
Abstract
Helicobacter pylori infection of the human stomach is associated with chronic gastritis, peptic ulcer disease or gastric carcinoma, and thus a high burden for the public health systems worldwide. Fortunately, only a small subfraction of up to 15–20% of infected individuals will develop serious complications. Unfortunately, it is not always known upfront, who will be affected by serious diesease outcome. For risk stratifications, it is therefore necessary to establish a common terminology and grading system, that can be applied worldwide to compare population data. The updated Sydney System for classification of gastritis with its semi-quantitative analogue scale is the system, that is currently used worldwide. Additionally, pathologists should always try to classify the etiology of the inflammatory infiltrates in the stomach to instruct the clinicians for choosing a proper treatment regime. Risk factors such as intestinal metaplasia, atrophy and scoring systems to classify these risk factors into a clinical context such as OLGA and OLGIM are discussed. Also, special forms of gastritis like lymphocytic gastritis, autoimmune gastritis and peptic ulcer disease are explained and discussed e.g. how to diagnose and how to treat. Extra-gastric sequelae of H. pylori infections inside and outside the stomach are shown in this chapter as well. Important host and bacterial risk factors such as pathogenicity islands are dicussed to draw a complete landscape around a H. pylori infection, that still can be diagnosed in patients. However, it needs to be noted that some countries have almost no H. pylori infection anymore, while others have still a very high frequency of infections with or without serious complications. The understanding and application of risk assessements may help to save money and quality of life. Extra-gastric H. pylori infections are rarely reported in the literature until today. The pathogenitiy is still under debate, but especially in the bile ducts and gallbladder, several pathological conditions may be also based on H. pylori infection, and will be also discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Helicobacter pylori Gastritis
1.1 The Sydney Classification System
H. pylori is a spiral-shaped, Gram-negative bacterium (Fig. 1a), that specifically colonizes the gastric epithelium in about 50% of the human world population (Kusters et al. 2006). This infection induces a state of chronic inflammation due to highly sophisticated mechanisms by H. pylori crosstalk with the host immune system (Pachathundikandi et al. 2016). For a long time, the classification and grading of gastritis was sort of an open field. Until a meeting in Sydney in 1986, and another meeting in Houston in 1994, there were mainly two ideas how to classify gastritis (Dixon et al. 1996). One concept was just to detect and describe inflammatory reactions and cells in various versions, whereas especially in Germany a system was proposed that focused more on the etiology of the inflammatory infiltrates (Fig. 2). The idea behind the second system was that clinicians can use a scheme of diagnosis and etiology of inflammation better than just the knowledge, that there are a certain numbers of neutrophils somewhere in the samples. This approach is coming from a background that the strength of a pathologist is to give etiological causes for given pathological changes, and that this increases the precision of diagnostics, and thus clinical impact for potential treatments and the clinical course. When the original Sydney System was proposed in 1986, there was also an endoscopic part, but it turned out that the classification of gastritis is a sole histological diagnosis and endoscopy cannot reliably generate reproducible descriptions or diagnoses. Thus, the endoscopic approach was dropped over the years and classification of gastritis became a sole histological diagnosis.


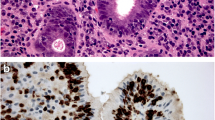
Microscopic analysis of different Helicobacter species (Warthin-Starry stain, 400x). (a) Helicobacter pylori are small bacteria (3–5 μm), often found attached to the foveolar epithelium (arrows). (b) Non-pylori Helicobacter are formerly known as Helicobacter heilmannii (circles) are longer and more spiraled than H. pylori and tend to stay in the foveolar lumen


Histology showing typical moderate, moderately active H. pylori gastritis. The band – like lymphoplasmocytic infiltrate can be found in the upper half of the mucosa. Neutrophilic infiltrates in the surface epithelium are marked with arrows
The really new proposals of the updated Sydney System were the introduction of mandatory 4-tiered semiquantitative analog scales for antrum and corpus mucosa, scoring the number of lymphocytes/plasma cells as expression of chronic inflammation, the number of neutrophils as expression of acute inflammation, the density of Helicobacter bacteria on the mucosal surfaces, assessing intestinal metaplasia and grading atrophy (Dixon et al. 1996). Traditionally, the semiquantitative grades are stated as “none”, “mild”, “moderate” or “severe”, respectively.
Bystanders such as lymphoid aggregates and follicles, mucous depletion and reactive epithelial changes were seen as optional criteria. It was recommended to name the etiology of the scored criteria after grading of the chronic and acute inflammatory infiltrates. Thus, the fathers of the Sydney System managed to create a combination of the two main scoring classifications used at that time. This helped to spread the use of the Sydney System worldwide and led to a uniform way to classify gastritis within a very short time period.
1.2 Intestinal Metaplasia and Atrophy
Intestinal metaplasia (IM) in the stomach is defined as goblet cell bearing metaplasia either of colonic type or small intestinal type, the latter including Paneth cells (Giroux and Rustqi 2017). This is seen in sites of mucosal damage of any type including chemical-reactive gastritis and autoimmune gastritis, and can be found both in the antrum and corpus mucosa. In many cases it does accompany gastric atrophy, which is defined as a loss of gastric glands, a hallmark of the pathological pattern. The goblet cells in IM are known to produce Mucin-2, which hampers the ability of H. pylori to adhere to the epithelium, and thus in cases with severe IM often no bound H. pylori can be detected anymore. The odds ratios (ORs) for IM and atrophy are to be seen around 5–7, whereas first degree relatives of patients with gastric cancer have an OR of 10, individuals with pan-gastritis with equal inflammatory infiltrates in the antrum and the corpus show an OR of about 15 to develop a gastric carcinoma. The 10 years risk for developing cancer in the stomach with IM and/or atrophy is less than 1.8% (Reddy et al. 2016). In contrast, the OR of a so-called corpus predominant gastritis with more severe inflammatory infiltrates in the corpus is believed to reach almost 35 for malignant transformation. The OR numbers show how the risk for patients with IM and/or atrophy has to be compared with other risk factors, that are much higher, and it is well known that the frequency of IM and atrophy decreased enormously since the 1970s as compared to data from today’s populations.
In 1994, an update of the Sydney System during the Houston workshop has shown that the biopsy protocol should be changed in a way that biopsies of the incisura are not obligate anymore (Dixon et al. 1996). The only shortcoming of the Sydney System may be that the requested biopsies from antrum and corpus may lead to a correct histological diagnosis in a certain high percentage of the cases, but patchiness of infiltrates can make it sometimes rather bothersome to identify the correct diagnosis, especially since the recommended sites for the biopsies are not those sites, where most of the IM and atrophy can be found. This leads to the idea that atrophy and IM should be graded separately, also because inter-and intraobserver variations of grading atrophy are somewhat poor with the Sydney System. Separate grading systems like operative link for gastritis assessment (OLGA) (Rugge et al. 2005) and operative link on IM assessment (OLGIM) (Capelle et al. 2010) were introduced in recent years, but made the situation somewhat more complicated, than it actually was. The shortcomings of poor inter-and intraobserver variations were not improved.
The observation that patients with multifocal atrophy and/or IM may have a higher risk for malignant transformation than those without is valid for H. pylori induced gastritis only. However, OLGA and OLGIM are applied to all various etiologies of gastritis leading to the situation that young patients with chemical reactive gastritis and foci of IM and/or atrophy get high scores in OLGA and/or OLGIM, suggesting a high risk for malignant transformation and regular endoscopic follow-up. It is also known for a long time that patients with duodenal ulcer during the course of H. pylori infection have a very low risk of malignant transformation, but can end up in high OLGA and OLGIM scores. A vice versa effect is known for patients with autoimmune gastritis in corpus and normal antrum mucosa showing low scores in OLGA and OLGIM leading to the recommendation that no follow-up is required, although they have higher prevalence of malignancy, especially for gastric neuroendocrine tumors (NET), squamous cell carcinoma of the esophagus and distal gastric adenocarcinoma (Ye and Nyren 2003).
Unfortunately, protagonists of OLGA and OLGIM seem to have dominated guideline meetings and forced OLGA and OLGIM into most European guidelines. The problem with OLGA and OLGIM is, however, that they are overrating atrophy and IM in a way that mixes up high risk and low risk individuals and are underrating the etiology of gastritis. This leads to frightened patients in the end and has been shown no benefit in form of number of saved lives so far, and probably never will. Besides this, there is also some criticism concerning the methods used against OLGA and OLGIM, evaluating the risk for gastric cancer development. It is also not clear why OLGA and OLGIM use four stages to conclude two risk groups, low risk and high risk.
Additionally, the widely used term “atrophic gastritis” is used in the literature for H. pylori induced gastric atrophy as well as autoimmune gastritis associated atrophy, in some papers there isn’t even a distinction between the two. Sometimes even atrophy during the course of or due to chemical reactive gastritis may have been included in those studies. This situation leads to reports of cancer risks and survival rates of a mixed patient cohort since etiology and thus risk for malignant transformation of atrophy due to H. pylori infection and autoimmune gastritis may differ.
In recent studies, growing skepticism was raised towards the impact of IM on gastric cancer, due to the fact that it’s not the IM that progresses to gastric cancer, but the destabilization of a gastric stem cell, which causes malignant transformation and growth of malignant cells (Graham and Zou 2018).
In conclusion, the Sydney System should still be used for all assessments of gastric specimen and the etiology of gastritis, always seen as integral part of the histological diagnosis. All further classifications such as OLGA and OLGIM should not be used in routine cases. The latter is against European and national guidelines, but makes a lot of sense due to the shortcomings of these additional scoring systems.
1.3 The Individuality of H. pylori Gastritis
H. pylori infection leads to a combined active and chronic inflammation in the gastric mucosa. The density of acute and chronic inflammatory infiltrates and their distribution throughout the stomach shows high variability of changes depending on H. pylori presence, duration of the infection, but also modifications due to medications like acetylsalicylic acids (ASA) or proton pump inhibitors (PPI). Both groups of drugs lower the number of H. pylori on the epithelium both in antrum and corpus. PPI medication often normalizes the antrum mucosa, whereas the status of the corpus mucosa switches to a corpus predominant gastritis with severe active inflammatory infiltrates within the corpus mucosa. This is the rationale why in long term PPI treatment a H. pylori infection should be excluded since there is an elevated risk for malignant transformation in individuals with predominant corpus gastritis, even when there is no such case described under PPI therapy until now in the literature (Sipponen and Stolte 1997). In ASA medication, the antrum can also normalize, but the corpus doesn’t reveal more severe inflammatory infiltrates. This shows why always two biopsies from the antrum and the corpus are recommended by the updated Sydney System to ensure a high probability of a correct histological assessment of the status of the gastric mucosa with the correct given etiology in case there are any inflammatory infiltrates.
Besides the variability of immune reactions in different patients, the variability of the disease can also be explained by the presence of H. pylori strains that contain different factors of virulence. As discussed in this book, not all H. pylori strains possess the cagA gene, whereas all have the vacA gene, but not all isolates express the more virulent vacA s1/m1 allele product. The presence of the cagA gene usually is a marker for cag pathogenicity island, which leads to the translocation of the CagA protein (Backert et al. 2015). Injected CagA can then be tyrosine-phosphorylated by Src and Abl kinases, which induces a change in cell morphology and provokes pro-inflammatory and mitogenic responses (Naumann et al. 2017). These genes are expressed in differing combinations in the various H. pylori strains throughout the world (Mueller et al. 2012). In eastern Asia nearly all Helicobacter strains express the cagA gene, regardless of developing certain diseases. So this alone is not the solution to the individually different reactions to H. pylori infection.
There are also differences in the CagA protein, for example the number and configuration of the tyrosine phosphorylation motifs (EPIYA) at the carboxy-terminal end of the protein (Lind et al. 2014, 2016). Of the four known EPIYAs, the EPIYA-type C is predominantly found in Western countries, whereas EPIYA type D almost exclusively exists in East Asia. On the other hand, the vacA gene includes three alleles with slightly differing codes in different strains, forming proteins of varying virulence (Posselt et al. 2013).
Of course, this is only the tip of the iceberg. Many more genetic variations have been found in the different H. pylori strains across the world. Maybe these discriminations are leading to the eminently varying rates of gastric ulcer and gastric cancer and other complications of a H. pylori infection in different countries (da Costa et al. 2015), and should be investigated in more detail in future.
1.4 H. pylori vs. Other Helicobacter Species
It took until 1984 when Marshall and Warren discovered the pathogenic nature of H. pylori in the human stomach (Marshall and Warren 1984). Even that the WHO graded H. pylori as a carcinogen of class I, this didn’t lead to a complete stop of the discussion how pathogenic H. pylori really is. The arguments and facts against its cancerogenic potential are, however, rather minor.
It is now known for around two decades that mixed infections of the human stomach can occur by different Helicobacter species, most of them usually colonizing also cats and dogs, and thus could be seen as a zoonotic disease. Besides H. pylori, more spiral-shaped Helicobacter species have been sporadically detected in the human stomach, and were named Helicobacter heilmannii (Fig. 1b). Gene sequencing revealed that H. heilmannii does not represent one, but several different species. H. heilmanii type I is similar to Helicobacter suis, whereas H. heilmannii type II represents different species like Helicobacter felis, Helicobacter bizzozeronii, Helicobacter salomonis and Candidatus H. heilmannii (Kubota-Aizawa et al. 2017). Other non-pylori Helicobacter species have also been found in the human stomach, like Candidatus Helicobacter bovis, Candidatus H. suis, Helicobacter cinaedi and many more (Bauwens et al. 2018; Smet et al. 2018). Mixed infections with more than one Helicobacter species have been found by RNA analysis in up to 10% of the infections (De Groote et al. 2005).
Morphologically, it is difficult to detect these co-infections since Helicobacter harbors an enormous capacity of adapting its shape. Sole “Helicobacter heilmannii” infections can be identified on the base of longer shape and a more pronounced spiral form. But as shown above, H. heilmannii is more a morphological description than a species diagnosis and should rather be stated as “Helicobacter heilmannii-like organisms (HHLO)” (Goji et al. 2015) or “non-Helicobacter pylori Helicobacter (NHPH) (De Groote et al. 2005)”. Fortunately, this seems not to play any role for an eradication therapy, since all these Helicobacter species in the stomach are susceptible for the regular antibiotic treatment regimen, unless there are antibiotic resistances. One major difference between H. heilmannii and H. pylori, however, lies in the discussion whether H. heilmannii shows an increased risk for low grade MALT lymphoma compared to H. pylori, but this is still an ongoing discussion in the community.
It needs to be noted, however, that according to textbooks so called coccoid forms of Helicobacter should not be diagnosed without “normal” forms of Helicobacter bacteria in the gastric mucosa aside, since the coccoid forms are not believed to be viable especially with no “normal shaped” Helicobacter bacteria being around.
1.5 Special Forms of Gastritis
1.5.1 Atrophic Gastritis
As mentioned above, an ongoing H. pylori gastritis can lead to atrophy of both antrum and corpus mucosa, also known as gastritis with atrophy. The atrophic gastric mucosa can also show metaplastic changes such as IM and pancreatic-acinar metaplasia or fibrosis. In total corpus atrophy the elimination of the parietal cells leads to iron deficiency anemia first in the clinical course, hypochlorohydria and later within decades to Vitamin B12 deficiency. Multifocal atrophy of stomach mucosa is associated with higher risk for gastric adenocarcinoma (Rugge et al. 2005).
Eradication of Helicobacter can stop and sometimes reverse atrophy in the corpus mucosa, but not in antrum mucosa. IM also stays after eradication therapy and at least does not progress (Wang et al. 2011).
1.5.2 Autoimmune Gastritis
Autoimmune gastritis is caused by autoantibodies against parietal cells (PCAs) (Fig. 3). Those antibodies have been reported to be present in 19.5% of patients recruited during a general health check at a general practitioners’ office. The numbers are increasing with H. pylori infection and age (Kulnigg-Dabsch 2016). PCAs are reactive against the proton pump mechanism in gastric parietal cells. A molecular similarity of the proton pump and H. pylori antigens has been reported, but it is unclear, which role the bacterium really plays in the pathogenesis of autoimmune gastritis. Destruction of the parietal cells leads to atrophy of the corpus mucosa with IM (Fig. 4). Individuals with autoimmune gastritis have not an elevated risk for gastric carcinoma, but reveal an increased rate of neuroendocrine tumors. Autoimmune gastritis has a different risk profile, etiology and even microbiome than H. pylori induced gastritis with atrophy and should be distinguished from the latter (Parsons et al. 2017). Many recent studies on atrophy and IM combine often all kinds of atrophy and IM, and do not distinguish between the etiologies. Thus, this is the problem with the reporting systems OLGA and OLGIM, as mentioned above.


Histology of autoimmune gastritis. (a) Autoimmune gastritis with severe atrophy and focal remnants of oxyntic mucosa are indicated (stars). Intestinal metaplasia with Paneth cell metaplasia (blue arrows). (b) Autoimmune gastritis with active inflammation (black arrows) and destruction of oxyntic glands (star)


Histology of autoimmune gastrisis with complete atrophy and intestinal metaplasia (arrows). Neuroendocrine tumor nests are indicated (stars)
1.5.3 Ex-Helicobacter-Gastritis
A very controversial form of gastritis is the so-called Ex-H. pylori-gastritis or Post-H. pylori-gastritis (Fig. 5), aiming to charcterize the state after successful Helicobacter eradication therapy (Oberhuber and Haidenthaler 2000; Livzan et al. 2004). There are no convincing sets of histological datasets in the literature, giving precise criteria how to diagnose. Some pathologists even deny the existence of such a gastritis form. Many colleagues refuse to use any specific term for a stomach, that has undergone successful eradication since for the vast majority of patients no precise clinical information is available regarding whether an eradication therapy has been undertaken and when. Most pathologists also like to have an information about Helicobacter serology to be sure about the diagnosis. But even this request will always leave some individuals behind, since the so-called serological scar (with persistent anti-Heliocbacter antibodies in serum after a successful eradication therapy) as a marker of a prior infection is not present in every patient after successful eradication therapy, since the duration of the presence may vary individually. It needs also to be taken into account, that it is not clear when a stomach is completetly “healed” and with no pathological changes anymore after eradication therapy. What most pathologists deny is, that one can diagnose such a form of gastritis. On the other hand with some experience, pathologists can very well diagnose an Ex-Helicobacter-gastritis: the number of lymphocytes and plasma cells is still mildly elevated with basal lymphoid aggregates or remnants of lymphoid follicles in antrum and corpus. Antrum and corpus biopsies show a mild not active gastritis according to the Sydney System (Dixon et al. 1996). It also needs to be noted that Ex-Helicobacter-gastritis and chemical reactive gastritis and even stomachs with no pathological changes may be hard to differentiate. Here, the corpus mucosa helps: when there are basal lymphoid aggregates or follicles present in the corpus of adults the pathologist could go for Ex-Helicobacter-gastritis, whenever the corpus shows no pathological changes most likely a chemical reactive gastritis in the antrum, which can be diagnosed unless the antrum shows no pathological changes, also. In the latter case, we would diagnose a gastric mucosa with no pathological changes. Attention should be paid in children with lymphocytic aggregates in corpus mucosa since these are considered physiological. From our daily routine we know, that there are some individuals in whom the stomach normalizes very fast after succuessful eradication, whereas in some others the picture of Ex-Helicobacter-gastritis stays for decades. Thus, it seems that there is a very individual variance of the duration of the histological picture.


Histology of post-H. pylori gastritis. (a) Nearly complete atrophy in corpus mucosa. Focal remnants of oxyntic glands are marked (stars).(b) Severe atrophy in the antrum mucosa with intestinal metaplasia (black arrows) including metaplastic Paneth cells (blue arrow)
The rationale why a pathologist should always try to diagnose a Ex-Helicocater-gastritis lies in the fact, that there is a point of no return for the development of a gastric carcinoma, and thus the risk of developing gastric malignancy even after successful eradication still persists to a somewhat smaller degree, and thus is different from a gastric mucosa that never harbored an Helicobacter infection. The argument that it cannot be diagnosed in all potential cases with certainty and with no serum titres or much clinical information about a possible eradication therapy does not really count since antibiotic treatment given for other reasons than Helicobacter may have led to eradication of the bacteria in the stomach, also.
1.5.4 Lymphocytic Gastritis
Lymphocytic gastritis shows an increase of lymphocytes in the lamina propria of gastric mucosa and additionally elevated counts of intraepithelial lymphocytes (IEL) above 25 per 100 epithelial cells (Fig. 6). In about a third of patients lymphocytic gastritis is associated with celiac disease, another third with H. pylori infection and the other patients have various causes for lymphocytic gastritis like varioliform gastritis or Crohn’s disease. Unlike lymphocytic gastritis in celiac disease there is no antrum dominant inflammation reported in the H. pylori associated cases. The lymphocytes usually are positive for CD3 and CD8. Many patients with H. pylori-induced lymphocytic gastritis show only a low count of bacteria, in some there is only serological proof of the infection (Madisch et al. 2006).


Histology of lymphocytic gastritis. (a) Overview showing mononuclear cells in the mucosa. (b) Dense inflammatory infiltrates in the surface epithelium are marked with arrows. (c) Dense lymphocytic infiltration. The darker, variously shaped nuclei belong to lymphocytes, the lighter, oval shaped to epithelial cells. More than 25 lymphocytes per 100 epithelial cells are needed for the diagnosis
Together with the lymphocytosis patients commonly develop atrophy in antrum and corpus mucosa and foveolar hyperplasia. Those also show increased proliferation in the foveolar epithelium. After eradication therapy even patients who were initially negative for H. pylori have been reported to improve serologically and histologically with improvement of mucosal atrophy in the corpus but not in the antrum mucosa and lower epithelial proliferation (Mäkinen et al. 2003). Thus, lympocytic gastritis is seen as a special form of Helicobacter infection that should be treated even if the bacteria cannot be detected morphologically.
2 H. pylori-Induced Diseases Other Than Gastritis
2.1 Gastro-Esophageal Reflux Disease
It is known for a long time, that H. pylori is capable to buffer the gastric acid by its ammonium secretion leading to a higher pH than without Helicobacter. The whole discussion on positive effects of a H. pylori infection began, when Labenz and co-workers (1997) published a study that was never intended to answer the question whether eradication may provoke gastro-esophageal reflux disease. Basically, it was demonstrated that about 25.8% of the patients cured from H. pylori infection in case of a duodenal ulcer will develop reflux disease within 3 years and only 12.9% in case the infection was ongoing. Risk factors identified are male sex, severity of corpus gastritis, and weight gain. Several comments and studies were following, some of which were partly supporting, others partly denying it. Progressive studies even suggested not to treat any H. pylori infection since this may escape from the reflux disease-Barrett-adenocarcinoma-sequence. When looking unemotionally at the numbers, it becomes very clear, that the whole discussion makes no sense at all. It needs to be noted that H. pylori eradication doesn’t induce reflux disease, but may unmask a prior existing reflux disease.
In Germany, with a population of 85 million inhabitants, there is an annual incidence of approximately 21,000 gastric carcinomas compared to 2,500 Barrett adenocarcinomas. Even if an eradication therapy against H. pylori would somehow trigger the gastro-esophageal reflux disease, we theoretically could “save” 2,500 lives from Barrett’s carcinoma, but would rather lose 21,000 according to gastric carcinoma. As albeit gastro-esophageal reflux disease is only unmasked by eradication therapy, we theoretically have the chance to save not 21,000 people, only from gastric cancer but also 2,500 from Barrett’s carcinoma, because unmasked reflux disease can be treated easily by standard medications.
2.2 Gastric and Duodenal Ulcer
In the 1960s, it was well known, that the risk of peptic ulcer in the stomach and the duodenum was still rising, but it was also known that there was a cohort phenomenon e.g. in the British population. Those born between 1870 and 1890 had the highest risk for ulcer disease. Susser and co-workers (1962) speculated that the affected individuals grew older and since their proportion in the population declined over time, and they were able to predict correctly from their birth-cohort analysis of peptic ulcer mortality in England and Wales between 1900 and 1950. The future decline of peptic ulcer disease correctly appeared 10–20 years prior to its actual occurrence. A birth cohort phenomenon suggests an influence by acquired relevant risk factors of a disease that occurs early in life (besides genetic bases). The marked changes of gastric and duodenal ulcer disease over time indicate that their presence was influenced by exogenous risk factors. Comparing frequencies, age and birth dates from numerous countries in 1987 showed, that the environmental factors must have taken effect prior to the age of 15 (Sonnenberg 1987). At that time, H. pylori was not yet discussed but could explain the findings very well. Thus, a lot of the disease burden and effects of a Helicobacter infection were already known before H. pylori was first described in 1984 (Warren and Marshall 1984).
2.3 Extra-Gastric Helicobacter Infections
Recently, there were cases in our routine biopsy practice showing no pathological changes of the gastric mucosa, but detection of H. pylori bacteria in a stool test was noted. It is known that the stool test is very sensitive (Iannone et al. 2018). Since this was seen independently across several laboratories, these observations lead to the idea that Helicobacter species may also colonize the human body outside the stomach. The clinical implications and whether there is a need to treat the infection, and if yes, how, still remains unclear. Interestingly, numerous case reports mainly in Asian populations, but also in European, have described that H. plyori can be found in the gallbladder, sometimes in conjunction with or without Helicobacter in the stomach. A reason for these sparse reports may be that gallbladders are not routinely checked for H. pylori or colonization by other bacteria. In addition, it is known that Helicobacter may colonize gastric heterotopias in the esophagus and small bowel (Meckel’s diverticulum) and rectum (Dye et al. 1990), leading there to rare ulcerations and (very rarely) to malignant transformations (Pech et al. 2001).
Several reports on extra-gastric Helicobacter species associated infections, such as Helicobacter hepaticus, pointed out early to the possibility that Helicobacter species could also survive outside the stomach (Kawaguchi et al. 1996; Chen et al. 2007; Bansal et al. 2012; Zhou et al. 2013).
In recent years a preference for non-invasive Helicobacter tests, especially after Helicobacter eradication therapy, evolved. Serological tests harbor the disadvantage of the so-called serum scar. This means that positive antibodies in some individuals can be detected also after successful antibiotic treatment of a H. pylori infection (Backert et al. 2018). The false positive tests lead to the development of more sensitive tests, like Helicobacter stool tests (Vaira et al. 1999).
Finally, numerous cases for a successful Helicobacter eradication therapy have been documented in the stomach by means of histology, which showed positive findings in the stool test against H. pylori. It has to be noted that Helicobacter stool tests are rather sensitive and specific, and false positive tests are very rare (da Silva-Etto et al. 2017). The number of these cases is increasing to probably less than 3% of all cases with successful eradication therapy, where a stomach colonization by H. pylori can definitively be excluded (unpublished data). Various reports on Helicobacter species detection in the gallbladder led us also to screen cases with acute cholecystitis and subsequent cholecystectomy for Helicobacter species. Among more than 1,000 gallbladder specimen within the past 15 years we identified 1 case with H. pylori based on histology, immunohistochemistry, histochemistry, sequencing and positive culture among other concomitant bacteria in the gallbladder. In this specific case it turned out that a previously eradicated gastric H. pylori had the same fingerprint-pattern and cagA sequence compared to the strain found in the gallbladder (Backert et al. 2018). This finding is interesting for several reasons:
-
(a)
H. pylori is able to survive without the acidic environment outside the stomach
-
(b)
Extra-gastric H. pylori seems to be less sensitive for antibiotics eradication therapy than gastric H. pylori
-
(c)
It needs to be studied whether H. pylori infection of the gallbladder can cause or fuel acute cholecystitis
It is striking that despite case reports have been published on this topic, the topic has not drawn any wider clinical interest. Thus, more systematic studies on large patient cohorts are required in the future.
A possible relation of gastric H. pylori infection to extra gastric diseases is based on more or less reliable up to obscure data on breast and prostate diseases (Kast 2007). Some seem to have rather strong evidence such as thrombocytic purpura and iron deficiency anemia (Malfertheiner and Selgrad 2010) but others as sudden infant death don’t (Vieth et al. 2001). Dermatologists also have some interest in H. pylori due to the therapy of urticaria that can be caused by H. pylori infection (Rebora et al. 1995).
3 Concluding Remarks
In conclusion, there is strong evidence for the influence of Helicobacter infection on the course of gastric and certain extra-gastric diseases, but others lack such evidence and seem more or less like urban legends. In our present review, we gave deeper explanations and rationales behind risk factors for malignant transformation and the etiology of different forms of gastritis for the daily routine use. Up till now, substantial knowledge has been acquired for H. pylori effects on the gastric mucosa including precancerous conditions and risk factors. During the last decades, it became evident that also Helicobacter species other than H. pylori may colonize the stomach in humans and animals. Extra gastric colonization by H. pylori may gain more interest in the future since carcinomas may also arise on these colonization sites outside the stomach.
References
Backert S, Tegtmeyer N, Fischer W (2015) Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol 10(6):955–965. https://doi.org/10.2217/fmb.15.32
Backert S, Tegtmeyer N, Oyarzabal OA, Osman D, Rohde M, Grützmann R, Vieth M. (2018)Unusual manifestation of live Staphylococcus saprophyticus, Corynebacterium urinapleomorphum and Helicobacter pylori in the gallbladder with cholecystitis. Int J Mol Sci 19(7) pii:E1826. https://doi.org/10.3390/ijms19071826
Bansal VK, Misra MC, Chaubal G, Datta Gupta S, Da B, Ahuja V, Sagar S (2012) Helicobacter pylori in gallbladder mucosa in patients with gallbladder disease. Indian J Gastroenterol 31:57–60. https://doi.org/10.1007/s12664-012-0162-8
Bauwens E, Joosten M, Taganna J, Rossi M, Debraekeleer A, Tay A, Peters F, Backert S, Fox J, Ducatelle R, Remaut H, Haesebrouck F, Smet A (2018) In silico proteomic and phylogenetic analysis of the outer membrane protein repertoire of gastric Helicobacter species. Sci Rep 8(1):15453. https://doi.org/10.1038/s41598-018-32476-1
Capelle LG, de Vries AC, Haringsma J, Ter Borg F, de Vries RA, Bruno MJ, van Dekken H, Meijer J, van Grieken NC, Kuipers EJ (2010) The staging of gastritis with the OLGA system by using intestinal metaplasia as an accurate alternative for atrophic gastritis. Gastrointest Endosc 71(7):1150–1158. https//doi.org/https://doi.org/10.1016/j.gie.2009.12.029
Chen DF, Hu L Yi P, Liu WW, Fang DC, Cao H (2007) H pylori exist in the gallbladder mucosa of patients with chronic cholecystitis. World J Gastroenterol 13(10):1608–1611. https://doi.org/10.3748/wjg.v13.i10.1608
da Costa DM, Pereira Edos S, Rabenhorst SH (2015) What exists beyond cagA and vacA? Helicobacter genes in gastric diseases. World J Gastroenterol 21(37):10563–10572. https://doi.org/10.3748/wjg.v21.i37.10563
da Silva-Etto JMK, Mattar R, Villares-Lopes CA, Marques SB, Carrilho FJ (2017) Evaluation of diagnostic accuracy of two rapid stool antigen tests using an immunochromatographic assay to detect Helicobacter pylori. Clin Biochem 50(16–17):959–962. https://doi.org/10.1016/jclinbiochem.2017.05.005
De Groote D, van Doorn LJ, van den Bulck K, Vandamme P, Vieth M, Stolte M, Debongnie JC, Burette A, Haesebrouck F, Ducatelle R (2005) Detection of non-pylori species in “Helicobacter heimannii” infected humans. Helicobacter 10(5):398–402. https://doi.org/10.1111/j.1523-5378.2005.00347.x
Dixon MF, Genta RM, Yardley JH, Correa P (1996) Classification and grading of gastritis. The updated Sydney System. International workshop on the histopathology of gastritis, Houston 1994. Am J Surg Pathol 20(10):1161–1181
Dye KR, Marshall BJ, Frierson HF Jr, Pambianco DJ, McCallum RW (1990) Camplyobacter pylori colonizing heterotopic gastric tissue in the rectum. Am J Clin Pathol 93(1):144–147
Giroux V, Rustqi AK (2017) Metaplasia: tissue injury adaption and a precursor to the dysplasia-cancer sequence. Nat Rev Cancer 17(10):594–604. https://doi.org/10.1038/nrc.2017.68
Goji S, Tamujra Y, Sasaki M, Nakamura M, Matsui H, Murayama SY, Ebi M, Ogasawara N, Funaki Y, Kasugai K (2015) Helicobacter suis-infected nodular gastritis and a review of diagnostic sensitivity for Helicobacter heilmanii-like organisms. Case Rep Gastroenterol 9(2):179–197. https://doi.org/10.1159/000431169
Graham DY, Zou WY (2018) Guilt by association: intestinal metaplasia does not progress to gastric cancer. Curr Opin Gastroenterol 34(6):458–464. https://doi.org/10.1097/MOG.000000000000472
Iannone A, Giorgio F, Russo F, Riezzo G, Girardi B, Pricci M, Palmer SC, Barone M, Principi M, Strippoli GF, Di Leo A, Ierardi E (2018) New fecal test for non-invasive Helicobacter pylori detection: a diagnostic accuracy study. World J Gastroenterol 24(27):3021–3029. https://doi.org/10.3748/wjg.v24.i27.3021
Kast RE (2007) Some fibrocystic breast change may be caused by sexually transmitted H. pylori during oral nipple contact: supporting literature and case report of resolution after gut H. pylori eradication treatment. Med Hypotheses 68(5):1041–1046. https://doi.org/10.1016/j.mehy.2006.09.050
Kawaguchi M, Saito T, Ohno H, Midorikawa S, Sanji T, Handa Y, Morita S, Yoshida H, Tsurui M, Misaka R, Hirota T, Saito M, Minami K (1996) Bacteria closely resembling Helicobacter pylori detected immunohistologically and genetically in resected gallbladder mucosa. J Gastroenterol 31:294–298. https://doi.org/10.1007/BF02389534
Kubota-Aizawa S, Ohno K, Fukushima K, Kenemoto H, Nakashima K, Uchida K, Chambers JK, Goto-Koshino Y, Watanabe T, Sekizaki T, Mimuro H, Tsujimoto H (2017) Epidemiological study of gastric Helicobacter spp. in dogs with gastrointestinal disease in Japan and diversity of Helicobacter heilmanii sensu stricto. Vet J 225:56–62. https://doi.org/10.1016/j.tvjl.2017.04.004
Kulnigg-Dabsch S (2016) Autoimmune gastritis. Wien Med Wochenschr 166:424–430. https://doi.org/10.1007/s10354-016-0515-5
Kusters JG, van Vliet AH, Kuipers EJ (2006) Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev 19(3):449–490. https://doi.org/10.1128/CMR.00054-05
Labenz J, Blum AL, Bayerdörffer E, Meining A, Stolte M, Börsch G (1997) Curing Helicobacter pylori infection in patients with duodenal ulcer may provoke reflux esophagitis. Gastroenterology 112(5):1442–1447. https://doi.org/10.1016/S0016-5085(97)70024-6
Lind J, Backert S, Pfleiderer K, Berg DE, Yamaoka Y, Sticht H, Tegtmeyer N (2014) Systematic analysis of phosphotyrosine antibodies recognizing single phosphorylated EPIYA-motifs in CagA of Western-type Helicobacter pylori strains. PLoS One 9(5):e96488. https://doi.org/10.1371/journal.pone.0096488
Lind J, Backert S, Hoffmann R, Eichler J, Yamaoka Y, Perez-Perez GI, Torres J, Sticht H, Tegtmeyer N (2016) Systematic analysis of phosphotyrosine antibodies recognizing single phosphorylated EPIYA-motifs in CagA of East Asian-type Helicobacter pylori strains. BMC Microbiol 16(1):201. https://doi.org/10.1186/s12866-016-0820-6
Livzan MA, Kononov AV, Mozgovoĭ SI (2004) Ex-Helicobacter gastritis: is it a neologism or clinical reality? Eksp Klin Gastroenterol 148(5):55–59
Madisch A, Miehlke S, Neuber F, Morgner A, Kuhlisch E, Rappel S, Lehn N, Bayerdörffer E, Seitz G, Stolte M (2006) Healing of lymphocytic gastritis after helicobacter pylori eradication therapy–a randomized, double-blind, placebo-controlled multicentre trial. Aliment Pharmacol Ther 23(4):473–479
Mäkinen JM, Niemelä S, Kerola T, Lehtola J, Karttunen TJ (2003) Epithelial cell proliferation and glandular atrophy in lymphocytic gastritis: effect of H. pylori treatment. World J Gastroenterol 9(12):2706–2710
Malfertheiner P, Selgrad M (2010) Helicobacter pylori infection and current clinical areas of contention. Curr Opin Gastroenterol 26(6):618–623. https://doi.org/10.1097/MOG.0b013e32833efede
Marshall BJ, Warren JR (1984) Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1(8390):1311–1315
Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, Sgouras D, Wessler S, Torres J, Smolka A, Backert S (2012) c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest 122(4):1553–1566. https://doi.org/10.1172/JCI61143
Naumann M, Sokolova O, Tegtmeyer N, Backert S (2017) Helicobacter pylori: a paradigm pathogen for subverting host cell signal transmission. Trends Microbiol 25(4):316–328. https://doi.org/10.1016/j.tim.2016.12.004
Oberhuber G, Haidenthaler A (2000) Histopathology of Helicobacter pylori infections. Acta Med Austriaca 27(4):100–103
Pachathundikandi SK, Muller A, Backert S (2016) Inflammasome activation by Helicobacter pylori and its implications for persistence and immunity. Curr Top Microbiol Immunol 397:117–131. https://doi.org/10.1007/978-3-319-41171-2_6
Parsons BN, Ijaz UZ, D’Amore R, Burkitt MD, Eccles R, Lenzi L, Duckworth CA, Moore AR, Tiszlavicz L, Varro A, Hall N, Pritchard DM (2017) Comparison of the human gastric microbiota in hypochlorhydric states arising as a result of Helicobacter pylori- induced atrophic gastritis, autoimmune gastritis and proton pump inhibitor use. PLoS Pathog 13(11):e1006653. https://doi.org/10.1371/journal.ppat.1006653
Pech O, May A, Gossner L, Vieth M, Trump F, Stolte S, Ell C (2001) Early stage adenocarcinoma of the esophagus arising in circular heterotopic gastric mucosa treated by endoscopic mucosal resection. Gastrointest Endosc 54(5):658–658
Posselt G, Backert S, Wessler S (2013) The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun Signal 11:77. https://doi.org/10.1186/1478-811X-11-77
Rebora A, Drago F, Parodi A (1995) May Helicobacter pylori be important for dermatologists. Dermatology 191(1):6–8. https://doi.org/10.1159/000246470
Reddy KM, Chang JI, Shi JM, Wu BU (2016) Risk of gastric cancer among patients with intestinal metaplasia of the stomach in a US integrated health care system. Clin Gastroenterol Hepatol 14(10):1420–1425. https://doi.org/10.1016/j.cgh.2016.05.045
Rugge M, Genta RM, OLGA Group (2005) Staging gastritis: an international proposal. Gastroenterology 129(5):1807–1808. https://doi.org/10.1053/j.gastro.2005.09.056
Sipponen P, Stolte M (1997) Endoscopy 29(7):671–678. PMID: 9360882
Smet A, Yahara K, Rossi M, Tay A, Backert S, Armin E, Fox JG, Flahou B, Ducatelle R, Haesebrouck F, Corander J (2018) Macroevolution of gastric Helicobacter species unveils interspecies admixture and time of divergence. ISME J 12(10):2518–2531. https://doi.org/10.1038/s41396-018-0199-5
Sonnenberg A (1987) Causative factors in the etiology of peptic ulcer disease become effective before the age of 15 years. J Clin Epidemiol 40:193–202. https://doi.org/10.1016/0021-9681(87)90153-6
Susser M, Stein Z (1962) Civilisation and peptic ulcer. Lancet 1(7221):115–119
Vaira D, Malfertheiner P, Mégraud F, Axon AT, Deltenre M, Hirschl AM, Gasbarrini G, O’Morain C, Garcia JM, Quina M, Tytgat GN (1999) Diagnosis of Helicobacter pylori infection with a new non-invasive antigen-based assay. HpSA European study group. Lancet 354(9172):30–33. https://doi.org/10.1016/S0140-6736(98)08103-3
Vieth M, Stolte M, De Groote D, Deeg KH, Seitz G (2001) Death kisses for newborns? Arch Dis Child 84(6):525. PMID:11372081
Wang J, Xu L, Shi R, Huang X, Li SW, Huang Z, Zhang G (2011) Gastric atrophy and intestinal metaplasia before and after helicobacter pylori eradication: a meta-analysis. Digestion 83(4):253–260. https://doi.org/10.1159/000280318. Epub 2011 Feb 1. PMID: 21282951
Warren JR, Marshall B (1984) Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1(8336):1273–1275. https://doi.org/10.1016/S0140-6736(84)91816-6
Ye W, Nyren O (2003) Risk of cancers of the oesophagus and stomach by histology or subsite in patients hospitalised for pernicious anaemia. Gut 52(7):938–941
Zhou D, Guan WB, Wang JD, Zhang Y, Gong W, Quan ZW (2013) A comparative study of clinicopathological features between chronic cholecystitis patients with and without Helicobacter pylori infection in gallbladder mucosa. PLoS One 8(7):e70265. https://doi.org/10.1371/journal.pone.0070265
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Falkeis-Veits, C., Vieth, M. (2019). Non-malignant Helicobacter pylori-Associated Diseases. In: Kamiya, S., Backert, S. (eds) Helicobacter pylori in Human Diseases. Advances in Experimental Medicine and Biology(), vol 1149. Springer, Cham. https://doi.org/10.1007/5584_2019_362
Download citation
DOI: https://doi.org/10.1007/5584_2019_362
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-21915-4
Online ISBN: 978-3-030-21916-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)