
Abstract
In this chapter we describe the recent applications of well-defined oxide-supported metal alkyls/alkylidenes/alkylidynes and hydrides of group IV, V, and VI transition metals in the field of C–H and C–C bond activation. The activation of ubiquitous C–H and C–C bonds of paraffin is a long-standing challenge because of intrinsic low reactivity. There are many concepts derived from surface organometallic chemistry (SOMC): surface organometallic fragments are always intermediates in heterogeneous catalysis. The study of their synthesis and reactivity is a way to rationalize mechanism of heterogeneous catalysis and to achieve structure activity relationship. By surface organometallic chemistry one can enter any catalytic center by a reaction intermediate leading in fine to single site catalysts. With surface organometallic chemistry one can coordinate to the metal which can play a role in different elementary steps leading for example to C–H activation and Olefin metathesis. Because of the development of SOMC there is a lot of space for the improvement of homogeneous catalysis. After the 1997 discovery of alkane metathesis using silica-supported tantalum hydride by Basset et al. at low temperature (150oC) the focus in this area was shifted to the discovery of more and more challenging surface complexes active in the application of C–H and C–C bond activation. Here we describe the evolution of well-defined metathesis catalyst with time as well as the effect of support on catalysis. We also describe here which metal–ligand combinations are responsible for a variety of C–H and C–C bond activation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The activation of ubiquitous C–H and C–C bonds of paraffins is a long-standing challenge for chemists because of their presence in petroleum and natural gas and their intrinsic low reactivity. In 1997, our group reported the catalytic transformation of acyclic alkanes into their lower and higher homologues using silica-supported tantalum hydride(s) [1] in the absence of hydrogen at low temperature (150°C). This has resulted in the faster development of surface organometallic chemistry (SOMC), a discipline which has progressively emerged as a new area of heterogeneous and homogeneous catalysis where one can prepare relatively well-defined “single-site catalysts” [2]. The first results in the area of C–H and C–C bond activation came from the discovery of single-site catalysts, e.g., [(≡Si–O–)3Zr–H], which were able to catalyze the low-temperature hydrogenolysis of alkanes [3] and later of polyethylene [4].
What was interesting and new in the SOMC approach is the fact that it was bridging the two areas of homogeneous and heterogeneous catalysis which did not overlap enough in the past. During the last 60 years, homogeneous catalysis played an important role in the selective organic transformations of lower to higher value products [5, 6]. Clear understanding of the reaction mechanism at the molecular level and selective formation of the product by tuning the metal center and its ligands are the main reasons for the increasing use of homogeneous catalysts. This resulted from the parallel development of molecular organometallic chemistry [7–11]. In contrast, the heterogeneous catalysts, which are more commonly used in industry than homogeneous catalysis, did not lead to a clear understanding of reaction mechanisms (at least at the atomic and molecular level), although the parallel development of surface science could allow successful story in the identification of elementary steps (e.g., in ammonia synthesis) [12]. The main reason was the small amount of active sites and consequently the difficulty to fully characterize these active sites and to draw a reliable and predictive structure–activity relationship.
In this chapter, we try to describe SOMC strategy in the recent years to achieve alkane and cycloalkane metathesis with increasing TONs and selectivities. We will explore the surface organometallic chemistry of Group IV, V and VI metals on various supports and the properties of these single-site systems in the area of alkane and cycloalkane metathesis.
2 Surface Organometallic Chemistry (SOMC)
A heterogeneous catalyst is an ideal choice for the great variety of chemical transformations carried out in industry. Homogeneous catalysts, mostly because of their fragility, instability at higher temperature, and difficulty to separate them from the product(s) after reaction, are comparatively less used in industry (although the number of homogeneous processes is steadily increasing with time). However, selective utilization of the small number of active sites in heterogeneous catalyst makes their characterization quasi-impossible, and, as a result, structure–activity relationship is rarely reached which prevents further improvement of these catalysts.
In order to bring the concepts of homogeneous catalysis into heterogeneous catalysis, a new field of catalysis was developed called surface organometallic chemistry (SOMC) (Scheme 1). SOMC led progressively to the discovery of a new area of chemistry [13, 14]. It has been found that organometallic complexes react with surfaces of oxides in a very specific way leading to new materials having an extremely high electron deficiency. As a consequence a very strong reactivity to activate the C–H and C–C bonds of paraffin’s was observed. It was discovered when group IV metal alkyls were reacted with silica surfaces. The first discovery of the hydrides of group IV was made in the field of olefin polymerization [15]. These group IV metal alkyls (or hydrides) are tremendously effective for low-temperature hydrogenolysis of most alkanes and polyolefins (similar to Ziegler–Natta depolymerization), activation of methane, and coupling of methane into ethane and hydrogen. A series of new reactions were developed by using this approach [16].

Schematic presentation of surface organometallic compound
After its origin, SOMC has been extended to the full ensemble of metallic elements of the periodical table, to a huge variety of ligands, and to a huge variety of supports. In addition, SOMC approach is applied in the area of nanoparticles. In almost all cases, we could progressively understand reaction mechanisms and make a clear structure–activity relationship.
SOMC is purely a surface phenomenon where an organometallic complex binds selectively with the surface by covalent (or sometimes ionic or both) bonds. One can then access to its electronic configuration and oxidation state, and this leads to a better understanding of the reaction mechanism. In SOMC, the surface acts as a ligand, which means one can tune the catalytic activity of the organometallic with the surface ligand. Surface can play the role of acid–base ligand as well as redox site. Besides this, steric constraints in a porous surface also play an important role in catalysis.
Surface organometallic fragments can also be considered as reaction intermediates in heterogeneous catalysis. Thus, the study of their stoichiometric reactivity allows identification of the elementary steps in a precise way. Thus, as short statement, SOMC is making a strong creative bridge between homogeneous and classical heterogeneous catalysis.
2.1 Various Oxide Supports and Their Functionalities Used in SOMC
To graft organometallic complexes in SOMC, various inorganic materials have been used as a support such as silica, silica–alumina, alumina, magnesia, MCM 41, SBA-15, amino-modified SBA-15, etc. Depending on the nature of the reactive sites on the surface of these materials, different behaviors were observed, leading to sometimes completely different catalytic activity. Before considering the reactivity of metal alkyls with, for example, silica surface, we have to understand the functional groups present on such surface. First, we will consider flame silica Aerosil® from Degussa partially dehydroxylated at various temperatures. This solid has a surface area of ca. ≈200 m2g−1 which contains isolated, vicinal, and geminal hydroxyl groups (Scheme 2).

Various surface silanols and siloxane bridges present on partially dehydroxylated silica
It was generally believed that reactivity of metal alkyls with the partially dehydroxylated silica surface occurs by protonolysis of the metal–alkyl bond by the remaining surface silanols. However, progressively it appeared that this is a very narrow description of a complex phenomenon. In a partially dehydroxylated surface, the dehydroxylation produces ≡Si–O–Si≡ groups for which the strain is the result of the pretreatment temperature. Thus, at very high temperature, more strained ≡Si–O–Si≡ groups were produced on silica surface and vice versa.
These strained groups exhibit also a typical reactivity with metal alkyls or metal hydrides
To understand the reaction mechanism for a given reaction on surface and correlate structure–activity relationship, first of all a well-defined “single-site” system is needed. To have a well-defined “single-site” system the silanols must be sufficiently isolated from each other to behave independently. This is the necessary condition to reach the ultimate goal of making “single-site” catalysts. Based on the above concepts, flame silica which has a surface area of ≈200 m2g−1 is usually pretreated at 700, 500, and 200°C under high vacuum (10−5 mbar) for 16 h. The corresponding number of silanols is equal to 0.26, 0.42, and 0.86 mmolg−1, respectively (measured either by 1H NMR of simple titration method via MeLi) [17]. For such low values for surface silanol especially in the case of SiO2-700, one can presume that the hydroxyl groups are far away from each other and so well-defined grafted organometallic isolated species will be expected upon reaction with these hydroxyl groups. This is the key point of surface organometallic chemistry.
In Table 1, we have mentioned some examples of grafting of organometallic complexes on various supports by the use of surface organometallic chemistry.
3 Surface Organometallic Chemistry of Metal Alkyls/Alkylidene and Alkylidyne
3.1 Reactivity of Group IV (Zr, Hf, and Ti) Metal Alkyls on Oxide Surfaces
This area was really at the origin of alkane activation with surface metal hydrides. Those surface metal hydrides were obtained by treatment under hydrogen of silica-supported metal alkyls. The first one in this series was tris-neopentylzirconium surface complex [≡SiO–Zr(Np)3] 1.
Zirconium tris-neopentyl surface complex 1 was synthesized by the sublimation of tetra-neopentylzirconium complex onto the surface of partially dehydroxylated silica at 500°C [26, 27] and fully characterized by IR, NMR, EXAFS, elemental analysis, and gas quantification methods as well as chemical methods [3, 28, 29]. Furthermore, in order to confirm its surface structure, the grafting experiment was carried out with deuterated silica and tetra-neopentylzirconium. Evolution of 1 mol of deuterated neopentane per mole of grafted zirconium proved that there is a single bond between zirconium and oxygen. This monopodal surface structure was further confirmed by EXAFS experiment with 1.
While zirconium and titanium lead to a monopodal species on SiO2-500 [30], in the case of [Hf(Np)4] the surface reaction produces a mixture of mono- and bipodal species (Scheme 3) [31]. The mono- and bipodal (70:30) mixture was confirmed by the evolution of gas during the reaction associated with the surface microanalysis: the lower the C/Hf ratio in elemental analysis, the higher the percentage of bipodal species. Further, it was confirmed by solid-state NMR: in 13C NMR, two peaks were found at 106 and 95 ppm corresponding to CH2 of neopentyl for monopodal surface complex (3) and bipodal surface complex (4). However, with SiO2-800, Hf(NP)4 gives only monopodal species which was proved by elemental analysis, gas quantification, and NMR.

Synthesis of mono- and bis-grafted group (IV) metal alkyls on SiO2-500
The grafting of Zr(Np)4 was not only restricted to silica surface but also to other well-defined oxide surfaces. Recently, amino-modified SBA-15 surfaces were prepared by the reaction of partially dehydroxylated SBA-15 at 1100°C with ammonia at 200 and 500°C to generate [(≡Si–NH2)(≡Si–OH)] (5) and [(≡Si–NH2)2] (6). The advantage of this method is the preparation of adjacent groups in close vicinity. The surface organometallic complexes were prepared by the reaction of [Zr(Np)4] with [(≡Si–NH2)(≡SiOH)] and [(≡Si–NH2)2] in pentane for 8 h at room temperature (Scheme 4) [20].

Grafting of Zr(Np)4 on modified SBA-15
The surface complexes [(≡SiNH–)(≡SiO–)Zr(Np)2] (7) and [(≡SiNH–)2Zr(Np)2] (8) were fully characterized by solid-state NMR, IR, and TEM. It was also observed in BET experiment that although the surface area decreases slightly due to the grafting of the bulky [Zr(Np)3] fragment, this fragment did not block the opening and the pores of the material. Additionally, bright-field transmission electron microscopy (BF-TEM) obtained with high-resolution TEM (HRTEM) confirms the preservation of the hexagonally ordered mesophase structure [20].
Similarly, Zr(Np)4 and Ti(Np)4 react with silica–alumina partially dehydroxylated at 500°C (SiO2–Al2O3-500) to form a 100% monopodal species in case of zirconium [4] and a mixture of 40% mono- and 60% bipodal species in case of titanium [32]. Similarly, when Ti(CH2–Ph)4 and Zr(CH2–Ph)4 were grafted onto SiO2-200 and SiO2-700, one could generate, respectively, bipodal and monopodal surface complex [33].
3.1.1 Hydrides of Group IV Metal Alkyls
After synthesis of [(≡SiO–Zr(Np)3] (1) and species [(≡SiO–Ti(Np)3] (2) [30], the efforts were made to synthesize and identify the corresponding surface organometallic hydride which we believed to be the active catalyst for various types of C–H bond activation reaction. 1 and 2 generate tri-podal monohydride (9, 10) as major component when reacted with H2 at 150°C [30, 34]. However, 3 generates bipodal bis-hydride (11) as major component under H2 atmosphere at temperature lower than 100°C (Scheme 5) [35]. Similar hydrides were observed when silica–alumina and alumina-supported Ti(Np)4 and Zr(Np)4 were heated at 150°C in the presence of hydrogen [32]. On the other hand under similar conditions, 7 and 8 generated various other hydrides (Scheme 5) [20].

Metal hydrides of group IV obtained when group IV metal alkyls react with hydrogen
3.1.2 Reactivity of Group V (Ta) Alkyls and Alkylidene on Oxide Surfaces
One of the important aspects of surface organometallic chemistry was achieved when [Ta(CH3)5] 12, which is known to be very unstable at room temperature in solution, was stabilized upon grafting on SiO2-700 surface (a similar observation was observed with [W(Me)6]/SiO2-700 and will be discussed separately in next section) [36]. [Ta(CH3)5] reacts with SiO2-700 at −20°C and generates [≡SiO–Ta(CH3)4] (13) monopodal species. The formation of 13 was confirmed by solid-state NMR, gas quantification methods, as well as elemental analysis (Scheme 6). 13 can easily be transformed into a mixture of mono- and bipodal tantalum–carbene surface complex (14) upon heating at 150°C for 4 h. The structure of 14 was precisely confirmed by advanced solid-state NMR, elemental analysis, and gas quantification methods.

Grafting of Ta(CH3)5 on SiO2-700 and formation of mono- and bipodal tantalum-methyl-methylidene surface complex
Similarly in previous literature reports, [Ta(=CtBu)(CH2 tBu)3] (15) was grafted on SiO2-500 [37] and SiO2-700 [22], respectively. While in SiO2-700, it gives exclusively monopodal surface organometallic, but in the case of SiO2-500 it gives a mixture of mono- and bipodal surface complex (17) via an intermediate 16 (Scheme 7) [37, 38]. The mechanism for the reaction between 15 and SiO2-500 was understood when deuterium labeled SiO2-500 was used. Evolution of more than one mole of neopentane per grafted tantalum (neopentane/Ta = 1.35) [37] indicated the formation of a mixture of mono- and bipodal species on the silica surface (Scheme 7).

Formation of mono- and bipodal tantalum-neopentyl-neopentylidene on the surface of SiO2-500
The reaction of 15 with deuterated (>90%) silica followed by hydrolysis with D2O produced 2.6 equiv. of neopentane with the major species as mono-deuterated neopentane (54.4%), followed by 36.7% as bis-deuterated and 5.5% tris-deuterated neopentane along with 3.3% neopentane. The evolution of tris-deuterated neopentane confirmed that the incorporation of deuterium to tantalum carbene occurs during the grafting of ≡SiOD with 15. Additionally, to confirm the presence of tantalum–carbene species on the surface, 17 was treated with excess of acetone (Scheme 8). Formation of 1 equiv. of 18 per grafted Ta complex confirms the presence of one carbene center per grafted tantalum complex.

Pseudo-Wittig reaction between 17 and acetone
Again to confirm the presence of tantalum carbene on silica surface 13C-enriched tantalum–carbene complex, [Ta(=C*HtBu)(CH2 tBu)3] was grafted on silica−(500). The 13C CP NMR showed a peak at 246 ppm which corresponds to (=C*HtBu) along with other peaks confirming the presence of tantalum carbene on the supported complex [39].
MCM-41 partially dehydroxylated at 500°C was also used to understand the behavior of the [Ta(=CtBu)(CH2 tBu)3] complex on oxide support. Interestingly, NMR, EXAFS, elemental analysis, and gas quantification results support the formation of monopodal species, whereas in the case of SiO2-500, it produces a mixture of mono and bipodal species [40, 41].
3.1.3 Reactivity of Group V (Ta) Hydride on Oxide Surfaces
Interestingly, clearly distinct results were observed when the MCM-41-supported [≡SiOTa(=CtBu)(CH2 tBu)2] and silica-supported 17 were treated with hydrogen at 150°C (Scheme 8). In the case of silica-supported tantalum monohydride is formed via the intermediacy of tantalum polyhydride which was proved by EXAFS [42], whereas in the case of MCM-41-supported tantalum complex, a mixture of tantalum monohydride (major) and tris-hydride (minor) was formed via tantalum polyhydride [40] (Scheme 9). Upon further heating from 150 to 500°C under hydrogen atmosphere, progressive decrease of the Ta–H peak was observed in IR spectra, and a new surface complex corresponding to [(≡SiO)3Ta] was formed. This can be explained by the fact that at higher temperature, a hydride transfer from tantalum to silicon and a siloxy transfer from silicon to tantalum were observed [40].

Evolution of surface tantalum hydride upon heating under hydrogen atmosphere (bipodal structure in the case of SiO 2-500 was omitted for clarity)
3.1.4 Reactivity of Group VI (W) Alkyls and Alkylidene on Oxide Surfaces
For the exploration of better catalyst for C–H bond activation especially, in the case of alkane metathesis, the focus was shifted from group V to group VI metal catalysts as these metals are well known for olefin metathesis as well as for C–H bond activation. They also were found to make low-temperature hydrogenolysis of alkanes and polymerization of olefin [19, 43]. Similar to group V metal alkyls, group VI metal alkyls can undergo reaction with dehydroxylated silica. Similar to Scheme 7, [W(≡CtBu)(CH2 tBu)3] (19) [Mo(≡CtBu)(CH2 tBu)3] (20) was employed for grafting of group VI metal alkyl on SiO2-700. In general, a pentane solution of an excess of 19 or 20 was added to SiO2-700 at room temperature to obtain [(≡SiO–)W(≡CtBu)(CH2 tBu)2] [44] (21) or [(≡SiO–)Mo(≡CtBu)(CH2 tBu)2] (22) (Scheme 10). The IR spectrum shows a decrease of the ν(≡Si–O–H) band at 3,747 cm−1 with the formation of two new series of bands at 3,000–2,700 and 1,500–1,300 cm−1 assigned to ν(CH) and δ(CH) vibrations.

Grafting of group VI metal alkylidyne on the surface of SiO2-700
However, in the case of [Ta(=CtBu)(CH2 tBu)2], the reaction proceeds with the addition of Si–OH bond onto the Ta=C bond followed by α-H abstraction (Scheme 7), but in the case of [W(≡CtBu)(CH2 tBu)3] or [Mo(≡CtBu)(CH2 tBu)3], the addition of Si–OH on W≡C or Mo≡C was not observed when grafted on SiO2-700 (Scheme 10). It forms a monopodal carbyne species on SiO2-700. Gas quantification and elemental analysis confirmed that there are nearly three neopentyl groups per tungsten atom which again corroborates the monopodal structure of the above grafted complex. Furthermore, formation of a carbyne ligand on silica surface was confirmed by 1H, 13C, and HETCOR solid-state NMR with the observation of characteristic peak at 318 ppm for the C (carbyne). The supported complex 21 was found to be very active in olefin metathesis but exhibited no activity in alkane metathesis. The corresponding hydride [(≡SiO–)W(≡CtBu)(CH2 tBu)2] was prepared by treating 21 with the excess of hydrogen at 150°C and found to be much less active in alkane metathesis. To generate more electrophilic metal center for better catalytic activity, alumina partially dehydroxylated at 500°C (PDA) was chosen as solid support. Under similar condition, 19 was grafted on Al2O3-500 at room temperature (Scheme 11).

Grafting of [W(≡CtBu)(CH2 tBu)3] on the surface of Al2O3-500 (for simplicity only Td Al is shown)
The supported organometallic complexes were characterized by IR, solid-state NMR, EXAFS, and elemental analysis. All these data taken together confirm that it is a mixture of monopodal carbyne (major) (Scheme 10) along with a minor amount of cationic tungsten with the migration of neopentyl group from tungsten to nearby electrophilic aluminum center. The corresponding polyhydride was prepared from 23 by the treatment of excess of hydrogen at 150°C (Scheme 12). IR spectra showed partial consumption of the Al–OH with simultaneously formation of Al–H bond, and it indicates the formation of bis-aluminoxy hydride species on the surface of alumina (Scheme 12) [45].

Formation of tungsten polyhydride on Al2O3-500
Initial attempts to generate W(carbene)(hydride) from a partial hydrogenation of 21 and 23 failed. W(carbene)(hydride) was believed to be an active catalytic species for alkane metathesis reaction. In 2010, Basset et al. reported direct methylation of tungsten polyhydride with methane at higher temperature followed by α-H abstraction generated tungsten carbene species along with tungsten carbyne species (Scheme 13) [46]. However, 25 was found to be very less active in alkane metathesis reaction.

Proposed mechanism for the non-oxidative coupling of methane catalyzed by the W–H supported onto SiO2–Al2O3 and Al2O3
On the other hand, in an important observation, it was found that highly unstable [W(CH3)6] (26) [47] can easily be grafted on the oxide support to generate the corresponding stable grafted surface organometallic complex (Scheme 14). When a pentane solution of 26 was allowed to react with SiO2-700 at −50 to −30°C for a required time period, golden yellow solid power was formed [23].

Grafting of [W(CH3)6] on SiO2-700
The supported complex [(≡SiO–)W(CH3)5] (27) was fully characterized by advanced solid-state NMR, IR, elemental analysis, and gas quantification method (Fig. 1). 13C solid-state NMR shows a peak at 82 ppm which auto correlates with peak at 2.0 ppm obtained from 1H NMR confirming the formation of 27. Furthermore, double-quantum (DQ) and triple-quantum (TQ) experiments confirmed the formation of 27 [23]. All combined experimental data along with NMR are in favor of formation of a monopodal complex on silica surface (Fig. 1).

(A) One-dimensional (1D) 1H MAS solid-state NMR spectrum of 27. (B) Two-dimensional (2D) 1H–1H double-quantum (DQ)/single-quantum (SQ) and (C) 1H–1H triple-quantum (TQ)/SQ NMR spectra of 27, (D) 13CCP/MAS NMR spectrum of 27, and (E) 2D 1H–13C CP/MAS dipolar HETCOR spectrum of 27
A 13C-enriched sample of 27 was heated in the NMR probe from 25 to 72°C. Maintaining temperature at 72°C for 12 h revealed several NMR signals in the region of 298 and 40–48 ppm. HETCOR spectra showed several correlation cross peaks. Specifically, the 13C peak at 298 ppm correlates with the 1H peak at 7.6 ppm in 1H NMR which confirms the formation of the tungsten carbyne (W≡CH) (Scheme 15).

Formation of tungsten methylidyne (W≡C) species
NMR results also confirm that along with all the peaks corresponding to tungsten-methyl-methylidyne, a peak at −0.5 ppm is also observed which auto correlates in double quantum (DQ) and triple quantum (TQ), confirming either the formation of CH4 while heating the species 27 or methyl migration from W to Si (Fig. 2). Finally, 29Si NMR proves that a peak at −12 ppm is due to methyl migration from W to Si which again confirms that the structure of the decomposed species is a mixture of mono- and bipodal (Scheme 15) [23].

(A) 1D 1H spin-echo MAS solid-state NMR spectrum of [(≡SiO) x W(≡CH)Me y ], (B) 2D 1H–1H DQ and (C) 1H–1H TQ, (D) 13CCP/MAS NMR spectrum, and (E) 2D CP/MAS HETCOR NMR spectrum
To understand the activity of the [W(CH3)6] (26) with other oxide supports, the synthesis was extended from silica to silica–alumina. In a similar way, like in silica, [W(CH3)6] was grafted on silica–alumina partially dehydroxylated at 500°C. Grafting experiments of [W(CH3)6] carried out in pentane at −50 to −30°C resulted in a brown solid 28 (Scheme 16).

Grafting of W(CH3)6 on silica–alumina partially dehydroxylated at 500°C
The resulting solid 28 was fully characterized using advance solid-state NMR techniques (Fig. 3) along with elemental analysis and gas quantification methods. Solid-state NMR shows two peaks in the 13C NMR: one at −17 ppm belongs to [Al–CH3] peak which is likely resulting from methyl migration from W to Al [48], and the other at 82 ppm is ascribed to a[W–CH3] which correlates with the 1H NMR in HETCOR: therefore, all peaks belong to same complex (Fig. 3) [49]. Based on the experimental evidence and characterization data, it was believed that the grafting of 26 on SiO2–Al2O3-500 leads to the formation of a mixture of mono- and bipodal surface complex.

HETCORE, DQ, and TQ spectra of surface complex 28
In conclusion, all the surface organometallic complexes synthesized and characterized so far are extremely electron deficient and between eight electrons to 12 electrons in the Green formalism [50]. The resulting complexes either alkyls or hydrides have exhibited very specific properties in catalysis related to alkanes but also olefins.
4 Metathesis of Alkane
Alkane metathesis is a catalytic reaction involving successive breaking and formation of C–H and C–C bonds of alkanes to give lower and higher alkanes homologues [51]. Alkane metathesis reaction can be described by the following general equation:
where n = 2…n − 1 and i = 1, 2, 3…n − 1.
In 1997, Basset et al. introduced catalytic transformation of acyclic alkanes into their lower and higher homologues using silica-supported tantalum hydrides [1] in the absence of hydrogen at low temperature (150°C).
Since in the alkane metathesis, one or more C–C bonds can be broken and reformed, it lacks the selectivity in the formation of products, in contrast to the olefin metathesis where only one type of C=C is cleaved and recombined. With certain exceptions, the observed product selectivity in alkane metathesis has been C n + 1 > C n + 2 ≫ C n + 3…; C n − 1 > C n − 2 ≫ C n − 3….
Later it was found that to get a successful activity in alkane metathesis, catalysts need to have a multifunctionality, i.e., (i) activation of the C–H bond resulting in a metal alkyl, (ii) α-H elimination leading to a metallocarbene, (iii) β-H elimination leading to an olefin, (iv) olefin metathesis, and (v) finally successive hydrogenations of the olefins or the carbenes leading to alkanes. The selectivity of products is a consequence of the relative stabilities of metallacyclobutanes intermediates formed during the olefin metathesis [52].
4.1 Mechanism for Alkane Metathesis Reaction
It took a long time to establish the mechanism of this fascinating reaction after its discovery: it was known that silica-supported tantalum hydride reacted with methane at very reasonable temperature (ca. 50°C) to give a tantalum methyl and hydrogen by sigma bond metathesis [51]. A first hypothesis was advanced in which the Ta-alkyl would react directly with the C–C bond of the alkane to give a redistribution of alkyl group by analogy with the redistribution of alkylidene in olefin metathesis and the redistribution of alkylidyne in alkyne metathesis [53]. However, there was no evidence (experimental or theoretical) of such redistribution. Progressively, the surface organometallic chemistry of tantalum and tungsten allowed the observation of primary products in alkane metathesis. It is only recently that all the elementary steps have been isolated with a tantalum tetramethyl linked to silica [36].
Presently, one can summarize as follows the various observations that we made on the mechanism of alkane metathesis:
As already mentioned, it was observed that one mole of hydrogen is liberated when methane is reacted with the tantalum hydride with the formation of tantalum methyl. The reaction with methane above 150°C leads to the formation of the Ta-methyl, Ta-methylene, and Ta-methylidyne species plus H2 (M = Ta) [40–42, 54]. These observations are a proof that the first step of alkane metathesis is the formation of metal alkyl intermediate via cleavage of the C–H bond of the alkane likely by sigma bond metathesis. Further, detailed mechanistic [22, 55] and experimental kinetic studies revealed that the alkenes and hydrogen are the primary products [56]. Initially, it was believed that the active site was a bis-siloxy tantalum-monohydride, but progressively, evidence came in favor of an equilibrium between bis-siloxy tantalum-monohydride d2 and bis-siloxy-tantalum-tris-hydride d0 [57], and the mechanism would fit much better with a bis-siloxy-tantalum-tris-hydride [58].
With this knowledge, a possible mechanism was proposed where the metal hydride activates the C–H bond of alkane to form H2 and alkyl-M surface species, e.g., in the case of propane using Ta-hydride, it forms n- and iso-propyl-Ta. The respective alkyl-Ta species either undergo α-H transfer [59, 60] leading to the two carbene–hydride complexes Ta(H)(=C(CH3)2) and Ta(H)(=CH–CH2–CH3) or β-H transfer [60, 61] forming an olefin–hydride complex Ta(H)(η 2-CH2=CH–CH3) (Scheme 17). The resulting propene then leaves the coordination sphere of the Ta(H)(η 2-CH2=CH–CH3) and undergoes a homologation process via cycloaddition with the carbenic species to form four differently substituted metallacyclobutanes with methyl or ethyl groups in [1,2] or [1,3] positions (Scheme 17a) [62–65]. These metallacyclobutanes undergo cycloreversion to give new olefins and new carbene–hydride species (Scheme 17b) [66]. This catalytic cycle further continues via hydride addition into the carbene as well as olefin insertion into the hydrides. Subsequently, the alkanes are liberated via a known process of hydrogenation and hydrogenolysis [67] or possibly via σ-bond metathesis [68].

Proposed mechanism for the propane metathesis (a) formation of linear alkanes and (b) formation of branched alkanes
The product selectivity in the case of propane for the formation of linear alkanes was found to be butane (C n + 1) in higher amount than pentane (C n + 2). This can be clearly explained based on the steric interactions between substituents in [1,2] or [1,3] positions in metallacyclobutane intermediates (Scheme 18) [22, 52].

Stability of metallacyclobutane intermediates in propane metathesis reaction
Very recently it was found that with 13C-labeled C2H6, it is possible to isolate the initiation products in metathesis of ethane into propane with [(≡SiO–)TaMe4] (13). This can be considered as a breakthrough because for the first time it was found that the reaction mechanism may proceed with a Ta-alkyl; previously, it is believed to proceed by a Ta–H (Scheme 19) ([36, 69]).

Proposed mechanism for metathesis of ethane by Ta(Me)4 – precatalyst
4.2 Metathesis of Linear Alkanes
4.2.1 Metathesis of Propane
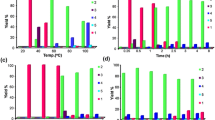
Synthesis and characterization of several catalyst precursors of [Ta] and [W] on different supports like silica [44], alumina [45], or silica–alumina [70] have been discussed in the previous section. These catalyst precursors were tested mainly in propane metathesis under similar condition in batch reactor at 150°C for 120 h. Catalytic performances revealed that the [SiO2–Al2O3-500–W–H] and [Al2O3-500–W–H] have similar and better activity (TON) than the corresponding silica-supported tantalum-based catalyst precursors (Table 2). It was also observed that the product selectivity for tungsten hydrides is narrower than for tantalum hydrides [71].
Recently, in order to further improve catalytic activity, two new catalyst precursors were developed [(≡SiO–)WMe5] (27) [23] and [(≡SiO–)TaMe4] (13) [36]. Such polymethyl complexes possess no β-H and can easily generate in situ the corresponding surface M-methylidene species (M = W [23, 72], Ta [36]) which has been done with little success in the past [44, 73]. The improvement in activity is marginal in the case of 13 compared to that of earlier reported tantalum catalyst precursors. However, the catalyst precursor 27 showed notable improvement than previously reported catalyst precursors with TON of 127 (Table 2, Entry 14).
Further investigation by preparing tungsten complexes on different supports and their corresponding hydride complexes to test their catalytic performances is under progress.
4.2.2 Metathesis of Decane
After tremendous success in propane metathesis (lower alkane) reaction, catalyst precursor 27 was employed for metathesis of n-decane (higher alkane). The n-decane metathesis reaction carried out at 150°C produced a broad distribution of linear alkanes from methane to C30 (triacontane) with trace amount of branched alkanes without any olefinic or cyclic products [74]. Interestingly, the formation of lower alkane was found to be predominant compared to higher alkanes. This was in sharp contrast to the narrow distribution observed in the case of propane metathesis (C n+1 > C n − 1). Further experiments for metathesis of n-hexane to n-nonane also showed similar product distribution. The distribution of product was found to be independent of the alkane carbon number. However, such a product distribution was closer to that reported with fully heterogeneous tandem system (Fig. 4). We assume that we have an ISOMET-PARAFFIN process by opposition to ISOMET-OLEFIN [6, 75].

Product distribution of n-decane metathesis for the catalysts [(≡SiO)WMe5]Si-Al-500
The drastic improvement in the catalytic activity was observed when 27 is replaced by [SiO2–Al2O3-500–WMe5] (28) catalyst precursor [49]. It gave 350 TON as compared to 153 TON using 27 as a catalyst precursor. This clearly shows the effect of the support as a ligand on the activity of catalyst. It should be noted that the product distribution was similar for the catalyst precursors 27 and 28.
Further investigation on the metathesis of 1-decene (an expected olefin during the alkane metathesis of n-decane) and the hydrogenation of the products formed clearly demonstrated that the distribution resulting from alkane and olefin metathesis completely differs with the same catalyst. If there is no double-bond migration, 9-octadecene and ethylene are expected to be the major primary products. Indeed, these primary products are observed, as the temperature reaches 150°C. However, after just 15 min, C7 to C12 and C13 to C20 olefins are also observed, clearly indicating that some isomerization of double bond occurs leading to several competitive metatheses (Fig. 5).

Product distribution of 1-decene metathesis with [(≡SiO–)WMe5] catalyst
In the proposed mechanism, for the olefin metathesis, the W-bis-carbene is generated via hydrogen transfer from the methyl to the W-methylidyne in the presence of an olefin. This further undergoes [2+2] cycloaddition with external olefin followed by cycloreversion to give ethylene and W-alkylidene, which reacts with an additional olefin to release the final metathesis product (Scheme 20, a). Other olefin formations can be explained through the ethylene insertion and isomerization (Scheme 20, c).

Proposed mechanism for (a) olefin metathesis, (b) alkane metathesis, and (c) olefin isomerization via ethylene insertion
While in alkane metathesis mechanism (Scheme 20, b), the n-decane undergoes σ-bond metathesis to generate methane and the W-bis-decyl species which, upon β-H elimination, produces the W–H with a coordinated olefin. Further, the α-hydrogen transfer from the alkyl to alkylidyne forms the hydrido W-bis-carbene [55, 76]. This upon [2+2] cycloaddition and cycloreversion gives an internal olefin and hydrido W-bis-carbene. Successive insertion/elimination steps (by chain walking) [77] give the terminal alkene, which reacts to a new W-alkylidene. The CH activation of the pendant W-hydride with n-decane followed by β-H elimination provides 1-decene. A second metathesis between 1-decene and newly formed W-alkylidene followed by hydrogenolysis produces the alkane.
It is noteworthy that the double-bond isomerization step is faster than the overall elementary steps of alkane metathesis. Formation of lower alkanes is due to the tungsten hydride intermediate, favoring chain walking with double-bond migration followed by fast cross metathesis with coordinated ethylene leading to lower alkenes in turn giving lower alkanes on hydrogenation. This intramolecular reaction pathway, without formation of the free olefin, probably is the difference between alkane and olefin metathesis.
4.3 Metathesis of Cycloalkanes
Metathesis of acyclic alkane produces lower and higher homologues of the corresponding alkanes. With the recent improvements in the catalysis using the precatalyst [(≡SiO)WMe5] (27), it was interesting for us to apply similar strategy for acyclic alkanes, e.g., cyclooctane expecting to have easy access to lower and higher homologues of cyclic alkanes.
Recently, metathesis of cyclooctane was reported using the tandem system having the pincer-ligated iridium complexes for hydrogenation/dehydrogenation and Schrock-type Mo-alkylidene complexes for olefin metathesis [78]. However, this system showed >80% formation of polymeric products with cyclic oligomers (C16, C24, C3, and C40) of cyclooctane.
4.3.1 Metathesis of Cyclooctane
Precatalyst 27 has shown surprising results in the metathesis of cyclooctane and cyclodecane with broad distribution of lower and higher macrocyclic alkanes. Cyclopentane, cyclohexane, and cycloheptane were found to be inactive under similar condition [79].
For example, the cyclooctane metathesis using 27 has shown exceptional distribution of macrocyclic alkanes in the range of C12 to C40 without any polymeric products [79], wherein the cyclooctane undergoes similar mechanism with C–H bond activation followed by β-H elimination to give W-methylidene hydride and cyclooctene. The cyclooctene undergoes ring-opening–ring-closing metathesis reactions (RO-RCM) via the backbiting of terminal double bond to give 1,9-cyclohexadecadiene. This on hydrogenation gives the cyclohexadecane (Scheme 21). In a similar manner, macrocyclic alkanes (C24, C32, C40) are generated via RO-RCM of cyclooctene.

Proposed mechanism for metathesis of cyclooctane into cyclohexadecane
The formation of other macrocyclic alkanes and lower cyclic alkanes (C5, C6, and C7) is clearly due to the double-bond isomerization before RCM. This was further proved by the reaction of macrocyclic alkane (C12 to C40) at 150°C for 48 h, which did not produce any ring contraction cyclic products (C5, C6, and C7) clearly, indicating that the lower cyclic alkanes are not formed by the secondary metathesis of macrocyclic alkanes (Scheme 22).

Proposed mechanism for ring expansion and ring contraction with some selected cyclic and macrocyclic alkanes formation from cyclooctane metathesis. ROM ring-opening metathesis, RCM ring-closing metathesis, Iso double-bond isomerization
The absence of polymeric products is definitely due the low steady-state concentration of cyclooctene formed during the reaction.
4.4 Branched Alkanes Metathesis: Metathesis of 2-Methylpropane
The alkane metathesis of highly branched alkanes and product selectivity also follows the same mechanism with catalyst 24. A selective and catalytic conversion of 2-methylpropane into 2,3-dimethylbutane (42%), and ethane (41%) (Scheme 23) [80] was observed when 2-methylpropane was passed over the catalyst 24 at 150°C. Conversion was reached up to 8% and 37 TON was achieved over 43 h (Scheme 23).

Proposed mechanism for metathesis of 2-methylpropane
4.5 Cross Metathesis Between Two Different Alkanes
Cross metathesis between two different alkanes represent one of the most difficult challenges in organic chemistry [53]. In 2001, Basset et al. first demonstrated the possibilities of sigma bond metathesis between two different alkanes [55]. In 2004, this same group has reported the cross metathesis between ethane and toluene [81] and methane and propane [82]. Silica-supported tantalum hydride catalyst \( \left[{\left(\equiv \mathrm{S}\mathrm{i}\mathrm{O}\right)}_2\mathrm{T}\mathrm{a}\mathrm{H}\right]\rightleftharpoons \left[{\left(\equiv \mathrm{S}\mathrm{i}\mathrm{O}\right)}_2{\mathrm{TaH}}_3\right] \) was employed for cross-metathesis reaction between toluene and ethane at 250°C. Under static condition, it produced mainly ethyl benzene and xylenes as major product along with propane and methane (Scheme 24).

Cross metathesis of toluene and ethane with silica-supported tantalum hydride
In order to have better understanding, the same reaction was performed with 100% 13C-enriched methyl in toluene and found mainly 13C-mono-labeled ethylbenzene at the α-position (92%), while xylenes were 98% 13C-mono-labeled. Besides this isolation of stable intermediates, such as [(≡SiO)2Ta–C2H5], [(≡SiO)2Ta–CH2C6H5], or [(≡SiO)2Ta–C6H4CH3] by 13C CP MAS NMR spectroscopy, the mechanism of the reaction leading to various xylenes is given in Scheme 25.

Possible mechanism of cross metathesis of toluene and methane with silica-supported tantalum tris-hydride
In this scheme, we assume that in the equilibrium \( \left[{\left(\equiv \mathrm{S}\mathrm{i}\mathrm{O}\right)}_2{\mathrm{Ta}}^{\left(\mathrm{I}\mathrm{I}\mathrm{I}\right)}\mathrm{H}\right]\rightleftharpoons \left[{\left(\equiv \mathrm{S}\mathrm{i}\mathrm{O}\right)}_2{\mathrm{Ta}}^{\left(\mathrm{V}\right)}{\mathrm{H}}_3\right] \), it is [(≡SiO)2TaH3], d0, which is involved in the succession of sigma bond metathesis. The reaction of methane with the Ta(H)2(aryl) is a multistep which is omitted for clarity.
4.5.1 Cross Metathesis Using Methane as Reactant
Although methane is the most abundant hydrocarbon found on earth, until now there is not enough applications found for this important chemical (except the partial oxidation to syngas). Thus, cross metathesis of higher alkane with methane is interesting. This question was raised since the discovery of alkane metathesis reaction in 1997 where it was found that the two molecules of ethane can participate in self-metathesis and give one molecule of methane and one molecule of propane [1]. Thus, it is interesting to see whether it is possible to drive this reaction in the reverse direction, i.e., reacting methane with another alkane to give a mixture of alkanes with incorporation of methane (Scheme 26).

Cross metathesis of methane and propane in the presence of silica-supported tantalum catalyst
The reaction with propane and methane was investigated under dynamic conditions with very high methane/propane ratio to overcome thermodynamic barrier and favor kinetics. It was found that ethane was selectively produced. This result was further supported by isotope labeling with 13C-enriched methane. A possible mechanism is given for the better understanding of this reaction (Scheme 27).

Possible mechanism for cross metathesis of methane and propane with silica-supported tantalum hydride
4.6 Hydro-metathesis Reactions
Hydro-metathesis of propene under hydrogen atmosphere, in the presence of TaH/KCC-1 catalyst, proceeds smoothly under dynamic reaction condition at 150°C for 65 h with 750 TON [83]. In addition to the expected hydrogenation product, propane, ethane, and butane were formed as major products, and methane, isobutane, and isopentanes formed as minor products in case of propene. Similarly in the case of 1-butene, propane and hexanes were formed as major products, and ethane, propene, pentanes, and heptanes were formed as minor products. In case the of butene, the catalyst was found to be stable even after 75 h and cumulative TON up to 1,150 achieved after 75 h of the reaction [83]. The most important issue with this catalyst is the stability and reusability of this Ta–H/KCC-1 catalyst and the high turnover numbers reached compared with the turnover numbers reported for the Ta–H/SiO2 catalyst in alkane metathesis reaction.
As expected, this reaction was found to be faster in comparison to alkane metathesis because of the absence of C–H bond activation steps which are assumed to be the difficult step of alkane metathesis reaction. Besides thermodynamic factors, this could also explain the comparative ease for the hydro-metathesis because olefin hydrogenation is thermodynamically favored even at low temperatures.
Based on the experimental fact and following the Chauvin mechanism for alkane metathesis, a probable mechanism was proposed for hydro-metathesis of propene (Scheme 28).

Possible mechanism of hydro-metathesis of propene with silica-supported tantalum hydride
5 Conclusions
The rules of molecular organometallic chemistry apply when reacting organometallics with surfaces of oxides. A new field has emerged in catalysis called SOMC.
The reasons why the surface organometallic compounds of group IV and V exhibit a high reactivity toward C–H and C–C bonds of alkane are likely multiple:
-
1.
They are highly electron deficient (between eight and 12 electrons in the Green formalism).
-
2.
It was already known in organometallic chemistry that the transition metals of group IV and V can activate C–H bonds of alkanes by sigma bond metathesis, but the number of catalytic examples (see, e.g., the early works of P. Watson H/D exchange and CH4/CD4 exchange) [84] was not so high because in solution, the complexes may lose activity by dimerization or any other type of bimolecular interactions. In contrast, isolation on a support of a highly electron-deficient complex prevents any sort of bimolecular deactivation. This is a well-known concept, but it deserves to be repeated.
-
3.
The use of a support as a ligand brings a supplementary electronic and steric parameter.
-
4.
The surface organometallic complexes are thermally much more stable than their molecular counterpart. The example of [(≡Si–O) WMe5] on silica which is stable up to 100°C is a good example of the ability of a surface to stabilize a molecular compound which is explosive at room temperature! So reactions can be performed on organometallic compounds at very high temperature which is not possible in classical homogeneous catalysis. Such stability allows the observation of carbynes, carbenes, alkyls, amido, imido, and hydrides even at elevated temperatures.
The absence of bimolecular reactions avoids many deactivation processes and allows better lifetime of the catalysts. This is a well-known concept but repetition is a source of pedagogy.
Using a variety of single-site surface complexes bearing simultaneously several types of highly reactive ligands has been at the origin of the various concepts of single-site multifunctional catalysts:
-
Monofunctional with metal hydride(s): (Ziegler–Natta depolymerization, hydrogenolysis of waxes, olefin polymerization), metal carbenes (olefin metathesis, ROMP ADMET, etc.)
-
Bifunctional with metal hydrides (or alkyls) (conversion of butenes to propylene)
-
Trifunctional (conversion of ethylene to propylene, metathesis of alkanes, cycloalkanes, etc.)
References
Vidal V, Theolier A, Thivolle-Cazat J, Basset JM (1997) Science 276:99–102
Basset JM, Ugo R (2009) Modern surface organometallic chemistry. Wiley-VCH, Weinheim, pp 1–21
Corker J, Lefebvre F, Lecuyer C, Dufaud V, Quignard F, Choplin A, Evans J, Basset JM (1996) Science 271:966–969
Dufaud VR, Basset JM (1998) Angew Chem Int Ed 37:806–810
Goldman AS, Roy AH, Huang Z, Ahuja R, Schinski W, Brookhart M (2006) Science 312:257–261
Huang Z, Rolfe E, Carson EC, Brookhart M, Goldman AS, El-Khalafy SH, MacArthur AHR (2010) Adv Synth Catal 352:125–135
Herisson JL, Chauvin Y (1971) Makromol Chem 141:161–176
Schrock RR, Murdzek JS, Bazan GC, Robbins J, Dimare M, Oregan M (1990) J Am Chem Soc 112:3875–3886
Bazan GC, Oskam JH, Cho HN, Park LY, Schrock RR (1991) J Am Chem Soc 113:6899–6907
Schwab P, Grubbs RH, Ziller JW (1996) J Am Chem Soc 118:100–110
Nguyen ST, Grubbs RH, Ziller JW (1993) J Am Chem Soc 115:9858–9859
Ertl G (1983) J Vac Sci Technol A 1:1247–1253
Cariati E, Dragonetti C, Lucenti E, Roberto D, Ugo R (2009) Modern surface organometallic chemistry. Wiley-VCH, Weinheim, pp 639–683
Dragonetti C, Ceriotti A, Roberto D, Ugo R (2007) Organometallics 26:310–315
Ballard DGH, Courtis A, Holton J, Mcmeeking J, Pearce R (1978) J Chem Soc Chem Commun 994–995
Popoff N, Mazoyer E, Pelletier J, Gauvin RM, Taoufik M (2013) Chem Soc Rev 42:9035–9054
Nedez C, Theolier A, Lefebvre F, Choplin A, Basset JM, Joly JF (1993) J Am Chem Soc 115:722–729
Wolke SI, Buffon R, Rodrigues UP (2001) J Organomet Chem 625:101–107
Ajjou JAN, Scott SL, Paquet V (1998) J Am Chem Soc 120:415–416
Bendjeriou-Sedjerari A, Azzi JM, Abou-Hamad E, Anjum DH, Pasha FA, Huang KW, Emsley L, Basset JM (2013) J Am Chem Soc 135:17943–17951
Saint-Arroman RP, Chabanas M, Baudouin A, Copéret C, Basset JH, Lesage A, Emsley L (2001) J Am Chem Soc 123:3820–3821
Le Roux E, Chabanas M, Baudouin A, de Mallmann A, Copéret C, Quadrelli EA, Thivolle-Cazat J, Basset JM, Lukens W, Lesage A, Emsley L, Sunley GJ (2004) J Am Chem Soc 126:13391–13399
Samantaray MK, Callens E, Abou-Hamad E, Rossini AJ, Widdifield CM, Dey R, Emsley L, Basset JM (2014) J Am Chem Soc 136:1054–1061
Chabanas M, Baudouin A, Copéret C, Basset JM (2001) J Am Chem Soc 123:2062–2063
Berthoud R, Rendon N, Blanc F, Solans-Monfort X, Coperet C, Eisenstein O (2009) Dalton Trans 5879–5886
Quignard F, Lecuyer C, Choplin A, Olivier D, Basset JM (1992) J Mol Catal 74:353–363
Niccolai GP, Basset JM (1998) Nato ASI 3 High Technol 44:111–124
Quignard F, Lecuyer C, Bougault C, Lefebvre F, Choplin A, Olivier D, Basset JM (1992) Inorg Chem 31:928–930
Saint-Arroman RP, Basset JM, Lefebvre F, Didillon B (2005) Appl Catal A Gen 290:181–190
Rosier C, Niccolai GP, Basset JM (1997) J Am Chem Soc 119:12408–12409
Tosin G, Santini CC, Taoufik M, De Mallmann A, Basset JM (2006) Organometallics 25:3324–3335
Larabi C, Merle N, Norsic S, Taoufik M, Baudouin A, Lucas C, Thivolle-Cazat J, de Mallmann A, Basset JM (2009) Organometallics 28:5647–5655
Popoff N, Espinas J, Pelletier J, Macqueron B, Szeto KC, Boyron O, Boisson C, Del Rosal I, Maron L, De Mallmann A, Gauvin RM, Taoufik M (2013) Chem Eur J 19:964–973
Rataboul F, Baudouin A, Thieuleux C, Veyre L, Copéret C, Thivolle-Cazat J, Basset JM, Lesage A, Emsley L (2004) J Am Chem Soc 126:12541–12550
Tosin G, Santini CC, Baudouin A, De Mallman A, Fiddy S, Dablemont C, Basset JM (2007) Organometallics 26:4118–4127
Chen Y, Abou-hamad E, Hamieh A, Hamzaoui B, Emsley L, Basset JM (2015) J Am Chem Soc 137:588–591
Dufaud V, Niccolai GP, Thivolle-Cazat J, Basset JM (1995) J Am Chem Soc 117:4288–4294
Lefort L, Chabanas M, Maury O, Meunier D, Copéret C, Thivolle-Cazat J, Basset JM (2000) J Organomet Chem 593:96–100
Chabanas M, Quadrelli EA, Fenet B, Copéret C, Thivolle-Cazat J, Basset JM, Lesage A, Emsley L (2001) Angew Chem Int Ed 40:4493–4496
Soignier S, Taoufik M, Le Roux E, Saggio G, Dablemont C, Baudouin A, Lefebvre F, de Mallmann A, Thivolle-Cazat J, Basset JM, Sunley G, Maunders BM (2006) Organometallics 25:1569–1577
Saggio G, Mallmann A, Maunders B, Taoufik M, Thivolle-Cazat J, Basset J-M (2002) Organometallics 21:5167–5171
Vidal V, Theolier A, Thivolle-Cazat J, Basset JM, Corker J (1996) J Am Chem Soc 118:4595–4602
Delley MF, Nunez-Zarur F, Conley MP, Comas-Vives A, Siddiqi G, Norsic S, Monteil V, Safonova OV, Copéret C (2014) Proc Natl Acad Sci U S A 111:11624–11629
Le Roux E, Taoufik M, Chabanas M, Alcor D, Baudouin A, Copéret C, Thivolle-Cazat J, Basset JM, Lesage A, Hediger S, Emsley L (2005) Organometallics 24:4274–4279
Le Roux E, Taoufik M, Copéret C, de Mallmann A, Thivolle-Cazat J, Basset JM, Maunders BM, Sunley GJ (2005) Angew Chem Int Ed 44:6755–6758
Szeto KC, Norsic S, Hardou L, Le Roux E, Chakka S, Thivolle-Cazat J, Baudouin A, Papaioannou C, Basset JM, Taoufik M (2010) Chem Commun 46:3985–3987
Galyer L, Mertis K, Wilkinson G (1975) J Organomet Chem 85:C37–C38
Marks TJ (1992) Acc Chem Res 25:57–65
Samantaray MK, Dey R, Abou-Hamad E, Hamieh A, Basset JM (2015) Chem Eur J 21:6100–6106
Green MLH (1995) J Organomet Chem 500:127–148
Basset JM, Copéret C, Soulivong D, Taoufik M, Thivolle-Cazat J (2006) Angew Chem Int Ed 45:6082–6085
Leconte M, Basset JM (1979) J Am Chem Soc 101:7296–7302
Basset JM, Copéret C, Soulivong D, Taoufik M, Cazat JT (2010) Acc Chem Res 43:323–334
Vidal V, Theolier A, Thivolle-Cazat J, Basset JM (1995) J Chem Soc Chem Commun 991–992
Copéret C, Maury O, Thivolle-Cazat J, Basset JM (2001) Angew Chem Int Ed 40:2331–2334
Basset JM, Copéret C, Lefort L, Maunders BM, Maury O, Le Roux E, Saggio G, Soignier S, Soulivong D, Sunley GJ, Taoufik M, Thivolle-Cazat J (2005) J Am Chem Soc 127:8604–8605
Avenier P, Taoufik M, Lesage A, Solans-Monfort X, Baudouin A, de Mallmann A, Veyre L, Basset JM, Eisenstein O, Emsley L, Quadrelli EA (2007) Science 317:1056–1060
Pasha FA, Cavallo L, Basset JM (2014) ACS Catal 4:1868–1874
Vanasselt A, Santarsiero BD, Bercaw JE (1986) J Am Chem Soc 108:8291–8293
Parkin G, Bunel E, Burger BJ, Trimmer MS, Vanasselt A, Bercaw JE (1987) J Mol Catal 41:21–39
Sharp PR, Astruc D, Schrock RR (1979) J Organomet Chem 182:477–488
Wallace KC, Dewan JC, Schrock RR (1986) Organometallics 5:2162–2164
Wallace KC, Liu AH, Dewan JC, Schrock RR (1988) J Am Chem Soc 110:4964–4977
Turner HW, Schrock RR (1982) J Am Chem Soc 104:2331–2333
Mclain SJ, Sancho J, Schrock RR (1979) J Am Chem Soc 101:5451–5453
Schinzel S, Chermette H, Copéret C, Basset JM (2008) J Am Chem Soc 130:7984–7987
Chabanas M, Vidal V, Copéret C, Thivolle-Cazat J, Basset JM (2000) Angew Chem Int Ed 39:1962–1965
Watson PL (1983) J Am Chem Soc 105:6491–6493
Chen Y, Ould-Chikh S, Abou-Hamad E, Callens E, Mohandas JC, Khalid S, Basset JM (2014) Organometallics 33:1205–1211
Le Roux E, Taoufik M, Baudouin A, Copéret C, Thivolle-Cazat J, Basset JM, Maunders BM, Sunley GJ (2007) Adv Synth Catal 349:231–237
Taoufik M, Le Roux E, Thivolle-Cazat J, Copéret C, Basset JM, Maunders B, Sunley GJ (2006) Top Catal 40:65–70
Callens E, Abou-Hamad E, Riache N, Basset JM (2014) Chem Commun 50:3982–3985
Buffon R, Leconte M, Choplin A, Basset JM (1994) J Chem Soc Dalton 1723–1729
Riache N, Callens E, Espinas J, Dery A, Samantaray MK, Dey R, Basset JM (2015) Catal Sci Technol 5:280–285
Kundu S, Choliy Y, Zhuo G, Ahuja R, Emge TJ, Warmuth R, Brookhart M, Krogh-Jespersen K, Goldman AS (2009) Organometallics 28:5432–5444
Rascon F, Coperet C (2011) J Organomet Chem 696:4121–4131
Domski GJ, Rose JM, Coates GW, Bolig AD, Brookhart M (2007) Prog Polym Sci 32:30–92
Ahuja R, Kundu S, Goldman AS, Brookhart M, Vicente BC, Scott SL (2008) Chem Commun 253–255
Riache N, Callens E, Samantaray MK, Kharbatia NM, Atiqullah M, Basset JM (2014) Chem Eur J 20:15089–15094
Merle N, Stoffelbach F, Taoufik M, Le Roux E, Thivolle-Cazat J, Basset JM (2009) Chem Commun 2523–2525
Taoufik M, Schwab E, Schultz M, Vanoppen D, Walter M, Thivolle-Cazat J, Basset JM (2004) Chem Commun 1434–1435
Soulivong D, Copéret C, Thivolle-Cazat J, Basset JM, Maunders BM, Pardy RBA, Sunley GJ (2004) Angew Chem Int Ed 43:5366–5369
Polshettiwar V, Thivolle-Cazat J, Taoufik M, Stoffelbach F, Norsic S, Basset JM (2011) Angew Chem Int Ed 50:2747–2751
Watson PL, Parshall GW (1985) Acc Chem Res 18:51–56
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Samantaray, M.K., Dey, R., Kavitake, S., Basset, JM. (2015). New Concept of C–H and C–C Bond Activation via Surface Organometallic Chemistry. In: Dixneuf, P., Doucet, H. (eds) C-H Bond Activation and Catalytic Functionalization II. Topics in Organometallic Chemistry, vol 56. Springer, Cham. https://doi.org/10.1007/3418_2015_139
Download citation
DOI: https://doi.org/10.1007/3418_2015_139
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-24802-8
Online ISBN: 978-3-319-29319-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)