
Abstract
The transformation of N2 into NH3 (nitrogen fixation) on transition metal complexes generally involves complicated elementary reaction steps and a number of possible reaction intermediates because at least six pairs of proton and electron (or six hydrogen atoms) must take part in this process. Mechanistic details of nitrogen fixation will be disclosed by close liaison between theory and experiment. In this chapter, recent advances in the mechanistic understanding of the catalytic transformation of N2 to NH3 on mono- and dinuclear Mo–N2 complexes are overviewed from a theoretical perspective. In particular, catalytic mechanisms of nitrogen fixation by dinitrogen-bridged dimolybdenum complexes bearing pincer ligands are discussed in detail based on density-functional-theory calculations corroborated by experimental findings.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
As a part of the global nitrogen cycle on the earth, biological nitrogen fixation is efficiently accomplished by nitrogenases in certain bacteria. The Mo-containing nitrogenase has a double cubane-type iron–molybdenum–sulfur complex MoFe7S9C called FeMo-cofactor at the active site, and the multinuclear transition-metal complex binds dinitrogen (N2) and reduces it to ammonia (NH3) at ambient temperature under atmospheric pressure (Fig. 1a) [1–4]. According to the optimal reaction, eight pairs of proton and electron are utilized for the formation of two molecules of NH3 and the obligatory evolution of one molecule of H2. Although the exact reaction mechanism of the biological nitrogen fixation has not been fully understood, a compelling mechanism is proposed on the basis of a series of excellent spectroscopic studies [5]. In the proposed mechanism, the binding of N2 occurs at the E4 intermediate, where four pairs of proton and electron are accumulated in the FeMo cofactor and two of the hydrogens bridge between iron atoms to form [Fe–H–Fe] moieties (Fig. 1b).

(a) Biological nitrogen fixation by the Mo-containing nitrogenase. (b) A possible structure of the E4 intermediate
The biological nitrogen fixation system is an attractive model for designing an alternative to the industrial Haber–Bosch process, in which NH3 is produced from N2 and H2 under harsh conditions. It should be noted that more than 90% of the total energy for the industrial NH3 production is consumed for the production of H2 (and CO2) from fossil fuels [6]. From an environmental point of view, much attention has been paid to novel nitrogen fixation systems that operate without the use of H2 under ambient conditions in the homogeneous phase. Since the discovery of the first transition-metal–N2 complex in 1965 [7], a large number of metal–N2 complexes have been synthesized and the stoichiometric reactivity of coordinated N2 has been thoroughly examined [8–12]. At present, however, very limited transition metal complexes are known to work as catalysts for the direct formation of NH3 from N2 in the presence of proton and electron sources (Fig. 2) [13–22]. Interestingly, all the catalysts ever reported contain Mo and Fe at their metal centers, similar to the FeMo cofactor. Quite recently, two research groups have independently demonstrated a cobalt-catalyzed transformation of N2 into NH3 [23, 24]. Unfortunately, the turnover numbers of all the catalysts mentioned above are still low compared with heterogeneous Fe catalysts utilized for the Haber-Bosch process. Relevant to the biological nitrogen fixation by the Mo-containing nitrogenase, it should be noted that Fe and Co complexes bearing an anionic PNP pincer ligand catalyzes the formation of NH3 and hydrazine N2H4 [22, 24], where N2H4 is known as an intermediate during the biological N2 reduction [1]. Very recently, Ashley and coworkers reported a selective catalytic reduction of N2 to N2H4 using an Fe(0) complex Fe(depe)2(N2) (depe=Et2PCH2CH2PEt2) [25].

Mo and Fe-based synthetic catalysts for nitrogen fixation with their turnover numbers of NH3 production per metal atom
To develop more effective nitrogen fixation catalysts under ambient conditions in the homogenous phase, we must extract as much information as possible from the limited successful examples. Computational chemistry will be able to provide useful mechanistic insights into the complicated catalytic cycles involving short-lived intermediates that are often difficult to detect or characterize experimentally. In particular for the Mo-catalyzed nitrogen fixation systems in Fig. 2, quantum chemical calculations by density-functional-theory (DFT) methods have been applied for elucidating detailed mechanisms of the transformation of N2 based on experimental findings, and also been exploited for designing more effective catalysts [15, 26]. In this chapter, we overview recent advances in the mechanistic understanding of catalytic transformation of N2 to NH3 by mono- and dimolybdenum complexes from a theoretical perspective.
2 Nitrogen Fixation Catalyzed by an Mo–Triamidoamine Complex: The Yandulov–Schrock Cycle
In 2003, Yandulov and Schrock showed a pioneering example of the direct formation of NH3 from N2 under ambient conditions using an Mo–triamidoamine complex, [HIPTN3N]Mo(N2) 1 (HIPT = hexaisopropylterphenyl). In the presence of 2,6-lutidinium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate ([LutH]BArF 4) as a proton source and decamethylchromocene (CrCp*2) as a reducing reagent, 1 served as the catalyst for nitrogen fixation at room temperature, where up to 8.2 equiv of NH3 was produced based on 1 (Scheme 1) [16]. Unfortunately, the turnover number of the catalyst has not been updated despite various modifications of the original complex 1. The preparation and characterization of intermediates relevant to the catalytic reaction led them to propose a catalytic mechanism involving a stepwise addition of protons and electrons, the so-called Yandulov–Schrock cycle [16, 26–29]. Their proposal based on concrete experimental evidence received much interest from theoretical researchers, and the validity of the proposed catalytic mechanism has been advocated by intensive computational studies on the energetics of possible reaction pathways conducted by Cao et al. [30], Studt and Tuczek [31], Magistrato et al. [32], and Reiher and coworkers [33–38].

Transformation of N2 into NH3 catalyzed by a Mo–triamidoamine complex 1
Figure 3 describes the Yandulov–Schrock cycle for nitrogen fixation catalyzed by 1, where [Mo] represents [HIPTN3N]Mo. The catalytic formation of NH3 in the Yandulov–Schrock cycle involves a successive protonation of the distal N atom of coordinated N2, which is the one far from Mo. Each protonation step is followed by one-electron reduction step. Addition of three pairs of proton/electron to 1 affords a nitride intermediate [Mo(≡N)] and the first molecule of NH3 via the formation of diazenide [Mo(N=NH)], hydrazide(2-) [Mo(NNH2)], and hydrazidium [Mo(NNH3)] intermediates. Protonation of the proximal N atom of the N2 ligand, which is the one directly connected with Mo, is not considered in this catalytic cycle because diazene (HN=NH) and hydrazine (H2N–NH2) were not observed experimentally. The remaining nitride N atom in [Mo(≡N)] is then protonated and reduced to give an ammine intermediate [Mo(NH3)] via imide [Mo(NH)] and amide [Mo(NH2)] intermediates. Finally the NH3 ligand is replaced by an incoming N2 molecule to complete the catalytic cycle. For the first protonation of 1, Reiher and coworkers theoretically suggested an alternative pathway in which protonation of an amide N atom of the HIPTN3N ligand precedes protonation of the N2 ligand [33, 35]. Later, Schrock and coworkers confirmed this computational suggestion by ENDOR spectroscopy [28].

The Yandulov–Schrock cycle for the transformation of dinitrogen into ammonia catalyzed by 1. [Mo] represents [HIPTN3N]Mo
3 Nitrogen Fixation Catalyzed by Dinitrogen-Bridged Dimolybdenum Complexes
3.1 Dimolybdenum Complexes Bearing PNP-Type Pincer Ligands
In 2011, Nishibayashi and coworkers reported a new nitrogen fixation system operating under mild conditions using a dinitrogen-bridged dimolybdenum complex bearing a pyridine-based PNP-type pincer ligand, [{Mo(N2)2(PNP)}2(μ-N2)] (2: PNP=2,6-bis(di-tert-butylphosphinomethyl)pyridine), as a catalyst (Scheme 2) [17]. In the presence of [LutH]OTf (OTf=CF3SO3) as a proton source and cobaltocene (CoCp2) as a reducing reagent, up to 23 equiv of NH3 based on the catalyst was produced from N2 gas. In this first report, they postulated a catalytic mechanism of the transformation of N2 into NH3 by reference to the Yandulov–Schrock mechanism, i.e., mononuclear Mo–N2 complexes separated from 2, such as [Mo(N2)3(PNP)], work as key reactive intermediates in the catalytic cycle (Fig. 4a). Batista and coworkers pointed out a remarkable role of dinuclear Mo complexes in the catalytic activity of 2 based on theoretical calculations from a thermodynamic aspect [39]. To get a mechanistic insight into nitrogen fixation in the dimolybdenum system, Yoshizawa, Nishibayashi, and coworkers demonstrated a synergetic relationship between theory and experiment [40].

Transformation of N2 into NH3 catalyzed by a dimolybdenum complex bearing PNP-type pincer ligands 2

A theoretically plausible catalytic cycle described in Fig. 4b is corroborated by the following experimental findings: [17, 40] (1) Dimolybdenum complex 2 contains five N2 ligands, four terminal and one bridging. The terminal N2 ligands can be replaced by CO molecules to afford [{Mo(CO)2(PNP)}2(μ-N2)]. The CO-substituted dimolybdenum complex produced less than a stoichiometric amount of NH3. (2) A six-coordinate mononuclear Mo–N2 complex, trans-[Mo(N2)2(PMe2Ph)(PNP)], showed no catalytic activity. (3) The catalytic activity of 2 strongly depends on the nature of the counter anion of the proton source (LutH+). The amount of produced NH3 was drastically decreased when [LutH]Cl and [LutH]BArF 4 were adopted as proton sources. (4) Two Mo–nitride complexes, [Mo(≡N)(PNP)Cl] and [Mo(≡N)(PNP)Cl]OTf exhibited almost the same catalytic activity as 2. (5) Fragments assignable to [Mo(≡N)(OTf)(PNP)–N≡N–Mo(N2)(PNP)] and [Mo(≡N)(OTf)(PNP)] were observed by mass spectrometry from the stoichiometric reaction of 2 with 2 equiv of [LutH]OTf in toluene at room temperature. (6) A mononuclear Mo–imide complex [Mo(=NH)(C5H5N)(PNP)Cl]OTf, in which a pyridine molecule occupies the equatorial coordination site, was isolated. (7) A fragment assignable to [Mo(NH3)(PNP)–N≡N–Mo(N2)2(PNP)] or [Mo(NH3)(N2)(PNP)–N≡N–Mo(N2)(PNP)] was observed by mass spectrometry from a reaction mixture of the catalytic reaction of 2 with excess amounts of [LutH]OTf and CoCp2. These experimental findings were exploited for developing a hypothesis in the theory-based proposal: (a) Terminal N2 ligands are essential for the catalytic formation of NH3. (b) Dimolybdenum complexes with the bridging N2 ligand must be involved in the catalytic cycle. (c) Triflate (OTf−) as the counter anion of LutH+ would play a role in the catalytic cycle. (d) A dinuclear Mo complex is separated into mononuclear ones to afford [Mo(≡N)(OTf)(PNP)] as a key intermediate at a certain stage in the catalytic cycle. (e) Six-coordinate mononuclear Mo-imide complexes such as [Mo(=NH)(OTf)(N2)(PNP)] and its cation, in which N2 occupies the equatorial site, are formed in the course of protonation and reduction of [Mo(≡N)(OTf)(PNP)]. This process is prepared for regeneration of 2 to complete the catalytic cycle.
The proposed catalytic cycle shown in Fig. 4b involves hydrogenations of an N2 ligand in 2 leading to generation of the first NH3 molecule without separation of the dinuclear Mo–N≡N–Mo structure. The numbering of intermediates is given in Fig. 5. The first protonation should occur at one of the terminal N2 ligands in 2 and is followed by exchange of the N2 ligand trans to the protonated N2 ligand for the counter anion of the proton source, OTf− (2 → C). Addition of two proton/electron pairs to C leads to the formation of the first NH3 molecule. The protonation and reduction of the NNH group in C prompts a facile dissociation of a dative Mo–N2 bond between an Mo atom and the bridging N2 ligand to give two mononuclear Mo complexes, a nitride complex G and a dinitrogen complex H. Addition of three proton/electron pairs to G accompanied by the coordination of N2 at the equatorial position results in the formation of a six-coordinate ammine complex L. Addition of the sixth electron to L liberates the OTf− ligand to form a five-coordinate ammine complex M, which couples with H to regenerate a dimolybdenum complex. The regeneration of the Mo–N≡N–Mo structure is linked with the replacement of the NH3 ligand by an incoming N2 molecule to complete the catalytic cycle.

(a) Protonation of a terminal N2 ligand. (b) First NH3 formation and separation into mononuclear Mo complexes. (c) Second NH3 formation. (d) Regeneration of dinuclear structure and ligand exchange. A possible reaction pathway for the transformation of N2 into NH3 starting from 2. L3 represents the PNP pincer ligand. Energy changes and activation energies (in parenthesis) for individual reaction steps were calculated at the B3LYP*/BS2 level of theory (units in kcal mol−1). NB means that the corresponding reaction has no activation barrier. Protons are supplied by lutidinium. For reduction steps, energy difference between protonated and neutral species is presented. Values in italics correspond to the reaction energies obtained with cobaltocene
Figure 5 presents the energetics of detailed reaction pathways for the transformation of N2 into NH3 catalyzed by 2. Alternating protonation/reduction steps were assumed in the calculated pathways. Activation energies of the proton transfer from LutH+ were evaluated for all protonation steps. Energy profiles of the reduction steps were obtained as the energy difference between protonated and neutral intermediates. The values in italics correspond to the reaction energies calculated based on the equation [XH]+ + [CoCp2] → [XH] + [CoCp2]+, where [XH]+ is a protonated intermediate. The latter values should be carefully treated because each species in the equation is separately calculated at infinite distance [35]. Energy changes (ΔE) and activation energies (E a in parenthesis) were calculated at the B3LYP* level of theory [41, 42]. For optimization, the LANL2DZ and 6-31G(d) basis sets were chosen for the Mo atom and the other atoms, respectively (BS1). To determine the energy profiles of individual reaction steps, single-point energy calculations were performed at the optimized geometries by BS1 using the Stuttgart-Dresden pseudopotentials (SDD) and 6-311+G(d,p) basis sets (BS2) in place of the LANL2DZ and 6-31G(d) basis sets, respectively. In this review, we concentrate on the three important steps in the catalytic cycle, (1) the protonation of a terminal N2 ligand in 2 leading to C, (2) the formation of mononuclear Mo–nitride complex G, and (3) the ligand exchange of NH3 for N2 at the final step of the catalytic cycle.
To start the catalytic cycle for the transformation of N2, one of the N2 ligands in 2 must be protonated. Dinitrogen complex 2 has at least two possible sites for protonation, four equivalent terminal N2 ligands and one bridging N2 ligand. The N≡N stretching frequencies of 2 in THF were experimentally observed at 1944 cm−1 (terminal N2) and 1890 cm−1 (bridging N2), indicating that the bridging N2 ligand is more strongly activated [17]. However, as shown in Fig. 6a, the protonation of the bridging N2 ligand is not likely to occur at room temperature due to its extremely high activation energy (40.7 kcal mol−1). In contrast, the activation energy for the protonation of a terminal N2 ligand is relatively low (8.4 kcal mol−1) although the reaction is endothermic by 6.5 kcal mol−1. A space-filling model of 2 will give a clue for the difference in reactivity of coordinated N2 with LutH+ (Fig. 6b). Eight tert-butyl groups in 2 makes LutH+ inaccessible to the bridging N2 ligand without a large distortion in the Mo–N≡N–Mo moiety. On the other hand, the distal N atom of terminal N2 ligands that sticks out from the molecular surface is likely to accept a proton from LutH+. It should be noted that the separation of 2 into two mononuclear Mo–N2 complexes [Mo(N2)3(PNP)] H and trans-[Mo(N2)2(PNP)] is endothermic by 24.9 kcal mol−1. The calculated results strongly suggest that the formation of mononuclear Mo–N2 complexes from 2 is an unacceptable reaction pathway for the transformation of N2 into NH3.

(a) Energy profiles of protonation of terminal and bridging N2 ligands by LutH+. Energy changes and activation energies (in parenthesis) are given in kcal mol−1. (b) A space-filling model of 2. The bridging N2 ligand is sterically protected by tBu groups in the pincer ligands
As shown in Fig. 5a, the proton detachment from A, which is the backward reaction of the protonation of 2, can easily proceed due to a very small activation energy of 1.9 kcal mol−1. On the other hand, the protonation of 2 dramatically activates the Mo–N2 bond between the Mo atom and the N2 ligand trans to the NNH group. The elimination of the N2 ligand is endothermic by 14.7 kcal mol−1 (E a = 20.0 kcal mol−1) for 2 and exothermic by 2.7 kcal mol−1 (E a = 4.4 kcal mol−1) for A. After the N2 elimination, the counter anion of LutH+ (OTf−) will attack the vacant coordinate site in B to compensate electric charges. This speculation is reasonable because OTf− can exist in the vicinity of B after the protonation of a terminal N2 ligand in 2. The coordination of OTf− to B is exothermic by 15.6 kcal mol−1, and thereby the NNH moiety is thermodynamically stabilized for the following protonation/reduction steps. Totally, the first protonation process (2 → C) is exothermic by 11.8 kcal mol−1.
In the proposed catalytic cycle, mononuclear Mo–nitride complex [Mo(≡N)(OTf)(PNP)] G is regarded as a key reaction intermediate (Fig. 4b). It is reasonable to consider the formation of dinuclear Mo–nitride complex F, [Mo(≡N)(OTf)(PNP)–NN–Mo(N2)2(PNP)], based on the experimental observation of a fragment assignable to [Mo(≡N)(OTf)(PNP)–NN–Mo(N2)(PNP)] by mass spectrometry. Actually, the dissociation energy of the Mo–N2 bond between the Mo(≡N) atom and the bridging N2 ligand in F is calculated to be only 4.4 kcal mol−1 (Fig. 5b). The small bond dissociation energy indicates that F should be separated into the corresponding mononuclear complexes G and H. It should be emphasized here that [Mo(N2)3(PNP)] H does not accept a proton from LutH+. All efforts to obtain a product complex consisting of the protonated H and Lut resulted in failure, and instead, only a reactant complex consisting of H and LutH+ was optimized. The computational result suggests that a dimolybdenum complex with the Mo–N≡N–Mo structure must be regenerated to start the next catalytic cycle.
As described in Fig. 5d, the regeneration of dimolybdenum complex 2 as the final step of the catalytic cycle involves the exchange of the coordinated NH3 for a newly incoming N2 molecule (N → O → 2). For Schrock’s Mo–triamidoamine system, interconversion of Mo–NH3 and Mo–N2 complexes has been investigated both experimentally [26, 27, 43] and theoretically [34–36] in great detail. In this system, the Mo–NH3 complex readily reacted with N2 to yield the Mo–N2 complex, and 1–2 h was required to establish the thermal equilibrium between Mo–NH3, Mo–N2, N2, and NH3 [43]. Reiher and coworkers reported that energy change (ΔE) in the exchange of NH3 for N2 is calculated to be −9.8 kcal mol−1 by using full [HIPTN3N]Mo complexes [35]. Very recently, Tuczek and coworkers reported ΔE = −7.6 kcal mol−1 [44]. Reiher and coworkers investigated the ligand exchange step with Car-Parrinello molecular dynamics simulations followed by stationary DFT calculations [36]. They found that N2 can access the Mo center through an entrance channel in the plane of three amide N atoms of [HIPTN3N]Mo(NH3) to form a six-coordinate intermediate [HIPTN3N]Mo(N2)(NH3).
For the exchange of NH3 for N2 in the dimolybdenum system, DFT calculations predicted a dissociation–addition mechanism via the formation of a five-coordinate intermediate [Mo(N2)(PNP)–NN–Mo(N2)2(PNP)] O (Fig. 5d). Elimination of the NH3 ligand in [Mo(NH3)(N2)(PNP)–NN–Mo(N2)2(PNP)] N is endothermic by only 2.7 kcal mol−1 with an activation energy of 7.1 kcal mol−1. Addition of N2 to O, the final step toward the regeneration of 2, is exothermic by 14.7 kcal mol−1 with a low activation barrier of 5.3 kcal mol−1. Totally, the ligand exchange is exothermic by 12.0 kcal mol−1 and it will be smoothly attained at room temperature via the dissociation–addition mechanism.
Mononuclear Mo–N2 complexes such as [Mo(N2)3(PNP)] and [Mo(N2)2(PNP)] do not serve as active catalytic species since their N2 ligands cannot be protonated by LutH+. This is the reason why the regeneration of dinuclear complex 2 is considered as the final step of the catalytic cycle in Fig. 4b. Cooperation between the two Mo units in 2 apparently plays an essential role in exhibiting the catalytic activity. The origin of the catalytic activity can be explained in terms of atomic charge of N2 in the dinuclear and mononuclear Mo–N2 complexes and their protonated ones at the first protonation step. Table 1 summarizes calculated differences (Δq) in the NPA atomic charge (q) [45] between the Mo–N2 and Mo–NNH+ complexes for the dinuclear (2 and A) and mononuclear (H and P) Mo complexes. The values of Δq obtained for the Mo atoms, N2, NNH and PNP indicate how the positive charge of the added proton is redistributed in the Mo–NNH+ complexes. In the mononuclear system, the protonation of an axial N2 ligand varies the NPA charges of the Mo atom, the NNH ligand, the remaining axial N2 ligand, the equatorial N2 ligand, and the pincer ligand by +0.38, +0.07, +0.17, +0.09, and +0.29, respectively (Δq = +1.00 in total). The Δq values of the dinuclear system were obtained for units A and B separately, where unit A is identical to H. Comparison between Δq (A–2) of unit A and Δq (P–H) provides insight into the function of unit B in the protonation of 2. The total Δq values of units A (+0.66) and B (+0.34) indicate that a large amount of electron (0.34e−) is donated from unit B to unit A upon protonation. The donated electron mainly distributes on the NNH ligand and bridging N2 ligand. Large differences in Δq between unit A of A–2 and P–H are observed for the NNH ligand (−0.10) and the bridging N2 (−0.14). The electron transfer between the two Mo units enhances the Brønsted basicity of the terminal N2 ligand attacked by LutH+, and an unprotonated Mo unit (unit B in this case) works as a mobile ligand in the first protonation step.

In general the degree of N2 activation in a metal–N2 complex is experimentally judged from elongation of the N≡N bond and red-shift of the N≡N stretching frequency relative to free N2. Deeth and Field [46], and Studt and Tuczek [47] reported that atomic charges on coordinated N2 is also a useful criterion for judging the degree of N2 activation. In a biomimetic nitrogen fixation system, a catalytic cycle starts with protonation of N2 on a metal center (reduction may occur first). Therefore, the reactivity of a metal–N2 complex with a proton donor should closely correlate with the amount of negative charge on the coordinated N2. Yoshizawa and coworkers reported that the gross NPA charge on an N2 ligand in M–N2 (M = Mo, Ru, and W) complexes showed a good correlation with activation energies of protonation by LutH+ [48]. As shown in Table 1, the gross NPA charges on a terminal/axial N2 ligand are almost identical in the di- and mononuclear Mo–N2 complexes (−0.12 for 2 and −0.11 for H). From the criteria of atomic charge, the degree of N2 activation in 2 is not sufficient for the protonation by LutH+. The small negative charge on N2 in 2 implies that the terminal N2 ligand to be protonated is not “preactivated” but activated through an on-demand electron transfer between the two Mo units induced by the approach of LutH+. Synergy of the two Mo centers in 2 can be understood by looking at the HOMO (Fig. 7). The HOMO of 2 is highly delocalized between d orbitals of the two Mo atoms and a π orbital of the bridging N2 ligand. Their overlap is distorted due to the twisted connection between the two Mo units, where the dihedral angle of N2–Mo–Mo–N2 is −54.4°. The intermetallic electron transfer allows 2 to receive a proton from LutH+ at the first step toward the catalytic transformation of N2 into NH3.

Spatial distribution of the HOMO of 2
3.2 Dimolybdenum Complexes Bearing Substituted PNP-Type Pincer Ligands
The theory-based reaction pathways supported by the experimental findings (Fig. 4b) give useful information on the development of more effective catalysts for nitrogen fixation using dinitrogen-bridged dimolybdenum complexes. The computational results indicate that the protonation of a terminal N2 ligand in 2 is one of the most energetically unfavorable steps. As a strategy for the improvement of catalytic activity of 2, introduction of an electron-donating group to the pyridine ring in PNP is expected to reinforce the backdonating ability of the Mo atoms. Enhancing the electron-donating ability of PNP will lead to the increase of electron density on the N2 ligands to strongly attract a proton at the first protonation step.
Yoshizawa and coworkers theoretically assessed the impact of introduction of electron-donating groups to the 4-position of the pyridine ring in PNP [49]. Substituent effects of the electron-donating groups were evaluated based on the energetics of the first protonation process in Fig. 5a. In the presence of [LutH]OTf as a proton source, this process involves three elementary reaction steps: protonation of a terminal N2 ligand, elimination of the N2 ligand trans to the generated NNH ligand, and coordination of OTf− to prevent the Mo–NNH complex from proton detachment. Figure 8 shows energy profiles of the first protonation process for 2a (equivalent to 2), methyl-substituted 2b, and methoxy-substituted 2c. While the use of methyl- and methoxy-substituted PNP ligands does not significantly affect the trend of the energy profiles, the introduction of an electron-donating group lowers the activation barrier for the protonation and stabilizes the generated diazenide complex A. The activation energy for the protonation step is decreased from 8.4 kcal mol−1 (2a) to 7.4 kcal mol−1 (2b) and 7.0 kcal mol−1 (2c). In addition, the backward reaction from A to 2 will be suppressed by using 2b and 2c as catalysts. The corresponding activation energies are calculated to be 2.4 kcal mol−1 for 2b and 3.7 kcal mol−1 for 2c, both of which are higher than that of 2a (1.9 kcal mol−1). On the other hand, the introduction of the methyl and methoxy groups does not change the energetics of the N2 elimination step. The elimination of the trans N2 ligand is exothermic by 2.6–2.8 kcal mol−1 and requires activation energies of 4.4–4.5 kcal mol−1 for all complexes. The N≡N stretching frequency calculated for 2a (1960 cm−1 in THF) is slightly red-shifted by introducing the methyl group (2b, 1958 cm−1) and the methoxy group (2c, 1954 cm−1). The gross NPA charge on a terminal N2 ligand is slightly increased from −0.125 (2a) to −0.127 (2b) and −0.133 (2c). The characteristics of the N2 ligands indicate a higher degree of N2 activation in the Mo complexes bearing the substituted PNP-pincer ligands. DFT calculations have predicted that the introduction of an electron-donating group to PNP facilitated the protonation of 2 and the methoxy-substituted 2c would serve as an effective catalyst.

Energy profiles of the first protonation process of 2a-2c calculated at the B3LYP*/BS2//B3LYP*/BS1 level of theory. Energy changes and activation energies (in parenthesis) are presented in kcal mol−1. Proton is transferred from LutH+
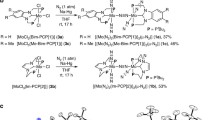
Nishibayashi and coworkers synthesized a series of substituted dinitrogen-bridged dimolybdenum complexes including 2b and 2c, and examined the catalytic transformation of N2 into NH3 using them as catalysts (Scheme 3) [49]. The N≡N stretching frequencies were observed at 1944 cm−1 (2a), 1939 cm−1 (2b), and 1932 cm−1 (2c) in THF, which reasonably agreed with the computational trend. In accordance with the theory-based design, the methoxy-substituted 2c produced the largest amount of NH3 (34 equiv) based on the catalyst. They eventually succeeded in producing 52 equiv of NH3 by using 2c in the presence of larger amounts of [LutH]OTf (480 equiv) and CoCp2 (360 equiv).

Transformation of N2 into NH3 catalyzed by dimolybdenum complexes bearing substituted PNP-type pincer ligands
3.3 Dimolybdenum Complexes Bearing PCP-Type Pincer Ligands
Experimental and theoretical investigations on a series of dimolybdenum complexes bearing PNP-type pincer ligands have provided three important clues to designing a new pincer ligand for the development of more effective catalysts for nitrogen fixation. (1) As mentioned in the former section, enhancement of the backdonating ability of the Mo centers will accelerate the protonation of a terminal N2 ligand, which is energetically less favorable. (2) The dinitrogen-bridged dimolybdenum (Mo–N≡N–Mo) structure must be retained because the intermetallic electron transfer via the bridging N2 ligand is essential for the protonation of a terminal N2 ligand. (3) The pincer ligand must be strongly coordinated to the Mo center in order to extend the lifetime of the catalyst. Dissociation of PNP from the Mo center was generally observed after the catalytic reaction, and it should lead to deactivation of the catalyst.
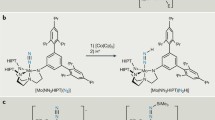
As a candidate pincer ligand that could meet the above requirements, Nishibayashi and coworkers adopted an N-heterocyclic carbene-based PCP-type pincer ligand [50]. N-heterocyclic carbene (NHC) is known to serve as a σ donor stronger than pyridine as well as a π acceptor [51–55], which is expected to coordinate to a metal center tightly. They newly synthesized two dinitrogen-bridged dimolybdenum complexes bearing two types of PCP pincer ligands containing 1,3-bis((di-tert-butylphosphino)methyl)benzimidazol-2-ylidene (Bim-PCP[1], 3a) and 1,3-bis(2-(di-tert-butylphosphino)ethyl)imidazol-2-ylidene (Im-PCP[2], 3b) (Scheme 4). These PCP ligands were prepared for investigating how the length of methylene linkers between the NHC framework and PtBu2 groups influences the catalytic activity for nitrogen fixation. The catalytic activities of 3a and 3b for nitrogen fixation were quite different. In the presence of 96 equiv of [LutH]OTf as a proton source and 72 equiv of decamethylchromocene (CrCp*2) as a reducing agent, 3a worked as a catalyst for nitrogen fixation to produce 18 equiv of NH3 based on the catalyst in toluene at room temperature. On the other hand, 3b produced only 3.5 equiv of NH3 under the same reaction conditions. Surprisingly, the amount of generated NH3 reached 2.0 × 102 equiv by using 3a as a catalyst in the presence of larger amounts of [LutH]OTf (1920 equiv) and CrCp*2 (1440 equiv), which is about an order of magnitude greater than the Mo–PNP complex 2a. A sharp contrast between 3a and 3b was observed in the stability of the dimolybdenum structure. IR and 15N{1H} NMR measurements in THF as a solvent revealed that 3b was separated into two mononuclear Mo–N2 complexes, such as [Mo(N2)3(Im-PCP[2])] 3b′ at room temperature.

Transformation of N2 into NH3 catalyzed by dimolybdenum complexes bearing PCP-type pincer ligands
Figure 9 depicts structures of 3a and 3b optimized at the B3LYP* level of theory. The SDD and 6-31G* basis sets (BS3) were employed for the Mo atoms and the others, respectively. The Mo–N bond distance for the bridging N2 ligand is 2.108 Å (3a) and 2.133 Å (3b), indicating a weaker binding of the bridging N2 ligand to the Mo atom in 3b. In terms of the separation of a dimolybdenum complex into two mononuclear complexes, the bond dissociation energy (BDE) of the Mo–N2(bridging) bond of 3b is calculated to be 9.0 kcal mol−1, which is much lower than that of 3a (18.8 kcal mol−1). The low BDE of the Mo–N2(bridging) bond of 3b can be associated with the IR and 15N{1H} NMR spectra of 3b measured in solution. Moreover, the low reactivity of 3b for nitrogen fixation experimentally proves the importance of the dinuclear Mo–N≡N–Mo structure for exhibiting the catalytic activity. In fact, DFT calculations demonstrated that mononuclear Mo–N2 complexes such as [Mo(N2)3(Bim-PCP[1])] 3a′ and [Mo(N2)3(Im-PCP[2])] 3b′ cannot be protonated by LutH+ in a similar manner as [Mo(N2)3(PNP)] G presented in the former section.

Optimized structures of dimolybdenum complexes 3a, 3b, and 2 calculated at the B3LYP*/BS3 level of theory. Bond distances are given in Å
Optimized structures of 3a′ and 3b′ in Fig. 10 and their space-filling models in Fig. 11 explain how the length of CH2 linkers in the PCP ligands influences thermodynamic stability of the dimolybdenum structure. The Mo–N distances for the equatorial N2 ligand are 2.084 Å for 3a′ and 2.041 for 3b′, and they are increased by 0.024 Å (3a′ → 3a) and 0.092 Å (3b′ → 3b) upon formation of the corresponding dimolybdenum complexes. Contrary to the large difference in the BDE of the Mo–N2(bridging) bond between 3a and 3b, the BDEs of the Mo–N2(equatorial) bond are almost identical for 3a′ and 3b′, 21.2 and 21.5 kcal mol−1, respectively. These results would imply that steric hindrance between tert-butyl groups on the phosphorus atoms in the PCP ligands governs the thermodynamic stability of the Mo–N≡N–Mo structure. The bond angle of P–Mo–P in 3b′ (164.3°) is larger than that in 3a′ (151.2°) due to the longer CH2 linkers in Im-PCP[2]. As shown in space-filling models in Fig. 11, the tert-butyl groups in Im-PCP[2] stick out along the equatorial N2 ligand compared to those in Bim-PCP[1] so as to effectively protect the N2 ligand utilized for the connection with another Mo unit. As a result, the Mo–N≡N–Mo structure of 3b is destabilized by steric repulsions between the tert-butyl groups in two mononuclear Mo units facing each other.

Optimized structures of mononuclear Mo–N2 complexes 3a′, 3b′, and 2′ calculated at the B3LYP*/BS3 level of theory. Bond distances are given in Å

Space-filling models of 3a′ and 3b′. tert-Butyl groups in Im-PCP[2] stick out to destabilize a dimolybdenum structure connected through the equatorial N2 ligand
Reaction pathways for the transformation of N2 into NH3 using 3a as a catalyst are expected to be basically similar to those using 2 as a catalyst shown in Fig. 5. As the first step toward the mechanistic understanding of the nitrogen fixation catalyzed by 3a, theoretical investigations [50] have been particularly focused on the first protonation process because this process involves one of the most energetically unfavorable reaction steps in the catalytic cycle proposed for 2.
Figure 12 compares energy profiles of two possible reaction pathways of the first protonation process leading to intermediate C from 3a and 2. The pathway shown above (Path I) is the same one as shown in Fig. 5a, i.e. the transformation of N2 starts with the protonation of a terminal N2 ligand. The protonation of 3a by LutH+ (3a → A-PCP) is endothermic by 8.1 kcal mol−1 with an activation energy of 8.3 kcal mol−1. This energy profile indicates that the backward reaction to release H+ from N2 would occur very rapidly due to a very low activation energy of 0.2 kcal mol−1, which is much lower than that in the Mo–PNP system (1.9 kcal mol−1). On the other hand, the following N2 elimination step (A-PCP → B-PCP) is exothermic by 5.2 kcal mol−1 with a low activation energy of 4.0 kcal mol−1. The low reactivity of terminal N2 ligands in 3a with LutH+ appears to be strange because the NHC-based PCP ligands were expected to work as a strong electron donor compared with the pyridine-based PNP ones.

Two possible reaction pathways and energy profiles of the first protonation process on a terminal N2 ligand in 3a and 2 calculated at the B3LYP*/BS2//B3LYP*/BS3 level of theory. Energy changes and activation energies (in parenthesis) are presented in kcal mol−1. Proton is supplied by LutH+
Another possible pathway to C (Path II in Fig. 12) involves the elimination of a terminal N2 ligand in the initial complex 3a (2) followed by the protonation of the remaining terminal N2 ligand (3a (2) → Q → B → C). In the Mo–PCP system, the elimination reaction (3a → Q-PCP) proceeds in an endothermic way by 11.9 kcal mol−1 and requires an activation energy of 19.7 kcal mol−1, which is acceptable for a reaction occurring at room temperature. In the Mo–PNP system, this reaction is endothermic by 14.4 kcal mol−1 with an activation energy of 20.0 kcal mol−1. Since the energy changes calculated here correspond to the BDEs of the Mo–N2(terminal) bond in 3a and 2, the Mo–N2(terminal) bond in 3a is 2.5 kcal mol−1 weaker than that in 2. The weaker Mo–N2(terminal) bond as well as the lower reactivity of 3a with LutH+ in Path I can be associated with less activation of the terminal N2 ligands. The N≡N stretching frequencies of terminal N2 ligands in 3a and 2 were experimentally observed at 1978 and 1936 cm−1 in the solid state, respectively, indicating that the terminal N2 ligands in 3a are less activated than those in 2. The N≡N stretching frequencies calculated for 3a (2012 cm−1 in vacuo) and 2 (1993 cm−1 in vacuo) reproduced the experimental trend; 1978 cm−1 (KBr) for 3a [50] and 1936 cm−1 (KBr) for 2 [17]. The dissociation of the terminal N2 ligand in 3a markedly activates the remaining N2 ligand at the trans position. The negative NPA charge on the trans N2 ligand is increased from −0.10 (3a) to −0.19 (Q-PCP). The protonation of Q-PCP is exothermic by 1.6 kcal mol−1 with a very low activation energy of 1.2 kcal mol−1. This activation energy is much lower than that of the backward reaction yielding 3a from Q-PCP and N2 (7.8 kcal mol−1). A similar trend was observed for Path II in the Mo–PNP system. At present, unfortunately, it is difficult to determine which reaction pathway is a major pathway for the first protonation process in the Mo–PCP system because of the quick proton detachment from A-PCP in Path I and the high activation barrier for the N2 elimination of 3a in Path II.
The NHC-based PCP ligands were first expected to work as a strong electron donor so as to activate coordinated N2 through the enhanced π-backbonding from Mo to N2. Actually, the improvement of catalytic activity of the Mo–PNP system was achieved by introducing electron-donating groups to the PNP ligand, as described in the Sect. 3.2. However, the N≡N stretching frequencies indicate that the N2 ligands in 3a (Bim-PCP[1]) are less activated than those in 2 (PNP). In order to elucidate how the pincer ligands influence the coordination of N2 to Mo, structural and electronic properties of mononuclear Mo–N2 complexes [Mo(N2)3(Bim-PCP[1])] 3a′ and [Mo(N2)3(PNP)] 2′ have been investigated in detail.
Table 2 compares the electron-donating ability of Bim-PCP[1] and PNP in terms of the NPA charge. Changes in the NPA charge (Δq) between the Mo–N2 complexes and the free pincer ligands were calculated for estimating the amount of electron donated from the pincer ligands to the Mo center and three N2 ligands during complexation. The Δq values were independently calculated for four moieties in 3a′ (2′); the NHC (pyridine) moiety with the CH2 linkers, the PtBu2 groups, the Mo atom, and the N2 ligands. The NPA charges of the Mo and N2 ligands were set to be zero for the free ligands, and hence Δq of the Mo(N2)3 moiety is identical to the sum of the NPA charges assigned to this moiety. As shown in Table 2, Bim-PCP[1] and PNP donate 0.81e− and 0.70e− to the Mo(N2)3 moiety during complexation, respectively. From a viewpoint of atomic charge, the NHC-based pincer ligand surely works as a strong electron-donor compared with the pyridine-based one. The electron-donating ability of the pincer ligands is governed by the NHC and pyridine moieties because Δq of the PtBu2 groups are identical (+0.29) in both Bim-PCP[1] and PNP. The difference in the amount of the donated electron comes from the Δq values of the NHC moiety (+0.23) and the pyridine moiety (+0.12).

Optimized structures in Fig. 10 and BDEs of the Mo–N2(equatorial) and Mo–N2(axial) bonds also provide useful information on the evaluation of the electron-donating ability of the pincer ligands. Table 3 summarizes geometric parameters around the Mo center in 3a′ and 2′, together with the BDEs of the Mo–N2 bonds. The Mo–C(carbene) bond distance of 2.099 Å in 3a′ is much shorter than the Mo–N(pyridine) bond distance of 2.240 Å in 2′, indicating a stronger binding of Bim-PCP[1] to the Mo center. The Mayer bond orders (b.o.) [56] of the Mo–C(carbene) bond (0.91) and Mo–N(pyridine) bond (0.39) also suggest a strong bonding interaction between the Mo center and Bim-PCP[1]. The solid connection between Mo and Bim-PCP[1] well explains the experimental result that 3a has a long lifetime as a catalyst for nitrogen fixation with a high turnover number of NH3 production compared with 2. On the other hand, the Mo–N2(equatorial) bond is weakened by a strong trans influence of the NHC moiety. The Mo–N2(equatorial) bond distance of 2.084 Å (b.o. = 0.50) in 3a′ is much longer than that of 2.018 Å (b.o. = 0.62) in 2′. The elongated Mo–N2(equatorial) bond in 3a′ results in a low BDE of 21.2 kcal mol−1 compared with that of 2′ (30.1 kcal mol−1). It is notable that the coordination of Bim-PCP[1] influences not only the equatorial N2 ligand but also the axial N2 ligands so as to weaken all Mo–N2 bonds. The Mo–N2(axial) bond distances (b.o.) are 2.034 Å (0.53) for 3a′ and 2.024 Å (0.54) for 2′, while the BDEs are 12.5 kcal mol−1 for 3a′ and 14.0 kcal mol−1 for 2′. Dimolybdenum complexes 3a and 2 show a similar trend in the BDEs of the Mo–N2(bridging) and Mo–N2(terminal) bonds. The BDEs of the Mo–N2(bridging) and Mo–N2(terminal) bonds are 18.8 and 11.9 kcal mol−1 for 3a, respectively, both of which are lower than those for 2 (24.9 and 14.4 kcal mol−1).
The influence of coordination of the NHC-based PCP ligand to the Mo center can be rationalized by spatial distribution of frontier orbitals responsible for the Mo–C(carbene) and Mo–N2 bonds. The HOMO-6 (3a′) and HOMO-5 (2′) in Fig. 13a contribute to a σ bond between the Mo center and the carbene C atom (or the pyridine N atom). The large amplitude of the lobe observed between Mo and C implies a strong σ-donating ability of Bim-PCP[1] compared to PNP. The HOMO-1 in Fig. 13b shows π backdonation from an out-of-plane d orbital of Mo to a π* orbital of N2 ligands. In general the π backdonation from metal to N2 plays an essential role in the binding and activation of N2 upon formation of a metal–N2 complex. It should be emphasized here that both equatorial and axial N2 ligands are affected by occupation of the HOMO-1. A large difference between the HOMO-1 of 3a′ and 2′ is the presence of π backdonation from Mo to the vacant p orbital of the carbene C atom in 3a′. The weak Mo–N2 bonds in 3a′ can be ascribed to the π-accepting ability of the NHC that decreases the propensity of Mo for π backdonation to both equatorial and axial N2 ligands (Fig. 14). Actually, as presented in Table 2, Δq of the three N2 ligands in 3a′ (−0.26) is smaller than that in 3b′ (−0.31) despite the higher electron-donating ability of Bim-PCP[1]. Whereas the π backdonation to the NHC weakens the Mo–N2 bonding in 3a′, it also contributes to the solid connection between Mo and Bim-PCP[1].

Spatial distribution of frontier orbitals of 3a′ and 2′: (a) HOMO-6 (3a′) and HOMO-5 (2′) contributing to σ donation from the pincer ligand to Mo, and (b) HOMO-1 contributing to π backdonation from Mo to both equatorial and axial N2 ligands. An “extra” backdonation from Mo to the pincer ligand is observed only in 3a′ (dashed circle). The molecular structures in (b) are rotated by 90° along the axis containing the Mo atom and the equatorial N2 ligand from those in (a)

Schematic drawing of the π-bonding interaction between the Mo atom and ligands in the HOMO-1 of 3a′
On the other hand, the contribution of the π backdonation seems to be less significant for the bonding between Mo and Im-PCP[2] in [Mo(N2)3(Im-PCP[2])] 3b′. As shown in Fig. 10, the coordination of the NHC moiety of Im-PCP[2] to Mo is highly twisted due to the ethylene linkers; the dihedral angle of NA–C–Mo–NB in 3b′ (43.8°) is much smaller than that in 3a′ (69.6°) and the dihedral angle of C–NA–Mo–NB in 2′ (67.7°). Because the overlap between the vacant p orbital of the carbene C atom and the out-of-plane d orbital of the Mo atom is maximized at the NA–C–Mo–NB dihedral angle of 90°, the twisted coordination of the NHC moiety decreases the propensity of Mo for π backdonation to the NHC. As a result, Im-PCP[2] only exhibits a very strong σ-donating ability and donates 0.90e− to the Mo atom during complexation. The gross NPA charge on the three N2 ligands (−0.36) as well as a high BDE of the Mo–N2(axial) bond of 3b′ (14.3 kcal mol−1) indicates that the coordination of Im-PCP[2] effectively activates the N2 ligands compared with Bim-PCP[1]. However, the N2 ligands in the mononuclear Mo complex 3b′ are not sufficiently activated for the reaction with LutH+, as mentioned in Sect. 3.1. Again, the low catalytic activity of 3b for nitrogen fixation would stem from the thermodynamic instability of the Mo–N≡N–Mo structure caused by the ethylene linkers of Im-PCP[2].
4 Conclusions
In this chapter, we have overviewed recent topics in the mechanistic understanding of artificial nitrogen fixation catalyzed by mono- and dimolybdenum complexes from a theoretical point of view. The Yandulov–Schrock cycle proposed for the mononuclear Mo–triamidoamine system involves alternating protonation/reduction steps via the formation of a Mo–nitride (Mo≡N) intermediate. The validity of the Yandulov–Schrock cycle has been thoroughly investigated both experimentally and theoretically, and this cycle is now accepted as an established catalytic mechanism of nitrogen fixation at a single metal site.
In the dimolybdenum Mo–PNP system, a dinuclear Mo–NN–Mo structure plays a decisive role in the transformation of N2 into NH3, particularly in the protonation of coordinated N2. While the catalytic cycle deduced from experimental and theoretical findings is analogous to the Yandulov–Schrock cycle in that one of the terminal N2 ligands at an Mo unit of 2 is converted to two molecules of NH3 via the formation of a Mo–nitride intermediate, a main difference from the Mo–triamidoamine system is that the other Mo unit works as a mobile ligand to donate electron in the first protonation process. Intermetallic electron transfer between the two Mo units through the bridging N2 ligand efficiently activates the terminal N2 ligand to be protonated, and opens the path to further protonation and reduction steps leading to the generation of NH3. Based on the catalytic mechanism proposed for the Mo–PNP system, dimolybdenum complexes bearing substituted PNP-pincer ligands were designed to achieve higher catalytic activity than the original complex. As predicted by theoretical calculations, the introduction of electron-donating groups to the pyridine moiety of the pincer ligand improved the turnover number of NH3 production from 23 (2a) to 52 (2c) equiv/catalyst.
As an alternative strategy for enhancing the electron-donating ability of pincer ligands, dimolybdenum complexes bearing N-heterocyclic carbene-based PCP-type pincer ligands (Bim-PCP[1] and Im-PCP[2]) have been newly designed and synthesized. Dimolybdenum complex 3a bearing Bim-PCP[1] exhibited high catalytic activity for nitrogen fixation, where up to 200 equiv of NH3 was produced on the catalyst. On the other hand, 3b bearing Im-PCP[2] exhibited very low catalytic activity. Separation of 3b into mononuclear Mo–N2 complexes observed in solution was rationalized by a low bond dissociation energy calculated for the Mo–N2(bridging) bond of 3b. The large differences in catalytic activity and behavior in solution testify the theory-based proposal that the dinuclear Mo–NN–Mo structure must be preserved at a certain stage of the nitrogen fixation in the Mo–PCP system (probably also the Mo–PNP system). Calculated reaction pathways of the first protonation process toward the transformation of N2 into NH3 by 3a demonstrated that a weak Mo–N2(terminal) bond of 3a may be associated with the high catalytic activity. The strong σ donation as well as π backdonation between Mo and Bim-PCP[1] provides a solid connection between the metal center and pincer ligand in 3a, and would contribute to the long lifetime of 3a as a catalyst. The catalytic activity of dimolybdenum complexes bearing pincer ligands is exquisitely controlled by both geometric and electronic factors. The former mainly influences the thermodynamic stability of the dimolybdenum structure through steric hindrance stemmed from the length of the phosphorus arms. The latter affects the binding of a pincer ligand to Mo through the σ donation and π backdonation. A pincer ligand with a strong σ-donating ability binds to Mo tightly, but its pronounced trans influence can decrease the thermodynamic stability of the dimolybdenum structure. The π-accepting ability of a pincer ligand should also be considered in tuning the catalytic activity of a dimolybdenum complex because the backdonation from Mo to the pincer ligand can weaken all the Mo–N2 bonds.
The mechanism of nitrogen fixation is quite complicated because at least six pairs of proton and electron must take part in the transformation of N2 into NH3. Therefore, the catalytic reaction pathway should compete with unexpected side reactions that limit the turnover number of NH3 production, but they are often difficult to observe and characterize experimentally. Automated screening techniques can be applied for locating such undesired pathways leading to deactivation of catalysts. For example, a heuristics-guided protocol for the automatic exploration of chemical reaction spaces developed by Reiher and coworkers [38] has recently been exploited for finding possible elementary reactions in the Mo–triamidoamine system. In their study, about 10,000 intermediates and more than 2000 transition states were optimized using a simplified model complex of 1. Computational and experimental researches in close coordination become increasingly important for accelerating the development of more effective nitrogen fixation catalysts working under ambient reaction conditions.
References
Burgess BK, Lowe DJ (1996) Chem Rev 96:2983–3011
Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC (2002) Science 297:1696–1700
Spatzal T, Aksoyoglu M, Zhang L, Andrade SLA, Schleicher E, Weber S, Rees DC, Einsle O (2011) Science 334:940
Lancaster KM, Roemelt M, Ettenhuber P, Hu Y, Ribbe MW, Neese F, Bergmann U, DeBeer S (2011) Science 334:974–977
Hoffman BM, Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC (2014) Chem Rev 114:4041–4062
Liu H (2013) Ammonia synthesis catalysts: innovation and practice. Chemical Industry Press & World Scientific, Singapore
Allen AD, Senoff CV (1965) Chem Commun 24:621–622
Chatt J, Dilworth JR, Richards RL (1978) Chem Rev 78:589–625
Hidai M, Mizobe Y (1995) Chem Rev 95:1115–1133
MacKay BA, Fryzuk MD (2004) Chem Rev 104:385–401
Nishibayashi Y (2012) Dalton Trans 41:7447–7453
Tanabe Y, Nishibayashi Y (2013) Coord Chem Rev 257:2551–2564
Nishibayashi Y (2015) Inorg Chem 54:9234–9247
Khoenkhoen N, de Bruin B, Reek JNH, Dzik WI (2015) Eur J Inorg Chem 567–598
Tanaka H, Nishibayashi Y, Yoshizawa K (2016) Acc Chem Res 49:987–995
Yandulov DV, Schrock RR (2003) Science 301:76–78
Arashiba K, Miyake Y, Nishibayashi Y (2011) Nat Chem 3:120–125
Anderson JS, Rittle J, Peters JC (2013) Nature 501:84–87
Del Castillo TJ, Thompson NB, Peters JC (2016) J Am Chem Soc 138:5341–5350
Ung G, Peters JC (2015) Angew Chem Int Ed 54:532–535
Arashiba K, Kinoshita E, Kuriyama S, Eizawa A, Nakajima K, Tanaka H, Yoshizawa K, Nishibayashi Y (2015) J Am Chem Soc 137:5666–5669
Kuriyama S, Arashiba K, Nakajima K, Matsuo Y, Tanaka H, Ishii K, Yoshizawa K, Nishibayashi Y (2016) Nat Commun 7:12181
Del Castillo TJ, Thompson NB, Suess DLM, Ung G, Peters JC (2015) Inorg Chem 54:9256–9262
Kuriyama S, Arashiba K, Tanaka H, Matsuo Y, Nakajima K, Yoshizawa K, Nishibayashi Y (2016) Angew Chem Int Ed 55:14291–14295
Hill PJ, Doyle LR, Crawford AD, Myers WK, Ashley AE (2016) J Am Chem Soc 138:13521–13524
Schrock RR (2008) Angew Chem Int Ed 47:5512–5522
Yandulov DV, Schrock RR (2005) Inorg Chem 44:1103–1117
Kinney RA, McNaughton RL, Chin JM, Schrock RR, Hoffman BM (2011) Inorg Chem 50:418–420
Munisamy T, Schrock RR (2012) Dalton Trans 41:130–137
Cao Z, Zhou Z, Wan H, Zhang Q (2005) Int J Quantum Chem 103:344–353
Studt F, Tuczek F (2005) Angew. Chem Int Ed 44:5639–5642
Magistrato A, Robertazzi A, Carloni P (2007) J Chem Theory Comput 3:1708–1720
Reiher M, Le Guennic B, Kirchner B (2005) Inorg Chem 44:9640–9642
Le Guennic B, Kirchner B, Reiher M (2005) Chem Eur J 11:7448–7460
Schenk S, Le Guennic B, Kirchner B, Reiher M (2008) Inorg Chem 47:3634–3650
Schenk S, Kirchner B, Reiher M (2009) Chem Eur J 15:5073–5082
Schenk S, Reiher M (2009) Inorg Chem 48:1638–1648
Bergeler M, Simm GN, Proppe J, Reiher M (2015) J Chem Theory Comput 11:5712–5722
Tian Y-H, Pierpont AW, Batista ER (2014) Inorg Chem 53:4177–4183
Tanaka H, Arashiba K, Kuriyama S, Sasada A, Nakajima K, Yoshizawa K, Nishibayashi Y (2014) Nat Commun 5:3737
Reiher M, Salomon O, Hess BA (2001) Theor Chem Acc 107:48–55
Reiher M (2002) Inorg Chem 41:6928–6935
Schrock RR (2005) Acc Chem Res 38:955–962
Thimm W, Gradert C, Broda H, Wennmohs F, Neese F, Tuczek F (2015) Inorg Chem 54:9248–9255
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926
Deeth RJ, Field CN (1994) J Chem Soc Dalton Trans 1943–1948
Studt F, Tuczek F (2006) J Comput Chem 27:1278–1291
Tanaka H, Ohsako F, Seino H, Mizobe Y, Yoshizawa K (2010) Inorg Chem 49:2464–2470
Kuriyama S, Arashiba K, Nakajima K, Tanaka H, Kamaru N, Yoshizawa K, Nishibayashi Y (2014) J Am Chem Soc 136:9719–9731
Eizawa A, Arashiba K, Tanaka H, Kuriyama S, Matsuo Y, Nakajima K, Yoshizawa K, Nishibayashi Y (2017) Nat Commun 8:14874
Hopkinson MN, Richter C, Schedler M, Glorius F (2014) Nature 510:485–496
Trnka TM, Grubbs RH (2001) Acc Chem Res 34:18–29
Ohki Y, Seino H (2016) Dalton Trans 45:874–880
Comas-Vives A, Harvey JN (2011) Eur J Inorg Chem 5025–5035
Nelson DJ, Nolan SP (2013) Chem Soc Rev 42:6723–6753
Mayer I (1983) Chem Phys Lett 97:270–274
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Tanaka, H., Yoshizawa, K. (2017). Computational Approach to Nitrogen Fixation on Molybdenum–Dinitrogen Complexes. In: Nishibayashi, Y. (eds) Nitrogen Fixation. Topics in Organometallic Chemistry, vol 60. Springer, Cham. https://doi.org/10.1007/3418_2016_7
Download citation
DOI: https://doi.org/10.1007/3418_2016_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-57713-5
Online ISBN: 978-3-319-57714-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)