
Abstract
Granulomatosis with polyangiitis (GPA) is an antineutrophil cytoplasmic antibody (ANCA)-associated disorder with necrotic vasculitis of small- and medium-size arteries and veins. In the literature, there are many case reports of patients with GPA of different, sometimes unusual, clinical manifestations. In this paper, we present difficulties that accompanied the process of diagnosing GPA in a 54-year-old symptomatic patient who was. Computer tomography scans showed numerous tumor-like lesions of various and irregular sizes in both lungs. Positron emission tomography scans suggested a lymphoproliferative disease, otherwise failing to provide a clue concerning its nature or localization. After a series of diagnostic twists and turns, inclusive of bronchoalveolar lavage, cervical mediastinoscopy, paratracheal lymph biopsy, and histopathologic examinations, and other tests, the diagnosis of GPA was established as the most probable. The patient was acutely treated with loading doses of methylprednisolone and cyclophosphamide, gradually tapered off during the long-term follow-up. He was discharged from the hospital in a good condition. We conclude that GPA is an uncommon disease with indistinctive signs, which raises the risk of its being overlooked. A diagnostic algorithm is required for patients with suspected GPA. A timely diagnosis is essential as the disease may quickly progress into renal or multiorgan dysfunction, and ultimately lead to death if untreated. Pulmonary involvement may also suggest neoplastic changes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Granulomatosis with polyangiitis (GPA, Wegener’s granulomatosis) is an antineutrophil cytoplasmic antibody (ANCA)-associated disorder with necrotic vasculitis of small- and medium-size arteries and veins. The course of illness differs between the patients, but in most cases, it appears in the upper respiratory tract, lungs, and kidneys, although other tissues or organs may be affected. Each year, there are 2.1–14.4 new cases per million people diagnosed in Europe, but other sources report the prevalence is even five times higher (Yates and Watts 2017; Greco et al. 2016). According to the American College of Rheumatology, the diagnostic criteria for the classification of GPA are granulomatous inflammation on biopsy, oral ulcers, nasal discharge, abnormal findings on the chest radiograph like nodules, cavities, and fixed infiltrates, and abnormal urinary sediment consisting of red cells casts or more than five red blood cells per high power field (Leavitt et al. 2010). The presence of two or more abnormalities is associated with a high possibility of the disease. ANCA antibodies play a role in recognition but are not decisive in the diagnostic process due to relatively low sensitivity. Herein we report diagnosing difficulties concerning the GPA.
2 Case Report
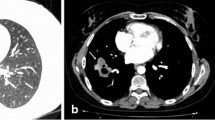
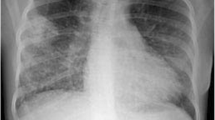
In March 2017, a 54-year-old male was admitted to the Department of Internal Medicine of the Fourth Military Teaching Hospital in Wroclaw, Poland due to cough, chronic fever of 38-40 °C accompanied by nausea, vomiting, and dripping sweat. The patient presented a history of 3 months of severe dry cough with breathlessness. He was treated empirically in an outpatient clinic with broad-spectrum antibiotics, which was of no avail. The patient was having type II diabetes, erosive gastropathy, colon diverticulosis, psoriasis, and a focal thyroid lesion in the right lobe. The ear-nose-throat (ENT) examination showed a left-sided polyp between the middle and inferior nasal concha. The patient’s general condition was poor at admission. A chest X-ray showed several round shadows of about 15 mm in diameter in the right lower pulmonary field (Fig. 1). Computed tomography (CT) examination showed numerous tumor-like lesions of irregular size in both lungs that suggested neoplastic infiltrations (Fig. 2), but without any diagnostic conclusion. The transthoracic needle biopsy and bronchoscopy remained diagnostically inconclusive. Bronchoalveolar lavage (BAL) revealed numerous neutrophils (71.0%), lymphocytes (18.0%), and an insignificant number of bacteria like Streptococcus oralis, Neisseria spp., Escherichia coli, and Streptococcus salivarius with a reduced number of macrophages (11.0%). Mycobacterium tuberculosis was not present in the culture.

Chest X-ray showing round bilateral opacities in the lungs

Computed tomography (CT) scan of the chest showing nodular shadows in lung parenchyma (marked by arrows)
The antinuclear antibody (ANA) panel was negative. Unfortunately, ANCA tests were not performed at this stage. Positron emission tomography (PET) confirmed the presence of an active proliferative process, but it failed to localize the primary neoplasm. The scans suggested an extension of diagnostics toward the lymphoproliferative disorder. Therefore, cervical mediastinoscopy was performed under general anesthesia. The paratracheal lymph nodes (station 3) and right lower paratracheal nodes (station 4R) were harvested. However, histopathological examination revealed normal lymphoid tissue without any malignancy. The open lung biopsy was considered, but the patient’s general condition was too poor to perform an invasive procedure.
As the patient was suspected of tuberculosis, having cough and general malaise, he was admitted to the Pulmonary Division at the end of April 2017. A repeated X-ray showed several tubercles in both lungs. Symptomatic treatment was implemented consisting of ibuprofen, dalteparin, megestrol, and paracetamol with tramadol. Additionally, metformin was given to control diabetes. Based on medical history, documentation, X-ray examinations, and a consultation by an experienced pulmonologist, the GPA was highly suspected.
The patient’s condition was deteriorating, and after a conclusive exclusion of neoplasm, he was referred to the Department of Rheumatology at Wroclaw Medical University. He had problems with keeping a vertical body position due to the progressing general malaise. C-reactive protein and the erythrocyte sedimentation rate were increased. Signs of renal impairment appeared consisting of protein 0.5 mg/dL (norm: 0.0–0.2 mg/dL), white blood cells 5–8 (norm: 0–5), and erythrocytes 9–13 (norm: 0–3) per high power field in the urine. ENT consultation and CT imaging showed polyps whose histopathological examination was unremarkable. Since the deterioration of the patient’s condition accelerated, steroid treatment was implemented. Methylprednisolone (1 g/day) and cyclophosphamide (1.2 g/day) were administered intravenously for 3 days accompanied by 2-mercaptoethanesulfonate sodium (Mesna; 0.72 g/day) for protection of the urinary bladder against inflammation. The patient’s condition and inflammatory biochemical indices improved. Then, oral methylprednisolone was continued in a dose of 52 mg/day, divided into 32 mg at 8 am, 16 mg at 11 am, and 4 mg at 1 pm. The total dose was gradually tapered off to 48 mg/day after 1 month and 40 mg/day after another month. Treatment consisting of six infusions of cyclophosphamide in a dose of 0.75 mg/kg every 4 weeks also was prescribed. However, steroid therapy worsened hyperglycemia, which required the introduction of insulin therapy. Additionally, trimethoprim-sulfamethoxazole in a dose of 480 mg bid was given for 5 months. Results of tests and cyclophosphamide courses are depicted in Table 1. Since the diagnosis of GPA was not confirmed by histopathology, the patient was scheduled for a monitoring chest CT examination 3 months after discharge.
3 Discussion
The GPA is a multisystem disease that has many faces, ranging from rhinosinusitis and cough to renal and lung involvement (Comarmond and Cacoub 2014). It is prevalent in Caucasians, with about equal gender distribution (Lutalo and D’Cruz 2014). The onset most often occurs at the age of 45–60 but may appear at another age as well (Filocamo et al. 2017). The pathophysiological mechanism involves necrotizing vasculitis of small- and medium-sized arteries and veins with intra- and extra-vascular granulomas that are most common in the upper and lower respiratory systems (Savage et al. 2000). Diagnostic criteria for GPA are based on clinical manifestations like inflammatory consolidations in chest X-ray, oral and nasal inflammatory lesions, and abnormal urinary sediment.
Herein, we presented the diagnostic workup of a patient whose clinical history at presentation was hardly suggestive of GPA. Additionally, it appears there is no distinct procedure algorithm for GPA-suspected cases, which complicates and delays the workup, notably when there is a need to differentiate a nature of tumor-like mass in the lungs visualized in X-rays. In such cases, tuberculosis, neoplasm, GPA, rheumatoid arthritis, and other illnesses should be considered. In the presented report, cough, breathlessness, enlarged mediastinal nodules, a chronic febrile state with accompanying nausea, vomiting, and dripping sweat were notable at the first admission. However, cough is present, on average, in a minority 34% of patients with GPA, which makes it hard of a diagnostic value. The X-ray and CT scans showed several round shadows in the lungs. To rule out tuberculosis, the bronchoalveolar lavage was microbiologically tested. However, no mycobacteria were cultivated. The PET scan gave a false positive outcome suggesting neoplasm, albeit giving no clue to its localization. Doctors became misled to search for the primary origin of neoplasm. Mediastinoscopy was performed and numerous specimens of lymphatic nodes were examined for histopathology but failed to conclusively set the diagnosis of neoplasm. The patient’s mistaken declaration that the polyp-like mass between nasal conchae detected at the ENT examination had been an inborn lesion further impeded the making of a correct diagnosis. Acquired abnormalities in the nasal cavity are often symptomatic for the GPA (Comarmond and Cacoub 2014; Masiak et al. 2017). The exclusion of neoplasm and nonconfirmation of tuberculosis has strengthened the probability of GPA, which was confirmed by positive cANCA antibody tests, and the patient was finally treated in the Rheumatology Ward as of May 2017. Abnormalities in the urine test were suggestive of kidney involvement in the form of glomerulonephritis, which is a frequent accompaniment of GPA. The immunosuppressive and anti-inflammatory treatment consisting of cyclophosphamide and methylprednisolone was commenced, leading to a prompt improvement in the patient’s condition. Trimethoprim-sulfamethoxazole was added to eliminate the microbes that could potentially underlie the disease (Hasan et al. 2019).
We conclude that GPA is an uncommon disease with symptoms and signs of little specificity, making the diagnosis difficult and raising the risk of overlooking. There is a strong need for a diagnostic algorithm for patients with suspected GPA, notably in cases suggesting pulmonary neoplastic changes. Time is of paramount essence in diagnosing GPA because the disease may rapidly progress into glomerulonephritis with renal failure or multiorgan dysfunction with the ultimate death.
References
Comarmond C, Cacoub P (2014) Granulomatosis with polyangiitis (Wegener): clinical aspects and treatment. Autoimmun Rev 13(11):1121–1125
Filocamo G, Torreggiani S, Agostoni C, Esposito S (2017) Lung involvement in childhood onset granulomatosis with polyangiitis. Pediatr Rheumatol Online J 15(1):28
Greco A, Marinelli C, Fusconi M, Macri GF, Gallo A, De Virgilio A, Zambetti G, de Vincentiis M (2016) Clinic manifestations in granulomatosis with polyangiitis. Int J Immunopathol Pharmacol 29(2):151–159
Hasan MR, Sakibuzzaman M, Tabassum T, Moosa SA (2019) A case of granulomatosis with polyangiitis (Wegener’s granulomatosis) presenting with rapidly progressive glomerulonephritis. Cureus 11(10):e5896
Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, Arend WP, Alabrese LH, Fries JF, Lie JT, Lightwood RW Jr (2010) The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum 33(8):1101–1107
Lutalo PM, D’Cruz DP (2014) Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis). J Autoimmun 48-49:94–98
Masiak A, Zdrojewski Z, Pęksa R, Smoleńska Ż, Czuszyńska Z, Siemińska A, Kowalska B, Stankiewicz C, Rutkowski B, Bułło-Piontecka B (2017) The usefulness of histopathological examinations of non-renal biopsies in the diagnosis of granulomatosis with polyangiitis. Reumatologia 55(5):230–236
Savage CO, Harper L, Cockwell P, Adu D, Howie AJ (2000) ABC of arterial and vascular disease: vasculitis. BMJ 320(7245):1325–1328
Yates M, Watts R (2017) ANCA-associated vasculitis. Clin Med (Lond) 17(1):60–64
Acknowledgments
This work was funded by the 2019-2022 program “Regional Initiative of Excellence” financed by the Ministry of Science and Higher Education in Poland; the project #016/RID/2018/19.
Conflicts of Interest
The authors declare no conflicts of interest concerning this article.
Ethical Approval
All procedures performed in this study involving a human participant followed the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The study was approved by the Ethics Committee of Wroclaw Medical University in Wroclaw, Poland.
Informed Consent
Written informed consent was obtained from the patient presented in the article.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Tomczyk, B., Janeczko, Z., Kruczkowska, A., Maciążek-Chyra, B., Tański, W., Chabowski, M. (2020). Thoracic Manifestation of Granulomatosis with Polyangiitis: A Case Report. In: Pokorski, M. (eds) Medical Research and Innovation. Advances in Experimental Medicine and Biology(), vol 1324. Springer, Cham. https://doi.org/10.1007/5584_2020_600
Download citation
DOI: https://doi.org/10.1007/5584_2020_600
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-70205-2
Online ISBN: 978-3-030-70206-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)