
Abstract
Endo-lysosomes are membrane-bound acidic organelles that are involved in endocytosis, recycling, and degradation of extracellular and intracellular material. The membranes of endo-lysosomes express several Ca2+-permeable cation ion channels, including two-pore channels (TPC1-3) and transient receptor potential mucolipin channels (TRPML1-3). In this chapter, we will describe four different state-of-the-art Ca2+ imaging approaches, which are well-suited to investigate the function of endo-lysosomal cation channels. These techniques include (1) global cytosolic Ca2+ measurements, (2) peri-endo-lysosomal Ca2+ imaging using genetically encoded Ca2+ sensors that are directed to the cytosolic endo-lysosomal membrane surface, (3) Ca2+ imaging of endo-lysosomal cation channels, which are engineered in order to redirect them to the plasma membrane in combination with approaches 1 and 2, and (4) Ca2+ imaging by directing Ca2+ indicators to the endo-lysosomal lumen. Moreover, we will review useful small molecules, which can be used as valuable tools for endo-lysosomal Ca2+ imaging. Rather than providing complete protocols, we will discuss specific methodological issues related to endo-lysosomal Ca2+ imaging.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Endo-lysosomes are membrane-bound acidic organelles that are involved in endocytosis, recycling, and degradation of extracellular as well as intracellular material (Luzio et al. 2007; van Meel and Klumperman 2008; Huotari and Helenius 2011).
The membranes of endo-lysosomes harbor several ion channels and transporters (summarized in Fig. 1). These intracellular transmembrane proteins are essential for the maintenance of several endo-lysosomal functions, including homeostasis of ions and pH, resting membrane potential, transport of amino acids, and trafficking and fusion of vesicles (Dong et al. 2008; Dong et al. 2010; Grimm et al. 2012; Cang et al. 2013; Grimm et al. 2014; Cang et al. 2015; Grimm et al. 2017; Jentsch and Pusch 2018; Chen et al. 2021; Rosato et al. 2021).

Ion channels in the endo-lysosomal system. The illustration shows an overview of different cation channels or transporters identified in intracellular endo-lysosomal membranes (light yellow circles), using the patch-clamp method. In lysosomes, the acidic lumen has a pH value of ~4.5, which is maintained with the help of the vacuolar-type H+-ATPase, Cl− channels, K+ channels, as well as Na+/K+ ATPase, Na+/H+ exchanger, and passive H+ leaks (not illustrated) (Grabe and Oster 2001; Morgan et al. 2011). Abbreviations: BK, large conductance Ca2+-activated K+ channels; CAX, vacuolar Ca2+/H+ exchanger; ClC, Cl− channel; P2X4, P2X purinoreceptor subunit 4; TMEM175, transmembrane protein 175; TPC, two-pore channels; TRPML, transient receptor potential mucolipin channel; V-ATPase, vacuolar-type H+-ATPase (modified from Chen et al. 2017b)
Genetic mutations and abnormal expression levels of endo-lysosomal ion channels have been associated with impaired cellular metabolism (Cang et al. 2013; Rosato et al. 2021) and various diseases, including congenital lysosomal storage disorders (Grimm et al. 2012; Chen et al. 2014; Xu and Ren 2015; Grimm et al. 2017), cancer (Dong et al. 2008; Grimm et al. 2018; Xu and Dong 2021; Wu et al. 2021; Abrahamian and Grimm 2021), neurodegenerative disorders, such as Alzheimer’s and Parkinson’s disease (Hockey et al. 2015; Jentsch and Pusch 2018; Bose et al. 2021), viral infections including HIV (Chao et al. 2020), Ebola (Sakurai et al. 2015; Grimm et al. 2017; Penny et al. 2019), SARS-CoV2 (Grimm and Tang 2020; Chao et al. 2020), non-alcoholic fatty liver disease, and hyperlipoproteinemia (Grimm et al. 2014).
2 Calcium-Permeable Ion Channels in Endo-lysosomes
2.1 TPCs
In mammals, two-pore channels (isoforms TPC1-3) represent a small family of non-selective cation channels that are specifically expressed in endo-lysosomal membranes and belong to the superfamily of voltage-gated ion channels. TPCs are composed of two subunits building a functional pore, and each TPC subunit contains two homologous Shaker-like six-transmembrane-domain repeats (She et al. 2018; She et al. 2019; Dickinson et al. 2020). Considering the number of transmembrane domains (TMs), mammalian TPCs (12 TMs) thus resemble the structure of transient receptor potential channels (6 TMs) and voltage-gated calcium as well as sodium channels (24 TMs) (Ishibashi et al. 2000; Grimm et al. 2017; Galione 2019).
The mammalian TPC1 channel has been first identified in rat kidney (Ishibashi et al. 2000). TPC1 is permeable for Na+, K+, and Ca2+, showing a higher selectivity for Na+ (She et al. 2018). The activation of TPC1 depends on membrane voltage depolarization and binding of the endolysosome-specific lipid phosphatidylinositol 3,5-bisophosphate (PI(3,5)P2) (She et al. 2018). PI(3,5)P2 has been detected on early endosomes, late endosomes, and lysosomes, and it regulates endo-lysosomal functions, such as formation of large vacuoles, acidification of endo-lysosomes, and traffic of cell surface receptors to lysosomes (de Lartigue et al. 2009; McCartney et al. 2014).
Using electrophysiological methods, several recent studies have demonstrated that PI(3,5)P2 application leads to Na+ release from endo-lysosomes through TPC1 channels (Wang et al. 2012; Cang et al. 2014; Lagostena et al. 2017; She et al. 2018) as well as through TPC2 channels (Boccaccio et al. 2014; Guo et al. 2017; Penny et al. 2019; Gerndt et al. 2020).
On the other hand, numerous studies using calcium imaging have shown that the intracellular messenger nicotinic acid adenine dinucleotide phosphate (NAADP) triggers Ca2+ release through TPC1 (Brailoiu et al. 2009; Ruas et al. 2015; Faris et al. 2019; Moccia et al. 2021; Hu et al. 2021) and TPC2 channels (Calcraft et al. 2009; Zong et al. 2009; Schieder et al. 2010; Pitt et al. 2010; Brailoiu et al. 2010; Grimm et al. 2014; Ruas et al. 2015; Gerndt et al. 2020; Zhang et al. 2021). NAADP is a potent Ca2+-mobilizing messenger that is synthetized from NADP in presence of the enzymes ADP-ribosyl cyclases (CD38), resulting in exchange of the nicotinamide moiety of NADP with nicotinic acid (Malavasi et al. 2008). This pathway of NAADP biosynthesis requires acidic pH (pH 4–5). Recently, a novel pathway for rapid formation and degradation of NAADP was identified in T cells, in which NAADP is generated from its reduced form, NAADPH, by a redox cycle involving NADPH oxidases (NOX) or dual NADPH oxidases (DUOX2) (Gu et al. 2021). NAADP is well known to evoke Ca2+ release from acidic lysosome-related organelles in sea urchin eggs (Lee and Aarhus 1995; Genazzani and Galione 1996; Churchill and Galione 2001; Churchill et al. 2002) as well as in several types of mammalian cells, including pancreatic acinar and β-cells, ventricular cardiac myocytes, pulmonary arterial smooth muscle cells, T lymphocytes, and hippocampal neurons (Cancela et al. 1999; Mitchell et al. 2003; Yamasaki et al. 2004; Kinnear et al. 2004; Macgregor et al. 2007; Davis et al. 2012; Capel et al. 2015; Lin et al. 2017; Foster et al. 2018). There is also evidence that NAADP-evoked Ca2+ release trough TPC2 channels can be modulated by PI(3,5)P2, Mg2+, and the mitogen-activated protein kinases MAPK, c-Jun N-Terminal Kinase (JNK), and p38 (Jha et al. 2014).
The NAADP-evoked Ca2+ rise depends on TPC2 in pancreatic beta cells (Calcraft et al. 2009), but is also abolished in TPC1/TPC2 double-deficient mouse embryonic fibroblasts (MEF) (Ruas et al. 2015) and recovered by the reintroduction of TPC1 and TPC2, rather than TRPML1 or a pore-dead mutant of TPC2 (Ruas et al. 2015) (see also chapter “NAADP-Dependent TPC Current” in this volume). Nevertheless, in TRPML1-deficient MEFs NAADP-mediated Ca2+ release was also largely reduced (Zhang et al. 2011), and the initial Ca2+ rise in microdomains in T cells evoked by T cell receptor stimulation, that depends on NAADP, was markedly reduced upon deletion of RYR1 (Diercks et al. 2018). However, TPC1 and TPC2 channel proteins are not required for NAADP binding in liver cells (Ruas et al. 2015). Two new NAADP binding proteins, JPT2/HNL1 and Lsm12, were described to be essential for NAADP-mediated Ca2+ release in T cells (Roggenkamp et al. 2021) and HEK293 or MEF cells (Zhang et al. 2021), but their relative contribution for NAADP-mediated Ca2+ release in other cell types and their interaction with NAADP targets in individual cell systems need to be demonstrated.
In addition, bioactive lipid sphingosine induces cytosolic Ca2+ release from acidic stores through TPC1 channels, and this effect is independent of calcium channels in the plasma membrane and endoplasmic reticulum (Höglinger et al. 2015).
2.2 TRPMLs
Transient receptor potential mucolipin channels (TRPML1-3) are non-selective cation channels that show specific expression in the membranes of late endosomes and lysosomes (Cheng et al. 2010; Grimm et al. 2012; Xu and Ren 2015). A recent electrophysiological study in macrophages has revealed that the TRPML1 isoform is expressed specifically on late endosomes and lysosomes, whereas TRPML3 has been recorded not only on late endosomes and lysosomes, but also on early endosomes (Chen et al. 2017a).
Functional TRPML complexes represent homotetramers, and each TRPML subunit is composed of six transmembrane domains (Cheng et al. 2010; Grimm et al. 2012; Schmiege et al. 2021). Electrophysiological studies have shown that TRPML1 is responsible for the transport of various cations from the endo-lysosomal lumen to the cytosol, including Ca2+, Na+, Fe2+, K+, and Zn2+ ions (Dong et al. 2008; Xu and Ren 2015).
Genetic mutations in MCOLN1, the gene encoding TRPML1, are associated with mucolipidosis type IV (MLIV), a neurodegenerative lysosomal storage disorder with a progressive time course. The patients suffer from neurological and ophthalmological symptoms, such as psychomotor delay, hypotonia, retinal degeneration, and corneal clouding (Sun et al. 2000; Bassi et al. 2000; Bargal et al. 2001; Mirabelli-Badenier et al. 2015; Saijo et al. 2016; Shiihara et al. 2016; Hayashi et al. 2020).
TRPML1 plays an important role in Ca2+-dependent lysosomal trafficking processes, including maturation of late endosomes, formation of autophagosomes, retrograde trafficking from late endosomes, and lysosomes to the trans-Golgi network (TGN), and lysosomal exocytosis (Chen et al. 1998; LaPlante et al. 2006; Thompson et al. 2007; Vergarajauregui et al. 2008; Dong et al. 2009; Curcio-Morelli et al. 2010; Shen et al. 2011; Medina et al. 2011; Wong et al. 2012; Xu and Ren 2015).
Furthermore, mitochondrial reactive oxygen species (ROS) can activate TRPML1-mediated Ca2+ release from lysosomes, which in turn has been associated with induction of autophagy, biogenesis of autophagosomes and lysosomes, and elimination of elevated ROS levels (Medina et al. 2015; Zhang et al. 2016). This pathway includes activation of the phosphatase calcineurin and subsequent calcineurin-mediated dephosphorylation and translocation of the transcription factor TFEB to the cell nucleus (Zhang et al. 2016).
3 Approaches for Imaging of Lysosomal Calcium
Here, we will present useful state-of-the-art approaches well suited to characterize the function of endo-lysosomal cation channels using four different Ca2+ imaging approaches: (1) global cytosolic Ca2+ measurements, (2) peri-endo-lysosomal Ca2+ imaging using genetically encoded Ca2+ sensors, which are directed to the cytosolic endo-lysosomal membrane surface, (3) Ca2+ imaging of endo-lysosomal cation channels, which are engineered in order to redirect them to the plasma membrane in combination with approaches 1 and 2, and (4) Ca2+ imaging by directing Ca2+ indicators to the endo-lysosomal lumen. In the following, we will present these methods, provide methodological details, and discuss these approaches in comparison with the current literature. Furthermore, we will provide information on useful small molecules, which can be used as valuable tools for endo-lysosmal Ca2+ imaging, most importantly novel compounds which activate or inhibit endo-lysosomal cation channels, small molecules, which differentially enlarge individual endo-lysosomal subpopulations as well as substances, which deplete endo-lysosomal Ca2+ stores. Rather than providing a complete protocol, we will discuss specific issues which are related to endo-lysosomal Ca2+ imaging. For details of more general Ca2+ imaging application, we refer to individual chapters.
3.1 Global Cytosolic Ca2+ Measurements
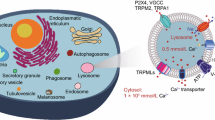
This method is intended to measure the Ca2+ signals that are evoked by ion channels in the endo-lysosomes and to differentiate these signals from those that originate in the cytosol. There are a variety of potential sources of Ca2+ and several ion channels that may contribute to these signals at the same time (Fig. 1). To analyze the input of these signals, an activator that opens endo-lysosomal Ca2+ channels, such as NAADP or PI(3,5)P2 (Table 1), is used to release Ca2+ from the endo-lysosomes into the cytosol. The resulting cytosolic signal is then measured both in the presence and absence of a given pharmacological (or genetic) blockade of a specific Ca2+ source (e.g., by depleting the Ca2+ store, by using a Ca2+ channel blocker, or by silencing a gene through RNAi). Since both chemicals, NAADP and PI(3,5)P2, are unable to cross the plasma membrane, they are commonly administered through a patch pipette. By comparing the signals obtained in the absence and presence of inhibitors that target a specific Ca2+ source, it is possible to determine the contribution of that store to the overall signal. For instance, the cytosolic Ca2+ signal displayed in Fig. 2 is a combination of two components: one originating from the endo-lysosomal Ca2+ store, and another resulting from the subsequent release of Ca2+ from the endoplasmic reticulum, triggered by the endo-lysosomal Ca2+ signal. By using Thapsigargin, it is possible to deplete the ER of Ca2+. By administering NAADP after ER-mediated depletion, it is possible to identify the endo-lysosomal contribution to the cytosolic Ca2+ signal. On the other hand, depleting the endo-lysosomal Ca2+ store eliminates both the endo-lysosomal and ER-dependent Ca2+ signals.

Mechanisms underlying calcium release from endo-lysosomes and the endoplasmic reticulum. Endo-lysosomal vesicles and the endoplasmic reticulum serve as Ca2+-storing organelles in the cell. (A) Application of NAADP activates endo-lysosomal TPC channels, allowing an efflux of calcium ions from the endo-lysosomal lumen to the cytosol and a slow, small increase in intracellular Ca2+ concentration (initial, slow phase of Ca2+ signal in B1). This NAADP-induced Ca2+ release leads to the subsequent activation of ER-specific IP3Rs or SR-specific RyRs, which mediate a steep, large rise in intracellular Ca2+ concentration (large peak of Ca2+ signal in B1). The SERCA-selective antagonist thapsigargin inhibits uptake of Ca2+ into sarcoplasmic reticulum, resulting in depletion of ER Ca2+ stores. After pre-treatment with thapsigargin, NAADP induces only a small, initial Ca2+ signal (B2). Bafilomycin A1 disturbs the uptake of H+ ions via the endo-lysosomal V-ATPase. Monensin and nigericin act as protonophores that disturb the H+ gradient in lysosomes by releasing H+ from the lumen. GPN causes the rupture of lysosomal membranes via cathepsin C-mediated cleavage. Application of NAADP fails to induce a Ca2+ response after depletion of endo-lysosomal Ca2+ stores using pre-treatment with bafilomycin, monensin, and GPN (B3). Abbreviations: ATP, adenosine triphosphate; Baf, bafilomycin A1; GPN, glycyl-L-phenylalanine 2-naphthylamide; IP3R, inositol-1,4,5-trisphosphate receptor; Mon, Monensin; NAADP, nicotinic acid adenine dinucleotide phosphate; Nig, Nigericin; SERCA, sarco-endoplasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum; TG, thapsigargin; TPC, two-pore channels; V-ATPase, vacuolar-type H+-ATPase

Approaches for monitoring of calcium release mediated by endo-lysosomal calcium channels. Schematic illustration of cells showing imaging of cytosolic Ca2+ released via endogenous cation channels in endo-lysosomes (a) or after channel expression in the plasma membrane (b). For this purpose, cells are loaded with chemical fluorescent Ca2+ indicators, such as Fura-2 and Fluo-4. Further, GECIs can be expressed in the cytosol, thus labeling the released calcium. Specific expression of GECIs coupled to ion channels in endo-lysosomal membranes (c) or expression of GECIs coupled to lysosomal channels retargeted to plasma membrane (d) represent useful approaches for lysosomal or whole-cell patch-clamp studies, respectively. Finally, imaging of luminal calcium is performed using chemical indicators conjugated to dextrans (e), such as Fura-dextran, Texas Red dextran, and rhod dextran. Note that large dextrans have a large molecular weight and are taken up by the endosomes via endocytosis. Abbreviations: GECI, genetically encoded calcium indicators; TPC, two-pore channel; TRPML, transient receptor potential mucolipin channel
The method outlined above can also be employed to indirectly evaluate the concentration of Ca2+ within endo-lysosomes. In this scenario, it is essential to quickly and thoroughly release the Ca2+ content from the endo-lysosomal store. This can be done by activating ion channels within the endo-lysosomes, or alternatively, by using a set of pharmacological agents that inhibit the loading of Ca2+ into lysosomes or disrupt endo-lysosomal membranes (Morgan et al. 2011). Then, the temporal profile and magnitude of the cytosolic Ca2+ signal are monitored using conventional Ca2+ dyes. A higher concentration of Ca2+ inside an organelle leads to a greater amplitude of the cytosolic Ca2+ spike and a more rapid rise time of the signal.
For endo-lysosomal Ca2+ imaging, we need to consider several technical aspects.
-
1.
Intracellular Ca2+ signals can be recorded with different fluorescent Ca2+ indicators, such as Fluo-4 or Fura-2, or by expression of GCaMP as a genetically encoded cytosolic Ca2+ indicator (Fig. 3a, b).
-
2.
Pilot experiments are necessary for each specific cell type to empirically determine whether the Ca2+ indicator used is loaded into the cytosol exclusively, as artifacts will arise, if the dye is loaded into other organelles. Therefore, it is recommended to load the cells with an AM-ester at room temperature, to reduce compartmentalization. Optimal loading times and dye concentration must be experimentally determined for each series of experiment, as they can considerably vary depending on the cell type.
-
3.
Due to the qualitative readout, it is possible to investigate the contribution of endo-lysosomal ion channels to Ca2+ storage and Ca2+ release.
-
4.
Ca2+ indicator experiments can be performed at different scales, such as on cell populations using plate readers, FACS or cuvettes, or on single cells using fluorescence microscopy techniques like epifluorescence or confocal laser scanning. The only requirement for these experiments is the availability of appropriate excitation and emission wavelengths, and filters for the fluorophore(s) used.
-
5.
This approach is well-suited to compare endo-lysosomal Ca2+ signals and content under different conditions, such as the effects of a drug, changes in protein expression, or a disease state.
-
6.
One potential limitation of this method is that depleting Ca2+ stores can be a major artificial alteration and may result in unintended effects. It is important to consider this when interpreting the results. Additionally, it is worth noting that different stores are often interlinked and targeting one organelle may inadvertently affect neighboring organelles.
-
7.
A further limitation of this approach is that it cannot be used to determine the absolute concentration of Ca2+ in the lumen of the endo-lysosomes.
So far, two primary activators for TPCs have been identified: NAADP and PI(3,5)P2. A recent drug screening process involving 80,000 small molecule compounds led to the discovery of two new TPC agonists, TPC2-A1-N and TPC2-A1-P (Gerndt et al. 2020). Furthermore, tetrandrine has been described as a TPC antagonist and two novel tetrandrine-derived TPC antagonists have been generated, SG-005 and SG-094 (Table 1) (Müller et al. 2021). At a concentration of 10 μM, both SG-005 and SG-094 were able to inhibit the activation of TPC1 and TPC2. Additionally, SG-005 was found to block the lysosomal cation channel TRPLM1 (Müller et al. 2021). Regarding TRPML1, there are several agonists and antagonists that have been reported in the literature, such as EVP-169 and EDME, which are also summarized in Table 1. Additionally, there are a range of compounds available for releasing Ca2+ from endo-lysosomal stores, such as Glycyl-L-Phenylalanine 2-Naphthylamide (GPN), bafilomycin A1, nigericin, and monensin. GPN is widely used to deplete lysosomal Ca2+ stores by disrupting lysosomes, and it has been extensively studied (Berg et al. 1994; Churchill et al. 2002; Coen et al. 2012; Davis et al. 2012; Dionisio et al. 2011; Fois et al. 2015; Garrity et al. 2016; Haller et al. 1996; Jadot et al. 1984; Kilpatrick et al. 2013; Li et al. 2012; Melchionda et al. 2016; Morgan and Galione 2007; Morgan et al. 2011; Penny et al. 2015; Penny et al. 2014; Ruas et al. 2015). GPN is a synthetic, membrane-permeable dipeptide that mimics a substrate and is broken down by the lysosome-specific enzyme Cathepsin C (Rao et al. 1997). GPN enters the lysosomal membrane through diffusion and is subsequently hydrolyzed by intraluminal cathepsin C (also known as dipeptidyl peptidase 1), releasing free amino acids. Due to their polarity, the amino acids build up in the lysosomal lumen, leading to reversible permeabilization of the lysosomal membrane by osmotic swelling (Berg et al. 1994; Haller et al. 1996). Notably, recent research suggests that GPN treatment results in membrane pores in lysosomes, allowing leaks of small molecules with a molecular mass <10 kDa (Penny et al. 2014), and that this is the mechanism of how GPN evokes Ca2+ release from lysosomes. However, this mechanism of action was questioned in a recent report demonstrating that GPN increases [Ca2+] in the cytoplasm by increasing its pH values, which then directly stimulates Ca2+ release from the ER (Atakpa et al. 2019). Nevertheless, GPN seems to be “rehabilitated” according to data from a subsequent study demonstrating that “GPN-evoked Ca2+ signals were better correlated with associated pH changes in the lysosome compared to the cytosol, and were coupled to lysosomal Ca2+ release” (Yuan et al. 2021).
To prevent loading of acidic Ca2+ stores, the macrolide antibiotic bafilomycin A1 (Baf-A1), an inhibitor of the lysosomal vacuolar-type H+-ATPase (V-ATPase) has been used in many cell types. The V-ATPase plays a role in acidifying the lumen of endo-lysosomes (Bowman et al. 1988; Dröse and Altendorf 1997; Huss et al. 2011), and Baf-A1 can inhibit V-ATPase with high affinity at nanomolar concentrations (Bowman et al. 1988). The idea is that inhibiting the H+ uptake into the lumen via the V-ATPase leads to passive loss of H+ through leaks, which in turn dissipates the H+ gradient across the lysosomal membrane and causes the lumen to become alkaline. Since this H+ gradient is vital for the proper functioning of Ca2+ uptake via Ca2+/H+ exchangers, bafilomycin and other V-ATPase inhibitors can indirectly block the accumulation of Ca2+ in endo-lysomes, promoting the release of Ca2+ into the cytosol (Christensen et al. 2002; Churchill et al. 2002; Yamasaki et al. 2004; Kinnear et al. 2004; Gerasimenko et al. 2006; Lloyd-Evans et al. 2008; Brailoiu et al. 2009; Vasudevan et al. 2010; Rah et al. 2010; Morgan et al. 2015; Kelu et al. 2017).
The release of Ca2+ from acidic stores can also be triggered by the application of electroneutral cation-exchanging ionophores, such as nigericin and monensin (Yagodin et al. 1999; Srinivas et al. 2002; Churchill et al. 2002; Ramos et al. 2010). Nigericin acts as a K+/H+ exchanger, allowing the transport of H+ out of the lysosome at the expense of K+ influx, thereby collapsing the H+ gradient across the lysosomal membrane. This results in luminal alkalinization, reducing the accumulation of Ca2+ via the lysosomal Ca2+/H+ exchanger and leaving Ca2+ leak uncompensated. Monensin, a polyether antibiotic, operates in a similar manner, but instead it translocates Na+ from the cytosol into the lysosome (Inabayashi et al. 1995). Additionally, monensin has been shown to reverse the mode of the Na+/Ca2+ exchanger (Asano et al. 1995). It is important to note that nigericin can also act on mitochondria, where its application leads to hyperpolarization of the mitochondrial inner membrane (Robb-Gaspers et al. 1998).
3.2 Peri-Vesicular Ca2+ Imaging
In recent years, a variety of genetically encoded calcium indicators (GECIs) have become available. One of the key advantages of these indicators is that they can be selectively expressed and retained in organelles by fusing organelle-specific targeting sequences to the indicator molecule. For example, an endoplasmic reticulum (ER)-targeted GECI, called “cameleon,” was generated by adding the calreticulin signal sequence 5′ to CFP and an ER-retention sequence to the 3′ end of citrine (Palmer et al. 2004). Similarly, the low-affinity GCaMP3 variant (GCaMPer 10.19) coding sequence was fused downstream of calreticulin and an ER-retention sequence was fused to the carboxy-terminus for imaging of the ER calcium stores (Henderson et al. 2015). GCaMP-Type GECIs were also successfully subcellularly targeted in neurons (Mao et al. 2008).
In the past decade, many GECIs have been targeted to specific organelles and used successfully for organellar calcium imaging (for review, see Suzuki et al. (2016)). In addition to the ER, endo-lysosomal Ca2+ stores play an important role in Ca2+-signaling in various cell types. However, the low pH environment of the lysosome lumen and high pH-sensitivity of most GECIs make it very challenging to measure [Ca2+] accurately in acidic organelles. To determine changes in lysosomal pH, Chin et al. generated FIRE-pHLy, a fluorescent-based ratiometric pH biosensor that specifically targets lysosomes (Chin et al. 2021). This genetically encoded pH sensor can detect pH levels between 3.5 and 6.0, making it suitable for determining the luminal pH of acidic lysosomes.
By placing GECIs on the cytosolic surface of endo-lysosomal vesicles, researchers can develop sophisticated methods for detecting Ca2+ release from endo-lysosomes in a more localized way around the peri-endo-lysosomal surface of the vesicle, without disrupting lysosomal pH. To record peri-vesicular Ca2+ release, Shen et al. (2011) fused GCaMP3 to the cytosolic N-terminus of TRPML1 to anchor the sensor to the cytosolic surface of vesicles. The idea is to monitor cytosolic Ca2+ while simultaneously activating endo-lysosomal channels or rapidly discharging intracellular Ca2+ stores with agents that selectively target endo-lysosomal stores, as described above (GPN, bafilomycin A1, monensin, and nigericin). These authors demonstrated that the GCaMP3 fluorescence responds preferentially and reliably to reagents that selectively mobilize Ca2+ from endo-lysosomal vesicles, including GPN and bafilomycin A1. In most cells, thapsigargin failed to significantly increase GCaMP3 fluorescence, indicating that slow and small ER Ca2+ release was not detected by GCaMP3-TRPML1. This construct was also used by other researchers to study lysosomal Ca2+ signaling mechanisms (Medina et al. 2015; Tsunemi et al. 2019; Gerndt et al. 2020). Using the same construct, Garrity et al. (2016) developed a robust lysosomal Ca2+ refilling assay to study release and uptake mechanisms of acidic Ca2+ stores (for a review, see Yang et al. (2019)). Gerndt et al. fused TPC2 to the genetically encoded Ca2+ indicator, GCaMP6(s) both with (TPC2-GCaMP) and without (TPC2-GCaMP/L265P) an intact pore (Fig. 3c) to measure global Ca2+ signals (Gerndt et al. 2020). TPC2-A1-N and TPC2-A1-P evoked Ca2+ signals in cells expressing intracellular TPC2-GCaMP but not TPC2-GCaMP/L265P. Another research group generated a lysosomally targeted Ca2+-sensitive FRET probe consisting of the lysosomal-resident membrane protein LAMP1 fused to the YCaM3.6 cameleon (LAMP1–YCaM) (McCue et al. 2013). They demonstrated that this genetically encoded ratiometric sensor efficiently targets and detects Ca2+ in close proximity to the cytoplasmic face of lysosomes. They also showed in HeLa cells that responses to the physiological agonist histamine persist in cells with depleted ER-Ca2+ content.
Recently, Davis et al. fused G-GECO1.2 to TPC1/TPC2 channels to measure peri-lysosomal Ca2+ nanodomains around TPCs in macrophages (Davis et al. 2020). Usage of this low-affinity GECI fusion constructs enables discrimination between local and global calcium events. This approach has been used to reveal that endosomes and lysosomes act as platforms for a novel phagocytic signaling pathway. Subsequently promoted NAADP activates TPC channels. It is noteworthy that this phagocytic pathway was driven by local calcium domains at the endo-lysosomes rather than global cytosolic calcium events.
The sensitivity (Kd, dynamic range, brightness) requirements for local calcium sensors can vary greatly in different cell types because the distance between different calcium sources is determined by the different morphological peculiarities. For example, GCaMP6m fused to TPC2 protein was used for the visualization of Ca2+ transients around the dense granules of platelets (Ambrosio et al. 2015). These dense granules represent endo-lysosome-related acidic calcium stores in platelets. Using TPC2-GCaMP6m sensor, these researchers were able to reveal and visualize in real time organelle “kiss-and-run” events. They reported also the presence of membranous tubules transiently connecting PDGs and demonstrated the critical role of Ca2+ flux through TPC2 in this process.
We used GCaMP6m, attached to the C-terminus of TPC2 channels to expose it at the cytoplasmic face of acidic organelles and secretory granules, and created transgenic mice ubiquitously expressing this sensor. In highly polarized pancreatic acinar cells, TPC2-GCaMP6m was localized exclusively at the apical pole (Fig. 4a-left, right), and the colocalization with the marker of secretory vesicles RAB-27B was observed (Fig. 4a-middle, right). Stimulation with cholecystokinin was used for the cells, as it is known to generate nicotinic acid adenine dinucleotide phosphate (NAADP) (Petersen and Tepikin 2008). The measurements were carried out in isolated acinar clusters expressing TPC2-GCaMP6m construct. An application of the cholecystokinin analogue CCK-8 (2 pM) in the absence of extracellular Ca2+ ions led to the Ca2+-oscillations (Fig. 4b). The fact that the Ca2+ sensor GCamP6m was targeted to the C-terminal end of TPC2 channel proteins (TPC2-GCaMP6m) suggests that these Ca2+ oscillations are arising at the cytosolic face of TPC2-containing organelles. TPC2 channels are localized in endosomes and lysosomes in many cell types investigated so far (Galione et al. 2009; Grimm et al. 2017; Patel and Kilpatrick 2018). From these recordings it is still not clear whether the detected Ca2+ release occurs via the same TPC2 channels fused to the GCamP6m sensor or via natively expressed TPC/TRPML channels located in close vicinity to these TPC2-GCaMP6m sensor molecules. To clarify this GCaMP6m fused to pore mutant TPC2 variant should be used in future experiments. Nevertheless, these recordings suggest that targeting GECIs such as GCaMP6m to endo-lysosomal TPC channels is a useful approach to study local Ca2+ signals triggered by NAADP-dependent Ca2+ release in primary acinar cell clusters. The specificity of the Ca2+ signals, measured by the TPC2-GCaMP6m sensors, for stimulation with the NAADP-generating CCK needs to be tested in detail in future experiments. It could not be excluded whether it can also be evoked by stimuli triggering Ca2+ release from other Ca2+ stores, e.g. via engagement of IP3 receptors following stimulation of muscarinic receptors. In this case, the Ca2+ sensitivity and local spatial selectivity of the peri-endo-lysosomal targeted Ca2+ indicator could be adjusted by the use of different GCaMP variants with distinct Ca2+ affinity (Henderson et al. 2015) or by GECO protein variants (Wu et al. 2014).

(a) Pancreatic acinar cells cluster co-stained with antibody against GCaMP6m (green, left panel) and RAB-27B (red, middle panel); an overlay picture of the transmission light, green and red channels (right panel). Images were taken using a confocal microscope (x63). (b) Representative recording of the TPC2-GCaMP6m fluorescent signal measured in 4 representative pancreatic acinar cells stimulated with 2 pM of Cholecystokinin in the absence of extracellular Ca2+
Although peri-endo-lysosomal Ca2+ signaling events are highly localized and small, their high functional relevance for exocytosis, vesicle fusion, and trafficking is well documented. The usage of high-affinity Ca2+ sensors enables that such small and localized Ca2+ signals can be detected, which are otherwise negligible among other cytosolic signals. Moreover, high-affinity Ca2+ indicators are indispensable if the signal-to-noise ratio is otherwise too low. Of note, the endo-lysosomal Ca2+ pool is small compared to the significant Ca2+ storage capacity of the ER or the extracellular milieu.
Genetically encoded Ca2+ sensors have several benefits and allow overcoming some technical limitations of small molecule chemical Ca2+ dyes. In case of problems with dye loading into the cells or too profound compartmentalization, GECIs could offer a plausible alternative in a given cell type. Finally, intracellular targeting/tethering also allows overcoming signal distortions evoked by the rapid diffusion of unattached Ca2+ indicators.
Intraluminal Ca2+ imaging inside the endo-lysosomes: The main technical limitation of such measurements is high intraluminal acidity of these organelles in combination with weak fluorescence of GECIs at low pH values. Nevertheless, the development of new Ca2+-sensors working at low pH values observed in secretory granules and endosomes was reported in several studies. Dickson et al. adapted the “D1-endoplasmic reticulum” probe (Palmer et al. 2004) to measure calcium in secretory granules by attaching D1 to tissue plasminogen activator (Dickson et al. 2012). This indicator responds to Ca2+ at the expected pH values of secretory granules, but simultaneous knowledge of the secretory granule pH is necessary to interpret corresponding measurements. To enable intra-endosomal Ca2+ imaging the pH-resistant ratiometric Ca2+-biosensor GEM-GECO1 was developed and validated in a fusion construct with tetanus-insensitive vesicle-associated membrane protein (Albrecht et al. 2015). A principally new approach was used to develop the fluorescent DNA-based reporter CalipHluor that can be targeted to specific organelles (Narayanaswamy et al. 2019). CalipHluor functions as a pH-correctable Ca2+ reporter, since it allows simultaneous ratiometric measurement of luminal pH and [Ca2+]. The usability of this sensor was demonstrated by targeting of CalipHluor to the endo-lysosomal pathway that enabled mapping of luminal Ca2+ changes during endosomal maturation and allowed detecting a surge in luminal [Ca2+] specifically in lysosomes.
Critical Points and Controls
-
1.
It is important to verify that targeted sensors co-localize in healthy cells with established markers of endo-lysosomes such as lysosomal associated membrane protein-1 (Lamp1), whereas no colocalization with markers for the ER, early endosomes, or mitochondria should be observed (Garrity et al. 2016).
-
2.
Another important aspect is the affinity of the utilized GECI. Ideally, for the selective measurements of local [Ca2+] in close vicinity of endo-lysosomal channels, the sensor must exhibit a low micromolar affinity, otherwise the tethered probe will also sense cytosolic [Ca2+]. To date, the GECIs that have been targeted to vesicles exerted rather high Ca2+ affinity. Despite the moderate selectivity for peri-vesicular Ca2+, they are useful for recording of small local [Ca2+] changes when global cytosolic Ca2+ signals are marginal.
-
3.
Of note, such probes allow an indirect assessment of vesicular Ca2+ release events, albeit an improvement over the recordings of global cytosolic Ca2+. It is clear that cytoplasmic microdomains adjacent to acidic calcium stores could have fluctuating pH values, which are potential sources of artifacts by usage of pH-sensitive Ca2+-sensors. However, further accumulation of knowledge about organellar ion homeostasis and further development of GECIs will allow overcoming these potential limitations.
-
4.
Importantly, for the confirmation of the spatial and Ca2+ specificity of GCaMP3-ML1 probe’s in HEK-GCaMP3-ML1 cells, GPN or BAPTA-AM pre-treatment should efficiently suppress the initial response to ML-SA1. Such pre-treatment is used to abolish the store refilling either in Ca2+-free solution or in the presence of La3+. Finally, the GPN effect should be reversible, and gradual recovery of ML-SA1 responses should be observed under GPN washout.
3.3 Ca2+ Imaging of Endo-Lysosomal Ca2+ Channels Redirected to the Plasma Membrane
Specific intracellular targeting of TPC and TRPML channels to endo-lysosomal compartments is frequently determined by conserved di-leucine motifs. Deletion of this sequence or mutation of these amino acid residues to alanine can efficiently target these channels to the plasma membrane (Fig. 2b and 2d). For example, human TPC2 channel was redirected to the plasma membrane after mutation of the N-terminal endo-lysosomal targeting motif (TPC2L11A/L12A). Our own measurements and the data published by another group (Brailoiu et al. 2009) validate this approach. In particular, Brailoiu et al. (2009) demonstrated that TPC2 proteins redirected to the plasma membrane mediated robust Ca2+ entry upon NAADP stimulation. We used this approach for the high-throughput identification of new TPC2 activators (Gerndt et al. 2020). To this end, we screened a cell line stably expressing (TPC2L11A/L12A) using an FLIPR-based Ca2+ assay with natural and synthetic small molecules. We used as control a “pore-dead” channel variant with disrupted pore (TPC2L11A/L12A/L265P) in order to increase the robustness of this assay. Despite high efficiency of this approach, our results need an additional confirmation using a Ca2+ imaging technique to demonstrate Ca2+ release from the endo-lysosomes (approaches 1 and 2). Furthermore, our results can be validated by electrophysiological recordings of ion channels redirected to the plasma membrane and by patch-clamp measurements of native endo-lysosomal channels. We successfully used similar approaches for TRPML channels. In this case, partial deletion of N- and C-termini containing lysosomal targeting motifs redirected human TRPML1 (TRPML1ΔNC) channels to the plasma membrane.
3.4 Intra-Endo-Lysosomal Ca2+ Imaging Using Fura-Dextran or Oregon Green 488 BAPTA-1 Dextran
Small molecule Ca2+ indicators can be targeted into the endo-lysosomal compartments using dextran-coupled conjugated forms of these indicators. The cells exhibiting endocytic activity can then effectively accumulate these conjugates in endo-lysosomal lumen. Applicability of this approach was demonstrated using Fura-Dextran and Oregon 488 BAPTA-1 dextran (OG-BAPTA-dextran) (Morgan et al. 2015). For pulse/chasing of these dextran conjugates, they are applied to the cells for a short time (≤15 min) at 37°C and then washed out (Fig. 2e). During this time, dextran-coupled indicators are taken up into the cells via endocytosis, and subsequently reach the endo-lysosome lumen (Christensen et al. 2002). This loading technique effectively works also with HEK293 cells stably or transiently expressing TPC2-mCherry or TRPML1-mCherry-tagged fusion proteins.
The main limitation of the abovementioned dyes is their pH sensitivity, and large pH elevations may cause dramatic changes in both Kd of OG-BAPTA-dextran and luminal Ca2+ buffering capability, preventing accurate determinations of [Ca2+]LY (Garrity et al. 2016). The pH stability could be evaluated using lysotracker or other more specific pH sensors. However, it was argued that ML-SA1 application induced Ca2+ release from the lysosome lumen in the Fura-Dextran-loaded ML1-mCherry-transfected HEK-293T cells, since the LysoTracker staining was not significantly reduced by ML-SA1, suggesting that the signals were primarily mediated by changes of intralysosomal Ca2+, but not by changes in the intralysosomal pH (Garrity et al. 2016).
4 Conclusions
This chapter together with the outlined method set is intended to provide a comprehensive toolbox from which appropriate methods can be used to characterize Ca2+ signals generated by endo-lysosomal Ca2+ channels. From this toolbox, individual methods can be picked and used in an appropriate format ranging from single cell microscopy and confocal microscopy to FACS and plate-reader-based assays.
References
Abrahamian C, Grimm C (2021) Endolysosomal cation channels and MITF in melanocytes and melanoma. Biomol Ther 11(7):1021. https://doi.org/10.3390/biom11071021
Albrecht T, Zhao Y, Nguyen TH, Campbell RE, Johnson JD (2015) Fluorescent biosensors illuminate calcium levels within defined beta-cell endosome subpopulations. Cell Calcium 57(4):263–274. https://doi.org/10.1016/j.ceca.2015.01.008
Ambrosio AL, Boyle JA, Di Pietro SM (2015) TPC2 mediates new mechanisms of platelet dense granule membrane dynamics through regulation of Ca2+ release. Mol Biol Cell 26(18):3263–3274. https://doi.org/10.1091/mbc.E15-01-0058
Asano S, Matsuda T, Takuma K, Kim HS, Sato T, Nishikawa T, Baba A (1995) Nitroprusside and cyclic GMP stimulate Na(+)-Ca2+ exchange activity in neuronal preparations and cultured rat astrocytes. J Neurochem 64(6):2437–2441. https://doi.org/10.1046/j.1471-4159.1995.64062437.x
Atakpa P, van Marrewijk LM, Apta-Smith M, Chakraborty S, Taylor CW (2019) GPN does not release lysosomal Ca2+ but evokes Ca2+ release from the ER by increasing the cytosolic pH independently of cathepsin C. J Cell Sci 132(3):jcs223883. https://doi.org/10.1242/jcs.223883
Bargal R, Avidan N, Olender T, Ben Asher E, Zeigler M, Raas-Rothschild A, Frumkin A, Ben-Yoseph O, Friedlender Y, Lancet D, Bach G (2001) Mucolipidosis type IV: novel MCOLN1 mutations in Jewish and non-Jewish patients and the frequency of the disease in the Ashkenazi Jewish population. Hum Mutat 17(5):397–402. https://doi.org/10.1002/humu.1115
Bassi MT, Manzoni M, Monti E, Pizzo MT, Ballabio A, Borsani G (2000) Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. Am J Hum Genet 67(5):1110–1120. https://doi.org/10.1016/S0002-9297(07)62941-3
Berg TO, Strømhaug E, Løvdal T, Seglen O, Berg T (1994) Use of glycyl-L-phenylalanine 2-naphthylamide, a lysosome-disrupting cathepsin C substrate, to distinguish between lysosomes and prelysosomal endocytic vacuoles. Biochem J 300(Pt 1):229–236. https://doi.org/10.1042/bj3000229
Boccaccio A, Scholz-Starke J, Hamamoto S, Larisch N, Festa M, Gutla PVK, Costa A, Dietrich P, Uozumi N, Carpaneto A (2014) The phosphoinositide PI(3,5)P2 mediates activation of mammalian but not plant TPC proteins: functional expression of endolysosomal channels in yeast and plant cells. Cell Mol Life Sci 71(21):4275–4283. https://doi.org/10.1007/s00018-014-1623-2
Bose S, He H, Stauber T (2021) Neurodegeneration upon dysfunction of endosomal/lysosomal CLC chloride transporters. Front Cell Dev Biol 9
Bowman EJ, Siebers A, Altendorf K (1988) Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A 85(21):7972–7976. https://doi.org/10.1073/pnas.85.21.7972
Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, Hooper R, Boulware MJ, Dun NJ, Marchant JS, Patel S (2009) Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol 186(2):201–209. https://doi.org/10.1083/jcb.200904073
Brailoiu E, Rahman T, Churamani D, Prole DL, Brailoiu GC, Hooper R, Taylor CW, Patel S (2010) An NAADP-gated two-pore channel targeted to the plasma membrane uncouples triggering from amplifying Ca2+ signals. J Biol Chem 285(49):38511–38516. https://doi.org/10.1074/jbc.M110.162073
Cai X, Xu Y, Cheung AK, Tomlinson RC, Alcázar-Román A, Murphy L, Billich A, Zhang B, Feng Y, Klumpp M, Rondeau J-M, Fazal AN, Wilson CJ, Myer V, Joberty G, Bouwmeester T, Labow MA, Finan PM, Porter JA, Ploegh HL, Baird D, De Camilli P, Tallarico JA, Huang Q (2013) PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chem Biol 20(7):912–921. https://doi.org/10.1016/j.chembiol.2013.05.010
Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang K-T, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, Zhu MX (2009) NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459(7246):596–600. https://doi.org/10.1038/nature08030
Cancela JM, Churchill GC, Galione A (1999) Coordination of agonist-induced Ca2+-signalling patterns by NAADP in pancreatic acinar cells. Nature 398(6722):74–76. https://doi.org/10.1038/18032
Cang C, Zhou Y, Navarro B, Seo Y-J, Aranda K, Shi L, Battaglia-Hsu S, Nissim I, Clapham DE, Ren D (2013) mTOR regulates lysosomal ATP-sensitive two-pore Na(+) channels to adapt to metabolic state. Cell 152(4):778–790. https://doi.org/10.1016/j.cell.2013.01.023
Cang C, Bekele B, Ren D (2014) The voltage-gated sodium channel TPC1 confers endolysosomal excitability. Nat Chem Biol 10(6):463–469. https://doi.org/10.1038/nchembio.1522
Cang C, Aranda K, Seo Y, Gasnier B, Ren D (2015) TMEM175 is an organelle K(+) channel regulating lysosomal function. Cell 162(5):1101–1112. https://doi.org/10.1016/j.cell.2015.08.002
Capel RA, Bolton EL, Lin WK, Aston D, Wang Y, Liu W, Wang X, Burton R-AB, Bloor-Young D, Shade K-T, Ruas M, Parrington J, Churchill GC, Lei M, Galione A, Terrar DA (2015) Two-pore channels (TPC2s) and nicotinic acid adenine dinucleotide phosphate (NAADP) at lysosomal-sarcoplasmic reticular junctions contribute to acute and chronic β-adrenoceptor signaling in the heart. J Biol Chem 290(50):30087–30098. https://doi.org/10.1074/jbc.M115.684076
Cerny J, Feng Y, Yu A, Miyake K, Borgonovo B, Klumperman J, Meldolesi J, McNeil PL, Kirchhausen T (2004) The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. EMBO Rep 5(9):883–888. https://doi.org/10.1038/sj.embor.7400243
Chao Y-K, Schludi V, Chen C-C, Butz E, Nguyen ONP, Müller M, Krüger J, Kammerbauer C, Ben-Johny M, Vollmar AM, Berking C, Biel M, Wahl-Schott CA, Grimm C (2017) TPC2 polymorphisms associated with a hair pigmentation phenotype in humans result in gain of channel function by independent mechanisms. Proc Natl Acad Sci U S A 114(41):E8595–E8602. https://doi.org/10.1073/pnas.1705739114
Chao Y-K, Chang S-Y, Grimm C (2020) Endo-lysosomal cation channels and infectious diseases. Rev Physiol Biochem Pharmacol. https://doi.org/10.1007/112_2020_31
Chen CS, Bach G, Pagano RE (1998) Abnormal transport along the lysosomal pathway in mucolipidosis, type IV disease. Proc Natl Acad Sci U S A 95(11):6373–6378. https://doi.org/10.1073/pnas.95.11.6373
Chen C-C, Keller M, Hess M, Schiffmann R, Urban N, Wolfgardt A, Schaefer M, Bracher F, Biel M, Wahl-Schott C, Grimm C (2014) A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat Commun 5:4681. https://doi.org/10.1038/ncomms5681
Chen C-C, Butz ES, Chao Y-K, Grishchuk Y, Becker L, Heller S, Slaugenhaupt SA, Biel M, Wahl-Schott C, Grimm C (2017a) Small molecules for early endosome-specific patch clamping. Cell Chem Biol 24(7):907–916.e4. https://doi.org/10.1016/j.chembiol.2017.05.025
Chen C-C, Cang C, Fenske S, Butz E, Chao Y-K, Biel M, Ren D, Wahl-Schott C, Grimm C (2017b) Patch-clamp technique to characterize ion channels in enlarged individual endolysosomes. Nat Protoc 12(8):1639–1658. https://doi.org/10.1038/nprot.2017.036
Chen C-C, Krogsaeter E, Grimm C (2021) Two-pore and TRP cation channels in endolysosomal osmo-/mechanosensation and volume regulation. Biochim Biophys Acta Mol Cell Res 1868(2):118921. https://doi.org/10.1016/j.bbamcr.2020.118921
Cheng X, Shen D, Samie M, Xu H (2010) Mucolipins: intracellular TRPML1-3 channels. FEBS Lett 584(10):2013–2021. https://doi.org/10.1016/j.febslet.2009.12.056
Chin MY, Patwardhan AR, Ang K-H, Wang AL, Alquezar C, Welch M, Nguyen PT, Grabe M, Molofsky AV, Arkin MR, Kao AW (2021) Genetically encoded, pH-sensitive mTFP1 biosensor for probing lysosomal pH. ACS Sens 6(6):2168–2180. https://doi.org/10.1021/acssensors.0c02318
Christensen KA, Myers JT, Swanson JA (2002) pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci 115(Pt 3):599–607. https://doi.org/10.1242/jcs.115.3.599
Churchill GC, Galione A (2001) NAADP induces Ca2+ oscillations via a two-pool mechanism by priming IP3- and cADPR-sensitive Ca2+ stores. EMBO J 20(11):2666–2671. https://doi.org/10.1093/emboj/20.11.2666
Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S, Galione A (2002) NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 111(5):703–708. https://doi.org/10.1016/s0092-8674(02)01082-6
Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W, Michiels C, Munck S, Baert V, Sugita S, Wuytack F, Hiesinger PR, Grinstein S, Annaert W (2012) Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol 198(1):23–35. https://doi.org/10.1083/jcb.201201076. Epub 2012 Jul 2. PMID: 22753898; PMCID: PMC3392942
Curcio-Morelli C, Charles FA, Micsenyi MC, Cao Y, Venugopal B, Browning MF, Dobrenis K, Cotman SL, Walkley SU, Slaugenhaupt SA (2010) Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol Dis 40(2):370–377. https://doi.org/10.1016/j.nbd.2010.06.010
Davis LC, Morgan AJ, Chen J-L, Snead CM, Bloor-Young D, Shenderov E, Stanton-Humphreys MN, Conway SJ, Churchill GC, Parrington J, Cerundolo V, Galione A (2012) NAADP activates two-pore channels on T cell cytolytic granules to stimulate exocytosis and killing. Curr Biol 22(24):2331–2337. https://doi.org/10.1016/j.cub.2012.10.035
Davis LC, Morgan AJ, Galione A (2020) NAADP-regulated two-pore channels drive phagocytosis through endo-lysosomal Ca2+ nanodomains, calcineurin and dynamin. EMBO J 39(14):e104058. https://doi.org/10.15252/embj.2019104058
de Lartigue J, Polson H, Feldman M, Shokat K, Tooze SA, Urbé S, Clague MJ (2009) PIKfyve regulation of endosome-linked pathways. Traffic Cph Den 10(7):883–893. https://doi.org/10.1111/j.1600-0854.2009.00915.x
Dickinson MS, Myasnikov A, Eriksen J, Poweleit N, Stroud RM (2020) Resting state structure of the hyperdepolarization activated two-pore channel 3. Proc Natl Acad Sci U S A 117(4):1988–1993. https://doi.org/10.1073/pnas.1915144117
Dickson EJ, Duman JG, Moody MW, Chen L, Hille B (2012) Orai-STIM-mediated Ca2+ release from secretory granules revealed by a targeted Ca2+ and pH probe. Proc Natl Acad Sci U S A 109(51):E3539–E3548. https://doi.org/10.1073/pnas.1218247109
Diercks B-P, Werner R, Weidemüller P, Czarniak F, Hernandez L, Lehmann C, Rosche A, Krüger A, Kaufmann U, Vaeth M, Failla AV, Zobiak B, Kandil FI, Schetelig D, Ruthenbeck A, Meier C, Lodygin D, Flügel A, Ren D, Wolf IMA, Feske S, Guse AH (2018) ORAI1, STIM1/2, and RYR1 shape subsecond Ca2+ microdomains upon T cell activation. Sci Signal 11(561):eaat0358. https://doi.org/10.1126/scisignal.aat0358
Dionisio N, Albarrán L, López JJ, Berna-Erro A, Salido GM, Bobe R, Rosado JA (2011) Acidic NAADP-releasable Ca(2+) compartments in the megakaryoblastic cell line MEG01. Biochim Biophys Acta 1813(8):1483–1494. https://doi.org/10.1016/j.bbamcr.2011.05.005. Epub 2011 May 13. PMID: 21601596
Dong X-P, Cheng X, Mills E, Delling M, Wang F, Kurz T, Xu H (2008) The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455(7215):992–996. https://doi.org/10.1038/nature07311
Dong X, Wang X, Shen D, Chen S, Liu M, Wang Y, Mills E, Cheng X, Delling M, Xu H (2009) Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J Biol Chem 284(46):32040–32052. https://doi.org/10.1074/jbc.M109.037184
Dong X, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H (2010) PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun 1(4):38. https://doi.org/10.1038/ncomms1037
Dröse S, Altendorf K (1997) Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J Exp Biol 200(Pt 1):1–8. https://doi.org/10.1242/jeb.200.1.1
Faris P, Pellavio G, Ferulli F, Di Nezza F, Shekha M, Lim D, Maestri M, Guerra G, Ambrosone L, Pedrazzoli P, Laforenza U, Montagna D, Moccia F (2019) Nicotinic acid adenine dinucleotide phosphate (NAADP) induces intracellular Ca2+ release through the two-pore channel TPC1 in metastatic colorectal cancer cells. Cancers 11(4):542. https://doi.org/10.3390/cancers11040542
Fois G, Hobi N, Felder E, Ziegler A, Miklavc P, Walther P, Radermacher P, Haller T, Dietl P (2015) A new role for an old drug: Ambroxol triggers lysosomal exocytosis via pH-dependent Ca2+ release from acidic Ca2+ stores. Cell Calcium 58(6):628–637. https://doi.org/10.1016/j.ceca.2015.10.002. Epub 2015 Oct 27. PMID: 26560688
Foster WJ, Taylor HBC, Padamsey Z, Jeans AF, Galione A, Emptage NJ (2018) Hippocampal mGluR1-dependent long-term potentiation requires NAADP-mediated acidic store Ca2+ signaling. Sci Signal 11(558):eaat9093. https://doi.org/10.1126/scisignal.aat9093
Galione A (2019) NAADP receptors. Cold Spring Harb Perspect Biol 11(11):a035071. https://doi.org/10.1101/cshperspect.a035071
Galione A, Evans AM, Ma J, Parrington J, Arredouani A, Cheng X, Zhu MX (2009) The acid test: the discovery of two-pore channels (TPCs) as NAADP-gated endolysosomal Ca(2+) release channels. Pflugers Arch 458(5):869–876. https://doi.org/10.1007/s00424-009-0682-y
Garrity AG, Wang W, Collier CM, Levey SA, Gao Q, Xu H (2016) The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife 5:e15887. https://doi.org/10.7554/eLife.15887
Genazzani AA, Galione A (1996) Nicotinic acid-adenine dinucleotide phosphate mobilizes Ca2+ from a thapsigargin-insensitive pool. Biochem J 315(Pt 3):721–725. https://doi.org/10.1042/bj3150721
Gerasimenko JV, Sherwood M, Tepikin AV, Petersen OH, Gerasimenko OV (2006) NAADP, cADPR and IP3 all release Ca2+ from the endoplasmic reticulum and an acidic store in the secretory granule area. J Cell Sci 119(Pt 2):226–238. https://doi.org/10.1242/jcs.02721
Gerndt S, Chen C-C, Chao Y-K, Yuan Y, Burgstaller S, Scotto Rosato A, Krogsaeter E, Urban N, Jacob K, Nguyen ONP, Miller MT, Keller M, Vollmar AM, Gudermann T, Zierler S, Schredelseker J, Schaefer M, Biel M, Malli R, Wahl-Schott C, Bracher F, Patel S, Grimm C (2020) Agonist-mediated switching of ion selectivity in TPC2 differentially promotes lysosomal function. Elife 9:e54712. https://doi.org/10.7554/eLife.54712
Grabe M, Oster G (2001) Regulation of organelle acidity. J Gen Physiol 117(4):329–344. https://doi.org/10.1085/jgp.117.4.329
Grimm C, Tang R (2020) Could an endo-lysosomal ion channel be the Achilles heel of SARS-CoV2? Cell Calcium 88:102212. https://doi.org/10.1016/j.ceca.2020.102212
Grimm C, Hassan S, Wahl-Schott C, Biel M (2012) Role of TRPML and two-pore channels in endolysosomal cation homeostasis. J Pharmacol Exp Ther 342(2):236–244. https://doi.org/10.1124/jpet.112.192880
Grimm C, Holdt LM, Chen C-C, Hassan S, Müller C, Jörs S, Cuny H, Kissing S, Schröder B, Butz E, Northoff B, Castonguay J, Luber CA, Moser M, Spahn S, Lüllmann-Rauch R, Fendel C, Klugbauer N, Griesbeck O, Haas A, Mann M, Bracher F, Teupser D, Saftig P, Biel M, Wahl-Schott C (2014) High susceptibility to fatty liver disease in two-pore channel 2-deficient mice. Nat Commun 5:4699. https://doi.org/10.1038/ncomms5699
Grimm C, Chen C-C, Wahl-Schott C, Biel M (2017) Two-pore channels: catalyzers of endolysosomal transport and function. Front Pharmacol 8
Grimm C, Bartel K, Vollmar AM, Biel M (2018) Endolysosomal cation channels and cancer – a link with great potential. Pharmaceuticals (Basel) 11(1):4. https://doi.org/10.3390/ph11010004
Gu F, Krüger A, Roggenkamp HG, Alpers R, Lodygin D, Jaquet V, Möckl F, Hernandez CLC, Winterberg K, Bauche A, Rosche A, Grasberger H, Kao JY, Schetelig D, Werner R, Schröder K, Carty M, Bowie AG, Huber S, Meier C, Mittrücker H-W, Heeren J, Krause K-H, Flügel A, Diercks B-P, Guse AH (2021) Dual NADPH oxidases DUOX1 and DUOX2 synthesize NAADP and are necessary for Ca2+ signaling during T cell activation. Sci Signal 14(709):eabe3800. https://doi.org/10.1126/scisignal.abe3800
Guo J, Zeng W, Jiang Y (2017) Tuning the ion selectivity of two-pore channels. Proc Natl Acad Sci U S A 114(5):1009–1014. https://doi.org/10.1073/pnas.1616191114
Haller T, Dietl P, Deetjen P, Völkl H (1996) The lysosomal compartment as intracellular calcium store in MDCK cells: a possible involvement in InsP3-mediated Ca2+ release. Cell Calcium 19(2):157–165. https://doi.org/10.1016/s0143-4160(96)90084-6
Hayashi T, Hosono K, Kubo A, Kurata K, Katagiri S, Mizobuchi K, Kurai M, Mamiya N, Kondo M, Tachibana T, Saitsu H, Ogata T, Nakano T, Hotta Y (2020) Long-term observation of a Japanese mucolipidosis IV patient with a novel homozygous p.F313del variant of MCOLN1. Am J Med Genet A 182(6):1500–1505. https://doi.org/10.1002/ajmg.a.61575
Henderson MJ, Baldwin HA, Werley CA, Boccardo S, Whitaker LR, Yan X, Holt GT, Schreiter ER, Looger LL, Cohen AE, Kim DS, Harvey BK (2015) A low affinity GCaMP3 variant (GCaMPer) for imaging the endoplasmic reticulum calcium store. PLoS One 10(10):e0139273. https://doi.org/10.1371/journal.pone.0139273
Hockey LN, Kilpatrick BS, Eden ER, Lin-Moshier Y, Brailoiu GC, Brailoiu E, Futter CE, Schapira AH, Marchant JS, Patel S (2015) Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J Cell Sci 128(2):232–238. https://doi.org/10.1242/jcs.164152
Höglinger D, Haberkant P, Aguilera-Romero A, Riezman H, Porter FD, Platt FM, Galione A, Schultz C (2015) Intracellular sphingosine releases calcium from lysosomes. Elife 4:e10616. https://doi.org/10.7554/eLife.10616
Hu W, Zhao F, Chen L, Ni J, Jiang Y (2021) NAADP-induced intracellular calcium ion is mediated by the TPCs (two-pore channels) in hypoxia-induced pulmonary arterial hypertension. J Cell Mol Med 25(15):7485–7499. https://doi.org/10.1111/jcmm.16783
Huotari J, Helenius A (2011) Endosome maturation. EMBO J 30(17):3481–3500. https://doi.org/10.1038/emboj.2011.286
Huss M, Vitavska O, Albertmelcher A, Bockelmann S, Nardmann C, Tabke K, Tiburcy F, Wieczorek H (2011) Vacuolar H(+)-ATPases: intra- and intermolecular interactions. Eur J Cell Biol 90(9):688–695. https://doi.org/10.1016/j.ejcb.2011.04.009. Epub 2011 Jun 2. PMID: 21640428
Inabayashi M, Miyauchi S, Kamo N, Jin T (1995) Conductance change in phospholipid bilayer membrane by an electroneutral ionophore, monensin. Biochemistry 34(10):3455–3460. https://doi.org/10.1021/bi00010a038
Ishibashi K, Suzuki M, Imai M (2000) Molecular cloning of a novel form (two-repeat) protein related to voltage-gated sodium and calcium channels. Biochem Biophys Res Commun 270(2):370–376. https://doi.org/10.1006/bbrc.2000.2435
Jadot M, Colmant C, Wattiaux-De Coninck S, Wattiaux R (1984) Intralysosomal hydrolysis of glycyl-L-phenylalanine 2-naphthylamide. Biochem J 219(3):965–970. https://doi.org/10.1042/bj2190965. PMID: 6743255; PMCID: PMC1153569
Jefferies HBJ, Cooke FT, Jat P, Boucheron C, Koizumi T, Hayakawa M, Kaizawa H, Ohishi T, Workman P, Waterfield MD, Parker PJ (2008) A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts endomembrane transport and retroviral budding. EMBO Rep 9(2):164–170. https://doi.org/10.1038/sj.embor.7401155
Jentsch TJ, Pusch M (2018) CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol Rev 98(3):1493–1590. https://doi.org/10.1152/physrev.00047.2017
Jha A, Ahuja M, Patel S, Brailoiu E, Muallem S (2014) Convergent regulation of the lysosomal two-pore channel-2 by Mg2+, NAADP, PI(3,5)P2 and multiple protein kinases. EMBO J 33(5):501–511. https://doi.org/10.1002/embj.201387035
Kelu JJ, Webb SE, Parrington J, Galione A, Miller AL (2017) Ca2+ release via two-pore channel type 2 (TPC2) is required for slow muscle cell myofibrillogenesis and myotomal patterning in intact zebrafish embryos. Dev Biol 425(2):109–129. https://doi.org/10.1016/j.ydbio.2017.03.031
Kilpatrick BS, Eden ER, Schapira AH, Futter CE, Patel S (2013) Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J Cell Sci 126:60–66
Kinnear NP, Boittin F-X, Thomas JM, Galione A, Evans AM (2004) Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J Biol Chem 279(52):54319–54326. https://doi.org/10.1074/jbc.M406132200
Lagostena L, Festa M, Pusch M, Carpaneto A (2017) The human two-pore channel 1 is modulated by cytosolic and luminal calcium. Sci Rep 7:43900. https://doi.org/10.1038/srep43900
LaPlante JM, Sun M, Falardeau J, Dai D, Brown EM, Slaugenhaupt SA, Vassilev PM (2006) Lysosomal exocytosis is impaired in mucolipidosis type IV. Mol Genet Metab 89(4):339–348. https://doi.org/10.1016/j.ymgme.2006.05.016
Lee HC, Aarhus R (1995) A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J Biol Chem 270(5):2152–2157. https://doi.org/10.1074/jbc.270.5.2152
Li S, Hao B, Lu Y, Yu P, Lee HC, Yue J (2012) Intracellular alkalinization induces cytosolic Ca2+ increases by inhibiting sarco/endoplasmic reticulum Ca2+-ATPase (SERCA). PLoS One 7(2):e31905. https://doi.org/10.1371/journal.pone.0031905. Epub 2012 Feb 27. PMID: 22384096; PMCID: PMC3288054
Lin WK, Bolton EL, Cortopassi WA, Wang Y, O’Brien F, Maciejewska M, Jacobson MP, Garnham C, Ruas M, Parrington J, Lei M, Sitsapesan R, Galione A, Terrar DA (2017) Synthesis of the Ca2+-mobilizing messengers NAADP and cADPR by intracellular CD38 enzyme in the mouse heart: Role in β-adrenoceptor signaling. J Biol Chem 292(32):13243–13257. https://doi.org/10.1074/jbc.M117.789347
Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM (2008) Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 14(11):1247–1255. https://doi.org/10.1038/nm.1876
Luzio JP, Pryor PR, Bright NA (2007) Lysosomes: fusion and function. Nat Rev Mol Cell Biol 8(8):622–632. https://doi.org/10.1038/nrm2217
Macgregor A, Yamasaki M, Rakovic S, Sanders L, Parkesh R, Churchill GC, Galione A, Terrar DA (2007) NAADP controls cross-talk between distinct Ca2+ stores in the heart. J Biol Chem 282(20):15302–15311. https://doi.org/10.1074/jbc.M611167200
Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S (2008) Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev 88(3):841–886. https://doi.org/10.1152/physrev.00035.2007
Mao T, O’Connor DH, Scheuss V, Nakai J, Svoboda K (2008) Characterization and subcellular targeting of GCaMP-type genetically-encoded calcium indicators. PLoS One 3(3):e1796. https://doi.org/10.1371/journal.pone.0001796
McCartney AJ, Zhang Y, Weisman LS (2014) Phosphatidylinositol 3,5-bisphosphate: low abundance, high significance. BioEssays News Rev Mol Cell Dev Biol 36(1):52–64. https://doi.org/10.1002/bies.201300012
McCue HV, Wardyn JD, Burgoyne RD, Haynes LP (2013) Generation and characterization of a lysosomally targeted, genetically encoded Ca(2+)-sensor. Biochem J 449(2):449–457. https://doi.org/10.1042/BJ20120898
Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, Palmieri M, Polishchuk R, Puertollano R, Ballabio A (2011) Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell 21(3):421–430. https://doi.org/10.1016/j.devcel.2011.07.016
Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17(3):288–299. https://doi.org/10.1038/ncb3114
Melchionda M, Pittman JK, Mayor R, Patel S (2016) Ca2+/H+ exchange by acidic organelles regulates cell migration in vivo. J Cell Biol 212(7):803–813. https://doi.org/10.1083/jcb.201510019. Epub 2016 Mar 21. PMID: 27002171; PMCID: PMC4810305
Mirabelli-Badenier M, Severino M, Tappino B, Tortora D, Camia F, Zanaboni C, Brera F, Priolo E, Rossi A, Biancheri R, Di Rocco M, Filocamo M (2015) A novel homozygous MCOLN1 double mutant allele leading to TRP channel domain ablation underlies mucolipidosis IV in an Italian child. Metab Brain Dis 30(3):681–686. https://doi.org/10.1007/s11011-014-9612-6
Mitchell KJ, Lai FA, Rutter GA (2003) Ryanodine receptor type I and nicotinic acid adenine dinucleotide phosphate receptors mediate Ca2+ release from insulin-containing vesicles in living pancreatic beta-cells (MIN6). J Biol Chem 278(13):11057–11064. https://doi.org/10.1074/jbc.M210257200
Moccia F, Zuccolo E, Di Nezza F, Pellavio G, Faris PS, Negri S, De Luca A, Laforenza U, Ambrosone L, Rosti V, Guerra G (2021) Nicotinic acid adenine dinucleotide phosphate activates two-pore channel TPC1 to mediate lysosomal Ca2+ release in endothelial colony-forming cells. J Cell Physiol 236(1):688–705. https://doi.org/10.1002/jcp.29896
Morgan AJ, Galione A (2007) NAADP induces pH changes in the lumen of acidic Ca2+ stores. Biochem J 402(2):301–310. https://doi.org/10.1042/BJ20060759. PMID: 17117921; PMCID: PMC1798430
Morgan AJ, Platt FM, Lloyd-Evans E, Galione A (2011) Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J 439(3):349–374. https://doi.org/10.1042/BJ20110949
Morgan AJ, Davis LC, Galione A (2015) Imaging approaches to measuring lysosomal calcium. Methods Cell Biol 126:159–195. https://doi.org/10.1016/bs.mcb.2014.10.031
Müller M, Gerndt S, Chao Y-K, Zisis T, Nguyen ONP, Gerwien A, Urban N, Müller C, Gegenfurtner FA, Geisslinger F, Ortler C, Chen C-C, Zahler S, Biel M, Schaefer M, Grimm C, Bracher F, Vollmar AM, Bartel K (2021) Gene editing and synthetically accessible inhibitors reveal role for TPC2 in HCC cell proliferation and tumor growth. Cell Chem Biol 28(8):1119–1131.e27. https://doi.org/10.1016/j.chembiol.2021.01.023
Narayanaswamy N, Chakraborty K, Saminathan A, Zeichner E, Leung K, Devany J, Krishnan Y (2019) A pH-correctable, DNA-based fluorescent reporter for organellar calcium. Nat Methods 16(1):95–102. https://doi.org/10.1038/s41592-018-0232-7
Ortega FG, Roefs MT, de Miguel PD, Kooijmans SA, de Jong OG, Sluijter JP, Schiffelers RM, Vader P (2019) Interfering with endolysosomal trafficking enhances release of bioactive exosomes. Nanomed Nanotechnol Biol Med 20:102014. https://doi.org/10.1016/j.nano.2019.102014
Palmer AE, Jin C, Reed JC, Tsien RY (2004) Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci U S A 101(50):17404–17409. https://doi.org/10.1073/pnas.0408030101
Patel S, Kilpatrick BS (2018) Two-pore channels and disease. Biochim Biophys Acta Mol Cell Res 1865(11 Pt B):1678–1686. https://doi.org/10.1016/j.bbamcr.2018.05.004
Penny CJ, Kilpatrick BS, Han JM, Sneyd J, Patel S (2014) A computational model of lysosome-ER Ca2+ microdomains. J Cell Sci 127(Pt 13):2934–2943. https://doi.org/10.1242/jcs.149047. Epub 2014 Apr 4. PMID: 24706947
Penny CJ, Kilpatrick BS, Eden ER, Patel S (2015) Coupling acidic organelles with the ER through Ca2+ microdomains at membrane contact sites. Cell Calcium 58(4):387–396. https://doi.org/10.1016/j.ceca.2015.03.006. Epub 2015 Mar 23. PMID: 25866010
Penny CJ, Vassileva K, Jha A, Yuan Y, Chee X, Yates E, Mazzon M, Kilpatrick BS, Muallem S, Marsh M, Rahman T, Patel S (2019) Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers. Biochim Biophys Acta Mol Cell Res 1866(7):1151–1161. https://doi.org/10.1016/j.bbamcr.2018.10.022
Petersen OH, Tepikin AV (2008) Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol 70:273–299. https://doi.org/10.1146/annurev.physiol.70.113006.100618
Pitt SJ, Funnell TM, Sitsapesan M, Venturi E, Rietdorf K, Ruas M, Ganesan A, Gosain R, Churchill GC, Zhu MX, Parrington J, Galione A, Sitsapesan R (2010) TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+. J Biol Chem 285(45):35039–35046. https://doi.org/10.1074/jbc.M110.156927
Rah S-Y, Mushtaq M, Nam T-S, Kim SH, Kim U-H (2010) Generation of cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate by CD38 for Ca2+ signaling in interleukin-8-treated lymphokine-activated killer cells. J Biol Chem 285(28):21877–21887. https://doi.org/10.1074/jbc.M109.066290
Ramos IB, Miranda K, Pace DA, Verbist KC, Lin F-Y, Zhang Y, Oldfield E, Machado EA, De Souza W, Docampo R (2010) Calcium- and polyphosphate-containing acidic granules of sea urchin eggs are similar to acidocalcisomes, but are not the targets for NAADP. Biochem J 429(3):485–495. https://doi.org/10.1042/BJ20091956
Rao NV, Rao GV, Hoidal JR (1997) Human dipeptidyl-peptidase I. Gene characterization, localization, and expression. J Biol Chem 272(15):10260–10265. https://doi.org/10.1074/jbc.272.15.10260. PMID: 9092576
Robb-Gaspers LD, Rutter GA, Burnett P, Hajnóczky G, Denton RM, Thomas AP (1998) Coupling between cytosolic and mitochondrial calcium oscillations: role in the regulation of hepatic metabolism. Biochim Biophys Acta 1366(1–2):17–32. https://doi.org/10.1016/s0005-2728(98)00118-2
Roggenkamp HG, Khansahib I, Hernandez CLC, Zhang Y, Lodygin D, Krüger A, Gu F, Möckl F, Löhndorf A, Wolters V, Woike D, Rosche A, Bauche A, Schetelig D, Werner R, Schlüter H, Failla AV, Meier C, Fliegert R, Walseth TF, Flügel A, Diercks B-P, Guse AH (2021) HN1L/JPT2: a signaling protein that connects NAADP generation to Ca2+ microdomain formation. Sci Signal 14(675):eabd5647. https://doi.org/10.1126/scisignal.abd5647
Rosato AS, Tang R, Grimm C (2021) Two-pore and TRPML cation channels: regulators of phagocytosis, autophagy and lysosomal exocytosis. Pharmacol Ther 220:107713. https://doi.org/10.1016/j.pharmthera.2020.107713
Ruas M, Davis LC, Chen C-C, Morgan AJ, Chuang K-T, Walseth TF, Grimm C, Garnham C, Powell T, Platt N, Platt FM, Biel M, Wahl-Schott C, Parrington J, Galione A (2015) Expression of Ca2+-permeable two-pore channels rescues NAADP signalling in TPC-deficient cells. EMBO J 34(13):1743–1758. https://doi.org/10.15252/embj.201490009
Rühl P, Rosato AS, Urban N, Gerndt S, Tang R, Abrahamian C, Leser C, Sheng J, Jha A, Vollmer G, Schaefer M, Bracher F, Grimm C (2021) Estradiol analogs attenuate autophagy, cell migration and invasion by direct and selective inhibition of TRPML1, independent of estrogen receptors. Sci Rep 11(1):8313. https://doi.org/10.1038/s41598-021-87817-4
Saijo H, Hayashi M, Ezoe T, Ohba C, Saitsu H, Kurata K, Matsumoto N (2016) The first genetically confirmed Japanese patient with mucolipidosis type IV. Clin Case Rep 4(5):509–512. https://doi.org/10.1002/ccr3.540
Sakurai Y, Kolokoltsov AA, Chen C-C, Tidwell MW, Bauta WE, Klugbauer N, Grimm C, Wahl-Schott C, Biel M, Davey RA (2015) Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347(6225):995–998. https://doi.org/10.1126/science.1258758
Samie M, Wang X, Zhang X, Goschka A, Li X, Cheng X, Gregg E, Azar M, Zhuo Y, Garrity AG, Gao Q, Slaugenhaupt S, Pickel J, Zolov SN, Weisman LS, Lenk GM, Titus S, Bryant-Genevier M, Southall N, Juan M, Ferrer M, Xu H (2013) A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell 26(5):511–524. https://doi.org/10.1016/j.devcel.2013.08.003
Schieder M, Rötzer K, Brüggemann A, Biel M, Wahl-Schott CA (2010) Characterization of two-pore channel 2 (TPCN2)-mediated Ca2+ currents in isolated lysosomes. J Biol Chem 285(28):21219–21222. https://doi.org/10.1074/jbc.C110.143123
Schmiege P, Fine M, Li X (2021) Atomic insights into ML-SI3 mediated human TRPML1 inhibition. Struct Lond Engl 29(11):1295–1302.e3. https://doi.org/10.1016/j.str.2021.06.003
She J, Guo J, Chen Q, Zeng W, Jiang Y, Bai X-C (2018) Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature 556(7699):130–134. https://doi.org/10.1038/nature26139
She J, Zeng W, Guo J, Chen Q, Bai X-C, Jiang Y (2019) Structural mechanisms of phospholipid activation of the human TPC2 channel. Elife 8:e45222. https://doi.org/10.7554/eLife.45222
Shen D, Wang X, Xu H (2011) Pairing phosphoinositides with calcium ions in endolysosomal dynamics: phosphoinositides control the direction and specificity of membrane trafficking by regulating the activity of calcium channels in the endolysosomes. BioEssays News Rev Mol Cell Dev Biol 33(6):448–457. https://doi.org/10.1002/bies.201000152
Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong X, Yu T, Lieberman AP, Showalter HD, Xu H (2012) Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 3:731. https://doi.org/10.1038/ncomms1735
Shiihara T, Watanabe M, Moriyama K, Maruyama Y, Kikuchi A, Arai-Ichinoi N, Uematsu M, Sameshima K (2016) Mucolipidosis IV: a milder form with novel mutations and serial MRI findings. Brain Dev 38(8):763–767. https://doi.org/10.1016/j.braindev.2016.02.009
Spector I, Shochet NR, Kashman Y, Groweiss A (1983) Latrunculins: novel marine toxins that disrupt microfilament organization in cultured cells. Science 219(4584):493–495. https://doi.org/10.1126/science.6681676
Spix B, Butz ES, Chen C-C, Rosato AS, Tang R, Jeridi A, Kudrina V, Plesch E, Wartenberg P, Arlt E, Briukhovetska D, Ansari M, Günsel GG, Conlon TM, Wyatt A, Wetzel S, Teupser D, Holdt LM, Ectors F, Boekhoff I, Boehm U, García-Añoveros J, Saftig P, Giera M, Kobold S, Schiller HB, Zierler S, Gudermann T, Wahl-Schott C, Bracher F, Yildirim AÖ, Biel M, Grimm C (2022) Lung emphysema and impaired macrophage elastase clearance in mucolipin 3 deficient mice. Nat Commun 13(1):318. https://doi.org/10.1038/s41467-021-27860-x
Srinivas SP, Ong A, Goon L, Goon L, Bonanno JA (2002) Lysosomal Ca(2+) stores in bovine corneal endothelium. Invest Ophthalmol Vis Sci 43(7):2341–2350
Sun M, Goldin E, Stahl S, Falardeau JL, Kennedy JC, Acierno JS, Bove C, Kaneski CR, Nagle J, Bromley MC, Colman M, Schiffmann R, Slaugenhaupt SA (2000) Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum Mol Genet 9(17):2471–2478. https://doi.org/10.1093/hmg/9.17.2471
Suzuki J, Kanemaru K, Iino M (2016) Genetically encoded fluorescent indicators for organellar calcium imaging. Biophys J 111(6):1119–1131. https://doi.org/10.1016/j.bpj.2016.04.054
Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP (1990) Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A 87(7):2466–2470. https://doi.org/10.1073/pnas.87.7.2466
Thompson EG, Schaheen L, Dang H, Fares H (2007) Lysosomal trafficking functions of mucolipin-1 in murine macrophages. BMC Cell Biol 8:54. https://doi.org/10.1186/1471-2121-8-54
Tsunemi T, Perez-Rosello T, Ishiguro Y, Yoroisaka A, Jeon S, Hamada K, Rammonhan M, Wong YC, Xie Z, Akamatsu W, Mazzulli JR, Surmeier DJ, Hattori N, Krainc D (2019) Increased lysosomal exocytosis induced by lysosomal Ca2+ channel agonists protects human dopaminergic neurons from α-synuclein toxicity. J Neurosci 39(29):5760–5772. https://doi.org/10.1523/JNEUROSCI.3085-18.2019
van Meel E, Klumperman J (2008) Imaging and imagination: understanding the endo-lysosomal system. Histochem Cell Biol 129(3):253–266. https://doi.org/10.1007/s00418-008-0384-0
Vasudevan SR, Lewis AM, Chan JW, Machin CL, Sinha D, Galione A, Churchill GC (2010) The calcium-mobilizing messenger nicotinic acid adenine dinucleotide phosphate participates in sperm activation by mediating the acrosome reaction. J Biol Chem 285(24):18262–18269. https://doi.org/10.1074/jbc.M109.087858
Vergarajauregui S, Connelly PS, Daniels MP, Puertollano R (2008) Autophagic dysfunction in mucolipidosis type IV patients. Hum Mol Genet 17(17):2723–2737. https://doi.org/10.1093/hmg/ddn174
Wang X, Zhang X, Dong X-P, Samie M, Li X, Cheng X, Goschka A, Shen D, Zhou Y, Harlow J, Zhu MX, Clapham DE, Ren D, Xu H (2012) TPC proteins are phosphoinositide-activated sodium-selective ion channels in endosomes and lysosomes. Cell 151(2):372–383. https://doi.org/10.1016/j.cell.2012.08.036
Wong C-O, Li R, Montell C, Venkatachalam K (2012) Drosophila TRPML is required for TORC1 activation. Curr Biol 22(17):1616–1621. https://doi.org/10.1016/j.cub.2012.06.055
Wu J, Prole DL, Shen Y, Lin Z, Gnanasekaran A, Liu Y, Chen L, Zhou H, Chen SRW, Usachev YM, Taylor CW, Campbell RE (2014) Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum. Biochem J 464(1):13–22. https://doi.org/10.1042/BJ20140931
Wu Y, Huang P, Dong X-P (2021) Lysosomal calcium channels in autophagy and cancer. Cancers 13(6):1299. https://doi.org/10.3390/cancers13061299
Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, Panayotou G (1996) Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol 16(4):1722–1733. https://doi.org/10.1128/MCB.16.4.1722
Xu M, Dong X-P (2021) Endolysosomal TRPMLs in cancer. Biomolecules 11(1):65. https://doi.org/10.3390/biom11010065
Xu H, Ren D (2015) Lysosomal physiology. Annu Rev Physiol 77:57–80. https://doi.org/10.1146/annurev-physiol-021014-071649
Yagodin S, Pivovarova NB, Andrews SB, Sattelle DB (1999) Functional characterization of thapsigargin and agonist-insensitive acidic Ca2+ stores in Drosophila melanogaster S2 cell lines. Cell Calcium 25(6):429–438. https://doi.org/10.1054/ceca.1999.0043
Yamasaki M, Masgrau R, Morgan AJ, Churchill GC, Patel S, Ashcroft SJH, Galione A (2004) Organelle selection determines agonist-specific Ca2+ signals in pancreatic acinar and beta cells. J Biol Chem 279(8):7234–7240. https://doi.org/10.1074/jbc.M311088200
Yang J, Zhao Z, Gu M, Feng X, Xu H (2019) Release and uptake mechanisms of vesicular Ca2+ stores. Protein Cell 10(1):8–19. https://doi.org/10.1007/s13238-018-0523-x
Yuan Y, Kilpatrick BS, Gerndt S, Bracher F, Grimm C, Schapira AH, Patel S (2021) The lysosomotrope GPN mobilises Ca2+ from acidic organelles. J Cell Sci 134(6):jcs256578. https://doi.org/10.1242/jcs.256578
Zhang F, Xu M, Han W-Q, Li P-L (2011) Reconstitution of lysosomal NAADP-TRP-ML1 signaling pathway and its function in TRP-ML1(−/−) cells. Am J Physiol Cell Physiol 301(2):C421–C430. https://doi.org/10.1152/ajpcell.00393.2010
Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, Gao Q, Yang M, Lawas M, Delling M, Marugan J, Ferrer M, Xu H (2016) MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun 7:12109. https://doi.org/10.1038/ncomms12109
Zhang J, Guan X, Shah K, Yan J (2021) Lsm12 is an NAADP receptor and a two-pore channel regulatory protein required for calcium mobilization from acidic organelles. Nat Commun 12(1):4739. https://doi.org/10.1038/s41467-021-24735-z
Zong X, Schieder M, Cuny H, Fenske S, Gruner C, Rötzer K, Griesbeck O, Harz H, Biel M, Wahl-Schott C (2009) The two-pore channel TPCN2 mediates NAADP-dependent Ca(2+)-release from lysosomal stores. Pflugers Arch 458(5):891–899. https://doi.org/10.1007/s00424-009-0690-y
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Wahl-Schott, C. et al. (2023). Characterization of Endo-Lysosomal Cation Channels Using Calcium Imaging. In: Wahl-Schott, C., Biel, M. (eds) Endolysosomal Voltage-Dependent Cation Channels. Handbook of Experimental Pharmacology, vol 278. Springer, Cham. https://doi.org/10.1007/164_2023_637
Download citation
DOI: https://doi.org/10.1007/164_2023_637
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-31522-0
Online ISBN: 978-3-031-31523-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)