
Abstract
Weber’s red-edge effect is formulated as follows: “In rigid and highly viscous environments the excited-state energy transfer producing depolarization of fluorescence emission in concentrated dye solutions stops to be observed when fluorescence is excited at the red (long wavelength) edge of absorption spectrum.” After its discovery, it led to finding of a number of new wavelength-selective effects in spectral shifts, quenching, anisotropy and lifetimes, and also in different excited-state reactions forming a new vision of structural disorder and molecular dynamics in condensed media. These effects were consistently explained based on a new paradigm that accounts for statistical distribution of fluorescence emitters on their interaction energy with the environment leading to static or dynamic inhomogeneous broadening of spectra and to directional excited-state energy homo-transfer. These phenomena can be modulated by the energy of the excitation quanta. Their description, optimal conditions for their observation, information that they carry, and overview of their different applications are the subject of this chapter.
Another area in which the interpretation of the data of fluorescence in terms of molecular properties is lacking is that of the red-edge effects . . . . Investigation of this spectral region is often important in biological samples because it offers the best possibilities of detecting compositional heterogeneities.
G. Weber (1997) Methods Enzymol. 278, 13.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Anisotropy-based assays
- Fluorescence polarization
- Gregorio Weber
- Inhomogeneous broadening
- Red edge
- Solvation dynamics
1 Historical Introduction
The story of this unusual phenomenon started in 1960 with the observation made by Gregorio Weber [1] studying concentrated solutions of tyrosine, tryptophan, and their analogs. He demonstrated an almost complete loss of depolarization of fluorescence emission existing both at room temperatures and on deep freezing at special excitation conditions. These conditions were the solid fluorophore environments (glass-forming solvents at low temperatures) and the shift of excitation at the red edge of absorption band (Fig. 1). In 1969 Weber reported that this effect can be observed for the already popular dye 1,8-ANS (1-anilino-8-naphthalene sulphonate) that was previously introduced by him as the fluorescent polarity-sensitive probe. It was observed not only in supercooled solvent glasses but also at room temperatures on binding to protein serum albumin [2]. These findings led to understanding on the generality of the observed effect.

Excitation polarization spectra of concentrated solutions of tyrosine, tryptophan, and their analogs [1]. (a) Cresol ( x) and tyrosine (•), (b) indole (•), N-methylindole (o), and tryptophan (©). In propylene glycol at −70°C. Concentrations 0.2–0.5 mM
The results of systematic studies of fluorescence polarization as a function of excitation wavelength appeared in 1970 [3]. The experiments were performed in conditions that excluded fluorophore rotation as the mechanism of depolarization, so that in highly concentrated solutions it should be due to the excited-state energy homo-transfer (the transfer between the same molecules). Migrating between differently located and oriented fluorophores, the emitted light loses its initial polarization. What was surprising, is that in solid solvent glasses the depolarization of fluorescence emission drops at the red edge of excitation band. In these conditions the polarization almost reached the values observed for highly diluted systems.
Special care was taken for excluding trivial reabsorption, optical artifacts, fluorescent contaminants, and evident aggregation as a possible cause of this phenomenon, but the effect existed. Moreover, the new effect was observed in dye dimers, polymers, and on their incorporation into micelles. The statement was made that “The failure of depolarization upon excitation at the long wave edge of the absorption spectrum was found to be a completely general phenomenon without a single exception among the aromatics investigated.”
By the time of Weber’s discovery the excited-state energy homo-transfer (homo-EET) had become a well-studied phenomenon. Being described for the first time by Gaviola and Pringsheim in 1924 for concentrated solutions of dyes dissolved in glycerol [4], in the middle of last century this phenomenon has attracted attention of different scientists, and Gregorio Weber was among them. Following Förster [5] and Vavilov [6] that treated the excitation energy transfer between the same molecules (homo-EET) as the resonance coupling between two oscillators, he derived an equation relating the fluorophore concentration in solution with the transfer efficiency [7]. Other researchers used this dependence for calculating molecular distances involved in the energy transfer process. These results were consistent and well understood. The dramatic situation with the discovery of red-edge effect was such that it could not be explained based on existing concepts.
In his classical paper [3] Weber tried to fit this new effect on the basis of the paradigm dominating at that time. A paradigm dominated in photophysics up to 1970 was based on two empirical principles that were considered fundamental [8, 9]. One of them is the Vavilov’s law stating an independence of emission energy on excitation energy within the absorption band or, in other words, an independence of fluorescence quantum yield on excitation wavelength [10, 11]. The other called the Kasha’s rule [12] states that the emission spectrum occupies the same position on energy scale irrespective of the wavelength of excitation so that the emission always proceeds from the lowest electronic and vibrational state of the same multiplicity. Being formulated in terms of Vavilov law and Kasha rule [9], it was inherently assumed that all fluorophore molecules in their studied ensemble were identical, so that the fast relaxational processes occurring on their electronic excitation always drive the system to the same lowest in energy excited state, from which the emission occurs (see, e.g., [13]). Based on this paradigm, the position of fluorescence emission spectra, quantum yield of this emission, and reactivity in excited-state processes including the energy homo-transfer should not depend on the excitation wavelength. Therefore the only possibility to explain the new phenomenon within that paradigm was to suggest that the excited state generated by excitation at the edge of the absorption band differs from the excited state achieved on excitation over the bulk of absorption. If it is so and since vibrational relaxation to the lowest excited state is an ultrafast process, then there must be at least two distinct lowest energy excited electronic states. The state excited by the quanta of lowest energy must have the lowest energy transfer probability.
The arguments that Weber presented for that were based on observations of wavelength-dependent differences in some fluorescence characteristics, such as quantum yield, lifetime, and sensitivity to collisional quenchers, which was also unusual. An obvious weak point in this interpretation was the generality of the red-edge effect. It was hard to assume that undetected electronic states that can be revealed only on excitation at low energies really exist in the cases of all studied aromatic compounds and of their associates. It became evident that here a new feature in photophysics was discovered that required a new paradigm for its explanation.
The new paradigm came with new discoveries. Two laboratories, of Bill Galley [14] in Canada and of Anatoliy Rubinov [15] in Belarus being a part of USSR, have reported on discovery of a new red-edge effect – the bathochromic shift of fluorescence spectra at the red-edge excitations. Both of these groups stated that the spectra of individual fluorophores in solutions shift differently because of differences in their intermolecular interactions with their environment. They form distributions on these interactions resulting in inhomogeneous broadening of spectra. Within such distribution, the photoselection of the fluorophores, the interactions of which with their environment deviates from their mean values, can be provided on the low-energy slope of excitation band (red edge). These photoselected fluorophores exhibit the wavelength-shifted emission. Of course, the molecular mobility in such systems should be slower than the excited-state lifetime, otherwise the local environments will be mixed and the effect has to disappear.
I can witness that the pathways to these findings by these two groups were really independent. At the time of cold war, such high barriers existed between Soviet and Western science that the exchange of information could be retarded for years. The Canadian group made an important observation that in the sequence of aromatic molecules with different polarities the emission band position dependence on exciting wavelength was the most prominent for the polar ones (indole, tryptophan, 13-naphthol, 9-aminoacridine cation, and proflavin), whereas small or even undetectable shifts were observed for nonpolar aromatic hydrocarbons, such as anthracene, perlyene, and naphthalene. Dependence on the solvent was also remarkable, the effect decreased as one passes from polar to nonpolar vitrified media. These results are quite understandable since the noncovalent dipole–dipole interactions between polar molecules provide the strong contributions to dielectric solvation.
The experiments of the Canadian group allowed providing direct connection between observation of red-edge effect and the dynamics of solvent molecules. They discovered the edge-excitation shifts not only for fluorescence but also for phosphorescence for the same dyes (Fig. 2). Studying the temperature dependences, they observed disappearance of red-edge effect on transition from cryogenic to room temperatures. For phosphorescence this transition was found to occur at much lower temperatures than for fluorescence, which correlated with much longer lifetimes providing larger time window for dynamic processes in the solvent.

Plot of the excitation-wavelength dependence for the indole (10−3 M) fluorescence and phosphorescence spectra versus the temperature of the 4:1 glycerol–water medium. Fluorescence and phosphorescence shifts were measured as the average of the red edge (295 nm excitation) and blue edge (280 nm excitation) differences between the recorded spectra [14]
The interpretation of these data was based on the following assumptions: (1) the electronic energies of chromophores in solution are a function of geometry-dependent solute–solvent interactions, (2) at any time instant there is an ensemble of interactions that gives rise to a distribution of electronic energies in the sample, and (3) the interactions of a solute molecule with its solvent environment, which rapidly fluctuate with time in fluid solution, become static if the system becomes rigid on a time scale of emission. The most essential conclusion from these data was that the dependence on the excitation wavelength of the emission spectra of polar aromatic molecules in rigid, polar solutions is a very general phenomenon and that its occurrence depends on both the excited-state lifetime of the chromophore and the degree of rigidity of the medium [16].
The conclusions made by Rubinov’s group [15] were quite similar. They observed the excitation-wavelength-dependent shifts of fluorescence spectra for different dyes in solvent glasses and reported on “bathochromic luminescence” as a new phenomenon of general significance. These results were confirmed in another laboratory of the same institution [17]. Weber’s effect of repolarization of emission in concentrated solid dye solutions at the red edge has also found its confirmation and a new interpretation based on inhomogeneous broadening was given [18]. It was then quite logical to look for the site-photoselection of emission quanta, and this effect was found at the “blue edge” of fluorescence bands [19].
New groups of researchers were attracted by these interesting phenomena, and original results on their correlation with solute–solvent molecular relaxations were confirmed in temperature-dependent studies [20]. The understanding that the excitation energy-dependent spectral shifts and the time-dependent spectral shifts have to be based on the same mechanism stimulated the time-resolved studies. Then it was found that in the systems performing solvent relaxations during the excited-state lifetime collisional or light-induced fluorescence quenching increases the red-edge effects and the motions of time-resolved spectra depend on excitation wavelength [21, 22]. This dependence was specific: the motions of spectra disappear at the red edge, and on shifting the excitation wavelength further to the far anti-Stokes region they can even proceed with the increase of excited-state energy [21]. This phenomenon was called “up-relaxation.” Instead of releasing the energy to the environment, it required absorbing the energy for achieving the relaxed state, providing the local cooling.
Among new site-selective effects that have been observed in time domain, one was quite unexpected but confirming the general concept. It was the rotation of fluorescent dye induced by excitation light and detected by time-resolved anisotropy [23]. It can be observed when the dye molecule is smaller than surrounding molecules, and the electric field formed by their dielectric environment activates the motion to equilibrium of the dye itself. Such situations can appear for the dyes incorporated into biomembrane [24].
Since in essence of Weber’s red-edge effect is the failure of excited-state energy transfer, it was of particular interest to observe it in connection with directed EET in solid environments. As a result of inhomogeneous broadening due to non-identical dye environments in their concentrated solutions the dyes do not simply exchange of their energies. The energy flows directly from the species displaying short-wavelength absorption and emission to those exhibiting long-wavelength absorption and emission, as a result of which the spectra move to longer wavelengths as a function of time [25]. This fact stimulated modeling the assembly of pigments in the natural systems of photosynthesis [26]. Since EET stops at the red edge, such motions of spectra discontinue also.
With these developments it became clear that all the effects observed on variation of excitation and emission wavelengths should originate not from the violation of fundamental principles, but from their operation in specific conditions, when the ensemble of excited molecules is distributed on interaction energy with molecules in their surrounding. To explain them, a physical theory and modeling started to play an increasingly important role. First of such models was developed by Gregorio Weber [27]. His analysis was based on a simple model considering the interaction of physical dipole (the dipole with distributed charge) with solvent dipoles when these interactions conformed to the Langevin distribution. Mazurenko [28] developed a quasi-thermodynamic method for the description of stochastic solvation of molecules in solutions. Gorbatsevich et al. [29] calculated the distribution function on the frequency of electronic transition in polar solvent using the Monte-Carlo technique. As a result of both experimental and theoretical studies the concept was developed on the exclusive role in all these phenomena of inhomogeneous broadening of spectra that arises from solute-solvent distribution in excitation energy. This broadening allows for providing site-photoselection by excitation and emission quanta with well-defined energies deviating from their average values. The observed deviating properties can be compared with their mean values.
With such a wealth of new knowledge on the dependence of new effects on structure and dynamics it was quite natural to start addressing the reverse problem – to gain the information on structure, dynamics, and interactions in unknown or poorly characterized systems on molecular level. The present author was the first who initiated such applications. The first objects were the protein molecules possessing tryptophan as an intrinsic probe [30]. The results obtained were summarized in the monograph of the author [31]. The application of fluorescence probes was also quite successful [30]. All these results allowed demonstrating that proteins in solutions on the time scale of nanoseconds behave as nanoscopic solids with the dynamics slower by several orders of magnitude than the surrounding solvent. In lipid analogs of biomembranes the probe depth dependence of the lipid segment dynamics was characterized [32]. Quantitative measure of dipole relaxation rate was introduced [33]. Many new applications came after demonstrating the great power of the approach and they were the subject of numerous reviews [34–37] some of them with particular focus of studying proteins [38–42], biomembranes [39, 43], ionic liquids [44], etc.
Thus, the major idea, which is behind different approaches used in site-photoselection spectroscopy, is the selection and observation of a small part of the fluorophore population together with studying the whole population that is responsible for an inhomogeneously broadened spectrum. The great number of recently published articles indicates that the field is blooming, and many more advancements are to be expected.
2 Modern Interpretation of Red-Edge Effects and of Related Phenomena
Operating with excitation light of definite energy and polarization one can excite exclusively those dye molecules, the energy and orientation of electronic transition of which match these excitation parameters. Thus, if a dye is excited by polarized light, its emission will be also highly polarized. Depolarization occurs only when the time correlation of these selectively excited species is lost due to their rotation or participation in some photophysical process, such as excitation energy transfer. Similarly, photoselection can be provided by variation of excitation energy. The dye molecule can absorb only the light quanta that correspond to its electronic transition energy. Being selected from the whole ensemble by the energy of electronic transition, this sub-ensemble can possess diverging features observed in fluorescence emission and also, as we will see below (Sect. 3), in photochemical reactivity. These basic considerations allow to explain consistently a group of phenomena that are known under a common name “Red-Edge effects.” The optimal conditions in which these phenomena are observed are now well understood [35, 45]. The dye should be solvatofluorochromic, that is, its fluorescence spectra should respond to the changes in interaction energy with its environment by significant shifts. In the case of recording the steady-state spectra the dye environment should be relatively polar but rigid or highly viscous, so that the relaxation times of its dipoles, τ R, should be comparable or longer than the fluorescence lifetime τ F. When time-resolved recording is applied, the relaxations should proceed slower or be on the same scale as the time scale of emission. Thus, these effects are coupled with molecular dynamics in condensed media and allow distinguishing and characterizing rigid, viscous, and highly mobile media.
2.1 Inhomogeneous Broadening and the Principle of Photoselection
When organic dyes are studied in any liquid or solid media their electronic absorption and fluorescence emission spectra do not represent a sequence of sharp lines corresponding to electronic-vibrational transitions. They usually display broadbands with vibrational structure smoothened or even entirely lost, so that cooling to cryogenic temperatures does not result in improvement of structural resolution. This means that in addition to common strongly temperature-dependent homogeneous broadening (which is mainly due to electron–lattice and electron–vibrational interactions), in the systems with molecular disorder there exists the so-called inhomogeneous broadening of the spectra [45]. The latter originates from non-equivalence of dye solute environments (sub-states) that results in the distribution on solute–solvent interaction energies. All types of intramolecular and intermolecular relaxations may contribute to the energy difference between the maxima of the absorption and emission spectra, the so-called Stokes shift. The contribution of dielectric relaxations is often the strongest, and the site-photoselection effects can be observed if they are frozen or not complete. As a result, for every species the electronic transition energies become distributed on the scale of energy and their superposition forms an inhomogeneously broadened contour. Each sub-state in this ensemble can possess a sharp maximum, but when their contributions are added, the broad-band absorption and emission in its spectrum are observed. This is the inhomogeneous broadening of spectra.
The extent of this broadening is determined by the energies of intermolecular interactions, which exhibit statistical variation in the ensemble. The estimated by Weber [27] distributions of ground-state energies were 200 cm−1. The width of inhomogeneous broadening function Δv depends on the polarity of the solvent (the number and the magnitudes of solvent dipoles) and, most importantly, on the change of fluorophore dipole moment on excitation, Δμ [45]. For its estimation one can use an expression obtained in the Onsager sphere approximation:
where
Here ε is the dielectric constant of the medium, and a is the Onsager sphere radius. According to these estimates for 3-aminophthalimide in ethanol, Δv at 20°C ranges from 400 to 500 cm−1, and it decreases to 300–400 cm−1 at the freezing point of solution. For the same dye in toluene these values are 250–300 and 200–250 cm−1 correspondingly. In contrast, for low-polar dye coronene in toluene Δv is 25–30 cm−1 only [45]. The solvent–solute hydrogen bonding may provide additional increase of the width of the distribution [46].
These values are in reasonable agreement with experimental data obtained in cryogenic site-selective experiments. However, they show that in organized environments such as protein molecules the distribution can be dramatically narrower [47], which is the result of smaller variation in the ensemble of fluorophores of electric field effects between structurally identical highly ordered protein molecules. The proteins with different structure exhibit different Δv values. Thus, for Mg-myoglobin it is around 100 cm−1, while for peroxydase it is much narrower, 40 cm−1. Upon denaturation of Zn-cytochrome c, when the porphyrin changes its environment from being inside the protein globule to become exposed to a polar solvent, Δv changes from 65 to 360 cm−1 [48]. The transformation of the resonance Raman excitation profiles for the π–π* Soret band of native heme proteins provides an estimate of 100–200 cm−1 [49].
In molecular spectroscopy it is a common way to present the electronic transitions generating absorption and emission spectra as the two-dimensional functions of vibrational and solvation coordinates. Meantime the main difference between these coordinates is the quantized origin of vibrational modes, achieved in a very fast Franck–Condon process. According to Kasha rule, they relax rapidly to the lowest energy level of the first excited state [50]. In contrast, solvation modes are intrinsically over-damped. This allows treating solvation coordinate as a classical coordinate with continuous availability of electronic states. Thus, in molecular ensemble at any finite temperature a Boltzmann distribution in population of different solvent configurations is responsible for the inhomogeneous broadening in the steady-state spectra [51]. Such broadening arises from the solute–solvent distribution in excitation energy that reflects the distribution in energy of dye interactions with its dielectric environment. The stronger will be these interactions, the broader the distribution. Thus, the contour of absorption band must contain valuable information on the extent of molecular disorder.
In condensed medium such distributions should always exist at the time of excitation. But its display in a variety of spectroscopic phenomena depends on how fast are the transitions between the species forming this ensemble of states. Depending on these conditions, the broadening of spectra can be either static or dynamic [45]. The signatures of static broadening are observed in rigid environments, when the dynamics described in terms of dipolar relaxation times τ R is slower than the rate of emission. The broadening is dynamic if the motions in the dye environment occur simultaneously or faster than the emission, so that the correlation time τ c or more frequently used relaxation time τ R, τ R ≤ τ F. The static effect that is integrated over the time of emission depends upon the time window. In viscous media (when τ R ≈ τ F) not only the freezing (increasing τ R) but also the fluorescence quenching (reduction of τ F) may cause the appearance of red-edge effects. Thus, the inhomogeneous broadening effects contain the information about the dynamic properties of condensed systems, and the rate of fluorescence emission provides the necessary time scale for these observations. The scheme presented below shows the correlation between different mechanisms of spectral broadening.

Thus, being the major factor that produces broadening of the spectra, inhomogeneous broadening originates from non-equivalence of dye environments in an ensemble of otherwise identical molecules resulting in the distribution in solute–solvent interaction energies [45]. In fact, every molecule is under the influence of different forces produced by configuration of surrounding molecules. Therefore the dye species become distributed on their electronic transition energy and their superposition forms inhomogeneously broadened contour. Excitation at the band edge selects a part of this distribution, the spectroscopic properties of which can be quite different from their mean values. At the long-wavelength edge of the absorption band only those species are excited, for which the excitation energy with the environment is the strongest, their excited-state energy level occupies the lowest position, and for them the emission spectrum becomes shifted to longer wavelengths.
One of the major goals in cryogenic site-selective spectroscopy (at liquid helium temperatures) is the dramatic improvement of spectral resolution by suppressing the homogeneous broadening [51–53]. This goal is not achievable at ambient temperatures because of unavoidable existence of a broad homogeneous component [45, 54]. Therefore the possibility of photoselection within the ensemble remains only from the side of low energies of absorption band (red excitation edge) and from the side of high energy in emission (blue emission edge). The essence of such photoselection can be explained based on the energy diagram presented in Fig. 3, see also [35].

Energy diagram of S0 and S1 electronic states, which takes into account the relaxation phenomena [30, 55]. Vibrational relaxation is shown for the S1 state only. Dipolar (dielectric) relaxation is described as the evolution of the excited-state distribution and a temporal loss of photoselection between different energy dipole-orientational states excited at the “red edge” (see text)
Absorption spectra reflect the transitions from the ground-state energy levels E g to excited-state levels E e (upward arrows). Their contour is formed by 0-0 transition plus transitions to different vibrational levels of the excited state of molecule, while fluorescence emission (downward arrows) is formed by 0-0 transitions and transitions from the lowest level of excited state (achieved by vibrational relaxation) to different vibrational sub-levels of the ground state. In condensed media the energy of any ground or excited state is modified by dipolar interaction energy with the environment, W dd, to a different extent for every member of molecular ensemble. This is shown by the distributions along the scale of energy – Ω(W dd). Since the fluorophore dipole moments μ differ in the ground and excited states, we have to observe two types of distributions – Ω(W gdd ) for the ground state and Ω(W edd ) for the excited state. The absorption of a light quantum with sufficiently high energy corresponding to mean of the distribution (\( h{v}_{{}_{\mathrm{a}}}^{\mathrm{mean}}\ \ge\ {E}_{\mathrm{e}}^{\mathrm{mean}}\hbox{--} {E}_{{}_{\mathrm{g}}}^{\mathrm{mean}} \)) causes excitation of fluorescence of all possible solute–solvent configurations that differ in interaction energy. The common case is the excitation at the band maximum, which corresponds to the center of excited-state distribution, and emission proceeding from the center of this distribution.
Now we consider the case when we excite the system with the quanta of energy that are so low that they cannot excite even the 0-0 transition for all members of the ensemble – hv edgea (at the “red” edge). Then only the fluorophores constituting a part of the distribution will be selectively excited. It will be those species that interact most strongly with the environment in the excited state (and form the lower part of the distribution Ω(W edd )) and the least strongly in the ground state (and constitute an upper part in the distribution Ω(W gdd )), see Fig. 3. If the fluorophore–environment interactions remain unchanged during the time of emission (no dielectric relaxations in the medium), then the emission energies of these fluorophores (hv edgeF ) will be also lower than for the mean of the distribution (hv meanF ). As a result, the emission spectra will be shifted towards longer wavelengths, compared to that excited at the band maximum. Often the vibrational structure is not resolved in absorption spectra. Since it propagates from 0-0 band to higher energies, the photoselection within its Ω(W gdd ) distribution (or within the distribution of most active long-wavelength vibronic band) is possible at its red edge only. So if we decrease substantially the energy of excitation quanta (shift the excitation to red edge on the wavelength scale), then this energy will become so small that it will not be able to excite all the species in dye ensemble but only those which can interact with the environment in the excited state much stronger than the average and which possess their individual excited-state levels shifted down along the energy scale, achieving photoselection in excitation energy. When excited, these selected species will emit fluorescence differently than that of the mean of the distribution. Their emission spectra will be shifted in the direction of low energies to longer wavelengths. This is the site-selective in excitation red-edge effect that is the most popular in many applications.
Thus, electronic transitions occur between ground and excited states of dyes participating in distributions on interaction energy with the environment existing both in the ground and excited states. These interactions involve molecular dipoles and the distributions of these dipolar interactions with the environment can be different. The stronger interactions always result in a broader distribution on the energy of these interactions. Typically for solvatochromic dyes the dipole moment is small in the ground state and it increases substantially in the excited state enhancing the broad distribution. Then the ground state can be approximated by a single energy level and the excited state as the state exhibiting a broad distribution on dye–environment interaction energy. In the other limiting case, when the chromophore dipole moment is high in the ground state but decreases substantially in the excited state, the ground state should exhibit a broad distribution and the excited state can be represented by a single energy level. Then by variation of the energy of the light quanta we can produce photoselection within the ground-state distribution. So if we decrease the energy of excited light quanta, these quanta will be absorbed mostly by species, the ground-state energy of which is higher than the mean (they interact weaker with the environment than the average species in the distribution), so that the separation of energy between ground and excited states for them is smaller than the mean. The energy of emitted quanta will also be small, and we will have the same red-edge effect – the shift of fluorescence spectra to longer wavelengths. In a more general case when the dye dipole moments are relatively high both in the ground and excited states and the distributions of interaction energies are broad but due to redistribution of electronic density in the excited state the dipole moment changes its orientation. Here the weakest ground-state interactions may become the strongest in the excited state and the shift to the red excitation edge will select from the whole ensemble the chromophore molecules that interact weaker in the ground state (upper part of the distribution) but stronger in the excited state (the lower part of its distribution). This will result in the long-wavelength shifting red-edge effect.
It is known that the most precise information about solute–matrix interactions can be obtained by site-selection spectroscopy at cryogenic conditions (5 K or less). Here the zero-phonon lines were spectrally distinguished and analyzed by probing of inhomogeneously broadened zero-phonon line performed by spectrally very narrow laser beam [56]. These methods got the names of energy-selection spectroscopy, such as hole-burning in absorbance [57] and line-narrowing in fluorescence [55], see [56] for recent review. They were designed to circumvent the problem of large inhomogeneous line width existing in common condensed media by selecting a narrow package of molecules absorbing light around a certain frequency within the inhomogeneous band via a narrow bandwidth laser excitation. By applying this technique, a strong variation of the electron–phonon coupling strength on excitation wavelength through the inhomogenously broadened absorption origin band was demonstrated [51]. Ultrafast dynamic hole-burning and hole-filling have been realized [58]. Though technically complicated, these methods have found different applications in chemistry and biology [59]. Meantime, featuring a highly improved resolution of spectra they are limited to cryogenic temperatures or ultra-short observation times, so that all the dynamic information on solute–solvent interactions is lost. And attractive feature of red-edge effects is that here observations are not limited to any temperature ranges and the information on molecular motions can be obtained.
Thus, the widely explored red-edge effect is the long-wavelength shift of fluorescence spectra at the red excitation edge. Exciting by monochromatic light and shifting the wavelength from of the band maximum further and further to the red edge, a smaller and smaller number of dye molecules are excited with correspondent reduction of light emission intensity. Out of total population of dyes those sub-populations are photoselected, which happen to have their light absorption energies fitting to the decreased energy of illuminating light and their emissive properties differ from the mean values more and more significantly. The experiment on shifting the excitation wavelength stops when the emission intensity becomes very low and the spectrum becomes indistinguishable from the background. This dependence becomes steep without reaching any limit at the far red edge (Fig. 4). Sometimes one can even reach the anti-Stokes region, where the excitation wavelength, λ ex, becomes longer than the position of the maximum of fluorescence spectrum, λ maxem , excited at the band maximum, λ maxex . In this far edge region the shift of emission spectrum approaches in value the shift of excitation wavelength. Such typical dependence of λ maxem on λ ex is observed only at the red edge, and no such dependence is detected at the excitation band maximum and shorter wavelengths.

Dependencies of positions of fluorescence band maxima, λ maxem , on excitation wavelength, λ ex, for different correlations between the dipole relaxation time, τ R, and fluorescence lifetime, τ F. When the relaxations are slow, the fluorescence band occupies extreme short-wavelength position and the red-edge effect is the most significant, and when they are faster than the emission rate, the spectrum is located at long wavelengths and the red-edge effect is absent. The excitation spectrum, F(λ ex), is also presented schematically. Δλ 0em and Δλ em are the magnitudes of red-edge effect, and λ *ex is the isorelaxation point (the excitation wavelength at which the position of fluorescence band does not depend on relaxations) [60]
2.2 Connection with Molecular Relaxations
In order to understand this connection, a short excursion to fluorescence spectroscopy of molecular relaxation is needed (Fig. 5). The energies of both ground and excited states are always influenced by intermolecular interactions: the stronger are the interactions the lower is the correspondent level on an energy scale. On excitation, the electronic distribution in dye molecule changes, so change the interactions with the surrounding. If they are stronger in the ground state, then on their increase the difference in energy between the states increases. It can be the opposite: increase of interactions in the excited state. Thus, the spectra can move to either direction. The rigid medium conditions are those, in which the interactions are strong but at the absence of solvent mobility they are not at equilibrium with excited dye molecule. Such dependence is typical for every system with static (τ R >> τ F) or slow dynamic (τ R ≥ τ F) inhomogeneous broadening. With an increase of relaxation rate the fluorescence spectra at the main-band excitation shift in time to longer wavelengths (the common relaxational shift of spectra) [61]. In liquid medium the fast mobility allows achieving the relaxed state during fluorescence lifetime [62]. Since the extent of this shift depends on solvent polarity, the dyes with most pronounced shift are used as polarity-sensitive probes.

Simplified Jablonski diagram of ground S0 and excited S1 energy levels and transitions between them. Vertical upward arrow shows the excitation and downward arrows show emissive (straight) and non-emissive transitions to the ground state. In condensed media, the energies of ground and excited states are decreased due to electronic interactions with the environment by solvation energies W g and W e correspondingly. In addition, in polar media there occurs an establishment of equilibrium in interactions of dye and surrounding dipoles (dipolar relaxation). As a result, the energy gap between S0 and S1 states decreases and the spectra shift to longer wavelengths
Thus, the time window for observing this relaxation is determined by the rate of fluorescence emission. Depending on molecular mobility in the medium the process of attaining a new equilibrium (relaxation) may be faster, slower, or occur simultaneously with the emission decay. In the case if it occurs simultaneously with the decay, complex emission-wavelength dependence should be observed for the decay kinetics, and the spectra should move as a function of time in the direction of lower energies.
If N 0 molecules are excited at an instant t = 0, the number dN of emitted quanta within a time interval dt and a frequency range dv can be determined from the formula:
where Q F is the quantum yield of emission and
The latter function determines the number of quanta emitted per unit time within a unit frequency interval. At a fixed time it can be regarded as an “instantaneous” emission spectrum, while at a fixed frequency v it represents a law of emission decay. ξ is the maximum or, more precisely, the center of gravity of the spectrum (in cm−1). It was shown [63] that within the Debye model of relaxation (single relaxation time τ R) the position of the spectrum ξ has to change exponentially with time:
Since the relaxation is the time-dependent loss of correlation between initial site distribution and the distribution at time t, a time-dependent correlation function C(t) for ξ = ξ(t) can be used for describing the process of relaxation:
It normalizes the spectral shifts to unity and allows comparison of the effects produced by different dyes. Such motions of spectra to longer wavelengths as a function of time are observed in experiment [64]. By assuming τ F to be unchanged in relaxation process the following expression can be obtained for the behavior of the steady-state spectra:
Here ξ St is the position of steady-state spectrum. The limiting values of ξ t→∞ and ξ t=0 and the variations of temperature are usually applied. The limit of slow relaxations ξ t=0 should be achieved at low temperatures (when τ R >> τ F) and the limit of fast relaxations ξ t→∞ – at high temperatures (when τ R << τ F).
It is hard to reach these conditions in real systems, especially in biophysical applications. Whereas reducing the temperature with maintenance of observed structure is frequently possible, subjecting to high temperatures often cannot be tolerated. Therefore in our earlier works [33, 35] we suggested an extension of this approach by incorporating the information obtained in the study of red-edge effect. When the relaxation is complete, it produces a new dynamic distribution of sites that becomes uncorrelated with initial distribution. Due to molecular motions, the excitation energies fluctuate in time causing redistribution within this ensemble, “mixing” different environments. Being selected at any wavelength, the sub-population of dyes is rapidly mixed within the whole population, so that the spectra become λ ex-independent and the red-edge effect has to vanish. If we assume that τ R and τ F at the red-edge excitation do not differ from their mean values, we obtain a very simple relation that allows us to obtain dynamic information from the steady-state spectra. By re-writing Eq. (7) for the mean and for the edge excitations and taking into account that when the relaxation is complete, \( {\xi}_{t\to \infty}^{\mathrm{mean}}={\xi}_{t\to \infty}^{\mathrm{edge}} \), we obtain
Equation (8) uses only the data on steady-state spectra and allows us to analyze the red-edge-excitation shifts of fluorescence maxima. ξ values on wavenumber scale can be easily transformed into wavelength λ values, ξ(cm−1) = 107/λ(nm) and the positions of wavelength maxima, λ maxem , can be used. Thus, dynamic information about molecular relaxations can be obtained in simple steady-state measurements using τ F as a time marker and analyzing the red-edge effects.
As a result of relaxation, two processes occur simultaneously: the shift of spectra to longer wavelengths and the decrease of excitation-wavelength dependence (see Fig. 4). In the dependence of the positions of fluorescence band maxima on λ ex there is one characteristic point, λ *ex , in which the energy of electronic transition corresponds to that of the relaxed state. At \( {\lambda}_{\mathrm{ex}}>{\lambda}_{\mathrm{ex}}^{*} \) the relaxation occurs with the decrease of energy, and the spectra have to move in time to longer wavelengths, while at \( {\lambda}_{\mathrm{ex}}>{\lambda}_{\mathrm{ex}}^{*} \) the relaxation results in increase in energy and in order to achieve the equilibrium the spectra move to shorter wavelengths (up-relaxation [21, 54]). We call λ *ex an isorelaxation point. Its presence allows us to introduce the quantitative characteristics of red-edge effect as the shift of fluorescence spectrum \( {\lambda}_{\mathrm{em}}^{\max}\left(\mathrm{mean}\right)-{\lambda}_{\mathrm{em}}^{\max}\left(\mathrm{edge}\right) \) on variation of λ ex from that at excitation band maximum λ meanex to λ *ex . The relaxational shift of emission spectra can be observed at any λ ex beside λ *ex . Thus, the inhomogeneous broadening effects contain information about the dynamic properties of condensed systems, and the rate of fluorescence emission provides the necessary time scale for the observations of red-edge effect [35].
One of the most intriguing properties of structurally disordered materials (liquid or solid) is the huge dispersion of structural relaxation rates [65, 66], therefore a simple model operating with single τ R and τ F values may not be applicable in all cases. However, being conceptually correct, it helps us to understand the basis of studied phenomena and the interpretation of many experimental data on quantitative level is quite satisfactory. One such result was was obtained for tryptophan, the major fluorescence emitter in proteins (Fig. 6). For tryptophan in glycerol in the lowest range of temperatures the fluorescence spectrum at the main-band excitation is the most significantly shifted to shorter wavelengths, and the red-edge effect is dramatic. On increase of temperature the spectrum at the main-band excitation shifts to longer wavelengths, and the red-edge effect decreases. At high temperatures, when the solvent becomes low-viscous, it becomes undetected. We observe, however, that all the curves λ maxem as a function of λ ex cross at the same point at about 307 nm.

Dependence of the maximum of fluorescence spectrum of tryptophan in glycerol on excitation wavelength at different temperatures: −196°C (1), −14°C (2); 20°C (3) and 50°C (4). Curve 5 is the excitation spectrum. The strong temperature dependence of spectra is observed at any wavelength beside the range 307–308 nm at the red edge [33]
2.3 Observations with Time Resolution
The new view on inhomogeneous distributions of light emitters in molecular ensembles required re-interpretation of many time-resolved spectroscopic data. Unlike radioactive isotope decay, which is strictly single-exponential, the fluorescence decays often display non-exponential, distributed character. The difference is that the nuclear processes responsible for radioactive decay do not depend on intermolecular interactions, but such dependence exists for light emitting dye molecules. Being in variable environments, the dyes may emit light with different rates, leading to non-exponentiality and site-selectivity on the population level. The spectral kinetics originated by dielectric relaxations and photochemical reactivity develops on this background.
Commonly, the spectroscopic observations of dielectric (dipolar) relaxation are provided by excitation at the band maximum and recording the shifts of emission spectra to longer wavelengths as a function of time [67]. As a result, when observed at the blue edge, the emission decay contains short-decaying positive component(s) due to fast temporal decrease of a number of emitters possessing higher energies. When observed at the red emission edge the decay contains a negative component due to increase with time of the number of excited-state species emitting at low energies. The major features of this process can be adequately described based on the Bakhshiev–Mazurenko model of dipolar relaxations [62, 63] that uses Eqs. (3–5). Such picture is often obscured by non-exponential decay functions recorded at different emission wavelengths that are used for constructing the time-resolved spectra. Moreover, apparent motions of spectra in time that derive from wavelength-dependent decay rates have been described that originate not from molecular relaxation but from heterogeneity of fluorescence emission [68].
The red-edge effects introduce a new dimension into this picture and allows probing the redistribution on emission energy as a function of time between the sites in an ensemble [21]. They allow decreasing or even eliminating this type of heterogeneity. Moreover, these studies bring in new concept of relaxation, which is the reorganization in ensemble of distributed states on the achievement of excited-state equilibrium [45]. In the conditions at which the major part of the excited dye population on achieving the equilibrium demonstrates the motion of spectra to longer wavelengths, the spectra of the sub-population selected at the red edge do not move [69], Fig. 7. This is because those species are photoselected, the interactions of which with the environment are close to the relaxed state. When the dye is excited by energy lower than that of the relaxed state, the spectra move to opposite direction, to higher energies (up-relaxation) [21].

Instantaneous fluorescence spectra of 1-phenylnaphthylamine in glycerol at 22°С for various excitation wavelengths: (a). Band maximum, λ ex = 337 nm, registration time t reg = 2 ns (1), 3 ns (2), 14 ns (3); (b) Red edge, λ ex = 416 nm, t reg = 2 ns (1), 8 ns (2) [69]
The changes at the red edge of time-dependent evolution of spectra can be observed in different ways. In the conditions of observing the time-dependent spectral shifts the time-resolved emission decays change dramatically at the red-edge excitation: its short components at the blue and red slopes of emission spectrum that reflect the relaxation, disappear [70]. The emission kinetics excited at the red edge becomes uni-modal and almost single-exponential. The evolution of fluorescence bandwidth in time-resolved spectra is much more pronounced at the red-edge than at the main-band excitation. The spectra are initially more narrow (since a part of the distribution is selected) and are broadened in the course of relaxation due to redistribution to a broader number of different sites. If fluorescence is selectively excited by a narrow-band pulse, then a time-dependent broadening (spectral diffusion) is observed [71], and due to temporal depopulation of ‘selected’ fluorophores, a selective decrease of τ F is observed at the frequency of excitation [72]. Site-photoselection at the red edge results in disappearance of these effects.
Thus, the time-resolved fluorescence methods can be easily extended to experiments with site-selective excitation. These results allowed achieving better understanding the molecular relaxation phenomena. Relaxation can be viewed as not only the decrease in time of the energy of the average (or most probable) species of molecular ensemble but also reorganization in this ensemble, the loss of time correlation and site-selected species as a function of time.
2.4 Diversity of Wavelength-Selective Effects
Since in absorption spectra the band corresponding to the 0-0 electronic transition is observed at the lowest energies (longest wavelengths), and the vibrational structure (often not observable) propagates in the direction of higher energies, the best site-selectivity can be achieved at the low-energy slope of absorption spectra, the “red edge.” In some organic dyes the 0-0 absorbance is weak and other Franck–Condon active vibronic bands can participate in this site-selectivity. In the cases when vibrational structure is relatively well resolved, the site-photoselection to some extent can be achieved at the red edge of every electron-vibrational band [73]. This depends on fluorophore structure and, together with fluorophore–environment interaction, determines the dynamic range of observed effects.
With these facts in mind let us summarize all the possibilities in observing the site-selective effects manipulating with excitation and emission wavelengths. Excitation proceeds from thermally relaxed ground state to different unrelaxed vibrational modes of excited state. Vibrational relaxation and thermalization on intramolecular and intermolecular level occur much faster than the emission [74] and the red-edge effects should be observed only if dielectric relaxations in intermolecular interactions proceed slower or in comparable rate with the emission. If these rates are comparable, then the red-edge effects will depend on the following factors:
-
Variation of temperature. Both τ R and τ F depend on temperature, so the correlation between them, τ R/(τ R + τ F), expressed by Eq. (6), should be temperature-dependent. Usually the relaxation rate increases with temperature faster than the emission rate. In some cases structural changes in the system with the change of τ R can be detected, but there may be the cases when the relaxation rates change without conformational change [75].
-
The effects of fluorescence quenchers. The result of collisional quenching is the change of fluorescence lifetime τ F that shortens the time window for relaxations and increases the red-edge effects [45].
-
The time-resolved observations. The spectra move to longer wavelengths as a result of dielectric relaxation with the decrease of energy (the common case). They stop to move if by shifting the wavelength the isorelaxation point is achieved, and they start to move to shorter wavelengths at the far red edge, when the relaxed state is of higher energy (the case of “up-relaxation”) [21]. Instead of heat release as a result of relaxation, a local cooling in the dye environment can be detected [76].
Photoselection can be observed also in emission spectra. The emissive electronic transitions extend from vibrationally relaxed excited state to different vibrational sub-levels of the ground state, so the vibrational progression extends from the 0-0 band located at higher energies in the direction of lower energies, to longer wavelengths. Emission proceeds to different vibronic high energy ground-state levels, which makes impossible the photoselection by collecting the low-energy emission quanta. Therefore excitation spectra measured by setting the emission wavelength longer than the band maximum will be always emission-wavelength-independent. The site-photoselection by probing the emission wavelengths is possible only from the side of high energies of emitted quanta, when the selected species possess the ground-state solvation (stabilization) energies higher than the mean, while their excited state should be less stabilized than for the average of the ensemble. This situation can be realized only at the blue (short-wavelength) edge of emission band. As a result, the excitation spectrum will gradually shift to the blue at the “blue edge” of emission band, i.e., at the high energy side of 0-0 transition [19]. Thus, in emission spectrum the site-selectivity leads to another site-selective effect – the dependence of excitation spectra on emission wavelength. Similarly to the other red-edge effects, this effect disappears as a result of relaxation.
Figure 8 illustrates the correlation of motions of excitation and emission spectra: red-edge effect by photoselecting the excitation and blue edge effect by photoselecting the emission wavelengths. The shift of excitation spectra to the blue is observed when fluorescence emission is collected at wavelengths shorter than fluorescence band maximum. The dependence of excitation spectra on emission wavelength appears if we collect the emitted quanta that possess higher energies than the mean, since they correspond to lower part of the distribution in the ground state and higher part of the distribution in the excited state. As a result, the excitation spectrum will shift to the blue. This dependence increases with the further shift to the blue edge, and no saturation point is observed.

Typical dependences of positions of fluorescence band maxima on excitation wavelength, λ maxF (λ ex), and positions of excitation band maxima, λ maxem (λ em), on emission wavelength for the case of inhomogeneous broadening of spectra [60]
Light has a selective power to excite exclusively those dyes, the properties of which match the energy and polarization of their electronic transitions. Thus, if the dye excitation is produced by polarized light, its emission will be also highly polarized. Depolarization occurs only when the time correlation in the excited state is lost due to the dye rotation or its participation in some photophysical process, such as excitation energy transfer [77]. Often the decay of time-resolved anisotropy, r(t), is non-exponential due to asymmetry of dye molecule and anisotropy of its interactions. Also, since in the course of relaxation τ F becomes shorter at the blue and longer at the red slopes of emission spectrum, a higher value of anisotropy is observed in the blue and lower in the red parts of the spectrum [78]. Such effects are produced due to variation of time window, in which fluorophore rotations are observed. Excitation at the red edge suppresses the relaxational shift of spectra and makes the emission decay more homogeneous [79, 80].
The spectral and temporal inhomogeneity of emission probed by the dependence on excitation and emission wavelengths may produce additional influence on r(t), even without EET in dilute solutions. Because of release of excessive vibrational and intermolecular energy, the local heating can occur [79, 80]. This effect is called a “light-induced rotation,” it is suppressed when λ ex is shifted far to the red edge where λ ex > λ *ex (λ *ex is the isorelaxation point, explained in Fig. 4), since in this case we excite the solutes with the strongest intermolecular energy in the excited state [80]. The local heating depends on the excess of configurational energy of selectively excited solvates and leads to specific dependencies of the kinetics of radiation anisotropy on the exciting light frequency and the frequency at which the emission is recorded [23]. It is clearly observed for fluorescence probes in biomembranes [24].
There is one more possibility of introducing time domain into spectrally selective molecular relaxation technique. It is the transient hole-burning spectroscopy [51, 81, 82]. Unlike persistent hole-burning [55–57] this method does not require cryogenic temperatures. By exciting the probe molecule with pulses shorter than the solvation time but longer than the dephasing time, a subset of the ground-state population is excited only and it can be recorded in a picosecond transient absorption spectrum. This subset corresponds to probe molecules experiencing similar interactions with the solvent. The rate of this broadening (hole-filling) is attributed to solvent dielectric relaxation kinetics resulting in a time-dependent disappearance of “selected” species. Thus, even at room temperature a transient spectrum initially shows a slightly sharp hole around the exciting energy, resulting in the time-dependent broadening of its shape.
Though in the background of this technique is a different methodology (the selection is produced within the ground-state distribution and the relaxation occurs within the ensemble of ground-state species), it has important common features with the time-dependent red-edge effects:
-
photoselection is produced within inhomogeneously broadened absorption band,
-
relaxation is observed as a time-dependent disappearance of “selected” species,
-
the time window for the observation of relaxation process is determined by the excited-state lifetimes. Usually it is the picosecond–nanosecond time range, but by populating the triplet state the time window can be extended to milliseconds.
3 Red-Edge Effects in Photochemical Transformations
Based on the interpretation presented above, of spectral broadening and molecular relaxations in solutions, it can be expected that any excited-state reaction, the rate of which depends on the energy and dynamics of weak noncovalent interactions with the environment will be modulated by site-selection effects. It is known that solvent-reorganizational coordinate is important for many excited-state reactions [83, 84], but not for all of them. There are reactions that are uncoupled with dipolar relaxations and that can occur on ultra-short time scale even at extremely low temperatures (for instance, intramolecular proton tunneling [85] and non-adiabatic electron transfer [86]). At the other extreme are slow reactions that occur in solution after attaining dielectric equilibrium, for instance, the reactions of diffusional bimolecular quenching. But there are many examples of those reactions that are coupled with these relaxations, and the study of this coupling may be used as an important clue for elucidating their mechanisms [87]. In unrelaxed states the distribution of excited-state species on their interaction energies with the environment may result in distributed reaction kinetics [88]. It was shown that the part of this distribution that interacts stronger with the environment may exhibit an extreme increase in reactivity in intramolecular electron transfer reaction and a decreased reactivity in proton transfer and energy transfer reactions [89, 90]. Site-photoselective spectroscopy allows not only to characterize the selective photochemical reactivity but also to provide the means to model the reactions occurring in the ground states, especially those of them which possess low intrinsic activational barriers and depend on dynamics in the environment. Those are many biocatalytic reactions.
3.1 Photoinduced Electron Transfer
The solvent effects influence the excited-state photoinduced electron transfer (PET) if the transition from the initial “locally excited” (LE) state to charge-transfer (CT) state is adiabatic, i.e. occurs in the conditions of strong electronic coupling between two states and proceeds continually along the reaction coordinate on a common adiabatic potential energy surface [84]. Quite often the PET reactions result in quenching, which complicates the observation of red-edge effects [91]. Bianthryl is a bright exception, in which PET reaction is intramolecular leading to brightly fluorescent product. It is an anthracene dimer demonstrating the excited-state electron switching between monomers in liquid polar media, and this reaction does not proceed if the solution is frozen.
This reaction in bianthryl was studied by variation of excitation wavelength (Fig. 9). It was found that PET, which requires dielectric relaxation and commonly does not occur in vitrified polar solutions, is dramatically facilitated when photoselection was provided at the red edge of excitation band [90].

Influence of the excitation wavelength on fluorescence spectra of bianthryl in propylene glycol [90]. I em(480)/I em(415) is the ratio of intensities at emission wavelengths 480 and 415 nm that reflects the relative contribution of CT and LE forms. The temperatures were −100°C (1), −53°C (2), and 18°C (3). Curve 4 and the left scale represent the excitation spectra at −53°C at emission velengths 395, 415, 445, and 480 nm, which are superimposed
The temperature range of disappearance of such red-edge effect correlates strongly with the solvent mobility. It occurs in the range of nanosecond dielectric relaxations in the solvent [89]. This connection with the solvent mobility is clearly demonstrated when the temperature dependences of spectral shift of LE band, the change of the band width, and the extent of PET reaction expressed as the relative ratio of LE and CT band intensities are displayed in the same plot (Fig. 10).

Temperature dependences of the bianthryl spectroscopic parameters in propylene glycol [90]. λ maxem is the position of fluorescence maxima at excitation wavelength 403 nm and Δλ is the half-maximum bandwidth of the spectrum, excitation wavelength 365 nm. The red-edge effect is displayed as the intensity ratio at two emission wavelengths, I em(480)/I em(415), the same parameter as in Fig. 9
The results obtained demonstrate the possibility of photoselection in the rigid environment of species, for which the solute–solvent configurations are close to the “relaxed” state. Because of that the solvent-reorganization barrier does not exist and the PET reaction proceeds easily. An attractive idea can be suggested to make organic solid-state optoelectronic devices switchable just by the energy of excitation light.
3.2 Intramolecular Charge Transfer and Excited-State Isomerizations
Electronic excitations may lead to intramolecular charge transfer (ICT) reactions that involve redistribution of electronic charge within the same molecule. These effects are typical for fluorophores that contain electron donor and electron acceptor groups incorporated into aromatic heterocycles. The intramolecular electronic charge redistribution is coupled with electronic and nuclear polarization of the medium resulting in dielectric relaxations. If the relaxations do not occur in rigid environments, the static distribution of sub-states may result in different ICT reactivity and the site-selective excitation may allow observing the switching of emission between initially locally excited (LE) and ICT forms. Dynamic solvent effects are observed when this reaction is adiabatic, i.e., occurs in the conditions of strong electronic coupling between two states and proceeds continually along the reaction coordinate on a common adiabatic potential energy surface [84]. Of special interest are the cases, in which the reactant and product forms are represented by separate emission bands. For the fluorescent dye Laurdan, popular in many applications, two fluorescence bands with maxima at 425 and 500 nm are observed in cooled glycerol [92]. Remarkably, together with redistribution of intensities between two bands, only the long-wavelength CT band shifts dramatically on transition of excitation energy to the red edge.
Charge redistribution is often coupled with rotations of molecular fragments. A number of fluorophore molecules that exhibit intramolecular flexibility and are planar in the ground state can rotate in the excited state to a perpendicular conformation (which can be energetically more favorable in polar environments). The reaction is easily observable when both forms are present in the emission and are represented by separate maxima. In these cases the fluorescence spectrum is usually shifted to longer wavelengths. Classical in this respect is N,N′-dimethylaminobenzonitrile (DMABN), for which the twisted intramolecular charge-transfer (TICT) state can be achieved even in rigid polymeric matrices. It was shown [93] that in polar polyvinyl alcohol (PVA) glass the contribution of the TICT form being small at the main-band excitation increases dramatically when fluorescence is excited at the red edge. Meantime, a strong deformation of an excitation spectrum as a function of emission wavelength indicated the significant site distribution within the ground-state species.
The barrierless stochastic staircase model of Bagchi was used for interpretation. Within this model, the long-wavelength excitation selects in the initial population the species that are closer on reaction coordinate to the region where the reaction occurs [94]. In related experiments on 1,4-diphenyl-1,3-butadiene the red-edge selective excited-state production of s-cis rotamers has been demonstrated [95]. Switching between LE and ICT emissions can be achieved with the shift of excitation wavelength in argon matrix for pyrrolyl benzonitrile [96]. These and other related experiments suggest that site-photoselection in excited state isomerizations coupled with ICT occur in the excited state, although their pre-existence in the ground state as minor forms is not excluded.
For organic dyes exhibiting intrinsic flexibility the possibility to form the distribution of sub-states due to formation of population of rotamers and the existence of variable free volumes in solid matrix was pointed out by different authors [97]. The cases are known, in which two rotamers are present in the ground state but only one of them is present in emission spectra at long-wave excitation, demonstrating the red-edge effect [98]. Recent experiments with DMABN point to this possibility. The red-edge effects reveal itself in the dependence of ratio LE to CT fluorescence bands on the wavelengths of excitation [99]. These features can be observed even in a liquid solvent acetonitrile within quite broad temperature interval from 0°C to 80°C [100], which may be connected both with slow interconversion between these sub-states and very short lifetimes narrowing the time window for observation.
Thus, the red-edge effects are important tools to study the excited-state dynamics and its coupling with the motions in reactant environment for ICT reactions and when these reactions are coupled with conformational changes. However, one has to keep in mind the possibility of selecting within the species already existing in the ground state. Photoselection in the latter case is quite different from that on existence of classical red-edge effect. This issue will be discussed in Sect. 5.
3.3 Excited-State Intramolecular Proton Transfer
Excited-state intramolecular proton transfer (ESIPT) reactions demonstrate very strong wavelength shifts and a great variety of rates, starting from ultrafast proton tunneling [101] to that coupled with solvation dynamics [92] and proceed under either thermodynamic or kinetic control [102]. In 3-hydroxyflavone (3HF) derivatives all these different conditions and mechanisms of photo-transformations can be realized. The initially excited LE and reaction product proton transfer (PT) excited-state forms are dramatically separated in emission energy. In 3HF derivatives possessing strong excited-state dipole moments, both spectral position of LE form and interplay of two forms in fluorescence spectrum depend strongly on solvent polarity [103, 104].
The site-selectivity of this reaction was first demonstrated in the complex of 4′-(diethylamino)-3-hydroxyflavone with the protein (serum albumin) [88]. It was found that while both LE and PT forms are present at the main-band excitation, the excitation at the red-edge results in elimination of the PT band from the spectrum (Fig. 11).

The scheme illustrating the appearance of red-edge effect in ESIPT reaction of 3HF derivatives (above) and experimental data on the disappearance of PT band (below) [83]. The notations in scheme are those as in Fig. 3. The site-photoselection of polar and highly solvated species in the LE state suppresses the PT reaction. The results on 3HF derivative demonstrate the spectral shift of LE band and disappearance of PT band in emission on shifting the excitation from 420 nm (1) to 440 nm (2) and further to 460 nm (3)
The scheme presented in Fig. 11 (above) illustrates the mechanism of site-photoselection modulating the PT reaction yield. The excitation results in the appearance of highly polar dipolar LE state that, interacting with the environment, generates an ensemble of sub-states of different energies. The reaction proceeds with the activation barrier to a low-polar PT state, in which the distribution in interaction energy with the environment is minimal. Since the reaction rate is much faster than any orientational relaxation of the environment dipoles, the distribution in LE state persists on the scale of reaction. The species on the lowest edge of this distribution need to overcome much higher activation energy, than in the middle of distribution, and for them the ESIPT reaction is suppressed.
Serum albumin was selected in our initial studies because this protein has a function to bind and transport in the blood of different substances with low solubility in water and its binding sites are well characterized. The high-affinity binding of 3HF derivatives to particular protein site was demonstrated [105]. The local environment formed by the binding site is rigid, so the distribution is maintained during the lifetime of LE state. The studies in polymer films, phospholipid membranes, and in complexes with proteins allow characterizing the static and dynamic disorder in these systems. This effect is easily observed as an excitation wavelength-dependent dramatic change of the emission band profile resulting in dramatic change of color [106].
The same effect of retardation of ESIPT reaction at the red-edge excitation is observed in viscous solvent triacetin [107], in ionic liquids [107, 108] and erythrocyte membranes [109]. Recently Tomin [110] demonstrated that it can be manifested even in liquid solvents due to strong shortening of excited-state lifetime. Thus, in the case of ESIPT reaction in excited-state dipolar 3-hydroxyflavones the site-photoselection at the red excitation edge results in stabilization of initially excited LE state and to disappearance of reaction product proton transfer band. This example of the red-edge effect in ESIPT reaction is a demonstration, how the coupling of site-photoselection with excited-state reaction can provide a significant amplification of spectroscopic signatures of molecular disorder.
Concluding this section, we derive that the red-edge effects have become the important tools for studying the mechanisms of different excited-state reactions, for which the solvent-reorganizational coordinate is important. They allow studying the coupling of these reactions with dielectric relaxations in the reaction site environments and the involvement of inhomogeneous reaction kinetics. These observations demonstrate how the coupling of site-photoselection with excited-state reaction can provide a significant amplification of spectroscopic signatures of molecular disorder. Strong site-selectivity is observed for different low-barrier excited-state reactions in the conditions of slow mobility in the environment of the excited reactant species. Some of them are very useful practically for characterization of the dynamic properties in particular systems and predicting their behavior in chemical and biochemical reactivity [83]. Because elementary rates in these reactions are also site-selective, their inhomogeneous kinetics should also be observed. Wavelength-selective reactivity was found in photochemistry for charge-recombination reactions [111]. In analogy, this type of reaction kinetics was suggested for biocatalytic reactions [83, 88]. Recently such reaction behavior was found in single-molecular studies [112].
4 Directional Excited-State Energy Transfer and Red-Edge Effects
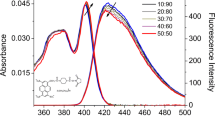
The discovered by Weber [1–3] red-edge effect of failure of energy homo-transfer between the dyes in highly concentrated solutions in rigid and highly viscous media (see Fig. 1) can be naturally explained by site-photoselection within the inhomogeneously contour of absorption band. Here the same molecules serve the role of both donors and acceptors. However, they are distributed being located in different environments and because of that their absorption and emission spectra are not identical. The species from upper part of the distribution (Fig. 3) can transfer their energy to another species on the same or lower energy level. In contrast, the species from the lower part of the distribution (their effective concentration is low) can transfer their energy only to other species of the lower part of this distribution. Because of low concentration of these selected species, such transfer is a low probable event. Thus, due to the presence of inhomogeneous broadening, the excited-state energy transfer between chemically identical molecules is not random, it is directed from those members of the ensemble which emit at shorter wavelengths to those which absorb at longer wavelengths [18].
As a result of this directed transfer the fluorescence spectra in concentrated dye solutions in rigid and highly viscous environments are shifted to longer wavelengths. This shift can be observed as a function of time [25], even if the environment is completely immobile. Such transfer stops at the red edge just because of low probability of site-photoselected species.
There is also an emission analog of this effect: in concentrated solid dye solutions the energy homo-transfer fails to occur at the short-wavelength edge of emission band [113, 114]. In this case the emission of the dyes serving as EET acceptors and emitting at lower energies is not recorded, and the emission of donors remains at high energies and is highly polarized. In this case also the failure of energy transfer occurs in rigid and highly viscous solutions or very short observation times in liquid solvents and can be recorded as highly polarized emission.
Figure 12 serves as illustration of the mechanism of failure of excited-state energy transfer leading to observed Weber red-edge effect.

The scheme explaining the red-edge effect as the site-photoselection within the population of fluorescence emitters and the disappearance of EET leading to increase of polarization at the red edge of excitation spectrum [115]. The system composed of similar molecules located within the distances of efficient homo-EET is shown. At shorter wavelengths (case a) and band maximum (case b) the emitters exhibiting different interactions with the environment (marked with different colors) have equal probability to absorb light and transfer the excitation energy to its neighbors, which dramatically depolarizes the emission. At long-wavelength edge (case c) the species absorbing and emitting the low-energy quanta are excited only. They do not exhibit EET and emit independently, so their emission is highly polarized
4.1 Spectral Dependence of Emission Polarization
Fluorescence polarization technique is one of the most popular and informative tools in the studies of molecular structure and dynamics [77, 116] and the Weber’s red-edge effect opens new dimension in these studies. One of the most important practical applications is to discriminate two generally occurring effects leading to fluorescence depolarization: excitation energy transfer and fluorophore rotations. Rotational depolarization of fluorescence commonly occurs as a diffusional motion in structurally relaxed environments, and the depolarized emission should not depend on the excitation or emission energy. On contrary, the energy transfer efficiency depends on the correspondence of excited-state energies of donor and acceptor and is influenced by photoselection. Therefore the red-edge effect allows introducing the reference point at which this process fails to occur providing the means to detect and characterize EET. In view that in homo-transfer systems the spectral changes may not be significant, emission anisotropy is probably the most convenient method for such analysis.
This tool is the most frequently used in protein and biomembrane studies. Thus, for the measurements of polarization of the protein Trp fluorescence the excitations at 295 and 310 nm (near the red edge) were chosen [117]. Rotational diffusion leads to depolarization of the emission excited at either 295 or 310 nm, but homo-transfer only contributes to depolarization upon excitation at 295 nm. Hence, the 310/295 polarization ratio gives an indication of Trp–Trp energy transfer.
This effect of wavelength shifting opens the ways of manipulating with emission anisotropy by creating or avoiding the EET conditions. It has got interesting application in fluorescence polarization microscopy for detecting the interactions between the molecules within the living cells [118]. In biomolecular studies, homo-EET allows single labeling in contrast to more difficult double labeling that is normally used for observing proximity effects in hetero-EET systems.
The study of the effect of excitation wavelength on energy switching among the dyes located in well-defined positions in supramolecular structures is of both theoretical and practical value since it allows evaluating the extent of molecular order in the studied systems. As an example, β-cyclodextrin with appended seven 2-naphthoyloxy dyes, CD7(6), was synthesized [119, 120], see Fig. 13. Because the rotational motions of the dyes are frozen in the rigid glass, the depolarization effect observed with CD7(6) as compared with free dye NAEt (which displays strongly polarized emission over the whole long-wavelength part of the spectrum) is attributed solely to energy transfer between the appended dyes. It was found that despite the attachment to rigid structures they have their emission transition moments oriented variably from that of the directly excited one. When increasing the excitation wavelength beyond 335 nm, the emission anisotropy drastically increases, thus indicating a gradual decrease in energy transfer efficiency, so that at the extreme red edge (~350 nm) there is a complete lack of energy transfer (the Weber red-edge effect).

Chemical structure and excitation polarization spectra (observation wavelength: 380 nm) of the multichromophoric cyclodextrin CD7(3) in comparison with the model chromophore NA-Et (ethyl naphthoate) in a mixture of ethanol and methanol (9:1 v/v) at 110 K [119]
4.2 Shifts in Emission Spectra
Whereas the increase of the degree of polarization of light emission known as the Weber red-edge effect is the most convenient for observation, the spectroscopic changes are no less spectaculous and important. Since both donors and acceptors in homo-EET are chemically identical molecules located in the same medium, their excited-state distributions (see Fig. 3) are identical, so that each donor can serve as acceptor and the reverse. However, as a result of this transfer directed from those members of the ensemble that emit at shorter wavelengths to those that absorb at longer wavelengths, in rigid and highly viscous environments the fluorescence spectra are shifted to longer wavelengths [18].
Directed transfer can be easily observed in rigid media by comparing the spectra at the main-band excitation for concentrated and dilute dye solutions (Fig. 14). Due to transfer from “blue” to “red” emitters occurring in time, the fluorescence spectra are already shifted at the main-band excitation. The transfer from the main-band excitation to the red excitation edge results in two effects operating in opposite directions. One is the failure of transfer that increases the population of short-wavelength emitting donors and the other is the red-edge photoselection of low-energy emitters within the population. As a result, the observed shift will be much smaller. Finally, in the case of absence of aggregation in concentrated solutions the spectra recorded in the red-edge wavelength range should occupy the same positions as in the absence of energy homo-transfer.

Illustration of typical transformations of fluorescence spectra on transition to the red excitation edge in rigid environments for dilute solutions (no EET) and concentrated solutions (efficient EET at the main-band excitation). In dilute solutions the spectral shift is maximal. In contrast, in concentrated solutions because of EET the steady-state spectra are already shifted (and this shift can be observed as a function of time from initial position shown as a dashed line). Because of that the shift on transition to the red edge is much smaller leading to the same spectral position as in dilute solutions
Such wavelength-dependent behavior of steady-state spectra is characteristic for all systems exhibiting homo-EET. As an example, the results of studies of tryptophan dimer Trp–Trp [89] are presented (Fig. 15). At the main-band excitation 280 nm the spectrum of Trp occupies the short-wavelength position and is dramatically shifted on the shift of excitation wavelength to the red edge (305 nm). For Trp–Trp dimer, due to homo-EET the spectrum is shifted already and therefore the shift on the change of excitation wavelength is much smaller. Essentially, the spectral positions of Trp and Trp–Trp at 305 nm excitation superpose.

Temperature dependence in the positions of fluorescence band maxima of tryptophan monomer (Trp) and the dimer (Trp–Trp) in glycerol on excitation wavelength at the band maxima (280 nm) and red edge (305 nm) [89]
Interesting result is observed when the temperature is increased over the solvent relaxation range (see Fig. 15). Excitation wavelength stops to be a tool for site-photoselection and EET becomes no longer directed. We observe that the failure of directed transfer correlates with temperature-dependent activation of relaxational dynamics in the solvent. Therefore in liquid environment the spectra of Trp and Trp–Trp are nearly identical and excitation-wavelength independent.
4.3 The Effects Observed in Time Domain
Excitation energy homo-transfer proceeds in time on the scale of picoseconds–nanoseconds that is available to time-resolved fluorescence technique. In solid environments, when both dielectric relaxations and dye rotational motions are retarded, it can be detected both by the temporal loss of anisotropy and by the shifts of fluorescence emission bands. As a result of this directed transfer the fluorescence spectra in concentrated dye solutions in rigid and highly viscous environments are shifted as a function of time to longer wavelengths [22, 25, 45]. The time-resolved technique makes possible to visualize the dynamics of the process and measure the required kinetic parameters. The higher the dye molecule concentration, the faster is the energy transfer.
Transition to the red edge of excitation suppresses the transfer and modifies dramatically the emission decay by decreasing its emission-wavelength dependence (see Fig. 15), which allows introducing an important “no-transfer” reference limit. At these wavelengths the fluorophores emit individually, their emission spectra are similar to that observed at low concentrations and the decay kinetics simplified.
4.4 Light Harvesting and Its Modulation at the Red Edge
Directed excitation energy hopping between chromophores is a fundamental process occurring in the antennae pigments of photosynthetic units. The studies of photosynthetic antenna pigments in solutions [26] suggested the directed energy transfer to be an important mechanism of light-harvesting of solar energy in photosynthesis. Here the idea is to accommodate the pigments in high density to provide optimal collection of sunlight. The energy migrating between them with gradual decrease become assembled at the site with the lowest energy where it will be transferred to the electron-generating site. It was shown that in isolated core complexes, the polarization of the emission increases smoothly as a function of the excitation wavelength, starting from the center of absorption band, whereas in membranes the increase is abrupt and occurs in the extreme red edge [121]. These facts demonstrating the Weber red-edge effect suggest the possibility of modulation in the system of natural pigments its efficiency by the energy of absorbed light.
In analogy with natural systems of photosynthesis the idea on energy collection and migration was implemented into artificial antenna-based photomolecular devices, such as artificial solar concentrators [122]. Different molecular ensembles and nanostructures were designed for that [123, 124]. An example of directed transfer in polymer films and of its absence for single molecules can be the result on poly(3-hexylthiophene) [125]. Essentially, in these systems just due to inhomogeneous broadening and site-photoselection effects the on–off switching of light harvesting can be achieved just by shifting the excitation energy.
5 Red-Edge Effects and Ground-State Heterogeneity
Excitation-wavelength guided site-selectivity can be achieved not only within inhomogeneous broadening profile of absorption band, which is in the background of red-edge effects. It can be observed in different structurally heterogeneous systems, since also different types of impurities as well as ground-state heterogeneity in dye state (ionization, charge-transfer complexes, and H-bonding) may be in the origin of photoselection by shifting the wavelength. Operating on the atomic scale of distances, the positional heterogeneity can arise from imprecise location and orientation of the dye, e.g. its distribution between the sites of different polarities or at the surface of molecule or nanoparticle. Apparent spectral broadening can be the result of all these factors, and this could result in preferable excitation of one of the species with respect to others, resulting in shifted fluorescence spectrum.
Thus there can exist the effects of ground-state heterogeneity that are usually discrete in nature and bring new site-photoselection effects that overlap those resulting from common inhomogeneous broadening. Many such cases were reported. For instance, two polypyridyl ruthenium complexes possessing an extended conjugation and strongly coupled electronic states had strong excitation dependent emission behaviors caused by mixing of different electronic states [126].Two or more conformers having different geometry existing in ground-state equilibrium and absorbing light at different wavelengths may display variations in positions of their emission bands [127]. In our work [128] we tried to introduce the criteria that distinguish the effects of inhomogeneous broadening and those that originate from positional (ground-state) heterogeneity.
The distinction between the cases of inhomogeneous broadening and ground-state heterogeneity can be observed when the excitation-wavelength dependencies of fluorescence spectra are analyzed on a broad scale (Fig. 16). The red-edge effects demonstrate a very characteristic shape with the absence of the shifts of fluorescence spectra as a function of excitation wavelength at the maxima and short-wavelength wing of excitation bands but an unlimited increase of effect at the red excitation edge [30, 31, 35]. In contrast, the ground-state heterogeneity comes from superposition of absorption (excitation) spectra of the dyes present in different forms or residing in different locations. Because the quantum yield, anisotropy, and lifetime of a dye in these forms or locations can differ, the shapes of measured excitation-wavelength dependences as a general case is variable, even sigmoid [129].

Schematic representation of spectroscopic effects of (a) ground-state heterogeneity and (b) slow dielectric relaxations leading to red-edge effects [128]. In the case (a) variation of excitation wavelength leads to photoselection between the species possessing difference in excitation spectra, so that the dye excited at shorter wavelengths (λ ex1 ) exhibits blue emission at λ em1 and, correspondingly, excited at λ ex2 exhibits red-shifted emission at λ em2 . The shifting of excitation wavelength leads to change of relative contributions of two emissions. In the case (b) there is a single ground-state form but the shift of excitation wavelength from band maximum, λ exmax , to the red edge of excitation leads to progressive shift of emission spectra (two of them, λ em1 and λ em2 are shown) to longer wavelengths
The measurements of the shifts in excitation spectra and their polarization as a function of excitation wavelength may also serve as a good criterion [98]. Such effect was demonstrated in 6-methoxyquinoline in different polymer matrices [130]. Excitation-wavelength dependence of the dual emission and the fluorescence decays show a non-exponential behavior throughout the emission profile. The results are interpreted in terms of two groups (normal and charge transfer species) of ground-state conformers that are inhomogeneously distributed and assume different geometries in the polymer matrices.
Sometimes it is hard to distinguish the ground-state heterogeneity, especially in the case of excited-state reactions. The fluorescence of dimethylaminobenzonitrile (DMABN) and similar molecules in polymer glass consists of two bands due to formation of twisted intramolecular charge-transfer (TICT) state, as in polar fluid solvents. The TICT band in emission being small at the mainband excitation increases dramatically when fluorescence is excited at the red edge. Meantime, a strong deformation of an excitation spectrum as a function of emission wavelength indicated the significant site distribution of the ground-state species [93]. Describing site-selectivity in the redistribution of intensities of these fluorescence bands, Tomin [100] explained it by the presence of rotational isomers. They may possess different orientations of the dimethylamino group with respect to the plane of the benzonitrile residue and display the TICT reaction with different rates.
Important criterion for distinguishing the red-edge effects can be based on time-resolved data. Commonly the inhomogeneous broadening leads to non-exponential fluorescence decays that can extend over several orders of magnitude. Approximating of such inhomogeneous (in other terms, polychromatic or dispersive) kinetics by two or more exponential functions usually do not fit the experimental data. It can be provided using Kohlrausch “stretched exponential” function [131]. The decay N(t) of initial number N o of reactant molecules can be described as follows:
Here β describes the deviation from the first-order kinetics and τ characterizes the time scale of the reaction [131–133]. Generally, a smaller value of β corresponds to a wider distribution of lifetimes, suggesting a more disordered system. At the site-photoselective excitations due to decrease of the distribution width the fluorescence decays should become closer to exponential.
The red-edge effects depend strongly on the properties of a fluorophore. The dyes with low dependence for absorption spectra on solvent polarity and a very strong such dependence for emission spectra could be ideal for this application. They demonstrate a strong increase of the dipole moment in the excited state and exhibit a broad excited-state distribution on interaction energies. These features will determine their properties in spectral, anisotropy, and time domains. Good in this respect is tryptophan, which is the constituent of many proteins, and therefore we observe many applications of this method in protein research [31, 35]. Regarding different fluorescence dyes used for probing, the situation is variable. For instance, Nile Red, with its strong dependence on the environment polarity variation of both excitation and emission spectra, can display both the red-edge effects and the ground-state heterogeneity [134], and the analysis of these effects is complicated by the strong variation of its lifetime.
Fluorescent nanoparticles and composites are the structures that may be heterogeneous in size and composition by their origin. If the positions of their excitation and emission bands are size-dependent, the site-photoselection effects can be observed in their mixtures [135]. By combining semiconductor quantum dots of different sizes the effect of directed energy transfer can be achieved [136]. Carbon dots may demonstrate strong excitation-wavelength dependence of emission spectra [137] and single-molecular studies demonstrated that this is the result of structural heterogeneity [138]. Therefore the reported “giant red-edge effect” observed in graphene oxide nanoparticles [139] is probably the sole result of their structural heterogeneity.
6 Connection with the Studies of Single Molecules
It is common for us to assume that molecules of the same chemical composition must have identical properties. In reality though, every molecule in a condensed medium is different. Unlike an ideal crystal, where only vibrational motions are allowed, real condensed matter systems display much greater possibilities for intermolecular configurations resulting in structural, energetic, and dynamic molecular disorders. This results in a variety of new properties: broadening or even disappearance of first-order phase transitions in macromolecules [140], non-exponential kinetics of photophysical and photochemical reactions [141], non-Arrhenius-type activated processes [142], and others. Therefore it is extremely important to characterize molecular disorder in both conceptual and quantitative terms. Heterogeneity arising on the level of weak, in physical terms, intermolecular interactions with the local dye environment and their fluctuations on a time scale may result in broad variation of photophysical properties and of photochemical reactivity, which can be studied by fluorescence techniques. As we discussed above, the power of red-edge effects in these studies is their ability to compare the static and dynamic spectroscopic information of the whole ensemble of innumerable number of molecules with its sub-ensemble of energy-selected species.
Further decrease of this ensemble leads us to a single molecular level. These studies required introduction of new methods enabling to achieve the absolute limit of sensitivity in fluorescence spectroscopy [143, 144]. Observing a single molecule removes the usual ensemble-averaged picture and allows obtaining the most valuable information on the intensity, excitation and emission spectra, polarization, fluorescence decay rate of chosen individual molecules and of their chemical and photochemical reactivity in comparison to average molecules in ensemble of presumably identical species [145]. Selection of individual molecules from the ensemble can be achieved by ultimate dilution of dye solutions, illumination of ultra-small sample volumes, and application of confocal or two-photon microscopy [146]. On a single molecular level the spectral shifts and intensity fluctuations can be revealed even if they are present as rare events allowing the exploration of hidden heterogeneity in complex condensed phases [147].
The single-molecule detection allows providing a conceptually important move from ensemble analysis to individual event analysis. Individual molecules can be fitted to statistical ensemble. The more informative histograms based on single-molecular studies can be recorded, in which the responses from the members of molecular ensemble are seen as distinct events [148].
In studies of single molecules the concept on broad distribution of solvation energies in ensembles of fluorophore molecules in condensed media has got final confirmation. It has become evident that the red-edge effects do not break the Kasha rule. The Kasha rule must be applied not to whole ensemble but to individual emitters forming their inhomogeneously broadened ensemble.
Thus, the role of red-edge effects in photophysical studies of dyes in condensed phase in comparison with common spectroscopic experiments and investigations of single molecules can be summarized as follows (see Fig. 17):

Illustration of comparative information that can be obtained in populational research in condensed phase, explorations of red-edge effects in site-selective studies and the reconstructing of molecular ensemble properties from single molecular data. In populational studies the properties of individual molecules or their sub-ensembles are not resolved. At the red edge the site-selective studies of specific sequence of states are possible. In single molecular research the members of the ensemble are studied one by one. They can be identical chemical species, which differ in interactions with the environment, and an inhomogeneous contour of the ensemble of molecules can be built up from their individual contributions
-
The standard spectroscopic measurements yield the information on ensemble of emitters. Those are the average values of studied parameters for a large number of molecules. These studies remain to be of great value, since in real systems the physical behavior and chemical reactivity is the property of whole molecular ensembles.
-
The red-edge effects allow the observations of inhomogeneous broadening and molecular dynamics based on comparison in behavior of molecular ensemble and its sub-ensemble. They have become “classical,” playing their important role as a part of standard spectroscopic techniques.
-
The level of single molecules allows studying the fluctuations and statistical distributions, and an inhomogeneous contour of the ensemble of molecules can be built from these individual contributions. By removing completely the ensemble averaging, the distribution of every measured parameter can be obtained and a frequency histogram of the actual distribution of values can be constructed. Such distributions contains more information than the average value alone [149].
7 Perspective
The observation and analysis of red-edge effects introduced by Gregorio Weber has developed into a powerful methodology for studying molecular disorder and its coupling with molecular dynamics. With years of exploration, variation of excitation wavelength has become an important spectroscopic tool generating informative response in all optical parameters: spectral shifts, quenching, anisotropy, and lifetimes. This methodology is simple in application; it can easily complement other fluorescence spectroscopic methods in studying microscopically heterogeneous systems of different kind, including biological macromolecules in solutions and complex nanoscale systems. New areas of research were opened for rapid development.
The red-edge effects gave rise to a new understanding of spectroscopy of organic molecules in condensed phase. They showed that variation in solute–solvent local interactions is a significant source of the broadening in the absorption and emission spectra. Even in a structurally homogeneous phase, solute molecules can occupy a broad population of solvation sites differing in interaction with the environment and generating an array of electronic transition energies. It is their summation that comprises the absorption or emission spectra. Selective excitation of some members of this population enables them to be studied and treated selectively, so that comparison with the results obtained for the whole population gives new knowledge. This field of research can be called the site-selective spectroscopy of organic molecules.
In molecular dynamics of condensed matter a new very productive concept was introduced: the light absorption and emission spectra being formed by inhomogeneous ensemble of molecules reflect the stochastic dynamics of their formation and re-arrangement in time. Therefore the possibility appears to extract dynamic information by comparing the data of spectroscopic, time-resolved and anisotropy studies obtained for the whole ensemble on comparison with its selected part. From the above discussion it is clearly seen that the relaxation in molecular ensembles not only changes the average energy of excited fluorophores (commonly, it decreases). It is also the loss of “molecular memory”, so the process in time develops from correlated to uncorrelated behavior.
In photochemical reaction dynamics, a direct connection was established of the reaction yield and rate with the interaction of reaction site with its environment and dynamics of this environment. Site-selective photochemistry is demonstrated for all types of these reactions: isomerizations, intramolecular charge transfer, electron and proton transfer. Studies of solvation dynamics are very important for a detailed understanding of chemical reaction dynamics, catalysis and biocatalysis in solutions. Many chemical reactions proceed with significant intramolecular charge redistribution, and when they occur in condensed media they may exhibit site-selective inhomogeneous behavior.
In designing new nanoscale and molecular devices, scientists are often interested in assembling fluorophores to their high density. Here these species can exchange the interaction energies due to mechanism of non-radiative energy transfer. Different compositions can be designed to modulate such transfer, including light harvesting and wavelength conversion, and the possibility to suppress such transfer at the red excitation edge can be used for such modulation.
Finally, we observe how the unusual phenomenon became classical with extremely broad range of applications. We came to recognition that molecular disorder is not a complication in spectroscopic studies if we know how to extract extremely important information regarding the behavior of condensed matter on molecular scale and on the scale of intermolecular interactions. This information is especially valuable if the observed systems are microscopically heterogeneous and exhibit hierarchical dynamic features.
References
Weber G (1960) Fluorescence-polarization spectrum and electronic-energy transfer in tyrosine, tryptophan and related compounds. Biochem J 75(2):335
Anderson SR, Weber G (1969) Fluorescence polarization of the complexes of 1-anilino-8-naphthalenesulfonate with bovine serum albumin. Evidence for preferential orientation of the ligand. Biochemistry 8(1):371–377
Weber G, Shinitzky M (1970) Failure of energy transfer between identical aromatic molecules on excitation at the long wave edge of the absorption spectrum. Proc Natl Acad Sci USA 65(4):823–830
Gaviola E, Pringsheim P (1924) Über den Einfluß der Konzentration auf die Polarisation der Fluoreszenz von Farbstofflösungen. Z Physik 24:24–36
Förster T (1948) Zwischenmolekulare energiewanderung und fluoreszenz. Ann Physik 437(1‐2):55–75
Vavilov VI, Galanin MD (1949) Emission and absorption of light in the system of inductively coupled molecules. Dokl Akad Nauk USSR 67:811–818
Weber G (1954) Dependence of the polarization of the fluorescence on the concentration. Trans Faraday Soc 50:552–555
Terenin AN (1967) Photonics of dye molecules. Nauka, Leningrad
Birks JB (1975) Organic molecular photophysics. Wiley, New York
Turro NJ (1991) Modern molecular photochemistry. University Science Books, Sausalito, CA
Turro NJ, Ramamurthy V, Scaiano JC (2009) Principles of molecular photochemistry: an introduction. University Science Books, Sausalito, CA
Kasha M (1950) Characterization of electronic transitions in complex molecules. Disc Faraday Soc 9:14–19
Lamola AA, Turro NJ (1969) Energy transfer and organic photochemistry, vol 14. Interscience Publishers, New York
Galley WC, Purkey RM (1970) Role of heterogeneity of the solvation site in electronic spectra in solution. Proc Natl Acad Sci USA 67(3):1116–1121
Rubinov A, Tomin V (1970) Bathochromic luminescence in solutions of organic dyes at low temperatures. Opt Spektrosk USSR 29(6):578
Milton JG, Purkey RM, Galley WC (1978) Kinetics of solvent reorientation in hydroxylated solvents from exciting-wavelength dependence of chromophore emission-spectra. J Chem Phys 68(12):5396–5404
Rudik K, Pikulik L (1971) Effect of exciting light on fluorescence spectra of phthalimide solutions. Opt Spektrosk USSR 30(2):147
Gulis I, Komyak A (1977) Peculiarities of inductive-resonance energy transfer in the conditions of organic molecule electronic levels inhomogeneous broadening. J Appl Spectrosc 27(5):841–845
Pavlovich V (1976) Dependence of the spectra of excitation of dipole molecule solutions on the recording wavelength. J Appl Spectrosc 25(3):1141–1147
Azumi T, Itoh KI, Shiraishi H (1976) Shift of emission band upon the excitation at the long wavelength absorption edge. III. Temperature dependence of the shift and correlation with the time dependent spectral shift. J Chem Phys 65(7):2550–2555
Nemkovich N, Matseyko V, Tomin V (1980) Intermolecular up-relaxation in phthalimide solutions at excitation by frequency tuned dye laser. Opt Spektrosk USSR 49(2):274–283
Rubinov AN, Tomin VI, Bushuk BA (1982) Kinetic spectroscopy of orientational states of solvated dye molecules in polar solutions. J Luminescence 26:377–391
Gakamsky D, Nemkovich N, Rubinov A (1992) Wavelength-dependent rotation of dye molecules in a polar solution. J Fluorescence 2(2):81–92
Nemkovich N, Rubinov A (1995) Spectral inhomogeneity and wavelength-dependent rotation of probe molecules in membranes. J Fluorescence 5(3):285–294
Nemkovich N, Rubinov A, Tomin V (1981) Kinetics of luminescence spectra of rigid dye solutions due to directed electronic energy transfer. J Luminescence 23(3):349–361
Rubinov A, Zen'kevich E, Nemkovich N, Tomin V (1982) Directed energy transfer due to orientational broadening of energy levels in photosynthetic pigment solutions. J Luminescence 26(4):367–376
Macgregor RB, Weber G (1981) Fluorophores in polar media: spectral effects of the Langevin distribution of electrostatic interactions. Ann N Y Acad Sci 366(1):140–154
Mazurenko YT (1983) Statistics of solvation and solvatochromy. Opt Spektrosk USSR 55(3):471–478
Gorbatsevich S, Gulis I, Komyak A (1982) Molecular distribution function over the 0-0 transition frequencies in polar solutions. J Appl Spectrosc 36(3):332–337
Demchenko AP (1982) On the nanosecond mobility in proteins. Edge excitation fluorescence red shift of protein-bound 2-(p-toluidinylnaphthalene)-6-sulfonate. Biophys Chem 15:101–109
Demchenko AP (1986) Ultraviolet spectroscopy of proteins. Springer Verlag, Berlin-Heidelberg-New York
Demchenko AP, Shcherbatska NV (1985) Nanosecond dynamics of charged fluorescent probes at the polar interface of a membrane phospholipid bilayer. Biophys Chem 22:131–143
Demchenko AP, Ladokhin AS (1988) Red-Edge-Excitation Fluorescence Spectroscopy of Indole and Tryptophan. Eur Biophys J 15(6):369–379
Demchenko AP (1991) Fluorescence and dynamics in proteins. In: Lakowicz JR (ed) Topics in fluorescence spectroscopy, vol 3. Plenum, New York, pp 61–111
Demchenko AP (2002) The red-edge effects: 30 years of exploration. Luminescence 17(1):19–42
Mulkherjee S, Chattopadhyay A (1995) Wavelength-selective fluorescence as a novel tool to study organization and dynamics in complex biological systems. J Fluorescence 5:237–246
Chattopadhyay A, Haldar S (2014) Dynamic insight into protein structure using red edge excitation shift. Ac Chem Res 47(1):12–19
Demchenko AP (1986) Fluorescence analysis of protein dynamics. Essays Biochem 22:120–157
Lakowicz JR (2000) On spectral relaxation in proteins. Photochem Photobiol 72:421–437
Haldar S, Chaudhuri A, Chattopadhyay A (2011) Organization and dynamics of membrane probes and proteins utilizing the red edge excitation shift. J Phys Chem B 115:5693–5706
Raghuraman H, Kelkar DA, Chattopadhyay A (2005) Novel insights into protein structure and dynamics utilizing the red edge excitation shift approach. In: Reviews in fluorescence 2005. Springer, New York, pp 199–222
Chattopadhyay A, Haldar S (2013) Dynamic insight into protein structure utilizing red edge excitation shift. Acc Chem Res 47(1):12–19
Chattopadhyay A (2003) Exploring membrane organization and dynamics by the wavelength-selective fluorescence approach. Chem Phys Lipids 122(1-2):3–17
Hu Z, Margulis CJ (2007) Room-temperature ionic liquids: slow dynamics, viscosity, and the red edge effect. Acc Chem Res 40(11):1097–1105
Nemkovich NA, Rubinov AN, Tomin VI (1991) Inhomogeneous broadening of electronic spectra of dye molecules in solutions. In: Lakowicz JR (ed) Topics in fluorescence spectroscopy, vol 2. Plenum, New York, pp 367–428
Bushuk B, Rubinov A, Stupak A (1987) Inhomogeneous broadening of spectra of dye solutions due to intermolecular hydrogen bonding. J Appl Spectrosc 47(6):1251–1254
Gafert J, Friedrich J, Vanderkooi JM, Fidy J (1995) Structural changes and internal fields in proteins: a hole-burning Stark effect study of horseradish peroxidase. J Phys Chem 99(15):5223–5227
Logovinsky V, Kaposi A, Vanderkooi J (1993) Native and denatured Zn cytochrome c studied by fluorescence line narrowing spectroscopy. Biochim Biophys Acta 1161(2):149–160
Schomacker K, Champion P (1986) Investigations of spectral broadening mechanisms in biomolecules: cytochrome-c. J Chem Phys 84(10):5314–5325
Klán P, Wirz J (2009) Photochemistry of organic compounds: from concepts to practice. Wiley, New York
Rätsep M, Pajusalu M, Freiberg A (2009) Wavelength-dependent electron–phonon coupling in impurity glasses. Chem Phys Lett 479(1):140–143
Jankowiak R, Reppert M, Zazubovich V, Pieper Jr, Reinot T (2011) Site selective and single complex laser-based spectroscopies: a window on excited state electronic structure, excitation energy transfer, and electron–phonon coupling of selected photosynthetic complexes. Chem Rev 111(8):4546–4598
Reppert M, Naibo V, Jankowiak R (2010) Accurate modeling of fluorescence line narrowing difference spectra: Direct measurement of the single-site fluorescence spectrum. J Chem Phys 133(1):014506
Tomin V, Rubinov A (1981) Spectroscopy of inhomogeneous configurational broadening in dye solutions. J Appl Spectrosc 35(2):855–865
Personov R, Al'Shits E, Bykovskaya L-A (1972) The effect of fine structure appearance in laser-excited fluorescence spectra of organic compounds in solid solutions. Optics Communs 6(2):169–173
Wagie HE, Geissinger P (2012) Hole-burning spectroscopy as a probe of nano-environments and processes in biomolecules: a review. Appl Spectrosc 66(6):609–627
Rebane L, Gorokhovskii A, Kikas J (1982) Low-temperature spectroscopy of organic molecules in solids by photochemical hole burning. Appl Phys B 29(4):235–250
Murakami H, Kinoshita S, Hirata Y, Okada T, Mataga N (1992) Transient hole‐burning and time-resolved fluorescence spectra of dye molecules in solution: Evidence for ground‐state relaxation and hole‐filling effect. J Chem Phys 97(11):7881–7888
Jankowiak R, Hayes J, Small G (1993) Spectral hole-burning spectroscopy in amorphous molecular solids and proteins. Chem Rev 93(4):1471–1502
Demchenko AP (2008) Site-selective red-edge effects. Chapter 4. In: Methods in enzymology, vol 450. Academic, New York, pp 59–78
Ware WR, Lee SK, Brant GJ, Chow PP (1971) Nanosecond time‐resolved emission spectroscopy: spectral shifts due to solvent‐excited solute relaxation. J Chem Phys 54(11):4729–4737
Bakhshiev NG, Mazurenko YT, Piterskaya IV (1966) On the emission decay in various regions of molecule luminescence spectra in viscous solutions. Opt Spektrosk USSR 21(5):550–554
Mazurenko YT, Bakhshiev NG (1970) The influence of orientational dipolar relaxation on spectral, temporal and polarization properties of luminescence in solutions. Opt Spektrosk USSR 28:905–913
Brand L, Gohlike JR (1971) Nanosecond time-resolved fluorescence of a protein-dye complex BSA+TNS. J Biol Chem 246:2317–2324
Yang M, Richert R (2001) Observation of heterogeneity in the nanosecond dynamics of a liquid. J Chem Phys 115(6):2676–2680
Richert R (2002) Heterogeneous dynamics in liquids: fluctuations in space and time. J Phys Condensed Matter 14(23):R703
Maroncelli M (1993) The dynamics of solvation in polar liquids. J Mol Liquids 57:1–37
Fee RS, Milsom JA, Maroncelli M (1991) Inhomogeneous decay kinetics and apparent solvent relaxation at low temperatures. J Phys Chem 95(13):5170–5181
Nemkovich N, Rubinov A (1996) Spectral and spatial heterogeneity of fluorescent probes in membranes. J Appl Spectrosc 63(4):522–529
Vincent M, Gallay J, Demchenko AP (1995) Solvent relaxation around the excited state of indole: analysis of fluorescence lifetime distributions and time-dependent spectral shifts. J Phys Chem 99:34931–34941
Richert R (2001) Spectral diffusion in liquids with fluctuating solvent responses: dynamical heterogeneity and rate exchange. J Chem Phys 115(3):1429–1434
Lëvshin L, Struganova I, Toleutaev B (1988) Dynamics of inhomogeneous broadening of fluorescence spectra of dye solutions. J Appl Spectrosc 49(1):695–699
Voropay E, Koyava V, Saechnikov V, Sarjevsky A (1980) Some effects in inhomogeneous level broadening at excitation energy transfer conditions. J Appl Spectrosc 32:457–463
Kovalenko S, Schanz R, Hennig H, Ernsting N (2001) Cooling dynamics of an optically excited molecular probe in solution from femtosecond broadband transient absorption spectroscopy. J Chem Phys 115(7):3256–3273
Demchenko AP, Ladokhin AS (1988) Temperature-dependent shift of fluorescence spectra without conformational changes in protein; studies of dipole relaxation in the melittin molecule. Biochim Biophys Acta 955(3):352–360
Clark J, Miller P, Rumbles G (1998) Red edge photophysics of ethanolic rhodamine 101 and the observation of laser cooling in the condensed phase. J Phys Chem A 102(24):4428–4437
Jameson DM, Ross JA (2010) Fluorescence polarization/anisotropy in diagnostics and imaging. Chem Rev 110(5):2685–2708
Mazurenko YT, Bakhshiev N, Piterskaya I (1968) Spectral dependence of the degree of rotational depolarization of the fluorescence of complex molecules in viscous solutions. Opt Spektrosk USSR 25:46
Gakamsky DM, Goldin AA, Petrov EP, Rubinov AN (1992) Fluorescence decay time distribution for polar dye solutions with time-dependent fluorescent shift. Biophys Chem 44(1):47–60
Gakamskii D, Nemkovich N, Rubinov A (1991) Molecular relaxation and rotation of dye molecules in polar solvents (A review). J Applied Spectrosc 54(2):99–111
Kinoshita S, Itoh H, Murakami H, Miyasaka H, Okada T, Mataga N (1990) Solvent relaxation effect on transient hole-burning spectra of organic dyes. Chem Phys Lett 166(2):123–127
Huang J, Ridsdale A, Wang J, Friedman JM (1997) Kinetic hole burning, hole filling, and conformational relaxation in heme proteins: direct evidence for the functional significance of a hierarchy of dynamical processes. Biochemistry 36(47):14353–14365
Demchenko AP (1994) Protein fluorescence, dynamics and function: exploration of analogy between electronically excited and biocatalytic transition states. Biochim Biophys Acta 1209:149–164
Maroncelli M, McInnis J, Fleming GR (1989) Polar solvent dynamics and electron transfer reactions. Science 243:1674–1681
Bader AN, Pivovarenko VG, Demchenko AP, Ariese F, Gooijer C (2004) Influence of redistribution of electron density on the excited state and ground state proton transfer rates of 3-hydroxyflavone and its derivatives studied by Spol’skii spectroscopy. J Phys Chem B 108(29):10589–10595
Marcus R, Sutin N (1985) Electron transfer in chemistry and biology. Biochim Biophys Acta 811:265–322
Demchenko AP, Tang K-C, Chou P-T (2013) Excited-state proton coupled charge transfer modulated by molecular structure and media polarization. Chem Soc Rev 42(3):1379–1408
Demchenko AP (1992) Does biocatalysis involve inhomogeneous kinetics? FEBS Lett 310(3):211–215
Demchenko AP, Sytnik AI (1991) Site-selectivity in excited-state reactions in solutions. J Phys Chem 95:10518–10524
Demchenko AP, Sytnik AI (1991) Solvent reorganizational red-edge effect in intramolecular electron transfer. Proc Natl Acad Sci USA 88(20):9311–9314
Letrun R, Vauthey E (2014) Excitation wavelength dependence of the dynamics of bimolecular photoinduced electron transfer reactions. J Phys Chem Lett 5(10):1685–1690
Tomin V, Heldt J (2003) The red-edge effects in Laurdan solutions. Z Naturforschung A 58(2–3):109–117
Al-Hassan KA, Rettig W (1986) Free volume sensing fluorescent probes. Chem Phys Lett 126(3–4):273–279
Braun D, Rettig W (1997) Excitation energy dependence of the kinetics of charge-transfer formation. Chem Phys Lett 268(1–2):110–116
Wallace-Williams SE, Møller S, Goldbeck RA, Hanson KM, Lewis JW, Lee WA, Kliger DS (1993) Excited-state s-cis rotamers produced by extreme red edge excitation of all-trans-1, 4-diphenyl-1, 3-butadiene. J Phys Chem 97(38):9587–9592
Schweke D, Baumgarten H, Haas Y, Rettig W, Dick B (2005) Charge-transfer-type fluorescence of 4-(1 H-Pyrrol-1-yl) benzonitrile (PBN) and N-phenylpyrrole (PP) in cryogenic matrixes: evidence for direct excitation of the CT band. J Phys Chem A 109(4):576–585
Al‐Hassan KA (1988) Edge‐excitation red shift of the fluorescence of flexible solute molecules in a poly (methyl methacrylate) polymer matrix. J Polym Sci B Polym Phys 26(8):1727–1733
Józefowicz M, Heldt JR, Bajorek A, Pączkowski J (2008) Red-edge and inhomogeneous broadening effects of the electronic spectra of ethyl 5-(4-aminophenyl)-3-amino-2, 4-dicyanobenzoate. J Photochem Photobiol A: Chem 196(1):38–43
Tomin V, Hubisz K (2006) Instantaneous emission spectra and molecular rotation of n-dimethylaminobenzonitrile fluorescing in the long-wavelength spectral range. Opt Spektrosk USSR 100(1):65–74
Tomin V, Wlodarkiewicz A (2013) The influence of temperature on red-edge excitation effects in liquid solutions of N,N′-dimethylaminobenzonitrile. Opt Spektrosk USSR 115(1):86–93
Bader AN, Pivovarenko VG, Demchenko AP, Ariese F, Gooijer C (2004) Excited state and ground state proton transfer rates of 3-hydroxyflavone and its derivatives studied by Shpol'skii spectroscopy: the influence of redistribution of electron density. J Phys Chem B 108(29):10589–10595
Tomin VI, Demchenko AP, Chou P-T (2015) Thermodynamic vs. kinetic control of excited-state proton transfer reactions. J Photochem Photobiol C: Photochem Rev 22:1–18
Klymchenko AS, Demchenko AP (2003) Multiparametric probing of intermolecular interactions with fluorescent dye exhibiting excited state intramolecular proton transfer. Phys Chem Chem Phys 5(3):461–468
Ghosh D, Batuta S, Das S, Begum NA, Mandal D (2015) Proton transfer dynamics of 4′-N,N-dimethylamino-3-hydroxyflavone observed in hydrogen-bonding solvents and aqueous micelles. J Phys Chem B 119(17):5650–5661
Ercelen S, Klymchenko AS, Demchenko AP (2003) Novel two-color fluorescence probe with extreme specificity to bovine serum albumin. FEBS Lett 538(1):25–28
Demchenko AP, Ercelen S, Klymchenko AS (2002) Site-selective red-edge spectroscopy of disordered materials and microheterogeneous systems: polymers, phospholipid membranes and proteins. SPIE Int Soc Optics Photonics 4938. doi:10.1117/12.486641
Suda K, Terazima M, Kimura Y (2012) Excitation wavelength dependence of photo-induced intramolecular proton transfer reaction of 4′-N, N-diethylamino-3-hydroxyflavone in various liquids. Chem Phys Lett 531:70–74
Suda K, Terazima M, Sato H, Kimura Y (2013) Excitation wavelength dependence of excited state intramolecular proton transfer reaction of 4′-N,N-diethylamino-3-hydroxyflavone in room temperature ionic liquids studied by optical Kerr gate fluorescence measurement. J Phys Chem B 117(41):12567–12582
Nemkovich N, Kruchenok J, Rubinov A, Pivovarenko V, Baumann W (2001) Site selectivity in excited-state intramolecular proton transfer in flavonols. J Photochem Photobiol A Chem 139(1):53–62
Tomin V, Jaworski R (2013) Modulation of the proton transfer rate by excitation photons. Opt Spektrosk USSR 114(5):729–736
Nicolet O, Banerji N, Pages S, Vauthey E (2005) Effect of the excitation wavelength on the ultrafast charge recombination dynamics of donor-acceptor complexes in polar solvents. J Phys Chem A 109(37):8236–8245
Engelkamp H, Hatzakis NS, Hofkens J, De Schryver FC, Nolte R, Rowan AE (2006) Do enzymes sleep and work? Chem Communs 9:935
Koyava V, Popechits V (1979) Directed energy transfer in solid polar dye mixtures. J Appl Spectrosc 31(6):1484–1488
Koyava V, Popechits V, Sarzhevskii A (1980) Concentration depolarization of fluorescence in systems with nonuniformly broadened electronic levels. J Appl Spectrosc 32(6):597–602
Demchenko AP (2013) Nanoparticles and nanocomposites for fluorescence sensing and imaging. Meth Appl Fluorescence 1(2):022001
Jameson DM, Croney JC (2003) Fluorescence polarization: past, present and future. Comb Chem High Throughput Screen 6(3):167–176
Moens PD, Helms MK, Jameson DM (2004) Detection of tryptophan to tryptophan energy transfer in proteins. Protein J 23(1):79–83
Squire A, Verveer PJ, Rocks O, Bastiaens PI (2004) Red-edge anisotropy microscopy enables dynamic imaging of homo-FRET between green fluorescent proteins in cells. J Struct Biol 147(1):62–69
Berberan-Santos MN, Pouget J, Valeur B, Canceill J, Jullien L, Lehn JM (1993) Multichromophoric cyclodextrins. 2. Inhomogeneous spectral broadening and directed energy hopping. J Phys Chem 97(44):11376–11379
Berberan-Santos MN, Canceill J, Gratton E, Jullien L, Lehn J-M, So P, Sutin J, Valeur B (1996) Multichromophoric cyclodextrins. 3. Investigation of dynamics of energy hopping by frequency-domain fluorometry. J Phys Chem 100(1):15–20
Monshouwer R, Visschers RW, van Mourik F, Freiberg A, van Grondelle R (1995) Low-temperature absorption and site-selected fluorescence of the light-harvesting antenna of Rhodopseudomonas viridis. Evidence for heterogeneity. Biochim Biophys Acta 1229(3):373–380
Harriman A (2015) Artificial light-harvesting arrays for solar energy conversion. Chem Communs 51(59):11745–11756
Andrews DL (2008) Energy harvesting: a review of the interplay between structure and mechanism. J Nanophotonics 2(1):022502–022525
Joly D, Delgado JL, Atienza C, Martın N (2015) Light-harvesting materials for organic electronics. Photonics Nanophotonic Struct Mat 2:311
Thiessen A, Vogelsang J, Adachi T, Steiner F, Bout DV, Lupton JM (2013) Unraveling the chromophoric disorder of poly (3-hexylthiophene). Proc Natl Acad Sci USA 110(38):E3550–E3556
Xiao L, Xu Y, Yan M, Galipeau D, Peng X, Yan X (2010) Excitation-dependent fluorescence of triphenylamine-substituted tridentate pyridyl ruthenium complexes. J Phys Chem A 114(34):9090–9097
Józefowicz M, Heldt JR (2011) Excitation-wavelength dependent fluorescence of ethyl 5-(4-aminophenyl)-3-amino-2, 4-dicyanobenzoate. J Fluorescence 21(1):239–245
Demchenko AP, Yesylevskyy SO (2011) Interfacial behavior of fluorescent dyes. In: Advanced fluorescence reporters in chemistry and biology III. Springer, Heidelberg, pp 3–62
Sharma VK, Sahare PD, Rastogi RC, Ghoshal SK, Mohan D (2003) Excited state characteristics of acridine dyes: acriflavine and acridine orange. Spectrochim Acta A 59(8):1799–1804
Mehata M, Joshi H, Tripathi H (2001) Edge excitation red shift and charge transfer study of 6-methoxyquinoline in polymer matrices. J Luminescence 93(4):275–280
Valeur B, Berberan-Santos MN (2012) Molecular fluorescence: principles and applications. Wiley, New York
Berberan-Santos M, Bodunov E, Valeur B (2005) Mathematical functions for the analysis of luminescence decays with underlying distributions 1. Kohlrausch decay function (stretched exponential). Chem Phys 315(1):171–182
Edholm O, Blomberg C (2000) Stretched exponentials and barrier distributions. Chem Phys 252(1-2):221–225
Datta A, Mandal D, Pal SK, Bhattacharyya K (1997) Intramolecular charge transfer processes in confined systems. Nile Red in reverse micelles. J Phys Chem B 101:10221–10225
Tintu R, Nampoori V, Radhakrishnan P, Unnikrishnan N, Thomas S (2012) Cluster size and excitation wavelength dependent photoluminescence behavior of nano colloidal Ge-Se-Sb-Ga chalcogenide glass solutions. J Optoelectronics Adv Mat 14(11):918
Kawazoe T, Kobayashi K, Ohtsu M (2005) Optical nanofountain: a biomimetic device that concentrates optical energy in a nanometric region. Appl Phys Lett 86(10):103102
Demchenko AP, Dekaliuk MO (2013) Novel fluorescent carbonic nanomaterials for sensing and imaging. Meth Appl Fluorescence 1(4):042001
Ghosh S, Chizhik AM, Karedla N, Dekaliuk MO, Gregor I, Schuhmann H, Seibt M, Bodensiek K, Schaap IA, Schulz O (2014) Photoluminescence of carbon nanodots: dipole emission centers and electron–phonon coupling. Nano Lett 14(10):5656–5661
Cushing SK, Li M, Huang F, Wu N (2013) Origin of strong excitation wavelength dependent fluorescence of graphene oxide. ACS Nano 8(1):1002–1013
Scharnagl C, Reif M, Friedrich J (2005) Stability of proteins: temperature, pressure and the role of the solvent. Biochim Biophys Acta 1749(2):187–213
Richert R, Elschner A, Bassler H (1986) Experimental-study of nonexponential relaxation processes in random organic-solids. Z Phys Chem Neue Folge 149:63–75
Demchenko A (1997) Breaks in Arrhenius plots for enzyme reactions: the switches between different protein dynamics regimes? Comments Mol Cell Biophys 9:87–112
Moerner W, Kador L (1989) Optical detection and spectroscopy of single molecules in a solid. Phys Rev Lett 62(21):2535
Ambrose W, Moerner W (1991) Fluorescence spectroscopy and spectral diffusion of single impurity molecules in a crystal. Nature 349:225–227
Moerner WE (2007) New directions in single-molecule imaging and analysis. Proc Natl Acad Sci USA 104(31):12596–12602
Tinnefeld P, Herten D-P, Sauer M (2001) Photophysical dynamics of single molecules studied by spectrally-resolved fluorescence lifetime imaging microscopy (SFLIM). J Phys Chem A 105(34):7989–8003
Xie XS, Trautman JK (1998) Optical studies of single molecules at room temperature. Ann Rev Phys Chem 49(1):441–480
Deniz AA, Mukhopadhyay S, Lemke EA (2008) Single-molecule biophysics: at the interface of biology, physics and chemistry. J Royal Soc Interface 5(18):15–45
Hohlbein J, Gryte K, Heilemann M, Kapanidis AN (2010) Surfing on a new wave of single-molecule fluorescence methods. Phys Biol 7(3):031001
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Demchenko, A.P. (2016). Weber’s Red-Edge Effect that Changed the Paradigm in Photophysics and Photochemistry. In: Jameson, D. (eds) Perspectives on Fluorescence. Springer Series on Fluorescence, vol 17. Springer, Cham. https://doi.org/10.1007/4243_2016_14
Download citation
DOI: https://doi.org/10.1007/4243_2016_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41326-6
Online ISBN: 978-3-319-41328-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)