control a number of bacterial behaviors including sporulation, competence, biofilm formation, pathogenesis, and antibiotic resistance across multiple bacterial species [88, 89]. TCS are activated by a variety of factors such as pH, nutrient level, redox state, osmotic pressure, quorum signals, and the presence of antibiotics. These systems are composed of a histidine kinase and a response regulator. In response to the external stimulus, the histidine kinase undergoes autophosphorylation at a conserved histidine residue. This phosphate group is then transferred to a conserved aspartate residue on the response regulator, inducing a conformational rearrangement of the protein and leading to DNA binding of the phosphorylated response regulator and subsequent alteration of gene expression [90]. Many histidine kinases can also act as phosphatases and dephosphorylate the response regulator, thus allowing precise control of gene expression in response to environmental change [88]. TCS are ubiquitous among bacteria and possess common structural motifs not found in higher eukaryotes, potentially allowing selective targeting by small molecules. Furthermore, most TCS are not essential for bacterial growth under normal conditions, and therefore small-molecule targeting of the TCS may place reduced selection pressure on the bacteria to acquire resistance to the action of the small molecule through mutation. The TCS systems are a potentially powerful, and thus far underexploited, antibiotic adjuvant target for small-molecule development [91].
One example of a TCS that plays an important role in antibiotic resistance is the VraRS system in MRSA. Inactivation of vraRS decreases methicillin resistance independently of mecA expression, supporting the hypothesis that the methicillin-resistant phenotype is influenced by factors other than PBP2a [92]. These factors are potential targets for the potentiation of methicillin and other β-lactams. The VraRS system has been proposed as a “sentinel” system capable of sensing perturbation of cell wall synthesis and coordinating a response that involves the mobilization of genes essential for high-level antibiotic resistance [93]. VraRS is unique among TCS involved with respect to resistance to cell wall-acting antibiotics in that it mediates the response to disruption of both the early and late steps of cell wall biosynthesis. VraRS senses cell wall damage and coordinates a general cell envelope stress response involving numerous genes necessary for cell wall synthesis that are referred to collectively as the cell wall stress stimulon (CWSS). VraRS is induced by the exposure of S. aureus to several antibiotics that act upon the cell wall, including β-lactams, glycopeptides, and bacitracin [90]. MRSA mutants that are deficient in vraRS are treatable with an oxacillin regimen in vivo, thus validating the potential of targeting this TCS as an antibiotic adjuvant strategy [94]. Recently, a third member of the vra operon encoded directly upstream of vraS and designated vraT was reported to be essential for optimal expression of methicillin resistance [92]. The vraT gene encodes a putative membrane protein VraT that has a regulatory role in the aforementioned VraRS-mediated cell wall stress stimulon. Similar to deletion of vraR and vraS, the deletion of vraT improved the outcome of oxacillin therapy in vivo [92]. VraT thus represents an additional target for the potentiation of β-lactam activity against MRSA. In addition to the role played by VraRS, it has been suggested that multiple TCSs might also be responsible for the variation in β-lactam resistance levels observed in clinical strains of MRSA [95]. Several 2-aminoimidazole compounds derived from the marine natural products oroidin and bromoageliferin suppress MRSA resistance to the β-lactams [96,97,98,99,100]. The lead compound from this series, compound 51 (Fig. 8), suppressed resistance in a number of MRSA clinical isolates by up to 512-fold via a mechanism that was dependent on the presence of VraRS [100]. The phenothiazine antipsychotic drug thioridazine 34 mentioned earlier also was reported to potentiate oxacillin and dicloxacillin against MRSA independently of the PBP2a-mediated resistance mechanisms and to repress transcription of several genes belonging to the vraRS regulon [101,102,103].
TCS also play a role in antibiotic resistance in Gram-negative bacteria. Another 2-aminoimidazole compound 52 (Fig. 8) suppresses resistance to colistin in both A. baumannii and K. pneumoniae [104]. Colistin resistance in A. baumannii is mediated by the PmrAB TCS, which controls the expression of the phosphoethanolamine transferase PmrC that catalyzes modification of the lipid A component of the outer membrane. This modification results in a reduction in the net negative charge of the membrane that subsequently leads to a reduced affinity for colistin and other cationic antimicrobials [105, 106]. Compound 52 downregulates the pmrCAB operon in A. baumannii. Mass spectrometry-based analysis of the lipid A fraction of bacteria treated with 52 showed a significant reduction in phosphoethanolamine modification, indicating that 52 potentiates colistin activity through a mechanism that involves the PmrAB TCS. Supporting the argument that targeting nonessential pathways may lead to a reduction in evolutionary pressure to develop resistance, it was reported that both colistin-susceptible and colistin-resistant bacteria that were serially passaged in the presence of colistin and 52 were unable to evolve resistance to the combination treatment [104]. The development of a second generation of analogs of compound 52 led to 53, which exhibits a greater degree of resistance suppression, lower inherent bacterial toxicity, and an expanded spectrum of activity that includes P. aeruginosa [107].
A fluorescence polarization displacement assay developed by the Carlson lab has facilitated high-throughput screening for compounds targeting the ATP-binding pocket that is specific to histidine kinases. Three representative histidine kinases were used with the aim of discovering inhibitors capable of targeting multiple histidine kinases. Nine compounds that inhibited at least two of the kinases were identified, including the aminobenzothiazole 54 (Fig. 8) which was subsequently shown to exhibit moderate antibiotic activity against E. coli and Bacillus subtilis [108]. Although the compounds identified in this screen were not investigated for their adjuvant activity, this high-throughput assay for the identification of broad-spectrum histidine kinases inhibitors is a significant step toward active scaffolds that potentiate antibiotic activity through the TCS. Thiophene 55 is another histidine kinase inhibitor, identified from a virtual screen against the essential B. subtilis histidine kinase WalK. It inhibits autophosphorylation of WalK and other histidine kinases in vitro. Compound 55 was selective for histidine kinases, as it lacked activity against the bacterial serine/threonine kinase IreK and exhibited moderate antibiotic activity against several bacterial species in addition to adjuvant activity at sub-MIC levels. Compound 55 potentiated the activity of β-lactam antibiotics against S. aureus and E. coli and of ofloxacin against one strain of E. coli [109].
The SOS DNA repair and mutagenesis pathway plays a role in antibiotic resistance by enabling adaptive resistance mutations and the acquisition of resistance genes and is thought to be induced by bactericidal antibiotics. The SOS pathway involves activation of the recombinase RecA, inactivation of the LexA repressor, and expression of SOS response genes that facilitate antibiotic resistance. RecA repairs DNA that has been damaged either directly by the antibiotic or by oxidative stress resulting from the action of the antibiotic, resulting in increased antibiotic tolerance. E. coli strains lacking recA exhibit increased sensitivity to bactericidal antibiotics. Further contributing to the role RecA has in antibiotic resistance, RecA-mediated repair induces a hypermutable state that promotes acquisition of antibiotic resistance. RecA is therefore a promising adjuvant target. A series of phthalocyanine tetrasulfonate (PcTs)-based inhibitors of RecA including iron(III) phthalocyanine-4,4′,4′′,4′′′-tetrasulfonic acid (Fe-PcTs) have recently been reported that prevent antibiotic-induced activation of the SOS pathway in E. coli and potentiate the activity of the bactericidal antibiotics ciprofloxacin, kanamycin, and ampicillin. The PcTs inhibitors also decreased the acquisition of resistance mutations in vitro, and both potentiated ciprofloxacin activity and reduced resistance acquisition in vivo in a neutropenic murine bacterial thigh infection model. Inhibitors of RecA such as Fe-PcTs have the advantage of being able to be combined with a wide range of antibiotics and could potentially offer a general strategy for prolonging the life span of antibiotics [110].
7 Targeting Nonessential Steps in Cell Wall Synthesis
Many cellular functions, including bacterial cell wall synthesis, are carried out not by discreet enzymatic transformations, but by multiple proteins that work together, some of which are interdependent and some of which are functionally redundant. In S. aureus deletion of seven of the nine genes that encode enzymes involved in peptidoglycan synthesis had no effect on cell growth or morphology in vitro, but did result in a marked increase in susceptibility to cell wall-acting antibiotics [111]. In practical terms, genes that are nonessential in certain environments can become essential upon changes in environmental conditions (such as the presence of antibiotics) making them an ideal adjuvant target. For example, PBP2 is essential in methicillin-sensitive S. aureus (MSSA) but not in MRSA due to the presence of PBP2a, which takes over transpeptidase activity. However, PBP2 becomes essential in MRSA in the presence of β-lactam antibiotics, as cooperation between the transglycosylase domain of PBP2 and the transpeptidase domain of PBP2a is required for survival [111]. The identification of nonessential steps in cell wall biosynthesis, and the effect that inhibiting these steps has upon the potency of cell wall-acting antibiotics, has been the subject of much investigation of late with regard to identifying potential adjuvant targets.
Wall teichoic acid (WTA) is a glycophosphate polymer that is cross-linked to peptidoglycan in the Gram-positive cell wall and has several functions including a role in cell growth and division. Despite this, bacteria lacking WTA are viable, and the genes encoding the proteins involved in the early stages of WTA synthesis are nonessential. Inactivation or alteration of WTA in MRSA results in an increase in susceptibility to β-lactam antibiotics [112, 113], particularly those that exhibit selectivity for PBP2 [114], making WTA a promising potential adjuvant target. One of the nonessential genes involved in the early stages of WTA synthesis is tarO, which encodes the N-acetylglucosamine-1-phosphate transferase TarO. Both genetic and pharmacological approaches show that inactivation of TarO, in combination with inactivation of native PBPs (PBPs other than PBP2a) by a β-lactam antibiotic, is synergistic. The natural product tunicamycin 56 (Fig. 9) inhibits the UDP-HexNAc:polyprenol-P HexNAc-1-P family of enzymes that includes TarO and the essential S. aureus peptidoglycan synthesis enzyme MraY. Tunicamycin exhibits selectivity for TarO over MraY, allowing it to be used as a probe to demonstrate the potential of TarO as an adjuvant target and to demonstrate synergy with oxacillin at subinhibitory concentrations of oxacillin. Inhibition of TarO by tunicamycin specifically increased susceptibility to β-lactam antibiotics, thought to be a result of mislocalization of PBP2 or PBP2a, and did not sensitize MRSA to other classes of antibiotics including those that inhibit peptidoglycan synthesis by other mechanisms, such as vancomycin [112]. Tunicamycin itself cannot be used clinically as it possesses eukaryotic toxicity as a result of its inhibition of protein glycosylation. Therefore, the identification of selective TarO inhibitors is necessary to realize WTA inhibition as a viable adjuvant strategy for potentiation of MRSA to β-lactam antibiotics. Screening of a library of over 2,000 previously approved drugs for compounds that potentiate the activity of the PBP2-selective β-lactam cefuroxime, which is highly sensitized upon deletion of tarO, identified the antiplatelet drug ticlopidine (Ticlid) 57 (Fig. 9). Ticlopidine did not exhibit growth inhibition alone but exhibited a strong synergistic effect with cefuroxime against wild-type MRSA, an effect that was abrogated in a tarO deletion strain. Ticlopidine 57 potentiated the activity of cefuroxime against a panel of MRSA clinical isolates and also demonstrated activity in vivo in a Galleria mellonella model of infection [114]. Another screen to identify inhibitors of TarO identified the benzimidazole tarocin B 58, which displayed synergy with imipenem against MRSA. Analog synthesis identified compound 59 (Fig. 9), which retained activity across a broad spectrum of clinical isolates of both MRSA and MRSE [115].


Adjuvants that target cell wall synthesis
Yet another small-molecule library screen for β-lactam potentiation against MRSA identified the steroid-like compound murgocil 60 (Fig. 9) showing synergy with imipenem, acting in a bactericidal manner in combination but exerting only a modest bactericidal effect alone. The β-lactam potentiation activity of murgocil resulted from inhibition of the glycosyltransferase MurG, which catalyzes the final step in peptidoglycan monomer synthesis (conversion of lipid I to lipid II, the substrate for PBPs). Murgocil impaired peptidoglycan synthesis as a result of its inhibition of the biosynthesis of lipid II. Lipid II is required for proper localization of PBP2. Murgocil effected delocalization of PBP2 from the cell division septum, explaining the synergy observed with β-lactam antibiotics [116].
The highly conserved cytoskeletal protein FtsZ is a prime target for antibacterial development due to the essential role it plays in cell division [117]. Its role in the recruitment of PBPs and other downstream components of the divisome means that inhibitors of FtsZ have the potential to enhance the activity of cell wall-acting antibiotics at sub-microbicidal concentrations [118]. For example, the thiazolopyridine PC190723 61 (Fig. 9), which had been previously developed through a medicinal chemistry program for anti-staphylococcal agents that inhibit FtsZ [119], exhibited synergy with imipenem and other β-lactam antibiotics against MRSA in vitro and in vivo. This synergy was postulated to result from the concomitant delocalization of FtsZ and PBP2 (the respective targets of PC190723 and imipenem) [118]. Another FtsZ inhibitor, the quinuclidine 62, was identified from a virtual screen and exhibited synergy with methicillin and imipenem against MRSA, in addition to moderate antibacterial activity against several species of bacteria [120]. A loss-of-viability screen identified DNAC-1 63 as potentiating the activity of oxacillin against MRSA [121]. Similar to PC190723, DNAC-1 elicited the mislocalization of FtsZ and PBP2 (and also PBP4). DNAC-1 63 exhibited in vivo activity in combination with oxacillin in a murine model of MRSA infection and also exhibited in vitro activity in combination with ceftriaxone against several Gram-negative pathogens including A. baumannii, P. aeruginosa, E. coli, and K. pneumoniae [121].
An antisense-interference screen in MRSA to identify auxiliary factors that are involved in resistance to β-lactam antibiotics recently identified several additional genes that were not previously known to play such a role and therefore are potential new targets for adjuvant design. Nva-FMDP 64 (Fig. 9) is an inhibitor of the enzyme encoded by one of these genes, GlmS. GlmS is a glucosamine-6-phosphate synthase involved in the synthesis of the peptidoglycan precursor [122]. Nva-FMDP had been demonstrated previously to possess antibacterial activity against B. subtilis and S. epidermidis, but not S. aureus. Nva-FMDP strongly potentiated the activity of several β-lactam antibiotics against MRSA [122].
The polyphenol (–)-epicatechin gallate (ECg) 65 (Fig. 9) found in green tea exhibits very little intrinsic antibacterial activity against MRSA but has long been known to suppress resistance to β-lactam antibiotics [123]. ECg does not affect PBP2a production and instead acts by binding to the cytoplasmic membrane and inducing a series of reorganization steps that increase the fluidity of the membrane, increase cell size, thicken the cell wall, elicit changes in production of lysyl phosphatidylglycerol, and culminate in either the delocalization of PBP2 and eventually PBP2a, or the eradication of the cooperation between the two enzymes that is required for cell wall synthesis in the presence of ß-lactam antibiotics [124, 125]. Synthesis of a series of analogs of ECg in which hydroxyl groups were removed in a stepwise manner led to the compound 66, which lacks two hydroxyl groups from the B ring of ECg. Compound 66 exhibited increased oxacillin potentiation. Removal of additional hydroxyl groups resulted in compounds possessing higher antibacterial activity and reduced resistance-modifying activity [126].
8 Conclusions
New antibiotics are not being developed at a fast enough rate to match increasing bacterial resistance. The development of small-molecule adjuvants offers an additional avenue to combat this significant problem. Several approaches to the identification of adjuvants have been discussed, from the well-known and clinically validated approach of inhibiting β-lactamase enzymes, to more indirect approaches such as inhibiting bacterial signaling and response systems that mediate antibiotic resistance. Also discussed are adjuvants that act by preventing efflux or increasing cellular uptake of antibiotics, adjuvants that inhibit modification of either the antibiotic or its target, and finally the identification of adjuvants that act upon less obvious targets such as nonessential steps in bacterial cell wall synthesis.
References
Wright GD (2016) Antibiotic adjuvants: rescuing antibiotics from resistance. Trends Microbiol 24:862–871. doi:10.1016/j.tim.2016.06.009
Dolgin E (2010) Sequencing of superbugs seen as key to combating their spread. Nat Med 16:1054–1054. doi:10.1038/Nm1010-1054a
Lewis II JS, Owens A, Cadena J, Sabol K, Patterson JE, Jorgensen JH (2005) Emergence of daptomycin resistance in Enterococcus faecium during daptomycin therapy. Antimicrob Agents Chemother 49:1664–1665. doi:10.1128/AAC.49.4.1664-1665.2005
Gonzales RD, Schreckenberger PC, Graham MB, Kelkar S, DenBesten K, Quinn JP (2001) Infections due to vancomycin-resistant Enterococcus faecium resistant to linezolid. Lancet 357:1179. doi:10.1016/S0140-6736(00)04376-2
Pawlowski AC, Johnson JW, Wright GD (2016) Evolving medicinal chemistry strategies in antibiotic discovery. Curr Opin Biotechnol 42:108–117. doi:10.1016/j.copbio.2016.04.006
Gill EE, Franco OL, Hancock REW (2015) Antibiotic adjuvants: diverse strategies for controlling drug-resistant pathogens. Chem Biol Drug Des 85:56–78. doi:10.1111/cbdd.12478
Roemer T, Boone C (2013) Systems-level antimicrobial drug and drug synergy discovery. Nat Chem Biol 9:222–231. doi:10.1038/nchembio.1205
Rodriguez de Evgrafov M, Gumpert H, Munck C, Thomsen TT, Sommer MO (2015) Collateral resistance and sensitivity modulate evolution of high-level resistance to drug combination treatment in Staphylococcus aureus. Mol Biol Evol 32:1175–1185. doi:10.1093/molbev/msv006
Walsh C (2000) Molecular mechanisms that confer antibacterial drug resistance. Nature 406:775–781. doi:10.1038/35021219
Wright GD (2005) Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv Drug Delivery Rev 57:1451–1470. doi:10.1016/j.addr.2005.04.002
Ramirez MS, Tolmasky ME (2010) Aminoglycoside modifying enzymes. Drug Resist Updates 13:151–171. doi:10.1016/j.drup.2010.08.003
Volkers G, Palm GJ, Weiss MS, Wright GD, Hinrichs W (2011) Structural basis for a new tetracycline resistance mechanism relying on the TetX monooxygenase. FEBS Lett 585:1061–1066. doi:10.1016/j.febslet.2011.03.012
Jovetic S, Zhu Y, Marcone GL, Marinelli F, Tramper J (2010) β-Lactam and glycopeptide antibiotics: first and last line of defense? Trends Biotechnol 28:596–604. doi:10.1016/j.tibtech.2010.09.004
Drawz SM, Papp-Wallace KM, Bonomo RA (2014) New β-lactamase inhibitors: a therapeutic renaissance in an MDR world. Antimicrob Agents Chemother 58:1835–1846. doi:10.1128/AAC.00826-13
Papp-Wallace KM, Bonomo RA (2016) New β-lactamase inhibitors in the clinic. Infect Dis Clin North Am 30:441–464. doi:10.1016/j.idc.2016.02.007
Drawz SM, Bonomo RA (2010) Three decades of β-lactamase inhibitors. Clin Microbiol Rev 23:160–201. doi:10.1128/CMR.00037-09
Bush K (2015) A resurgence of β-lactamase inhibitor combinations effective against multidrug-resistant Gram-negative pathogens. Int J Antimicrob Agents 46:483–493. doi:10.1016/j.ijantimicag.2015.08.011
Ball P (2007) The clinical development and launch of amoxicillin/clavulanate for the treatment of a range of community-acquired infections. Int J Antimicrob Agents 30(Suppl 2):S113–S117. doi:10.1016/j.ijantimicag.2007.07.037
Walsh C (2003) Where will new antibiotics come from? Nat Rev Microbiol 1:65–70. doi:10.1038/nrmicro727
Shlaes DM (2013) New β-lactam-β-lactamase inhibitor combinations in clinical development. Ann N Y Acad Sci 1277:105–114. doi:10.1111/nyas.12010
Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL (2012) Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci U S A 109(29):11663–11668. doi:10.1073/pnas.1205073109
Levasseur P, Girard AM, Miossec C, Pace J, Coleman K (2015) In vitro antibacterial activity of the ceftazidime-avibactam combination against Enterobacteriaceae, including strains with well-characterized β-lactamases. Antimicrob Agents Chemother 59:1931–1934. doi:10.1128/AAC.04218-14
Klibanov OM, Phan D, Ferguson K (2015) Drug updates and approvals: 2015 in review. Nurse Pract 40:34–43. doi:10.1097/01.NPR.0000473071.26873.3c
Petersen PJ, Jones CH, Venkatesan AM, Bradford PA (2009) Efficacy of piperacillin combined with the penem β-lactamase inhibitor BLI-489 in murine models of systemic infection. Antimicrob Agents Chemother 53:1698–1700. doi:10.1128/AAC.01549-08
Bassetti M, Ginocchio F, Mikulska M (2011) New treatment options against Gram-negative organisms. Crit Care 15:215. doi:10.1186/cc9997
Paukner S, Hesse L, Prezelj A, Solmajer T, Urleb U (2009) In vitro activity of LK-157, a novel tricyclic carbapenem as broad-spectrum β-lactamase inhibitor. Antimicrob Agents Chemother 53:505–511. doi:10.1128/AAC.00085-08
Livermore DM, Mushtaq S (2013) Activity of biapenem (RPX2003) combined with the boronate β-lactamase inhibitor RPX7009 against carbapenem-resistant Enterobacteriaceae. J Antimicrob Chemother 68:1825–1831. doi:10.1093/jac/dkt118
Lapuebla A, Abdallah M, Olafisoye O, Cortes C, Urban C, Quale J, Landman D (2015) Activity of meropenem combined with RPX7009, a novel β-lactamase inhibitor, against Gram-negative clinical isolates in New York City. Antimicrob Agents Chemother 59:4856–4860. doi:10.1128/AAC.00843-15
Griffith DC, Loutit JS, Morgan EE, Durso S, Dudley MN (2016) Phase 1 study of the safety, tolerability, and pharmacokinetics of the β-lactamase inhibitor vaborbactam (RPX7009) in healthy adult subjects. Antimicrob Agents Chemother 60:6326–6332. doi:10.1128/AAC.00568-16
van Duin D, Bonomo RA (2016) Ceftazidime/avibactam and ceftolozane/tazobactam: second-generation β-lactam/β-lactamase inhibitor combinations. Clin Infect Dis 63:234–241. doi:10.1093/cid/ciw243
Walsh TR, Toleman MA, Poirel L, Nordmann P (2005) Metallo-β-lactamases: the quiet before the storm? Clin Microbiol Rev 18:306–325. doi:10.1128/CMR.18.2.306-325.2005
King AM, Reid-Yu SA, Wang W, King DT, De Pascale G, Strynadka NC, Walsh TR, Coombes BK, Wright GD (2014) Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature 510:503–506. doi:10.1038/nature13445
Cornaglia G, Giamarellou H, Rossolini GM (2011) Metallo-β-lactamases: a last frontier for β-lactams? Lancet Infect Dis 11(5):381–393. doi:10.1016/S1473-3099(11)70056-1
Nordmann P, Poirel L, Walsh TR, Livermore DM (2011) The emerging NDM carbapenemases. Trends Microbiol 19:588–595. doi:10.1016/j.tim.2011.09.005
Page MGP, Dantier C, Desarbre E, Gaucher B, Gebhardt K, Schmitt-Hoffmann A (2011) In vitro and in vivo properties of BAL30376, a β-lactam and dual β-lactamase inhibitor combination with enhanced activity against Gram-negative bacilli that express multiple β-lactamases. Antimicrob Agents Chemother 55:1510–1519. doi:10.1128/AAC.01370-10
Hinchliffe P, Gonzalez MM, Mojica MF, Gonzalez JM, Castillo V, Saiz C, Kosmopoulou M, Tooke CL, Llarrull LI, Mahler G, Bonomo RA, Vila AJ, Spencer J (2016) Cross-class metallo-β-lactamase inhibition by bisthiazolidines reveals multiple binding modes. Proc Natl Acad Sci U S A 113:E3745–E3754. doi:10.1073/pnas.1601368113
Labby KJ, Garneau-Tsodikova S (2013) Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med Chem 5(11):1285–1309. doi:10.4155/fmc.13.80
Gao F, Yan X, Shakya T, Baettig OM, Ait-Mohand-Brunet S, Berghuis AM, Wright GD, Auclair K (2006) Synthesis and SAR of truncated bisubstrate inhibitors of aminoglycoside 6'-N-acetyltransferases. J Med Chem 49:5273–5281. doi:10.1021/jm060732n
Lin DL, Tran T, Alam JY, Herron SR, Ramirez MS, Tolmasky ME (2014) Inhibition of aminoglycoside 6'-N-acetyltransferase type Ib by zinc: reversal of amikacin resistance in Acinetobacter baumannii and Escherichia coli by a zinc ionophore. Antimicrob Agents Chemother 58:4238–4241. doi:10.1128/Aac.00129-14
Li Y, Green KD, Johnson BR, Garneau-Tsodikova S (2015) Inhibition of aminoglycoside acetyltransferase resistance enzymes by metal salts. Antimicrob Agents Chemother 59:4148–4156. doi:10.1128/AAC.00885-15
Chiem K, Fuentes BA, Lin DL, Tran T, Jackson A, Ramirez MS, Tolmasky ME (2015) Inhibition of aminoglycoside 6'-N-acetyltransferase Type Ib-mediated amikacin resistance in Klebsiella pneumoniae by zinc and copper pyrithione. Antimicrob Agents Chemother 59:5851–5853. doi:10.1128/Aac.01106-15
Shakya T, Stogios PJ, Waglechner N, Evdokimova E, Ejim L, Blanchard JE, McArthur AG, Savchenko A, Wright GD (2011) A small molecule discrimination map of the antibiotic resistance kinome. Chem Biol 18:1591–1601. doi:10.1016/j.chembiol.2011.10.018
Suga T, Ishii T, Iwatsuki M, Yamamoto T, Nonaka K, Masuma R, Matsui H, Hanaki H, Omura S, Shiomi K (2012) Aranorosin circumvents arbekacin-resistance in MRSA by inhibiting the bifunctional enzyme AAC(6′)/APH(2″). J Antibiot 65:527–529. doi:10.1038/ja.2012.53
Hernick M (2013) Mycothiol: a target for potentiation of rifampin and other antibiotics against Mycobacterium tuberculosis. Expert Rev Anti-Infect Ther 11:49–67. doi:10.1586/Eri.12.152
Gutierrez-Lugo MT, Baker H, Shiloach J, Boshoff H, Bewley CA (2009) Dequalinium, a new inhibitor of Mycobacterium tuberculosis mycothiol ligase identified by high-throughput screening. J Biomol Screen 14:643–652. doi:10.1177/1087057109335743
Ramon-Garcia S, Ng C, Anderson H, Chao JD, Zheng XJ, Pfeifer T, Av-Gay Y, Roberge M, Thompson CJ (2011) Synergistic drug combinations for tuberculosis therapy identified by a novel high-throughput screen. Antimicrob Agents Chemother 55:3861–3869. doi:10.1128/Aac.00474-11
Pieren M, Tigges M (2012) Adjuvant strategies for potentiation of antibiotics to overcome antimicrobial resistance. Curr Opin Pharmacol 12:551–555. doi:10.1016/j.coph.2012.07.005
Maravic G (2004) Macrolide resistance based on the Erm-mediated rRNA methylation. Curr Drug Targets Infect Disord 4:193–202. doi:10.2174/1568005043340777
Clancy J, Schmieder BJ, Petitpas JW, Manousos M, Williams JA, Faiella JA, Girard AE, McGuirk PR (1995) Assays to detect and characterize synthetic agents that inhibit the ErmC methyltransferase. J Antibiot (Tokyo) 48:1273–1279. doi:10.7164/antibiotics.48.1273
Feder M, Purta E, Koscinski L, Cubrilo S, Maravic Vlahovicek G, Bujnicki JM (2008) Virtual screening and experimental verification to identify potential inhibitors of the ErmC methyltransferase responsible for bacterial resistance against macrolide antibiotics. ChemMedChem 3:316–322. doi:10.1002/cmdc.200700201
Webber MA, Piddock LJV (2003) The importance of efflux pumps in bacterial antibiotic resistance. J Antimicrob Chemother 51:9–11. doi:10.1093/jac/dkg050
Li XZ, Plesiat P, Nikaido H (2015) The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin Microbiol Rev 28:337–418. doi:10.1128/CMR.00117-14
Jang S (2016) Multidrug efflux pumps in Staphylococcus aureus and their clinical implications. J Microbiol 54:1–8. doi:10.1007/s12275-016-5159-z
Abreu AC, McBain AJ, Simoes M (2012) Plants as sources of new antimicrobials and resistance-modifying agents. Nat Prod Rep 29:1007–1021. doi:10.1039/c2np20035j
Markham PN, Westhaus E, Klyachko K, Johnson ME, Neyfakh AA (1999) Multiple novel inhibitors of the NorA multidrug transporter of Staphylococcus aureus. Antimicrob Agents Chemother 43:2404–2408
Markham PN, Neyfakh AA (1996) Inhibition of the multidrug transporter NorA prevents emergence of norfloxacin resistance in Staphylococcus aureus. Antimicrob Agents Chemother 40:2673–2674
Fujita M, Shiota S, Kuroda T, Hatano T, Yoshida T, Mizushima T, Tsuchiya T (2005) Remarkable synergies between baicalein and tetracycline, and baicalein and β-lactams against methicillin-resistant Staphylococcus aureus. Microbiol Immunol 49:391–396
Kalle AM, Rizvi A (2011) Inhibition of bacterial multidrug resistance by celecoxib, a cyclooxygenase-2 inhibitor. Antimicrob Agents Chemother 55:439–442. doi:10.1128/AAC.00735-10
Sabatini S, Gosetto F, Serritella S, Manfroni G, Tabarrini O, Iraci N, Brincat JP, Carosati E, Villarini M, Kaatz GW, Cecchetti V (2012) Pyrazolo[4,3-c][1,2]benzothiazines-5,5-dioxide: a promising new class of Staphylococcus aureus NorA efflux pump inhibitors. J Med Chem 55:3568–3572. doi:10.1021/jm201446h
Lepri S, Buonerba F, Goracci L, Velilla I, Ruzziconi R, Schindler BD, Seo SM, Kaatz GW, Cruciani G (2016) Indole-based weapons to fight antibiotic resistance: a SAR study. J Med Chem 59:867–891. doi:10.1021/acs.jmedchem.5b01219
Kaatz GW, Moudgal VV, Seo SM, Kristiansen JE (2003) Phenothiazines and thioxanthenes inhibit multidrug efflux pump activity in Staphylococcus aureus. Antimicrob Agents Chmother 47:719–726. doi:10.1128/Aac.47.2.719-726.2003
Mirza ZM, Kumar A, Kalia NP, Zargar A, Khan IA (2011) Piperine as an inhibitor of the MdeA efflux pump of Staphylococcus aureus. J Med Microbiol 60:1472–1478. doi:10.1099/jmm.0.033167-0
Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, Blais J, Cho D, Chamberland S, Renau T, Leger R, Hecker S, Watkins W, Hoshino K, Ishida H, Lee VJ (2001) Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob Agents Chemother 45:105–116. doi:10.1128/AAC.45.1.105-116.2001
Pages JM, Amaral L (2009) Mechanisms of drug efflux and strategies to combat them: challenging the efflux pump of Gram-negative bacteria. Biochim Biophys Acta 1794:826–833. doi:10.1016/j.bbapap.2008.12.011
Lomovskaya O, Bostian KA (2006) Practical applications and feasibility of efflux pump inhibitors in the clinic – a vision for applied use. Biochem Pharmacol 71:910–918. doi:10.1016/j.bcp.2005.12.008
Chalhoub H, Saenz Y, Rodriguez-Villalobos H, Denis O, Kahl BC, Tulkens PM, Van Bambeke F (2016) High-level resistance to meropenem in clinical isolates of Pseudomonas aeruginosa in the absence of carbapenemases: role of active efflux and porin alterations. Int J Antimicrob Agents 48:740–743. doi:10.1016/j.ijantimicag.2016.09.012
Bohnert JA, Schuster S, Kern WV, Karcz T, Olejarz A, Kaczor A, Handzlik J, Kiec-Kononowicz K (2016) Novel piperazine arylideneimidazolones inhibit the AcrAB-TolC pump in Escherichia coli and simultaneously act as fluorescent membrane probes in a combined real-time influx and efflux assay. Antimicrob Agents Chemother 60:1974–1983. doi:10.1128/Aac.01995-15
Lawler AJ, Ricci V, Busby SJW, Piddock LJV (2013) Genetic inactivation of acrAB or inhibition of efflux induces expression of ramA. J Antimicrob Chemother 68:1551–1557. doi:10.1093/jac/dkt069
Bailey AM, Paulsen IT, Piddock LJV (2008) RamA confers multidrug resistance in Salmonella enterica via increased expression of acrB, which is inhibited by chlorpromazine. Antimicrob Agents Chemother 52:3604–3611. doi:10.1128/AAC.00661-08
Kinana AD, Vargiu AV, May T, Nikaido H (2016) Aminoacyl β-naphthylamides as substrates and modulators of AcrB multidrug efflux pump. Proc Natl Acad Sci U S A 113:1405–1410. doi:10.1073/pnas.1525143113
Saw HT, Webber MA, Mushtaq S, Woodford N, Piddock LJV (2016) Inactivation or inhibition of AcrAB-TolC increases resistance of carbapenemase-producing Enterobacteriaceae to carbapenems. J Antimicrob Chemother 71:1510–1519. doi:10.1093/jac/dkw028
Piddock LJV, Garvey MI, Rahman MM, Gibbons S (2010) Natural and synthetic compounds such as trimethoprim behave as inhibitors of efflux in Gram-negative bacteria. J Antimicrob Chemother 65:1215–1223. doi:10.1093/jac/dkq079
Handzlik J, Szymanska E, Chevalier J, Otrgbska E, Kiec-Kononowicz K, Pages JM, Alibert S (2011) Amine-alkyl derivatives of hydantoin: new tool to combat resistant bacteria. Eur J Med Chem 46:5807–5816. doi:10.1016/j.ejmech.2011.09.032
Otrebska-Machaj E, Chevalier J, Handzlik J, Szymanska E, Schabikowski J, Boyer G, Bolla JM, Kiec-Kononowicz K, Pages JM, Alibert S (2016) Efflux pump blockers in Gram-negative bacteria: the new generation of hydantoin based-modulators to improve antibiotic activity. Front Microbiol 7:622. doi:10.3389/fmicb.2016.00622
Cox G, Koteva K, Wright GD (2014) An unusual class of anthracyclines potentiate Gram-positive antibiotics in intrinsically resistant Gram-negative bacteria. J Antimicrob Chemother 69:1844–1855. doi:10.1093/jac/dku057
Taylor PL, Rossi L, De Pascale G, Wright GD (2012) A forward chemical screen identifies antibiotic adjuvants in Escherichia coli. ACS Chem Biol 7:1547–1555. doi:10.1021/cb300269g
Mollmann U, Heinisch L, Bauernfeind A, Kohler T, Ankel-Fuchs D (2009) Siderophores as drug delivery agents: application of the “Trojan Horse” strategy. Biometals 22:615–624. doi:10.1007/s10534-009-9219-2
Livermore DM (1990) Antibiotic uptake and transport by bacteria. Scand J Infect Dis Suppl 74:15–22. doi:10.3109/inf.1990.22.suppl-74.01
Lambert PA (2002) Cellular impermeability and uptake of biocides and antibiotics in Gram-positive bacteria and mycobacteria. J Appl Microbiol 92(Suppl):46S–54S. doi:10.1046/j.1365-2672.92.5s1.7.x
Zabawa TP, Pucci MJ, Parr Jr TR, Lister T (2016) Treatment of Gram-negative bacterial infections by potentiation of antibiotics. Curr Opin Microbiol 33:7–12. doi:10.1016/j.mib.2016.05.005
Viljanen P, Vaara M (1984) Susceptibility of Gram-negative bacteria to polymyxin-B nonapeptide. Antimicrob Agents Chemother 25:701–705. doi:10.1128/AAC.25.6.701
Ofek I, Cohen S, Rahmani R, Kabha K, Tamarkin D, Herzig Y, Rubinstein E (1994) Antibacterial synergism of polymyxin-B nonapeptide and hydrophobic antibiotics in experimental Gram-negative infections in mice. Antimicrob Agents Chemother 38:374–377. doi:10.1128/AAC.38.2.374
Pages JM, Peslier S, Keating TA, Lavigne JP, Nichols WW (2016) Role of the outer membrane and porins in susceptibility of β-lactamase-producing Enterobacteriaceae to ceftazidime-avibactam. Antimicrob Agents Chemother 60:1349–1359. doi:10.1128/Aac.01585-15
Ejim L, Farha MA, Falconer SB, Wildenhain J, Coombes BK, Tyers M, Brown ED, Wright GD (2011) Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat Chem Biol 7:348–350. doi:10.1038/nchembio.559
Lamers RP, Cavallari JF, Burrows LL (2013) The efflux inhibitor phenylalanine-arginine β-naphthylamide (PAβN) permeabilizes the outer membrane of Gram-negative bacteria. PLoS One 8:e60666. doi:10.1371/journal.pone.0060666
Hider RC, Kong X (2010) Chemistry and biology of siderophores. Nat Prod Rep 27(5):637–657. doi:10.1039/b906679a
Boudreau MA, Fishovitz J, Llarrull LI, Xiao QB, Mobashery S (2015) Phosphorylation of BlaR1 in manifestation of antibiotic resistance in methicillin-resistant Staphylococcus aureus and its abrogation by small molecules. ACS Infect Dis 1(10):454–459. doi:10.1021/acsinfecdis.5b00086
Gotoh Y, Eguchi Y, Watanabe T, Okamoto S, Doi A, Utsumi R (2010) Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr Opin Microbiol 13:232–239. doi:10.1016/J.Mib.2010.01.008
Mejean V (2016) Two-component regulatory systems: the moment of truth. Res Microbiol 167(1):1–3. doi:10.1016/j.resmic.2015.09.004
Gardete S, Wu SW, Gill S, Tomasz A (2006) Role of VraSR in antibiotic resistance and antibiotic-induced stress response in Staphylococcus aureus. Antimicrob Agents Chemother 50:3424–3434. doi:10.1128/Aac.00356-06
Worthington RJ, Blackledge MS, Melander C (2013) Small-molecule inhibition of bacterial two-component systems to combat antibiotic resistance and virulence. Future Med Chem 5:1265–1284. doi:10.4155/fmc.13.58
Boyle-Vavra S, Yin SH, Jo DS, Montgomery CP, Daum RS (2013) VraT/YvqF is required for methicillin resistance and activation of the VraSR regulon in Staphylococcus aureus. Antimicrob Agents Chemother 57:83–95. doi:10.1128/Aac.01651-12
Belcheva A, Golemi-Kotra D (2008) A close-up view of the VraSR two-component system. A mediator of Staphylococcus aureus response to cell wall damage. J Biol Chem 283:12354–12364. doi:10.1074/jbc.M710010200
Jo DS, Montgomery CP, Yin S, Boyle-Vavra S, Daum RS (2011) Improved oxacillin treatment outcomes in experimental skin and lung infection by a methicillin-resistant Staphylococcus aureus isolate with a vraSR operon deletion. Antimicrob Agents Chemother 55:2818–2823. doi:10.1128/AAC.01704-10
Matsuo M, Kato F, Oogai Y, Kawai T, Sugai M, Komatsuzawa H (2010) Distinct two-component systems in methicillin-resistant Staphylococcus aureus can change the susceptibility to antimicrobial agents. J Antimicrob Chemother 65:1536–1537. doi:10.1093/jac/dkq141
Rogers SA, Huigens RW, Cavanagh J, Melander C (2010) Synergistic effects between conventional antibiotics and 2-aminoimidazole-derived antibiofilm agents. Antimicrob Agents Chemother 54:2112–2118. doi:10.1128/AAC.01418-09
Su Z, Peng L, Worthington RJ, Melander C Evaluation of 4,5-disubstituted-2-aminoimidazole-triazole conjugates for antibiofilm/antibiotic resensitization activity against MRSA and Acinetobacter baumannii. ChemMedChem 6:2243–2251. doi:10.1002/cmdc.201100316
Su ZM, Peng LL, Melander C (2012) A modular approach to the synthesis of 1,4,5-substituted-2-aminoimidazoles. Tetrahedron Lett 53:1204–1206. doi:10.1016/J.Tetlet.2011.12.090
Yeagley AA, Su Z, McCullough KD, Worthington RJ, Melander C (2013) N-substituted 2-aminoimidazole inhibitors of MRSA biofilm formation accessed through direct 1,3-bis(tert-butoxycarbonyl)guanidine cyclization. Org Biomol Chem 11:130–137. doi:10.1039/c2ob26469b
Harris TL, Worthington RJ, Melander C (2012) Potent small-molecule suppression of oxacillin resistance in methicillin-resistant Staphylococcus aureus. Angew Chem Int Ed 51:11254–11257. doi:10.1002/anie.201206911
Klitgaard JK, Skov MN, Kallipolitis BH, Kolmos HJ (2008) Reversal of methicillin resistance in Staphylococcus aureus by thioridazine. J Antimicrob Chemother 62:1215–1221. doi:10.1093/jac/dkn417
Bonde M, Hojland DH, Kolmos HJ, Kallipolitis BH, Klitgaard JK (2011) Thioridazine affects transcription of genes involved in cell wall biosynthesis in methicillin-resistant Staphylococcus aureus. FEMS Microbiol Lett 318:168–176. doi:10.1111/j.1574-6968.2011.02255.x
Poulsen MO, Jacobsen K, Thorsing M, Kristensen NR, Clasen J, Lillebaek EM, Skov MN, Kallipolitis BH, Kolmos HJ, Klitgaard JK (2013) Thioridazine potentiates the effect of a β-lactam antibiotic against Staphylococcus aureus independently of mecA expression. Res Microbiol 164:181–188. doi:10.1016/j.resmic.2012.10.007
Harris TL, Worthington RJ, Hittle LE, Zurawski DV, Ernst RK, Melander C (2014) Small molecule downregulation of PmrAB reverses Lipid A modification and breaks colistin resistance. ACS Chem Biol 9:122–127. doi:10.1021/cb400490k
Beceiro A, Llobet E, Aranda J, Bengoechea JA, Doumith M, Hornsey M, Dhanji H, Chart H, Bou G, Livermore DM, Woodford N (2011) Phosphoethanolamine modification of lipid A in colistin-resistant variants of Acinetobacter baumannii mediated by the pmrAB two-component regulatory system. Antimicrob Agents Chemother 55:3370–3379. doi:10.1128/AAC.00079-11
Arroyo LA, Herrera CM, Fernandez L, Hankins JV, Trent MS, Hancock RE (2011) The pmrCAB operon mediates polymyxin resistance in Acinetobacter baumannii ATCC 17978 and clinical isolates through phosphoethanolamine modification of lipid A. Antimicrob Agents Chemother 55:3743–3751. doi:10.1128/AAC.00256-11
Brackett CM, Furlani RE, Anderson RG, Krishnamurthy A, Melander RJ, Moskowitz SM, Ernst RK, Melander C (2016) Second generation modifiers of colistin resistance show enhanced activity and lower inherent toxicity. Tetrahedron 72:3549–3553. doi:10.1016/j.tet.2015.09.019
Wilke KE, Francis S, Carlson EE (2015) Inactivation of multiple bacterial histidine kinases by targeting the ATP-binding domain. ACS Chem Biol 10:328–335. doi:10.1021/cb5008019
Boibessot T, Zschiedrich CP, Lebeau A, Benimelis D, Dunyach-Remy C, Lavigne JP, Szurmant H, Benfodda Z, Meffre P (2016) The rational design, synthesis, and antimicrobial properties of thiophene derivatives that inhibit bacterial histidine kinases. J Med Chem 59:8830–8847. doi:10.1021/acs.jmedchem.6b00580
Alam MK, Alhhazmi A, DeCoteau JF, Luo Y, Geyer CR (2016) RecA inhibitors potentiate antibiotic activity and block evolution of antibiotic resistance. Cell Chem Biol 23:381–391. doi:10.1016/j.chembiol.2016.02.010
Reed P, Atilano ML, Alves R, Hoiczyk E, Sher X, Reichmann NT, Pereira PM, Roemer T, Filipe SR, Pereira-Leal JB, Ligoxygakis P, Pinho MG (2015) Staphylococcus aureus survives with a minimal peptidoglycan synthesis machine but sacrifices virulence and antibiotic resistance. PLoS Pathog 11:e1004891. doi:10.1371/journal.ppat.1004891
Campbell J, Singh AK, Santa Maria Jr JP, Kim Y, Brown S, Swoboda JG, Mylonakis E, Wilkinson BJ, Walker S (2011) Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem Biol 6:106–116. doi:10.1021/cb100269f
Wang H, Gill CJ, Lee SH, Mann P, Zuck P, Meredith TC, Murgolo N, She X, Kales S, Liang L, Liu J, Wu J, Santa Maria J, Su J, Pan J, Hailey J, McGuinness D, Tan CM, Flattery A, Walker S, Black T, Roemer T (2013) Discovery of wall teichoic acid inhibitors as potential anti-MRSA β-lactam combination agents. Chem Biol 20:272–284. doi:10.1016/j.chembiol.2012.11.013
Farha MA, Leung A, Sewell EW, D'Elia MA, Allison SE, Ejim L, Pereira PM, Pinho MG, Wright GD, Brown ED (2013) Inhibition of WTA synthesis blocks the cooperative action of PBPs and sensitizes MRSA to β-lactams. ACS Chem Biol 8(1):226–233. doi:10.1021/cb300413m
Labroli MA, Caldwell JP, Yang C, Lee SH, Wang H, Koseoglu S, Mann P, Yang SW, Xiao J, Garlisi CG, Tan C, Roemer T, Su J (2016) Discovery of potent wall teichoic acid early stage inhibitors. Bioorg Med Chem Lett 26:3999–4002. doi:10.1016/j.bmcl.2016.06.090
Mann PA, Muller A, Xiao L, Pereira PM, Yang C, Ho Lee S, Wang H, Trzeciak J, Schneeweis J, Dos Santos MM, Murgolo N, She X, Gill C, Balibar CJ, Labroli M, Su J, Flattery A, Sherborne B, Maier R, Tan CM, Black T, Onder K, Kargman S, Monsma Jr FJ, Pinho MG, Schneider T, Roemer T (2013) Murgocil is a highly bioactive staphylococcal-specific inhibitor of the peptidoglycan glycosyltransferase enzyme MurG. ACS Chem Biol 8:2442–2451. doi:10.1021/cb400487f
Hurley KA, Santos TM, Nepomuceno GM, Huynh V, Shaw JT, Weibel DB (2016) Targeting the bacterial division protein FtsZ. J Med Chem 59(15):6975–6998. doi:10.1021/acs.jmedchem.5b01098
Tan CM, Therien AG, Lu J, Lee SH, Caron A, Gill CJ, Lebeau-Jacob C, Benton-Perdomo L, Monteiro JM, Pereira PM, Elsen NL, Wu J, Deschamps K, Petcu M, Wong S, Daigneault E, Kramer S, Liang L, Maxwell E, Claveau D, Vaillancourt J, Skorey K, Tam J, Wang H, Meredith TC, Sillaots S, Wang-Jarantow L, Ramtohul Y, Langlois E, Landry F, Reid JC, Parthasarathy G, Sharma S, Baryshnikova A, Lumb KJ, Pinho MG, Soisson SM, Roemer T (2012) Restoring methicillin-resistant Staphylococcus aureus susceptibility to β-lactam antibiotics. Sci Transl Med 4:126ra135. doi:10.1126/scitranslmed.3003592
Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE, Heal JR, Sheridan JM, Aiwale ST, Chauhan PK, Srivastava A, Taneja A, Collins I, Errington J, Czaplewski LG (2008) An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 321:1673–1675. doi:10.1126/science.1159961
Chan FY, Sun N, Leung YC, Wong KY (2015) Antimicrobial activity of a quinuclidine-based FtsZ inhibitor and its synergistic potential with β-lactam antibiotics. J Antibiot (Tokyo) 68:253–258. doi:10.1038/ja.2014.140
Nair DR, Monteiro JM, Memmi G, Thanassi J, Pucci M, Schwartzman J, Pinho MG, Cheung AL (2015) Characterization of a novel small molecule that potentiates β-lactam activity against Gram-positive and Gram-negative pathogens. Antimicrob Agents Chemother 59:1876–1885. doi:10.1128/AAC.04164-14
Lee SH, Jarantow LW, Wang H, Sillaots S, Cheng H, Meredith TC, Thompson J, Roemer T (2011) Antagonism of chemical genetic interaction networks resensitize MRSA to β-lactam antibiotics. Chem Biol 18:1379–1389. doi:10.1016/j.chembiol.2011.08.015
Stapleton PD, Shah S, Anderson JC, Hara Y, Hamilton-Miller JM, Taylor PW (2004) Modulation of β-lactam resistance in Staphylococcus aureus by catechins and gallates. Int J Antimicrob Agents 23:462–467. doi:10.1016/j.ijantimicag.2003.09.027
Bernal P, Lemaire S, Pinho MG, Mobashery S, Hinds J, Taylor PW (2010) Insertion of epicatechin gallate into the cytoplasmic membrane of methicillin-resistant Staphylococcus aureus disrupts penicillin-binding protein (PBP) 2a-mediated β-lactam resistance by delocalizing PBP2. J Biol Chem 285:24055–24065. doi:10.1074/jbc.M110.114793
Rosado H, Turner RD, Foster SJ, Taylor PW (2015) Impact of the β-lactam resistance modifier (–)-epicatechin gallate on the non-random distribution of phospholipids across the cytoplasmic membrane of Staphylococcus aureus. Int J Mol Sci 16:16710–16727. doi:10.3390/ijms160816710
Palacios L, Rosado H, Micol V, Rosato AE, Bernal P, Arroyo R, Grounds H, Anderson JC, Stabler RA, Taylor PW (2014) Staphylococcal phenotypes induced by naturally occurring and synthetic membrane-interactive polyphenolic β-lactam resistance modifiers. PLoS One 9:e93830. doi:10.1371/journal.pone.0093830
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG, part of Springer Nature
About this chapter
Cite this chapter
Melander, R.J., Melander, C. (2017). Antibiotic Adjuvants. In: Fisher, J.F., Mobashery, S., Miller, M.J. (eds) Antibacterials. Topics in Medicinal Chemistry, vol 25. Springer, Cham. https://doi.org/10.1007/7355_2017_10
Download citation
DOI: https://doi.org/10.1007/7355_2017_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-68096-5
Online ISBN: 978-3-319-68097-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)