
Abstract
The kappa opioid receptor (KOR) is a G protein-coupled receptor (GPCR) that can signal through multiple signaling pathways. KOR agonists are known to relieve pain and itch, as well as induce dysphoria, sedation, hallucinations, and diuresis. As is the case with many other GPCRs, specific signaling pathways downstream of the KOR have been linked to certain physiological responses induced by the receptor. Those studies motivated the search and discovery of a number of KOR ligands that preferentially activate one signaling pathway over another. Such compounds are termed functionally selective or biased ligands, and may present a way of inducing desired receptor effects with reduced adverse reactions. In this chapter, I review the molecular intricacies of KOR signaling and discuss the studies that have used biased signaling through the KOR as a way to selectively modulate in vivo physiology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Functional selectivity
- G protein-coupled receptor
- G proteins
- Kappa opioid receptor
- Ligand bias
- Pain
- βarrestin
1 Introduction: Biased Signaling and the Kappa Opioid Receptor
The kappa opioid receptor (KOR) is a G protein-coupled receptor (GPCR) which is the main target of the endogenous opioid peptides dynorphins (Chavkin and Goldstein 1981). GPCRs are seven-transmembrane domain proteins that can activate multiple signaling pathways (Hanson and Stevens 2009; Lefkowitz and Shenoy 2005). From a drug discovery perspective, GPCRs (also termed seven-transmembrane domain receptors – 7TMRs) are extremely relevant, given that over one third of all FDA-approved drugs directly target a member of the GPCR protein superfamily (Santos et al. 2017). Moreover, a number of other therapies target GPCRs indirectly. Reuptake inhibitors, for instance, indirectly target GPCRs by increasing the availability of endogenous ligands of the receptors (Price and Brust 2019).
As indicated by the nomenclature, GPCRs activate heterotrimeric G proteins, which are composed of Gα, Gβ, and Gγ subunits (Birnbaumer 2007; Wootten et al. 2018). Activation of G proteins happens once the GPCR is activated, which causes a conformational change in the receptor that is transmitted to the G proteins (Nygaard et al. 2013; Venkatakrishnan et al. 2013). This leads to the replacement of guanosine diphosphate (GDP) by guanosine triphosphate (GTP) in the Gα subunit (Goricanec et al. 2016). Subsequently, Gα and the Gβγ dimer interact with their cellular targets (Rankovic et al. 2016; Birnbaumer 2007). Following G protein activation, the receptor is phosphorylated by G protein-coupled receptor kinases (GRKs) and other kinases that are activated by downstream signaling (such as protein kinase C [PKC] and protein kinase A [PKA]), events that allow for the recruitment of arrestin to the receptor (Komolov and Benovic 2018; Wootten et al. 2018; Yang et al. 2017). Arrestin recruitment to the GPCR causes receptor internalization and can lead to receptor degradation, recycling, or additional signaling events (Peterson and Luttrell 2017; Rankovic et al. 2016).
GPCRs can activate a number of different signaling pathways. Even within the same class of immediate receptor interacting partners there is room for an enormous heterogenicity of signaling responses. For instance, there are sixteen Gα, five Gβ, and thirteen Gγ subunits, in addition to four arrestins (in this chapter we focus on βarrestin2), PDZ-domain-containing scaffolds and numerous A kinase anchoring proteins (AKAPs) that directly interact with GPCRs (Dupre et al. 2009; Khan et al. 2013; Wootten et al. 2018). Notably, decades ago inconsistencies between molecular efficacies and functional physiological responses for GPCR ligands were reported (Jim et al. 1985; Portoghese 1965). And even though these types of inconsistencies can sometimes be explained by pharmacokinetic factors, it was later hypothesized (and since shown a number of times) that it was possible to design GPCR ligands that would selectively activate certain pathways over others (Kenakin et al. 1991; Rankovic et al. 2016; Roth and Chuang 1987). Those studies formed the basis for what we now know as biased signaling or functional selectivity.
Ligands that upon interaction with a GPCR favor activity in one signaling pathway over another are termed biased or functionally selective (Rankovic et al. 2016; Kenakin 2011). Numerous studies have suggested that activation of certain signaling pathways downstream of a GPCR may be related to desired therapeutic effects, while activation of other signaling pathways may be related to undesired adverse effects (Kenakin 2019). The therapeutic potentials of biased ligands are based on the hypothesis that these compounds can favor GPCR signaling towards a desired pathway over an undesired pathway and, therefore, improve therapeutic efficacy and reduce adverse effects (Kenakin 2018). In fact, a number of studies have suggested that to be indeed the case (Grim et al. 2020; Schmid et al. 2017; DeWire et al. 2013; Brust et al. 2016; Allen et al. 2011; DeWire and Violin 2011; Violin et al. 2010, 2014; Kenakin 2018; Luttrell et al. 2015). However, we are yet to see the full benefits of these compounds reflected clinically.
An important step in the discovery of biased ligands is determining how to identify such compounds. Considerations on this range from selecting the appropriate system (cell line, tissue, etc.) to choosing a method to quantify (or qualify) bias. Several different methods of quantifying ligand bias have been developed, compared, and reviewed in detail (Brust et al. 2015b; Kenakin 2014; Kenakin and Christopoulos 2013a, b; Kenakin et al. 2012; Rajagopal et al. 2011; Rankovic et al. 2016; Stahl et al. 2015; Hoare et al. 2020; Gundry et al. 2017; Zhu et al. 2019). The most commonly used methods of quantifying ligand bias compare the ability of a ligand to activate one signaling pathway over another, to that of a reference compound and result in a bias factor (Kenakin and Christopoulos 2013b; Rankovic et al. 2016). Therefore, ligand bias is relative to the reference compound used for the comparison. Some of the most used methods of quantifying ligand bias use the Black and Leff operational model to calculate transduction coefficients (Black and Leff 1983; Kenakin and Christopoulos 2013b; Kenakin et al. 2012; Rankovic et al. 2016). The transduction coefficients calculate the intrinsic ligand efficacy and the dissociation constant of agonist-receptor-signal transducer complex from functional data to generate bias factors that are independent of signal amplification and receptor reserve (Black and Leff 1983; Kenakin and Christopoulos 2013b). Bias factors can be used in drug discovery efforts to compare the levels of bias of different test ligands. This approach also allows for incorporation of structure activity relationship studies and molecular modeling approaches to pursue potent biased ligands (Lovell et al. 2015; Manglik et al. 2016; McCorvy et al. 2018).
As discussed below, biased signaling at the KOR has been studied in cell models, primary neurons, and animals. The KOR has been pursued as a target in therapies for treating pain and pruritus (itch) (Cowan et al. 2015; Kivell and Prisinzano 2010). However, adverse effects such as dysphoria, sedation, and diuresis have generally precluded the clinical use of these compounds (Brust et al. 2016; Knoll and Carlezon 2010; Mercadante and Arcuri 2004; Pfeiffer et al. 1986). Nalfurafine is the only KOR-selective agonist that is currently in clinical use for the treatment of pruritus (Kumagai et al. 2010). Notably, compared to opioid analgesics that target the mu opioid receptor for pain relief, KOR agonists are generally devoid of addictive and life-threatening side effects, such as respiratory depression (Bruijnzeel 2009; Brust et al. 2016). In this chapter I review the signaling pathways downstream of the KOR and the available studies on biased signaling at this receptor.
2 Signaling Cascades Downstream of the Kappa Opioid Receptor
2.1 G Proteins
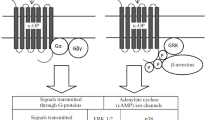
The KOR is coupled to inhibitory Gαi/o subunits (Fig. 1) (Meng et al. 1993; Simonin et al. 1995). The most studied effect of these subunits is the inhibition of adenylyl cyclases (Sunahara and Taussig 2002; Syrovatkina et al. 2016), which are enzymes that catalyze the conversion of adenosine triphosphate (ATP) into cyclic adenosine monophosphate (cAMP) (Price and Brust 2019). Therefore, KOR agonists generally lead to a reduction in cellular cAMP production. Even though specific consequences of KOR-induced decrease in cAMP levels have not yet been fully unveiled, certain adenylyl cyclase isoforms have functions that overlap with physiological roles that are attributed to the KOR. For instance, adenylyl cyclase 1 (AC1) has a role in pain perception and adenylyl cyclase 7 (AC7) appears to play a role in anxiety and depressive disorders (Brust et al. 2017; Price and Brust 2019).

Summary of the signaling pathways downstream of the KOR. Activation of the KOR leads to signaling events through heterotrimeric G proteins (G) and βarrestins (βARR). The figure depicts the pathways discussed in the chapter: G protein-mediated activation of GIRK, ERK, and JNK, and inhibition of adenylyl cyclases (AC), and βarrestin-mediated activation of ERK, JNK, and p38 MAPK, which inhibits JNK and increases membranous expression of the serotonin transporter (SERT). As noted in the figure, G protein activation downstream of the KOR has been linked to the receptor’s analgesic and antipruritic properties, while βarrestin recruitment to the KOR has been related to the sedative, dysphoric, and anhedonic properties of the receptor. Biased ligands are able to favor the activation of certain pathways over others
A better understanding of how the KOR modulates the activity of different adenylyl cyclase isoforms may enhance our predictions of desired functional responses downstream of the KOR. Especially as some adenylyl cyclase isoforms are not inhibited by Gαi/o and are conditionally activated by Gβγ subunits (AC2, AC4, and AC7) (Price and Brust 2019). KOR action on these isoforms would be expected to increase cAMP levels. Moreover, AC1, AC3, and AC8 are inhibited by Gβγ subunits, providing an additional layer of biased receptor modulation of tissue-specific cAMP signaling (Price and Brust 2019). The length of KOR activation is also crucial for determining the outcome of KOR modulation of cAMP production. Many studies have shown that through sensitization of adenylyl cyclase (also referred to as cAMP overshoot, heterologous sensitization, superactivation, or supersensitization of adenylyl cyclase), prolonged KOR activation followed by an antagonist can cause a robust increase in cAMP production, a phenomenon that adds a temporal aspect to KOR modulation of the cAMP pathway (Brust et al. 2015a; Nakagawa et al. 1999; Avidor-Reiss et al. 1995; Li et al. 2003; Wang et al. 2003). Therefore, KOR modulation of the cAMP signaling pathway is tissue and time-dependent, resulting in paradoxical inhibitory and excitatory responses.
The process of G protein activation also causes Gβγ subunits to become active. Gβγ subunits can lead to multiple signaling events, including modulation of adenylyl cyclase isoforms, activation of phosphoinositide 3 kinase, phosphorylation of extracellular signal-regulated kinase (ERK), activation of phospholipase C, activation of G protein-coupled inwardly rectifying potassium (GIRK) channels, modulation of calcium channels, and stabilization of microtubules (Dupre et al. 2009; Khan et al. 2013; Wootten et al. 2018). Activation of the KOR has classically been associated with Gβγ-mediated activation of GIRK and inhibition of voltage-gated calcium channels (Ho et al. 2018; Luscher and Slesinger 2010; Ikeda et al. 2002). These mechanisms would cause an overall inhibition of neurotransmission. We have also shown an enhancement of AC2 activity in cell lines that is consistent with the effects of Gβγ subunits (unpublished observations).
2.2 βarrestin2 Activation
Activation of the KOR leads to phosphorylation of the receptor by GRKs, an event that allows arrestin recruitment to the receptor (Bruchas and Chavkin 2010). Arrestins are proteins that were originally thought to serve only to terminate G protein signaling (Shenoy and Lefkowitz 2011). By interacting with the receptor, arrestins can first sterically prevent G proteins from coupling to the receptor (Gurevich and Gurevich 2019). Second, arrestins can internalize the receptor and remove it from the membrane, an event that can prevent additional agonist interactions with the GPCR (Gurevich and Gurevich 2019; Goodman et al. 1996). And third, arrestins can recruit phosphodiesterases and diacylglycerol kinases to the cellular membrane, proteins that act by degrading GPCR second messengers (Nelson et al. 2007; Perry et al. 2002). These actions would justify the name of arrestins and suggest an overall inhibitory role for arrestins on GPCR signaling.
It is now well understood that arrestins interact with a number of proteins that have important cellular signaling functions, such as components of the mitogen-activated protein kinase (MAPK) cascades, the Src family tyrosine kinases, and Akt (Chavkin et al. 2014; Luttrell et al. 1999; McLennan et al. 2008; Schmid and Bohn 2010). Arrestins appear to act as scaffolding proteins, bringing together several signaling proteins and facilitating their interactions (Gurevich and Gurevich 2019; Lefkowitz and Shenoy 2005). Therefore, in addition to “arresting” GPCR signaling, arrestins can also lead to GPCR-mediated signaling events. Downstream of the KOR, ERK phosphorylation can be induced through G proteins and βarrestin2 in immortalized astrocytes (McLennan et al. 2008). Sequestration of Gβγ subunits with βARKct (C terminus of GRK2), inhibition of G protein signaling with pertussis, and knockdown of βarrestin2 with siRNA, all reduce ERK phosphorylation (McLennan et al. 2008). Moreover, the proportion of G protein-dependent versus βarrestin2-dependent activation of ERK is ligand-dependent, indicating a role for signaling bias (McLennan et al. 2008). Another well-studied signaling pathway downstream of the KOR is the phosphorylation of p38 MAPK. Previous studies have shown that βarrestin2 recruitment to the receptor is required for KOR agonist-induced p38 MAPK phosphorylation (Schattauer et al. 2019; Bruchas et al. 2006). Inhibition of GRK3-mediated phosphorylation of the KOR or knockdown of βarrestin2 prevents p38 MAPK phosphorylation in neurons and astrocytes (Bruchas et al. 2006).
KOR agonists also cause c-Jun N-terminal kinase (JNK) phosphorylation (Jamshidi et al. 2016; Schattauer et al. 2019; Bruchas et al. 2007b). In cell lines, this process occurs in two distinct phases. Early JNK phosphorylation happens in an arrestin-independent fashion and involves G protein-mediated activation of PKC and the small G protein RAC (Schattauer et al. 2019). The late phase JNK phosphorylation happens through βarrestin2 and also involves the activation of RAC in addition to the RHO effector ROCK1 (Schattauer et al. 2019). Whereas the early phase JNK phosphorylation leads to the formation of reactive oxygen species (ROS) through peroxiredoxin 6 (PRDX6), the late phase does not induce significant ROS generation in cell lines (Schattauer et al. 2019). Notably, JNK is inhibited by p38 MAPK and inhibition of KOR-mediated phosphorylation of p38 MAPK causes an increase of KOR agonist-induced ROS generation (Schattauer et al. 2019).
3 Physiological Implications of Signaling Pathways Downstream of the Kappa Opioid Receptor
Studies with genetically modified animals and biased ligands at the KOR have helped to uncover the physiological functions of the different signaling pathways downstream of the receptor. Several studies have correlated the βarrestin2 pathway downstream of the KOR with the aversive and dysphoric properties (Bruchas et al. 2006, 2007a; Brust et al. 2016). This pathway involves the phosphorylation of the KOR by GRK3 followed by the recruitment of βarrestin2 and subsequent phosphorylation of p38 MAPK (Fig. 1). As a way of assessing aversive and dysphoric effects, many of these studies made use of animal models that are commonly used to mirror depressive and aversive states. For instance, repeated swim stress is a method that assesses stress-coping strategies and has been used for identifying compounds that can treat depression (Commons et al. 2017). In one study, repeated swim stress in mice was correlated with KOR phosphorylation and a KOR-dependent increase of p38 MAPK phosphorylation in GABAergic neurons of the nucleus accumbens of mice by immunohistochemistry and in the striatum by immunoblotting (Bruchas et al. 2007a). Notably, inhibition of p38 MAPK attenuates stress-induced immobility in mice, suggesting a reduction of depressive-like symptoms in the animals (Bruchas et al. 2007a).
The conditioned place aversion (CPA) is another model that is commonly employed to determine the aversive motivational properties of drugs (Cunningham et al. 2006). In this model the animal becomes conditioned to associate the experience elicited by the drug (or other stimulus) with the environment where the experience happened. As the animal is presented with a chance to choose between the drug- or vehicle-associated environment, it will avoid the environment where unpleasant experiences took place (Cunningham et al. 2006). KOR agonists cause aversion in this model and inhibition of p38 MAPK prevents KOR agonist-mediated CPA (Bruchas et al. 2007a). KOR-induced phosphorylation of p38 MAPK is dependent on the expression of GRK3 (Bruchas and Chavkin 2010). Therefore, it is not surprising that in GRK3 knockout mice, KOR agonist-induced CPA and swim stress-induced immobility were significantly decreased (Bruchas et al. 2007a). As KOR-induced phosphorylation of p38 MAPK is dependent on the recruitment of βarrestin2 to the receptor, these results indicate that KOR agonist-induced mouse behaviors that reflect aversion and dysphoria are mediated by the βarrestin2 pathway (Bruchas et al. 2006). However, in stark contrast with these data, KOR agonists still induce CPA in βarrestin2 knockout mice (White et al. 2015).
Dopamine neurons have long been associated with the rewarding and aversive properties of drugs (Volman et al. 2013). KOR activation causes a decrease in dopamine release in the nucleus accumbens, which receives dopaminergic input from the ventral tegmental area (VTA) (Crowley and Kash 2015; Karkhanis et al. 2016; Knoll and Carlezon 2010; Rose et al. 2015; Brust et al. 2016). This phenomenon has traditionally been linked to the aversive and dysphoric properties of the receptor (Knoll and Carlezon 2010). Notably, conditional deletion of the genes for the KOR or p38 MAPK in dopaminergic neurons prevents KOR agonist-induced CPA in mice (Ehrich et al. 2015). In addition, conditional expression of the KOR in VTA dopaminergic neurons of KOR knockout mice rescues KOR agonist-induced CPA, but expression of a mutant form of the KOR (KSA, which is not phosphorylated by GRK3 at serine 369) does not (Ehrich et al. 2015). Interestingly, while genetic or pharmacological inhibition/disruption of p38 MAPK prevents KOR agonist-induced CPA, it does not affect KOR agonist-induced reduction in dopamine release in the nucleus accumbens (Ehrich et al. 2015). These findings suggest that at least part of the aversive properties of KOR agonists may be independent of KOR-mediated inhibition of dopamine release.
The serotonergic system is also closely related with depressive symptoms and is commonly targeted in therapies to treat depressive disorders (Price and Brust 2019). Serotonin and its targets have also been implicated in the dysphoric and aversive effects that result from KOR activation (Schindler et al. 2012; Land et al. 2009). One study showed that conditional knockout of the KOR in serotonergic neurons blocks KOR agonist-induced CPA in mice (Ehrich et al. 2015). Another study showed that repeated forced swim stress increases membrane expression of the serotonin transporter (SERT) in a GRK3 and p38 MAPK-dependent manner in mice (Schindler et al. 2012). In addition, conditional expression of the KOR in serotonergic neurons of KOR knockout mice rescues KOR agonist-induced CPA (Land et al. 2009). Notably, the KOR antagonist norbinaltorphimine (norBNI) or KOR knockout prevents stress-induced increases in membranous SERT (Schindler et al. 2012). A number of antidepressant agents aim at increasing serotonin concentration at synaptic terminals, many of them act by inhibiting SERT (Price and Brust 2019). Therefore, increased membranous expression of SERT is consistent with depressive and dysphoric symptoms. Furthermore, KOR agonist-induced CPA is inhibited by the selective serotonin reuptake inhibitor (SSRI) citalopram and KOR agonist treatment increases serotonin uptake in whole brain samples from mice (Bruchas et al. 2011).
In contrast to the βarrestin2 pathway, the G protein pathway downstream of the KOR has been associated with antinociceptive and antipruritic properties (Fig. 1). Pertussis toxin causes ADP ribosylation of Gαi/o subunits, resulting in an inhibitory effect on G protein signaling (Mangmool and Kurose 2011). In the tail flick assay, which is used as a measure of thermal nociception, intracerebroventricular (ICV) pertussis toxin treatment completely inhibits KOR agonist-induced antinociception in mice (Goicoechea et al. 1999). In rats, intrathecal pertussis toxin treatment also abolishes KOR agonist-induced antinociception in the tail flick assay (Hernandez et al. 1995; Przewlocki et al. 1987). Moreover, KOR agonists retain their antinociceptive properties in βarrestin2 knockout mice, indicating that the βarrestin2 pathway is not necessary for KOR-induced pain relief (White et al. 2015). Notably, intrathecal injections with a cell-permeable cAMP analog (dibutyryl-cAMP) had no effects on KOR agonist-induced antinociception in the tail flick assay (Hernandez et al. 1995), suggesting that Gαi/o-mediated inhibition of the cAMP pathway is not involved in the antinociceptive properties of KOR agonists. This is consistent with studies showing that while inhibition of certain adenylyl cyclase isoforms relieves inflammatory and neuropathic pain, it has no effects on thermal nociception (Brust et al. 2017; Wang et al. 2011). KOR-mediated inhibition of AC1 has also been recently linked to a mechanism to mask postoperative latent pain sensitization in rodents (Custodio 2019).
G protein activation by the KOR has also been linked to the antipruritic properties. KOR agonists are antipruritic and KOR antagonists cause pruritus (Morgenweck et al. 2015). While βarrestin2 knockout in mice reduced slightly the pruritic effects of KOR antagonists, it did not affect the antipruritic effects of KOR agonists (Morgenweck et al. 2015). These data suggest that βarrestin2 recruitment to the KOR is not required for the antipruritic effects. In addition, the relative potencies of KOR agonists for activating G proteins ([35S]GTPγS binding assay) and for receptor internalization correlate with their antipruritic potencies (Wang et al. 2005). However, as discussed below, the use of biased KOR ligands indicates a G protein-mediated mechanism. KOR agonists also induce diuresis and hallucinations (Mercadante and Arcuri 2004; Pfeiffer et al. 1986). KOR-induced diuresis happens through the suppression of antidiuretic hormone (ADH or vasopressin) release from the posterior pituitary gland (Kapusta 1995). However, the precise molecular mechanisms underlying KOR-induced diuretic and psychotomimetic effects still remain to be fully detailed.
4 Biased Ligands at the Kappa Opioid Receptor
Several groups have reported biased KOR ligands. Here I will discuss the in vitro and in vivo data available for the compounds that displayed the largest bias factors (Fig. 2) (Mores et al. 2019; Gupta et al. 2016; Schmid et al. 2013; Rives et al. 2012; Zheng et al. 2017; Kivell et al. 2018; Zhou et al. 2013; White et al. 2014; Schattauer et al. 2017; Spetea et al. 2017; Bedini et al. 2020). Important factors to be considered when interpreting the results from those studies are the systems used to report bias, the methods used for quantifying bias, and the pharmacological effects of those compounds in vivo. The last of these factors may also be dependent on the pharmacokinetic properties and the route of administration.

Chemical structures of reference ligands U50,488 and U69,593 and the biased compounds discussed in the chapter
Triazole 1.1 (2-(4-(furan-2-ylmethyl)-5-((4-methyl-3-(trifluoromethyl)benzyl)thio)-4H-1,2,4-triazol-3-yl)pyridine) is a G protein-biased ligand at the KOR that retains potency and efficacy for activating G proteins in the [35S]GTPγS binding assay and is considerably less potent for recruiting βarrestin2 to the receptor in comparison with reference ligands (U50,488 and U69,593) in cell lines (Brust et al. 2016; Zhou et al. 2013). Upon ligand bias quantification, triazole 1.1 had a bias factor of 28 for [35S]GTPγS binding over βarrestin2 recruitment in comparison with U50,488 using the transduction coefficient method (Brust et al. 2016). This bias profile was maintained in transfected primary striatal neurons when comparing [35S]GTPγS binding to KOR internalization, with the apparent difference of triazole 1.1 being a partial agonist for KOR internalization in neurons versus the full efficacy for βarrestin2 recruitment in cell lines (Ho et al. 2018). Triazole 1.1 also presents a significant bias (factor equal to 23) for [35S]GTPγS binding over inhibition of cAMP accumulation in Chinese hamster ovary (CHO) cells (Ho et al. 2018). In addition, the compound displays a bias factor of 26 for GIRK activation over cAMP accumulation in cell lines. These data would suggest a bias among G proteins (Gβγ over Gα), a phenomenon that has been observed for other GPCRs (Brust et al. 2015c; Ho et al. 2018). However, this bias was absent in neuronal cells, further highlighting the importance of choosing an appropriate cell model in studies of functional selectivity (Ho et al. 2018). These results also demonstrate that ligands that appear biased in cell lines may not be biased endogenously.
In vivo, triazole 1.1 reaches the brain of rodents when administered subcutaneously or intraperitoneally (Brust et al. 2016). The compound also displays similar antinociceptive and antipruritic properties to U50,488 in the tail flick assay and to relieve chloroquine phosphate-induced scratching in mice, respectively (Brust et al. 2016). However, in contrast to U50,488, triazole 1.1 does not reduce locomotor activity or decrease dopamine release in the nucleus accumbens of mice. The compound also does not decrease intracranial self-stimulation (ICSS) in rats, a method that is used to measure anhedonia (Brust et al. 2016). In addition to compounds that induce anhedonia and dysphoric states, acute pain can also disrupt ICSS. Notably, triazole 1.1 is able to recover visceral pain-induced (through intraperitoneal lactic acid injection) decreases in ICSS. The effects of triazole 1.1 were also examined in rhesus monkeys (Huskinson et al. 2020). In that study, a series of behaviors were evaluated in response to the KOR agonists triazole 1.1, U50,488, salvinorin A, and nalfurafine. All compounds decreased scratching, however, in contrast to the other KOR agonists tested, triazole 1.1 did not decrease species-typical activity, increase passive visual (immobility), cause lip droop (muscle relaxation), or induce rest/sleep postures (Huskinson et al. 2020). It should be noted that all compounds were more potent for reducing scratching than for inducing any other behavior studied, indicating some level of separation of desired and undesired effects even for the reference ligands (Huskinson et al. 2020). Moreover, from the KOR agonists tested, triazole 1.1 was the least potent for reducing scratching (Huskinson et al. 2020). Together, these results indicate that at the doses tested, triazole 1.1 retains the antinociceptive and antipruritic properties of KOR agonists, but lacks the sedative and anhedonic characteristics that are commonly associated with these ligands.
RB-64 (22-thiocyanatosalvinorin A) is another G protein-biased ligand at the KOR (White et al. 2014, 2015). Compared to U69,593 and salvinorin A, RB-64 retains potency for G protein activation (luciferase-based cAMP assay) and is less potent for the recruitment of βarrestin2 in cell lines (White et al. 2014). RB-64 presents a bias factor of 35 for cAMP inhibition over βarrestin2 recruitment in comparison to salvinorin A using the transduction coefficient method (White et al. 2014). In mice, RB-64 is antinociceptive in the hotplate assay (used to measure thermal nociception), but the compound also induces CPA. Notably, RB-64 does not impair performance in the rotarod assay nor does it affect novelty-induced locomotor activity in mice. In ICSS studies in mice, RB-64 (1 mg/kg) also caused a small rightward shift in the rate-frequency curve, but did not diminish the maximal response rate compared to vehicle. These data also provide evidence that a G protein-biased ligand at the KOR is antinociceptive and does not induce sedation or as much anhedonia as reference compounds. However, the study also indicates that KOR ligands that are G protein-biased in cell lines still retain the aversive properties of the receptor, a finding that is consistent with the presence of KOR-induced CPA in βarrestin2 knockout mice (White et al. 2015). It is noteworthy that the method used to measure G protein activation for RB-64 was inhibition of cAMP accumulation, which is in contrast to [35S]GTPγS binding that was used for triazole 1.1 (White et al. 2014; Brust et al. 2017). The fact that triazole 1.1 was biased against inhibition of cAMP in cell lines indicates that RB-64 may engage the receptor in a different way and responses that are downstream of the immediate receptor effectors may differ between those two compounds (Ho et al. 2018).
Nalfurafine, the only selective KOR ligand that is clinically used, has also been reported to be biased. The compound is more potent for inducing ERK phosphorylation than for p38 MAPK phosphorylation in cell lines (Schattauer et al. 2017). Nalfurafine has a bias factor equal to 300 for ERK phosphorylation over p38 MAPK phosphorylation in comparison to U50,488 using the transduction coefficient method (Schattauer et al. 2017). Defining bias using downstream phosphorylation cascades is a valuable strategy that may lead to a more specific correlation with physiological effects. However, as ERK phosphorylation can be mediated both by G protein and arrestin pathways, this bias may not represent a G protein bias (Bruchas and Chavkin 2010; Lovell et al. 2015; McLennan et al. 2008). As the study timepoint for measuring ERK phosphorylation was 5 min, a Gβγ-mediated mechanism would be predicted. However, later studies found that nalfurafine is not biased for G protein ([35S]GTPγS binding) over βarrestin2 recruitment to the KOR in comparison to U50,488, or even that nalfurafine is biased for βarrestin2 recruitment over [35S]GTPγS binding in comparison to U69,593, both using the equiactive comparison (Liu et al. 2019; Dunn et al. 2019). This method of quantifying bias uses ratios of relative efficacies by potencies from standard four-parameter Hill equations to generate a bias factor, which is comparable to the transduction coefficients (Brust et al. 2015b; Ehlert 2008; Griffin et al. 2007). These discrepancies may be reconciled if nalfurafine induces bias among G proteins. In fact, nalfurafine has been shown to be biased for cAMP inhibition over βarrestin2 recruitment in comparison to U50,488 using the equiactive comparison (Kaski et al. 2019). In addition to the different methods used to measure G protein activity ([35S]GTPγS binding vs. cAMP inhibition), these studies also used different cell lines to assess KOR signaling. These cell lines likely present a distinct repertoire of G protein subunit and adenylyl cyclase isoforms, which should also have an impact on the calculated bias factors.
In vivo, nalfurafine is antipruritic and analgesic (formalin-induced inflammatory pain) and (at antipruritic and analgesic doses) does not induce CPA, decrease locomotor activity, or change the baseline threshold of ICSS in mice (Liu et al. 2019). In rhesus monkeys nalfurafine increases passive visual, causes lip droop, and induces rest/sleep postures at doses that are higher than those required for its antipruritic effects (Huskinson et al. 2020). Together, these studies show an interesting correlation of the compound’s bias against p38 MAPK phosphorylation and the lower potency for causing aversive effects in animals. It also shows how the activation of downstream signaling pathways may differ from the commonly assessed immediate receptor effectors. The different assay- and cell line-dependent bias factors are also noteworthy, and once more highlight the importance of choosing the appropriate model in studies of functional selectivity. It would be interesting to determine if the bias against p38 MAPK phosphorylation is a feature shared by other biased KOR ligands, as that may be a valuable strategy for screening for aversive properties of KOR ligands.
Additional G protein-biased KOR ligands, HS665 (3-(2-((Cyclobutylmethyl)(phenethyl)amino)ethyl)phenol) and HS666 (3-(2-((cyclopropylmethyl)(phenethyl)amino)ethyl)phenol) display bias factors equal to 389 and 62, respectively ([35S]GTPγS binding over βarrestin2 recruitment to the KOR in cell lines), in comparison to U69,593 using the transduction coefficients method (Spetea et al. 2017). These compounds have the distinction of being partial agonists. In cell lines, HS665 is a partial agonist for βarrestin2 recruitment and a full agonist for [35S]GTPγS binding and HS666 is a partial agonist in both assays. In mice, these compounds were administered ICV (thus circumventing certain potential pharmacokinetic issues) and caused antinociception in the tail flick assay, did not induce incoordination (although, data for a positive control was not shown for this assay), and HS665, but not HS666, induced CPA at the dose tested (Spetea et al. 2017). Another study tested HS665 and found that the compound increases serum prolactin levels (a common effect of KOR agonists) and that at 30 mg/kg it causes incoordination in mice (Dunn et al. 2018). As partial agonists also behave as partial antagonists, it would be interesting to determine if these compounds can inhibit KOR agonist-induced adverse effects in vivo (such as sedation, anhedonia, and CPA). Especially as a recent study that examined certain signaling pathways downstream of the KOR made a correlation of KOR ligand efficacy for βarrestin2 recruitment with the efficacy for causing incoordination (Dunn et al. 2019). In this context, KOR ligands that are partial agonists for βarrestin2 recruitment would be desirable, particularly those that retain full efficacy for G protein activation. However, as molecular efficacy is system-dependent, it would be interesting to determine if G protein over βarrestin2 bias factors correlate with improved therapeutic windows as previously reported for ligands of the mu opioid receptor (Schmid et al. 2017).
A recently discovered peptide-derived KOR agonist, LOR17 (c[Phe-Gly-(β-Ala)-D-Trp]) displays a bias factor equal to 853 for inhibition of cAMP over βarrestin2 recruitment in comparison to U50,488 in cell lines using the transduction coefficients method (Bedini et al. 2020). It should be noted that the efficacy of the doses of LOR17 tested (up to 10 μM) for βarrestin2 recruitment to the KOR was very low. Therefore, the extremely high bias factor calculated may not be accurate. Another recently developed method could be used to provide a more accurate bias factor for this compound (Stahl et al. 2019). Alternatively, higher compound doses could be used to provide more accurate measures of transduction ratio and functional affinity for the operational model. Nevertheless, the bias from LOR17 is evident from the concentration response curves (Bedini et al. 2020). The compound also displays differential kinase signaling. LOR17 induces early phase (15 min) ERK phosphorylation, contrasting with U50,488, which induces ERK phosphorylation in early and late phases in cell lines (15 and 60 min) (Bedini et al. 2020). Notably, an increase in ERK phosphorylation at an even earlier time point of 5 min was only observed for U50,488 in HEK293 cells (not in U87-MG cells or human astrocytes and not for LOR17 in any of the cells tested). In contrast to U50,488, LOR17 induces neither p38 MAPK phosphorylation nor astrocyte proliferation. Additionally, LOR17 inhibits both U50,488-induced p38 MAPK phosphorylation and cell proliferation (Bedini et al. 2020). In mice, LOR17 is antinociceptive in the tail flick assay, relieves visceral pain in the writhing assay, and is more effective than U50,488 for reducing thermal hypersensitivity caused by oxaliplatin-induced neuropathy. Contrasting to U50,488, LOR17 did not cause incoordination in the rotarod test or decrease exploratory activity in the hole-board test. The compound also did not reduce locomotor activity or diminish mobility time in the forced swim test (Bedini et al. 2020). As LOR17 inhibits U50,488-mediated p38 MAPK phosphorylation, it would also be of interest to determine if the compound can also reduce KOR agonist-mediated adverse effects, such as CPA, sedation, and anhedonia.
5 Conclusions
Overall, there is good agreement between the studies using genetic manipulations and the studies with biased ligands to suggest that G protein activation over βarrestin recruitment to the KOR may be beneficial for therapies to treat pain and pruritus. However, it is notable that βarrestin2 knockout mice still display CPA to KOR agonists (White et al. 2015). This is in contrast to other studies that showed that knockout of GSK3 (which allows for βarrestin2 recruitment to the KOR) and inhibition (genetic and pharmacological) of p38 MAPK phosphorylation (a signaling event that is downstream of βarrestin2 recruitment to the KOR) prevents KOR agonist-induced CPA (Bruchas et al. 2006, 2007a, 2010). Perhaps those studies could be reconciled if another arrestin (or other proteins) possesses a compensatory function for the lack of βarrestin2 in the βarrestin2 knockout mice. Nevertheless, the G protein-biased RB-64 still induces CPA in mice (White et al. 2015). In contrast, HS666, which is also G protein-biased does not induce CPA in mice (Spetea et al. 2017). However, HS666 has the notable distinction of being a partial agonist. It would be interesting to compare those two compounds (and others) side by side for pathway activation and behavioral tests, as subtle differences in methodological approaches may sometimes lead to different results. Testing additional doses of HS666 would also unveil the complete potential of that compound causing aversion. It should also be noted that certain studies used [35S]GTPγS binding while others used inhibition of cAMP accumulation as an endpoint for G protein activation. However, the two are not interchangeable and bias between the two pathways has been observed (Ho et al. 2018). Moreover, the pharmacokinetic profile of biased ligands should also be considered. As different compounds may have distinct absorption and distribution rates, it would be of interest to test their in vivo pharmacology at time points that reflect their peak plasma and brain concentrations. The different protocols that are used for in vivo studies may result in inappropriate time points for measuring behavior and, therefore, provide inaccurate results. The pharmacokinetic properties may also be affected by ligand binding kinetics. Different binding affinities and off rates are also likely to have an impact on the timing for behavior monitoring as ligands may remain bound to the receptor for different periods of time. This may also influence bias, as the kinetics of G protein and βarrestin recruitment (as well as other signaling pathways) are different (Hoare et al. 2020).
It is also desirable to find compounds that display the reverse type of bias downstream of the KOR (βarrestin2-biased ligands). Those compounds would help to confirm the role of G proteins and βarrestin2 for physiological responses of the receptor. In a similar way to the studies recently done with the mu opioid receptor, it would be interesting to determine if bias factors correlate with therapeutic windows for KOR ligands (Schmid et al. 2017). Future studies should also test the activation of pathways that are downstream of the KOR. Most studies focusing on ligand bias through the KOR were bidimensional and compared G protein activation (using the [35S]GTPγS binding or cAMP inhibition) to βarrestin2 recruitment. However, GPCR signaling is multidimensional and there are numerous signaling outcomes that may have physiological implications and may not be identified by G protein vs βarrestin2 bias comparisons. Recent studies have started to shift in that direction and present promising avenues for the discovery of new multidimensional bias profiles (Bedini et al. 2020; Ho et al. 2018; Liu et al. 2019). And as discussed above, it will also be important to consider the time variable for many of those signaling pathways.
While promising, it is advisable to be conservative with the expectations regarding the translational potential of these compounds, as this particular area of study is still in its early years. As an example, for other GPCRs contrasting hypotheses and results have been presented regarding the role of bias in improved therapeutic windows and the specific function of signaling proteins (Bohn et al. 1999, 2000; Raehal et al. 2005; Kliewer et al. 2019; Gillis et al. 2020). Nevertheless, the progress described in this chapter is remarkable. It shows how hypotheses generated from signaling studies can guide the pursuit of specific ligands, which present improved pre-clinical outcomes. It is also noteworthy that for the studies discussed, the bias observed in cell models appears to translate to a bias in in vivo pharmacological responses. The use of primary tissue or techniques that measure signaling in vivo is still very attractive and desired. Moreover, ligand binding kinetics and compound pharmacokinetic properties are also expected to play an important role in in vivo experimentation. The discovery of the pharmacological tools described in this chapter is certain to be informative and guide the field. Considering (and pursuing) ligand bias is becoming a norm in GPCR drug discovery and, hopefully, future studies will enlighten us on the clinical potential of these compounds.
References
Allen J et al (2011) Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci U S A 108:18488–18493
Avidor-Reiss T et al (1995) kappa-opioid receptor-transfected cell lines: modulation of adenylyl cyclase activity following acute and chronic opioid treatments. FEBS Lett 361:70–74
Bedini A et al (2020) Functional selectivity and antinociceptive effects of a novel KOPr agonist. Front Pharmacol 11:188
Birnbaumer L (2007) Expansion of signal transduction by G proteins. The second 15 years or so: from 3 to 16 alpha subunits plus betagamma dimers. Biochim Biophys Acta 1768:772–793
Black JW, Leff P (1983) Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220:141–162
Bohn LM et al (1999) Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286:2495–2498
Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG (2000) Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408:720–723
Bruchas MR, Chavkin C (2010) Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl) 210:137–147
Bruchas MR, Macey TA, Lowe JD, Chavkin C (2006) Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem 281:18081–18089
Bruchas MR et al (2007a) Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci 27:11614–11623
Bruchas MR et al (2007b) Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem 282:29803–29811
Bruchas MR, Land BB, Chavkin C (2010) The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res 1314:44–55
Bruchas MR et al (2011) Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron 71:498–511
Bruijnzeel AW (2009) kappa-opioid receptor signaling and brain reward function. Brain Res Rev 62:127–146
Brust TF, Conley JM, Watts VJ (2015a) Galphai/o-coupled receptor-mediated sensitization of adenylyl cyclase: 40 years later. Eur J Pharmacol 763:223–232
Brust TF, Hayes MP, Roman DL, Burris KD, Watts VJ (2015b) Bias analyses of preclinical and clinical d2 dopamine ligands: studies with immediate and complex signaling pathways. J Pharmacol Exp Ther 352:480–493
Brust TF, Hayes MP, Roman DL, Watts VJ (2015c) New functional activity of aripiprazole revealed: robust antagonism of D2 dopamine receptor-stimulated Gbetagamma signaling. Biochem Pharmacol 93:85–91
Brust TF et al (2016) Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci Signal 9:ra117
Brust TF et al (2017) Identification of a selective small-molecule inhibitor of type 1 adenylyl cyclase activity with analgesic properties. Sci Signal 10(467):eaah5381
Chavkin C, Goldstein A (1981) Specific receptor for the opioid peptide dynorphin: structure--activity relationships. Proc Natl Acad Sci U S A 78:6543–6547
Chavkin C, Schattauer SS, Levin JR (2014) Arrestin-mediated activation of p38 MAPK: molecular mechanisms and behavioral consequences. Handb Exp Pharmacol 219:281–292
Commons KG, Cholanians AB, Babb JA, Ehlinger DG (2017) the rodent forced swim test measures stress-coping strategy, not depression-like behavior. ACS Chem Nerosci 8:955–960
Cowan A, Kehner GB, Inan S (2015) Targeting itch with ligands selective for kappa opioid receptors. Handb Exp Pharmacol 226:291–314
Crowley NA, Kash TL (2015) Kappa opioid receptor signaling in the brain: circuitry and implications for treatment. Prog Neuropsychopharmacol Biol Psychiatry 62:51–60
Cunningham CL, Gremel CM, Groblewski PA (2006) Drug-induced conditioned place preference and aversion in mice. Nat Protoc 1:1662–1670
Custodio L (2019) Spinal kappa opioid receptor activity inhibits adenylyl cyclase-1 dependent mechanisms of chronic postoperative pain. Dissertation, University of Kentucky
Dewire S, Violin J (2011) Biased ligands for better cardiovascular drugs: dissecting G-protein-coupled receptor pharmacology. Circ Res 109:205–216
Dewire SM et al (2013) A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther 344:708–717
Dunn AD et al (2018) Structurally-related kappa opioid receptor agonists with substantial differential signaling bias: neuroendocrine and behavioral effects in C57BL6 mice. Int J Neuropsychopharmacol 21(9):847–857
Dunn AD, Reed B, Erazo J, Ben-Ezra A, Kreek MJ (2019) Signaling properties of structurally diverse kappa opioid receptor ligands: toward in vitro models of in vivo responses. ACS Chem Nerosci 10:3590–3600
Dupre DJ, Robitaille M, Rebois RV, Hebert TE (2009) The role of Gbetagamma subunits in the organization, assembly, and function of GPCR signaling complexes. Annu Rev Pharmacol Toxicol 49:31–56
Ehlert FJ (2008) On the analysis of ligand-directed signaling at G protein-coupled receptors. Naunyn Schmiedebergs Arch Pharmacol 377:549–577
Ehrich JM et al (2015) Kappa opioid receptor-induced aversion requires p38 MAPK activation in VTA dopamine neurons. J Neurosci 35:12917–12931
Gillis A et al (2020) Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci Signal 13(625):eaaz3140
Goicoechea C, Ormazabal MJ, Abalo R, Alfaro MJ, Martin MI (1999) Calcitonin reverts pertussis toxin blockade of the opioid analgesia in mice. Neurosci Lett 273:175–178
Goodman OB Jr et al (1996) Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 383:447–450
Goricanec D et al (2016) Conformational dynamics of a G-protein alpha subunit is tightly regulated by nucleotide binding. Proc Natl Acad Sci U S A 113:E3629–E3638
Griffin MT, Figueroa KW, Liller S, Ehlert FJ (2007) Estimation of agonist activity at G protein-coupled receptors: analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. J Pharmacol Exp Ther 321:1193–1207
Grim TW et al (2020) A G protein signaling-biased agonist at the mu-opioid receptor reverses morphine tolerance while preventing morphine withdrawal. Neuropsychopharmacology 45:416–425
Gundry J, Glenn R, Alagesan P, Rajagopal S (2017) A practical guide to approaching biased agonism at G protein coupled receptors. Front Neurosci 11:17
Gupta A et al (2016) Collybolide is a novel biased agonist of kappa-opioid receptors with potent antipruritic activity. Proc Natl Acad Sci U S A 113:6041–6046
Gurevich VV, Gurevich EV (2019) GPCR signaling regulation: the role of GRKs and arrestins. Front Pharmacol 10:125
Hanson MA, Stevens RC (2009) Discovery of new GPCR biology: one receptor structure at a time. Structure 17:8–14
Hernandez A et al (1995) Intrathecal pertussis toxin but not cyclic AMP blocks kappa opioid-induced antinociception in rat. Int J Neurosci 81:193–197
Ho JH et al (2018) G protein signaling-biased agonism at the kappa-opioid receptor is maintained in striatal neurons. Sci Signal 11(542):eaar4309
Hoare SRJ, Tewson PH, Quinn AM, Hughes TE (2020) A kinetic method for measuring agonist efficacy and ligand bias using high resolution biosensors and a kinetic data analysis framework. Sci Rep 10:1766
Huskinson SL et al (2020) Quantification of observable behaviors induced by typical and atypical kappa-opioid receptor agonists in male rhesus monkeys. Psychopharmacology (Berl) 237:2075–2087
Ikeda K et al (2002) Molecular mechanisms of analgesia induced by opioids and ethanol: is the GIRK channel one of the keys? Neurosci Res 44:121–131
Jamshidi RJ et al (2016) Long-term reduction of kappa opioid receptor function by the biased ligand, norbinaltorphimine, requires c-Jun N-terminal kinase activity and new protein synthesis in peripheral sensory neurons. J Pharmacol Exp Ther 359:319–328
Jim KF, Macia RA, Matthews WD (1985) An evaluation of the ability of a series of full alpha-1 adrenoceptor agonists to release internal calcium in venous smooth muscle. J Pharmacol Exp Ther 235:377–381
Kapusta DR (1995) Opioid mechanisms controlling renal function. Clin Exp Pharmacol Physiol 22:891–902
Karkhanis AN, Rose JH, Weiner JL, Jones SR (2016) Early-life social isolation stress increases kappa opioid receptor responsiveness and downregulates the dopamine system. Neuropsychopharmacology 41(9):2263–2274
Kaski SW et al (2019) Preclinical testing of nalfurafine as an opioid-sparing adjuvant that potentiates analgesia by the Mu opioid receptor-targeting agonist morphine. J Pharmacol Exp Ther 371:487–499
Kenakin T (2011) Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther 336:296–302
Kenakin T (2014) What is pharmacological ‘affinity’? Relevance to biased agonism and antagonism. Trends Pharmacol Sci 35(9):434–441
Kenakin T (2018) Is the quest for signaling bias worth the effort? Mol Pharmacol 93:266–269
Kenakin T (2019) Emergent concepts of receptor pharmacology. Handb Exp Pharmacol 260:17–41
Kenakin T, Christopoulos A (2013a) Measurements of ligand bias and functional affinity. Nat Rev Drug Discov 12:483
Kenakin T, Christopoulos A (2013b) Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12:205–216
Kenakin TP, Ambrose JR, Irving PE (1991) The relative efficiency of beta adrenoceptor coupling to myocardial inotropy and diastolic relaxation: organ-selective treatment for diastolic dysfunction. J Pharmacol Exp Ther 257:1189–1197
Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem Nerosci 3:193–203
Khan SM et al (2013) The expanding roles of Gbetagamma subunits in G protein-coupled receptor signaling and drug action. Pharmacol Rev 65:545–577
Kivell B, Prisinzano TE (2010) Kappa opioids and the modulation of pain. Psychopharmacology (Berl) 210:109–119
Kivell BM et al (2018) Kappa opioid receptor agonist Mesyl Sal B attenuates behavioral sensitization to cocaine with fewer aversive side-effects than salvinorin A in rodents. Molecules 23(10):2602
Kliewer A et al (2019) Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat Commun 10:367
Knoll AT, Carlezon WA Jr (2010) Dynorphin, stress, and depression. Brain Res 1314:56–73
Komolov KE, Benovic JL (2018) G protein-coupled receptor kinases: past, present and future. Cell Signal 41:17–24
Kumagai H et al (2010) Effect of a novel kappa-receptor agonist, nalfurafine hydrochloride, on severe itch in 337 haemodialysis patients: a Phase III, randomized, double-blind, placebo-controlled study. Nephrol Dial Transplant 25:1251–1257
Land BB et al (2009) Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A 106:19168–19173
Lefkowitz R, Shenoy S (2005) Transduction of receptor signals by beta-arrestins. Science 308:512–517
Li JG, Zhang F, Jin XL, Liu-Chen LY (2003) Differential regulation of the human kappa opioid receptor by agonists: etorphine and levorphanol reduced dynorphin A- and U50,488H-induced internalization and phosphorylation. J Pharmacol Exp Ther 305:531–540
Liu JJ et al (2019) Phosphoproteomic approach for agonist-specific signaling in mouse brains: mTOR pathway is involved in kappa opioid aversion. Neuropsychopharmacology 44:939–949
Lovell KM et al (2015) Structure-activity relationship studies of functionally selective kappa opioid receptor agonists that modulate ERK 1/2 phosphorylation while preserving G protein over betaArrestin2 signaling bias. ACS Chem Nerosci 6:1411–1419
Luscher C, Slesinger PA (2010) Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 11:301–315
Luttrell LM et al (1999) Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283:655–661
Luttrell LM, Maudsley S, Bohn LM (2015) Fulfilling the promise of “Biased” G protein-coupled receptor agonism. Mol Pharmacol 88:579–588
Manglik A et al (2016) Structure-based discovery of opioid analgesics with reduced side effects. Nature 537:185–190
Mangmool S, Kurose H (2011) G(i/o) protein-dependent and -independent actions of pertussis toxin (PTX). Toxins (Basel) 3:884–899
Mccorvy JD et al (2018) Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat Chem Biol 14:126–134
Mclennan GP et al (2008) Kappa opioids promote the proliferation of astrocytes via Gbetagamma and beta-arrestin 2-dependent MAPK-mediated pathways. J Neurochem 107:1753–1765
Meng F et al (1993) Cloning and pharmacological characterization of a rat kappa opioid receptor. Proc Natl Acad Sci U S A 90:9954–9958
Mercadante S, Arcuri E (2004) Opioids and renal function. J Pain 5:2–19
Mores KL, Cummins BR, Cassell RJ, Van Rijn RM (2019) A review of the therapeutic potential of recently developed G protein-biased kappa agonists. Front Pharmacol 10:407
Morgenweck J, Frankowski KJ, Prisinzano TE, Aube J, Bohn LM (2015) Investigation of the role of betaarrestin2 in kappa opioid receptor modulation in a mouse model of pruritus. Neuropharmacology 99:600–609
Nakagawa T, Ozawa T, Watanabe T, Minami M, Satoh M (1999) Sensitization of the adenylyl cyclase system in cloned kappa-opioid receptor-transfected cells following sustained agonist treatment: a chimeric study using G protein alpha(i)2/alpha(q) subunits. Jpn J Pharmacol 81:353–361
Nelson CD et al (2007) Targeting of diacylglycerol degradation to M1 muscarinic receptors by beta-arrestins. Science 315:663–666
Nygaard R et al (2013) The dynamic process of beta(2)-adrenergic receptor activation. Cell 152:532–542
Perry SJ et al (2002) Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science 298:834–836
Peterson YK, Luttrell LM (2017) The diverse roles of arrestin scaffolds in G protein-coupled receptor signaling. Pharmacol Rev 69:256–297
Pfeiffer A, Brantl V, Herz A, Emrich HM (1986) Psychotomimesis mediated by kappa opiate receptors. Science 233:774–776
Portoghese P (1965) A new concept on the mode of interaction of narcotic analgesics with receptors. J Med Chem 8:609–616
Price T, Brust TF (2019) Adenylyl cyclase 7 and neuropsychiatric disorders: a new target for depression? Pharmacol Res 143:106–112
Przewlocki R, Costa T, Lang J, Herz A (1987) Pertussis toxin abolishes the antinociception mediated by opioid receptors in rat spinal cord. Eur J Pharmacol 144:91–95
Raehal KM, Walker JK, Bohn LM (2005) Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacol Exp Ther 314:1195–1201
Rajagopal S et al (2011) Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol 80:367–377
Rankovic Z, Brust TF, Bohn LM (2016) Biased agonism: an emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett 26:241–250
Rives ML, Rossillo M, Liu-Chen LY, Javitch JA (2012) 6′-Guanidinonaltrindole (6′-GNTI) is a G protein-biased kappa-opioid receptor agonist that inhibits arrestin recruitment. J Biol Chem 287:27050–27054
Rose JH et al (2015) Supersensitive kappa opioid receptors promotes ethanol withdrawal-related behaviors and reduce dopamine signaling in the nucleus accumbens. Int J Neuropsychopharmacol 19(5):pyv127
Roth B, Chuang D (1987) Multiple mechanisms of serotonergic signal transduction. Life Sci 41:1051–1064
Santos R et al (2017) A comprehensive map of molecular drug targets. Nat Rev Drug Discov 16:19–34
Schattauer SS, Kuhar JR, Song A, Chavkin C (2017) Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell Signal 32:59–65
Schattauer SS et al (2019) Reactive oxygen species (ROS) generation is stimulated by kappa opioid receptor activation through phosphorylated c-Jun N-terminal kinase and inhibited by p38 mitogen-activated protein kinase (MAPK) activation. J Biol Chem 294:16884–16896
Schindler AG et al (2012) Stress produces aversion and potentiates cocaine reward by releasing endogenous dynorphins in the ventral striatum to locally stimulate serotonin reuptake. J Neurosci 32:17582–17596
Schmid CL, Bohn LM (2010) Serotonin, but not N-methyltryptamines, activates the serotonin 2A receptor via a ss-arrestin2/Src/Akt signaling complex in vivo. J Neurosci 30:13513–13524
Schmid CL et al (2013) Functional selectivity of 6′-guanidinonaltrindole (6′-GNTI) at kappa-opioid receptors in striatal neurons. J Biol Chem 288:22387–22398
Schmid CL et al (2017) Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 171:1165–1175.e13
Shenoy SK, Lefkowitz RJ (2011) beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci 32:521–533
Simonin F et al (1995) kappa-Opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc Natl Acad Sci U S A 92:7006–7010
Spetea M et al (2017) Selective kappa receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice. Br J Pharmacol 174:2444–2456
Stahl EL, Zhou L, Ehlert FJ, Bohn LM (2015) A novel method for analyzing extremely biased agonism at G protein-coupled receptors. Mol Pharmacol 87:866–877
Stahl EL, Ehlert FJ, Bohn LM (2019) Quantitating ligand bias using the competitive model of ligand activity. Methods Mol Biol 1957:235–247
Sunahara RK, Taussig R (2002) Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv 2:168–184
Syrovatkina V, Alegre KO, Dey R, Huang XY (2016) Regulation, signaling, and physiological functions of G-proteins. J Mol Biol 428:3850–3868
Venkatakrishnan AJ et al (2013) Molecular signatures of G-protein-coupled receptors. Nature 494:185–194
Violin JD et al (2010) Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther 335:572–579
Violin JD, Crombie AL, Soergel DG, Lark MW (2014) Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci 35:308–316
Volman SF et al (2013) New insights into the specificity and plasticity of reward and aversion encoding in the mesolimbic system. J Neurosci 33:17569–17576
Wang Y, Li JG, Huang P, Xu W, Liu-Chen LY (2003) Differential effects of agonists on adenylyl cyclase superactivation mediated by the kappa opioid receptors: adenylyl cyclase superactivation is independent of agonist-induced phosphorylation, desensitization, internalization, and down-regulation. J Pharmacol Exp Ther 307:1127–1134
Wang Y et al (2005) Comparison of pharmacological activities of three distinct kappa ligands (Salvinorin A, TRK-820 and 3FLB) on kappa opioid receptors in vitro and their antipruritic and antinociceptive activities in vivo. J Pharmacol Exp Ther 312:220–230
Wang H et al (2011) Identification of an adenylyl cyclase inhibitor for treating neuropathic and inflammatory pain. Sci Transl Med 3(65):65ra3
White KL et al (2014) Identification of novel functionally selective kappa-opioid receptor scaffolds. Mol Pharmacol 85:83–90
White KL et al (2015) The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther 352:98–109
Wootten D, Christopoulos A, Marti-Solano M, Babu MM, Sexton PM (2018) Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol 19:638–653
Yang Z et al (2017) Phosphorylation of G protein-coupled receptors: from the barcode hypothesis to the flute model. Mol Pharmacol 92:201–210
Zheng Z et al (2017) Structure-based discovery of new antagonist and biased agonist chemotypes for the kappa opioid receptor. J Med Chem 60:3070–3081
Zhou L et al (2013) Development of functionally selective, small molecule agonists at kappa opioid receptors. J Biol Chem 288:36703–36716
Zhu X, Finlay DB, Glass M, Duffull SB (2019) An intact model for quantifying functional selectivity. Sci Rep 9:2557
Acknowledgements
The author would like to acknowledge Ms. Isabelle Verona Brust for preparing the figure in this chapter. This work was supported by the Lloyd L. Gregory School of Pharmacy and Palm Beach Atlantic University.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Brust, T.F. (2020). Biased Ligands at the Kappa Opioid Receptor: Fine-Tuning Receptor Pharmacology. In: Liu-Chen, LY., Inan, S. (eds) The Kappa Opioid Receptor. Handbook of Experimental Pharmacology, vol 271. Springer, Cham. https://doi.org/10.1007/164_2020_395
Download citation
DOI: https://doi.org/10.1007/164_2020_395
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-89073-5
Online ISBN: 978-3-030-89074-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)