
Abstract
Thermo- and pH-responsive microgels were prepared from solubilized native elastin by crosslinking of the elastin lysine residues with poly(ethylene glycol) diglycidyl ether (PEG-DGE) and with bis(sulfosuccinimidyl) suberate (BS3). In the first case, a peptide-PEG conetwork was obtained whereas, in the second case, the elastin peptides were interlinked with hydrophobic bridges. The structure of the microgels was controlled by the ratio of crosslinker to elastin and by performing the crosslinking reaction in an inverse minielemulsion, yielding particles with a diameter in the submicron range. Depending on the degree of crosslinking, the hybrid microgels exhibited a volume change transition at 37 and 35.5°C and pH responsivity in the range of 5–7 for microgels crosslinked with PEG-DE and BS3, respectively. This temperature- and pH-responsive behavior can be assigned to the well-investigated coacervation of elastin peptides, demonstrating that the elastin functionality is abolished only by rather dense crosslinking. In spite of the broad distribution in the molecular weight of the elastin molecules, the microgels remained soluble. Light scattering and sedimentation experiments demonstrated that the coacervation occurred predominantly intramolecularly, i.e., by collapse in the core while the corona stabilized the colloidal dispersion against precipitation. Preliminary experiments were conducted to evaluate the suitability of these microgels for use as a drug-release system and demonstrated cytocompatibility, enzymatic degradability by elastase, and entrapping and slow release of a water-soluble biopolymer (Texas Red-labeled dextran with M w = 70,000). In summary, we present an easy entry to functional biohybrid microgels, where the responsiveness to temperature and pH can be exploited further for application of the microgel as a drug carrier.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Covalently crosslinked hydrogel particles of size in the nanometer to micrometer range (i.e., microgels), have found increasing interest for controlled and targeted drug delivery [1–3]. These particles are soft colloids that are swollen by the aqueous medium in which they are dispersed. Typically, they have a gradient in the distribution of their crosslinks, such that the corona contains more dangling ends and the crosslinks are more densely distributed in the core. This gradient not only results in a gradient of the swelling but also in a gradient of the solubility/solubilization, mostly due to endgroup effects [4]. Thus, if the solvent quality is decreased, the hydrogel particles tend to collapse first in the core and maintain a solubilizing corona. In this aspect they can be regarded as a primitive analogue to proteins that can adopt a globular structure with the hydrophilic segments directed towards the outside.
In the medical application of micro- and nanoparticles for the therapeutic delivery of an active ingredient important aspects are:
-
Premature removal from the circulating blood must be avoided before the target is reached
-
Active ingredients must not be leached out
-
The particles must be taken up in the organ to be treated
-
The particle itself must be degraded afterwards so that the polymer does not remain in the body to exert unwanted long term effects.
Here, hydrophilic micro- and nanogels can provide certain advantages. Protein adsorption or opsonization makes nanoparticles recognizable for phagocytic cells, but is strongly reduced for hydrogels due to their hydrophilic nature and high water content [1, 5]. Active ingredients may be linked covalently or entrapped in the network and released by degradation. Targeting is promoted by the high mobility of the soft gel particles in the smallest capillaries. As for other nanoparticles, targeting can be effected by the EPR effect (enhanced permeability and retention) observed for tumor tissue [6] and by decoration with specific ligands, (e.g., peptides and glucosides). In contrast to solid nanoparticles, hydrophilic nanogels can be designed to respond to changes in temperature and pH by a change in the degree of swelling. Furthermore and due to their open structure, degradability can be designed to be triggered rather specifically and instantaneously by changes in the environment. The latter may be achieved by biohybrid nanogels or nano-conetworks, where the biological constituent ensures a specific responsiveness and degradability. Beyond the scope of this work, modern tools to design biopolymers open further perspectives to introduce specific and predictable bioactive functions, such as the ability to present receptor-binding ligands, cell-triggered proteolytic degradation, and natural remodeling.
Here we report on the synthesis and properties of micro-conetworks from solubilized elastin derivatives using poly(ethylenglycol) (PEG) prepolymers or bis(sulfosuccinimidyl)suberate (BS3) as crosslinker for the elastin. We demonstrate that such micro-conetworks can be prepared in an easy procedure from soluble elastin peptides obtained by partial hydrolysis of native elastin. Because of the biodegradability of the elastin peptide, such hydrogels are fully biodegradable. The molecular weight of the remaining PEG chains is chosen sufficiently low so that these can be removed from the body via the renal system. In addition to the biocompatibility and biodegradability, the elastin component provides thermal and pH-dependent transformation at 35.5 or 37°C and pH 5–7, depending on the crosslinker used, from a swollen hydrogel to a globular state. Potentially, the responsiveness of the elastin component to pH and temperature can be exploited to trigger endosomal uptake in cells and release of drugs bound to the network. Based on these findings, it appeared attractive to us to prepare hydrophilic micro-conetworks of soluble elastin, as prepared by standard methods [7]. We expected that such hydrogel colloids can be prepared even from solubilized elastin samples that undergo coacervation like the well-defined elastin-like peptides (ELPs) [8–11], and that such particles can serve as a carrier for drugs where loading and release as well as internalization by cells can be affected by the volume change phase transition.
Elastin is the primary structural component of elastic tissues, e.g., it accounts for about 50 wt% of the total vascular extracellular matrix [12, 13]. Primarily it is formed from tropoelastin molecules as a precursor that assemble to microfibrils and are covalently interlinked by enzymatic oxidation of four lysine residues to desomosine units. The primary structure comprises hydrophilic segments with the lysine units and repeating hydrophobic polypenta- and polyhexapeptide sequences [14] that act as an entropic spring [15, 16]. Furthermore, temperature- and pH-dependent coacervation has been demonstrated for tropoleastin and ELPs [17, 18]. This coacervation effect is thought to be related to the self-assembly mechanism for the formation of the microfibrils [12, 19]. The controlled coacervation of ELPs has been exploited for novel and intriguing drug delivery concepts, e.g., injectable depots of an ELP fusion protein [10]. Another example is ELP block copolymers fused to a protein, which assemble into thermoresponsive micelles [11]. This concept has been extended to enable specific uptake into tumor cells by temperature-controlled assembly only in the tumor, as is possible in hyperthermia therapy. ELP block copolymers were conjugated to pentaarginine blocks that were presented at the surface of the micelles and triggered cell penetration only when they were assembled by the micelle formation. The experiments indicated that endocytosis plays an important role in internalization [20].
2 Experimental Details
2.1 Materials
Soluble elastin ES12 was purchased from Elastin Company (USA) as a salt-free lyophilized powder. Elastin Company indicates a molecular weight of 60,000 Da, but that the sample contains lower molecular weight peptides and smears on PAGE. Coacervates are formed at pH 5 and 37°C. Samples were dissolved in borate buffer (pH 9). Poly(ethylene glycol) diglycidyl ether (PEG-DGE) (M W = 526 g/mol) and bis(sulfosuccinimidyl) suberate (BS3) (M W = 572.43 g/mol) were purchased from Sigma Aldrich (Germany) and Piercenet Biotechnology Inc (USA) respectively. N-Hexane (99%, VWR), Span 80 (Sigma), Tween 80 (Sigma), tetrahydrofuran (99% Sigma Aldrich), Texas Red dextran (M W = 70 KDa, Leunonostoc bacteria, Invitrogen) were used as received. Dialysis membranes (molecular weight cut-off 3.5, 25, and 100 kDa) were purchased from Spectrum Laboratories.
2.2 Elastin-PEG DGE and Elastin-BS3 Microgels
Microgels were prepared by the inverse miniemulsion method with molar ratios of elastin to crosslinker of 1:2, 1:1.5, 1:1 1:0.5, and 1:0.25. By agitation for 10 min, 25 mg (4.166 × 10−4 mmol) of elastin was dissolved in 125 μL of 0.04 M PBS buffer (pH 9) and dispersed in 1.25 mL n-hexane containing 37.5 mg surfactant (3:1 weight ratio of Span 80 and Tween 80). Subsequently, the dispersion was ultrasonicated under ice cooling for 60 s using a Branson sonifier W450 with a ¼״ horn at a duty cycle of 30% and output control of 90%. Crosslinking was effected by addition of PEG-DGE or BS3 followed by a further sonication for another 60 s. The dispersion was further agitated for 45 min at room temperature before the reaction was quenched by addition of 1.5 mL of acidic water (pH 3). Separation of the microgels was achieved by centrifugation at 10,000 rpm for 30 min with subsequent decantation of the supernatant. Microgels present in the aqueous layer were carefully washed with hexane (2 × 1.5 mL) and THF (4 × 2.5 mL) in order to remove the surfactants and unreacted elastin. The remaining organic solvents and acid were removed by dialysis. Purified microgels were stored in Millipore water at 4°C for further use.
2.3 XTT Cytotoxicity Test
Cytocompatibility of the BS3-elastin microgels with molar ratio of 1:0.5 was investigated by XTT-based cell proliferation assay (Roche XTT Cell Proliferation Kit II, catalog no. 1465015) according to the manufacturer’s guidelines. The gels were prepared on the bottom of 96-well polystyrene plates. Human umbilical vein endothelial cells (HUVECs) (10,000 per well) and 500 μL of 10% fetal bovine serum medium were added to the wells (1.77 cm2), and the mixtures were incubated in 5% CO2 at 37°C for 96 h. To halt the experiment, aliquots of XTT stock solution (5 mg/mL) were added to each experimental well group at a ratio of 50/100 μL/μL of medium. Experimental plates were incubated for 4 h, and samples (100 μL) from each well were transferred to 96-well plates and quantified using a TECAN reader at a λ of 490 nm and a reference λ of 655 nm.
2.4 Sedimentation Analysis
Sedimentation of microgels was investigated with a LUMiFuge 114 separation analyzer (L.U.M. GmbH, Germany). Measurements were performed in glass tubes at acceleration velocities of 500–3,000 rpm at 20°C. The slope of the sedimentation curves was used to calculate the sedimentation velocity, indicating the colloidal stability of the samples.
2.5 Field Emission Scanning Electron Microscopy
Field emission scanning electron microscopy (FESEM) analysis was performed with a HITACHI S-4800 instrument in a cryo-mode with secondary electron image resolution of 1.0–1.4 nm at voltages of 1–15 kV. For all measurements, aqueous solutions of nanogels with a concentration of 5 mg/mL were rapidly vitrified in liquid propane and transferred to the high vacuum Balzers BF freeze-etching chamber. Fracture surfaces were prepared by means of a lever and etched by sublimation of the vitrified water for 5–15 min.
2.6 UV–Visible Spectrophotometer
UV–visible spectra were determined using a Varian Cary 100 Bio-UV-Visible split-beam spectrophotometer running with Cary WinUV scan application with a capacity of measuring six samples at a time. Samples were scanned at 500 nm. A high-intensity Xe flash lamp was used as the source for UV light, which permits taking 80 data per second.
2.7 Zeta Potential Measurements
Measurements were performed at 20°C as a function of pH in the range of 3–10 by addition of 0.01 M HCl or 0.01 M NaOH using a Malvern Zetasizer Nano ZS. Microgel solutions with a concentration of 5 mg/mL were dialyzed in standard 1 mM KCl solution and measured in disposable polystyrene cuvettes. One hundred scans were made for each sample and the zeta potential was calculated using Henry’s equation. Expert System software was used for data interpretation.
2.8 Dynamic Light Scattering
Microgel solutions of about 1 mg/mL in double-distilled water were passed through a 5-μm poly(tetrafluoroethylene) membrane filter. The particle sizes were measured by photon correlation spectroscopy performed at an angle θ = 90°with a setup consisting of an ALV-SP8 goniometer, an ALV-SIPC photomultiplier, a multiple τ digital real-time ALV-7004 correlator, and a solid state laser (Koheras) with a wavelength of 473 nm as the light source. Sample cuvettes were immersed in a toluene bath and thermostatted within an error of ±0.1°C. Autocorrelation functions (ACF) of intensity fluctuations g 2 (q, t) in the self-beating mode were measured and expressed by the Siegert relation:
where t is the decay time, A is a measured baseline, β is the coherence factor, and g 1(q,t) is the normalized first-order electric field time correlation function related to the measured relaxation rate Γ according to Eq. (2):
Deconvolution of the measured intensity autocorrelation was achieved by the DTS software. For a pure diffusive relaxation, Γ is related to the translational diffusion coefficient D at q → 0 and c → 0 according to Eq. (3):
The hydrodynamic radius R h is calculated by the Stokes–Einstein equation as \( {R}_h=\raisebox{1ex}{${k}_BT$}\!\left/ \!\raisebox{-1ex}{$6\uppi \eta D$}\right. \) with q, k B, T and η being the scattering vector, the Boltzmann constant, absolute temperature, and solvent viscosity, respectively. A hydrodynamic radius distribution was calculated from the Laplace inversion of g 1(t) by using the CONTIN procedure. Thermoresponsivity of nanogels was investigated between 10 and 55°C. In order to check the reversibility of the hydrodynamic radius with temperature transition, five heating and subsequent cooling cycles were performed. Influence of pH was investigated at 20°C.
2.9 Circular Dichroism Spectroscopy
Circular dichroism (CD) measurements were carried out with an Olis 17 DSM; a Cary 17 monochromater was used with a spectral output of 184–260 nm. CD spectra were measured at protein concentrations of 0.5 mg/mL using a cell with 0.090 mm light path in the wavelength range of 190–260 nm, with a bandwidth of 2.00 nm and with number of increments of 50 with an integration time of 20 s at 20°C. CD spectra were expressed in terms of mean residue ellipticity (deg cm2 dmol−1). The depicted data represent an average of five scans.
2.10 Enzymatic Degradation
Degradation of microgels was investigated in 5 mM elastase in PBS buffer at 37°C and followed by measuring the hydrodynamic radius of the microgels by dynamic light scattering (DLS) after every 180 s for a total time of 24 h.
2.11 Model Drug Release
PEG-DGE-1:0.25, PEG-DGE-1:0.5, BS3-1:0.25, and BS3-1:0.5 crosslinked microgels loaded with Texas Red-dextran were prepared by adding the dextran (15 wt% of dextran with respect to elastin) to the aqueous phase in the inverse emulsion in which the microgels were synthesized, under the same conditions as the blank samples. In order to determine the amount of Texas Red-dextran taken up by the micro-conetworks, the extracted Texas Red-dextran in the separated water was quantified by measuring the absorbance at 596 nm and using a standard calibration curve. The difference from the feed was taken as the amount loaded to the microgels. To study the release of loaded dextran at 20 and 40°C, the microgels were taken up in 10 mL PBS buffer at pH 5 by shaking the cuvettes at 100 rpm. After leaving for 0, 1, 5, 10, 15, 25, 36, 48 or 72 h, the microgels were centrifuged. The release was monitored by measuring the extinction of the supernatants. The sedimented microgels were resuspended in fresh buffer solution. All studies were performed in triplicate and the cumulative release was normalized to the original uptake:
where D t is the actual amount of the dextran in the microgel at time t and D 0 is the initial amount of dextran in the microgel.
3 Results and Discussion
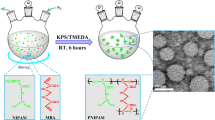
Solubilized native elastin with a molecular weight range of 10–60 kDa was dispersed in an inverse miniemulsion and crosslinked by reaction of PEG-DGE or BS3 with the ε-amino groups of lysines in the sequence of the elastin peptide fragments (Scheme 1).

Elastin chemical crosslinking reactions
PEG-DGE crosslinked microgels can be regarded as a conetwork of the hydrophilic PEGs and the thermoresponsive elastin. In contrast, the linking groups of the BS3 crosslinked microgels are hydrophobic and are expected to promote precipitation above the coacervation temperature. The crosslinking density was varied by adding different amounts of the telechelic linkers. In Table 1, the amount of linker is given as a molar ratio of elastin and linker based on the molecular weight of M n = 60,000. Ratios of 4–0.5 crosslinker per elastin molecule correspond to relatively low crosslinking degrees because of the lower molecular weight fraction in the elastin sample. It must be noted, however, that the microgels were fractionated by separation of very loosely crosslinked and very small particles during the isolation/sedimentation procedure. Thus, the crosslinker ratios can only be taken as an indication of the relative variation in crosslinking density. Figure 1 depicts the most densely crosslinked microgels (elastin to crosslinker ratio of 0.5) by scanning electron microscopy (SEM) cryo-electron micrographs as quenched from ambient temperature, i.e., below the coacervation temperature. Irrespective of the type of crosslinker, both samples demonstrate the formation of spheres with diameters in the range of 250 nm < d < 550 nm for PEG-DGE and 200 nm < d < 350 nm for BS3-based crosslinking. These sizes are in good agreement with the hydrodynamic radii determined by light scattering (see Table 1). In many cases, the electron micrographs demonstrate the presence of protrusions, indicating a gradient in density from the core towards the outside, as established for microgels prepared by precipitation polymerization [4].

Cryo-FESEM images showing (a) elastin-PEG-DGE and (b) elastin-BS3 microgels prepared with 0.5 equivalents of the crosslinker
In order to verify that the microgel samples are in general biocompatible we used a cell proliferation assay. All microgel samples were proven cytocompatible throughout the 6-day cell culture experiment, with no significant effect on cell proliferation. These results demonstrate that ES12 microgels may be used for further in vitro or in vivo experiments.
Circular dichroism spectroscopy allows one to follow conformational changes of the elastin peptides that are caused by the crosslinking. Solubilized elastin exhibits a negative band near 200 nm that is characteristic of unfolded proteins with segments in the PPII conformation [8, 21]. Upon coacervation, the CD spectra indicate a conformational change to an α-helix with two negative bands at 208 and 224 nm and a positive band at 193 nm [8]. Figure 2 displays the CD spectra of ES12 measured at 20°C and of samples crosslinked by different amounts of PEG-DGE and BS3. The negative band at 222 nm indicates the onset of the formation of α-helical conformations. In a first approximation, we calculated a helix content of 22%, and 11% for the samples with least PEG-DGE and BS3 respectively [22–24]. For high degrees of crosslinking, the CD spectra indicated a fully disordered elastin structure. The change in CD spectra suggests that the conformation-controlled coacervation becomes impeded by higher degrees of crosslinking of the microgels.

CD spectra of elastin before and after crosslinking in a PEG-DGE (left) and BS3 (right) microgel
All microgel samples were characterized by DLS regarding the hydrodynamic radius and the polydispersity at 20°C and at 50°C, the latter temperature being in the regime where the elastin ES12 undergoes coacervation, i.e., above the temperature of 37°. Results are summarized in Table 1. The light scattering data demonstrate a significant shrinkage of the particle size when the temperature was raised from 20 to 50°C. It must be noted that the crosslinking was achieved at room temperature, i.e., under conditions where the elastins were fully soluble.
Remarkably, all particles remained soluble or dispersable as single particles, irrespectively of whether they were compacted by a high degree of crosslinking or by raising the temperature above the volume change transition. This can be regarded as indicating preferential collapse in the core of the particles while hydrophilic segments and endgroups remain oriented towards the outside. Also, sedimentation experiments demonstrated an increase in the rate of sedimentation that corresponds to intramolecular collapse of the dispersed microgel particles, i.e., the rate of sedimentation changes in a rough approximation with 1/R h [25].
Figure 3 depicts the corresponding plots of the temperature versus the hydrodynamic radii. As expected from the CD experiments, the volume change transition became as less pronounced and even vanished with the strength of elastin crosslinking. At low degrees of crosslinking, the volume change transition was well developed, irrespectively of whether the gel particles were crosslinked by the hydrophilic PEG-DGE or the hydrophobic BS3. Correspondingly, the swollen hydrodynamic radii of the BS3-elastin microgels were in general smaller than those of the PEG-DGE-elastin microgels and also the volume change occurred at lower temperature. This is similar to the behavior of polyacrylamide microgels with amide residues that differ in their hydrophilicity.

Hydrodynamic radius (R h) of PEG-DGE (left) and BS3 (right) crosslinked microgels as a function of temperature (10–55°C)
For the least crosslinked microgels, with ES12 to crosslinker ratio equal to 4, DLS experiments and R h determinations were repeated within four heating and cooling cycles, alternating the temperature between 20 and 44°C (Fig. 4). The repeated change in R h was reversible and also the polydispersity index (PDI) of the microgels did not broaden, demonstrating that neither coagulation nor extraction of free chains occurred to a significant extent.

Hydrodynamic radii (R h) during changes upon repeated heating and cooling of PEG-DGE (1:0.25), PEG-DGE (1:0.5), BS3 (1:0.2), and BS3 (1:0.5) crosslinked microgels
Soluble elastin prepared by the method of Partridge [7] has an isoelectric point around pH 5. Correspondingly, we measured isoelectric points between pH 5 and 6 for ES12-PEG-DGE and around 6 for BS3 crosslinked microgels. For the elastin at low ionic strength, it has been reported that the coacervation temperatures exhibit a minimum around pH 5, i.e., coincident with the isoelectric point. Correspondingly, a pH-dependent collapse has been expected for the microgels. This was confirmed by the variation in the hydrodynamic radius, R h with pH, as shown in Fig. 5. For PEG-DGE microgels, R h decreased from pH 3–6 and increased again from pH 7–11. A minimum in size was also found for the BS3 crosslinked microgels, but shifted to pH 7. The collapse was in both cases fully reversible. Similarly to the temperature-dependent measurements of the hydrodynamic radius, the collapse was more pronounced for the loosely crosslinked samples.

Change in hydrodynamic radius (R h) as a function of pH (3–11) for (a) PEG-DGE (left) and (b) BS3 (right) crosslinked microgels

Degradability of PEG-DGE (1:0.25) and BS3 (1:0.25) crosslinked microgels, as measured by the change in hydrodynamic radius (R h) as a function of time using 5 mM elastase
Degradation of the elastin microgels is expected to be catalyzed by the enzyme elastase. Elastase is known to cleave peptidic bonds at the lysine units, i.e., in the hydrophilic segments. In order to evaluate the enzymatic degradation of the elastin microgels, the dispersed microgels were incubated with the enzyme at 37°C in a BPS buffered solution at 37°C and samples characterized by DLS for the decrease in hydrodynamic radius with time. At this temperature, the microgels are in the collapsed state and the enzymatic degradation is expected to occur as an erosion process from the surface. Figure 6 depicts the change in particle size within a period of 1 day for the least crosslinked elastin microgels. Initially, the hydrodynamic radius increased as the particles were further swollen when the first elastin bridges were split. Afterwards, the radius decreased nearly linearly to full degradation after about 16 h for the PEG-DGE-elastin microgel, while the BS3-elastin microgel clearly degraded in two steps, more quickly at the beginning and then with a significantly reduced degradation rate. Such an effect might be explained by a change in crosslinking density and swelling from the outside towards the core of the particles.
As an example of a water-soluble biopolymer that can be entrapped in and released from the elastin microgels we choose a dextran that has been used as a blood plasma expander. Loading of the microgels with the model drug dextran was achieved by addition of dextran to the inverse emulsion during microgel preparation. The particles were isolated by centrifugation and washed according to the procedure for particles prepared without dextran. The loading of the particles by the Texas Red-labeled dextran was corrected for the dextran washed out during the washing procedure. The microgel particles were taken up in water for a defined time and the release of dextran was determined after centrifugation by the calibrated extinction of the decanted aqueous solution. Sedimented particles were redispersed in fresh buffer solution and extracted again. The release was analyzed as the cumulative ratio of the original loading. The procedure was performed at 20°C with the non-collapsed particles and at 40°, above the coacervation temperature.
Table 2 summarizes data on the initial loading and the hydrodynamic radii of the microgels prepared at 20°C and characterized for their hydrodynamic radius at temperatures below and above the volume change transition. Regarding the fact that the dextran did not participate in the crosslinking reaction but serves just as another component in the solvent, it appears unsurprising that the particle sizes came out similar to those prepared without dextran (see Table 1). Figure 7 depicts the cumulative release curves for the two different crosslinking densities and at the two temperatures. Release was slower for the more densely crosslinked BS3-elastin microgels than for the hydrophilic PEG-DGE crosslinked gels. With the exception of the release experiments with the PEG-DGE-elastin microgels above the coacervation temperature, the release rates did not depend significantly on the degree of crosslinking. For both types of microgels, the release rate is higher above than below the coacervation temperature. Also, because the diffusion coefficient increases with temperature we regard this as an indication that the dextran is squeezed out by the collapsed gel structure.

Temperature-dependent release of dextran from PEG-DGE 1:0.25, 1:0.50 (left) and BS3 1:0.25, 1:0.50 (right) crosslinked microgels at 20 and 40°C
4 Conclusions
In summary, we have shown that rather well-defined thermo- and pH-responsive microgels with sizes in the submicron range can be prepared by an easy procedure with soluble elastin obtained by acidic hydrolysis of native elastin. The temperature- and pH-dependent collapse was most pronounced for low degrees of crosslinking and coincided rather well with the coacervation transition of free elastin. It can thus be concluded that the transition is related to the conformational transformation of the elastin peptide that causes the coacervation process. The fact that the biohybrid particles remained soluble and did not aggregate can be seen as evidence for an intramolecular collapse, whereby the elastin aggregates become stabilized against precipitation by a corona formed of water-soluble segments. As might be expected, this is more pronounced for the conetwork microgels with hydrophilic PEG segments; however, it was also observed when the elastin peptides were crosslinked by a hydrophobic diester segment. This can be explained by the rather broad molecular weight distribution of the elastin peptides, whereby only the longer peptides are expected to undergo coacervation while the shorter peptides remain fully solubilized. Regarding the temperature and pH response, the microgel particles resemble hydrophilic polyacrylamide microgels, which are also considered smart particles due to their volume change phase transition. However, the hybridization with defined proteins or peptides offers additional and very specific tools for the design of properties. In our example, the microgels exhibited a dual stimulus-response such that the thermal transition was shifted to higher temperatures below and above the isoelectric point of the peptide.
Because soluble elastin peptides are highly biocompatible, do not trigger a thrombogenic response, and show low immunogenicity elastin is suitable as a component of stealth-type microgel particles [26]. To begin with this aspect, we have concentrated on microgel use as a potential drug carrier by demonstrating the cytocompatibility and the biodegradability of the microgel. Furthermore, we have performed a model study on the release properties with dextran as a water-soluble biomacromolecule. The results demonstrate the ability of the concept to significantly retard release. However, elastin peptides are also known to influence signaling and proliferation by binding to several cell receptors, the most well investigated being the elastin binding protein (EBP) [27, 28]. Within this aspect, elastin is an example of a bioactive compound with an activity that can be expected to be manipulated by the collapse of the microgel particles. Recombinant or synthetic ELPs will offer new possibilities to tailor the responsive properties and to introduce switchable receptor affinities, also with other specific peptide segments as ligands [13].
References
Goncalves C, Pereira P, Gama M (2010) Materials 3:1420–1460
Singh S, Möller M, Pich A (2013) J Polym Sci Polym Chem 51:3044
Das M, Zhang H, Kumacheva E (2006) Ann Rev Mater Res 36:117
Balanceanu A, Demco DE, Möller M, Pich A (2011) Macromolecules 44:2161
Storm G, Belliot S, Daemen T, Lasic DD (1995) Adv Drug Deliv Rev 17:31
Matsumura Y, Maeda HA (1986) Cancer Res 46:6387
Partridge SM, Davis HF, Adair GS (1948) Biochem J 43:387
Urry DW, Starcher B, Partridge SM (1969) Nature 222:795
Volpin D, Urry DW, Cox BA, Gotte L (1976) Biochim Biophys Acta 439:253
Amiram M, Luginbuhl KM, Feinglos MN, Chilkoti A (2013) J Contr Release 172:144–151 doi:10.1016/j.jconrel.2013.07.021
Hassouneh W, Fischer K, MacEwan S, Branscheid R, Fu CL, Liu R, Schmidt M, Chilokoti A (2012) Biomacromolecules 13:1598
Patel D, Vandromme SE, Reid ME, Taite LJ (2012) Biomacromolecules 13:1420
Rodriguez-Cabello JC (2004) Smart elastin-like polymers. In: Hasirci N, Hasirci V (eds) Biomaterials: from molecules to engineered tissues. Advances in experimental medicine, vol 553. Kluwer Academic/Plenum, New York, p 45
Kumashiro KK, Ho JP, Niemczura WP, Keeley FW (2006) J Biol Chem 281:23757
Baldock C, Oberhauser AF, Ma L, Lammie D, Siegler V, Mithieux SM, Tu Y, Yuen J, Chow H, Suleman F, Malfois M, Rogers S, Guo L, Irving TC, Wess TJ, Weiss AS (2011) Proc Natl Acad Sci USA 108(11):4322
Conticello VP, Desai HC (2012) In: Langer R, Tirrell D (eds) Polymer science: a comprehensive reference, vol 9. Elsevier, Amsterdam, p 71
Maeda I, Fukumoto Y, Nose T, Shimohigashi Y, Nezu T, Terada Y, Kodama H, Kaibara K, Okamoto K (2011) J Pept Sci 17:735
Yeo GC, Keeley FW, Weiss AS (2011) Adv Colloid Interface Sci 167:94
Bochicchio B, Floquet N, Pepe A, Alix AJP, Tamburro AM (2004) Chem Eur J 10:3166
MacEvan SR, Chilkoti A (2012) Nano Lett 12:3322
Ferreon JC, Hilser VJ (2003) Protein Sci 12:447
Lin T, Quinn TP, Grandgenett D, Walsh MT (1989) Proteins 5:156
Greenfield N, Fasman GD (1969) Biochemistry 8:4108
Mattice WL, Lo JT, Mandelkern L (1972) Macromolecules 5:729
Vidakovic P, Rondelez F (1984) Macromolecules 17:418
Darnule T, Likhite V, Turino GM, Mandl I (1977) Connect Tissue Res 5:67
Privitera S, Prody CA, Callahan JW, Hinck A (1998) J Biol Chem 99:870
Daamen WF, Veerkamp JH, van Hest JCM, Kuppervelt TH (2007) Biomaterials 28:4378
Acknowledgements
F.T. thanks Marie Curie ITN project Hierarchy (contract: PITN-2007-215851) for the Ph.D. fellowship. The authors thank Mr. Arpit Bajpai for cell tests and Ms. Helin Li for support during microgel preparation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Singh, S., Topuz, F., Albrecht, K., Groll, J., Möller, M. (2013). Stimuli-Sensitive Microgels from Native Elastin: An Easy Approach for a Drug Release System. In: Percec, V. (eds) Hierarchical Macromolecular Structures: 60 Years after the Staudinger Nobel Prize II. Advances in Polymer Science, vol 262. Springer, Cham. https://doi.org/10.1007/12_2013_268
Download citation
DOI: https://doi.org/10.1007/12_2013_268
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-03718-9
Online ISBN: 978-3-319-03719-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)