
Abstract
This review considers interpolyelectrolyte complexes, with a particular emphasis on advanced macromolecular co-assemblies based on polyionic species with nonlinear topology and on polymer–inorganic hybrids formed by interpolyelectrolyte complexes containing metal ions and/or metal nanoparticles.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Interpolyelectrolyte complexes
- Interpolyelectrolyte reactions
- Macromolecular co-assembly
- Metal nanoparticles
- Metallo-containing interpolyelectrolyte complexes
- Nanostructures
- Polyelectrolytes
- Polymer–inorganic hybrids
1 Introduction
During recent decades, self-organization phenomena in macromolecular systems based on synthetic polyelectrolytes, as well as in complex systems containing natural macromolecules, have been extensively and successfully investigated. First of all, the studies have been aimed at gaining deeper understanding of self-organization mechanisms in biological systems. An example is the phenomenon of DNA compaction, which was revealed upon examination of the conformational behavior of double-helix DNA complexed with cationic surfactants in nonpolar solvents such as chloroform. The experimental results on such “null DNA,” whose charge is fully compensated, under conditions pronouncedly different from those typical for common aqueous media, suggested that DNA compaction can be considered as intrinsic property of an uncharged double helix. Experts both in physical chemistry of polymers and in molecular biology have positively evaluated this concept [1–3].
Interpolyelectrolyte complexes (IPECs) resulting from coupling oppositely charged (or getting charged) polyelectrolytes obviously take an important and significant part in a domain of self-organizing polymer systems. Such macromolecular co-assemblies built up from linear polyions have attracted the attention of experts in polymer science for a long time [4–9]. Nowadays, one can confidently relate IPECs to smart and intelligent functional complex polymers having a pronounced tendency toward self-organization. Also of interest is the response of IPECs to minor variations in their environment (particularly to changes in pH, ionic strength, temperature, etc.) via considerable conformational changes and the whole spectrum of their physico-chemical and mechanical characteristics. The unique simplicity of preparation of IPECs, together with an availability of linear polyelectrolytes, make such macromolecular co-assemblies and IPEC-based materials very promising for numerous applications.
IPECs have been demonstrated to act as effective flocculants and surface modifiers [10–13]. One can emphasize the successful application of IPECs as effective and available binders for soils and grounds in the aftermath of the accident in the Chernobyl atomic power station (Ukraine).These macromolecular co-assemblies were used to suppress erosion of soils and grounds and thus to prevent radionuclide contamination caused by spreading of radioactive particles [10, 14]. Twenty-five years later, IPECs have again attracted attention because of similar problems arising from the accident in the Fukushima Daiichi atomic power station (Japan) [15, 16]. Another attractive application of IPECs is for medical purposes in the design of pharmaceuticals for targeted drug delivery followed by controlled release, e.g., for a nonviral gene delivery into cells [17, 18].
Nowadays, we clearly see that the domain of interpolyelectrolyte interactions and IPECs is rapidly developing, involving relatively new areas of science and opening new possibilities for application of these macromolecular co-assemblies. With IPECs as templates, novel organic–inorganic complex systems and hybrid nanocomposites based on these are successfully being developed. During the last few years, studies on IPECs have been considerably extended as polyelectrolytes with new (nonlinear) topologies have become available. This becomes possible due to remarkable advances in controlled radical polymerization techniques, such as atom-transfer radical polymerization (ATRP), stable free-radical polymerization (SFRP), and reversible addition-fragmentation transfer (RAFT) polymerization [19, 20]. As a result, well-defined star-shaped polyelectrolytes and star-like polyionic species (micelles of ionic amphiphilic block copolymers) as well as cylindrical polyelectrolyte brushes can now be utilized as polymeric components to be involved in interpolyelectrolyte complexation. This opens new possibilities for design of a novel generation of IPECs having a distinct compartmentalized structure. These problems and points will be considered in this review.
2 Formation and Basic Properties of Interpolyelectrolyte Complexes
2.1 Peculiarities of IPEC Formation
During recent decades, considerable attention has been paid to interpolymer interactions and interpolymer complexes. This interest is motivated by a number of fundamental and application-oriented problems. In a fundamental aspect, interpolymer interactions are of significant importance because of the possibilities for design and fabrication of novel polymeric co-assemblies, structures, and materials. Such macromolecular co-assemblies (also referred to as interpolymer complexes) possess unique properties that are remarkably different from the properties of their polymeric components. In the simplest case, they result from “weak” mutual attraction of chemically and/or stereo-complementary macromolecules. Thus, “stereocomplexes” formed due to van der Waals interaction between stereoregular poly(β-propiolactone)s are well known [21]. Interpolymer complexes might also be stabilized via hydrogen bonds between monomer units of the polymeric counterparts [22, 23], for example, co-assemblies of poly(carboxylic acid)s with polyethers, i.e., poly[oxyethylene(propylene) glycol]s. Despite the low attraction energy between complementary monomer units, such interpolymer complexes are rather stable because of the cooperative character of interpolymer interaction. At the same time, they can reversibly dissociate to their polymeric components if certain conditions (temperature, pH, ionic strength, etc.) are met. This opens a unique opportunity for design and fabrication of new polymeric materials and structures of various scales and dimensions (ranging from nano- to macroscopic) with variable characteristics, which are determined by environmental conditions. Such materials and structures are referred to as functional, whereas the co-assemblies themselves are regarded as “smart.” Similar principles for construction of complex polymeric structures are widely exploited and exemplified in nature by production of the most important biological architectures, such as double helices (i.e., DNA) and fibrillar proteins (e.g., collagen). Thus, the modern physical chemistry of polymers follows fundamental principles, which are widely used in nature.
Among interpolymer complexes, IPECs, which result from the interaction between oppositely charged polyelectrolytes, are of a special interest. Such macromolecular co-assembly processes proceed with high rates, which are typical for diffusion-controlled reactions. As a result of collisions between oppositely charged macromolecular coils, contacts (referred to as ion–ion or salt bonds) are formed between their monomer units while low molecular weight counterions previously associated with charged groups of the polymeric components release into bulk solution (1). This release of low molecular weight counterions causes the entropy of the system to increase, i.e., ΔS > 0. Calorimetric studies point to athermic character of interpolyelectrolyte complexation (in aqueous media). This fact indicates that co-assembly processes in such systems are predominantly driven by a gain in entropy.

Upon the formation of IPECs, the concentration of a low molecular weight salt (a−, b+) in the system increases. The increasing salt concentration shifts equilibrium (1) to the left, that is, it favors the dissociation of interpolymer salt bonds. This phenomenon is observed at high concentrations of the polymeric components and/or upon addition of low molecular weight salts to mixtures of oppositely charged polyelectrolytes and has been reported in numerous works [8, 24–26]. It is widely used when IPECs are applied.
At sufficient content of low molecular weight salts, the equilibrium, which can be shifted to one or another side by the changing salt concentration, is settled. There are numerous reports on fundamental aspects of interpolyelectrolyte reactions, in which equilibrium (1) is considered. Such equilibria are analyzed in terms of chemical thermodynamics, using equilibrium constants. For (1), the equilibrium constant K eq is given by:
If, for simplicity, \( {[ \vdash {{\mathrm{ A}}^{\ominus }}{{\mathrm{ b}}^{+ }}]_0}\;{ = [} \vdash {{\mathrm{ B}}^{\oplus }}{{\mathrm{ a}}^{- }}{]_0} = {C_0} \), that is, considering mixtures of oppositely charged polyelectrolytes at the stoichiometric (1:1) ratio between their ionic groups, then dividing the numerator and the denominator in Eq. (2) by the initial concentration (C 0) yields:
Here, Θ is a conversion in the reaction described by (1), such that \( \varTheta = [ \vdash {{\mathrm{ A}}^{{\ominus \oplus }}}{\mathrm{ B}} \dashv ]/{C_0} \), which equals the ratio of the concentration of the formed interpolymer salt bonds to the initial concentrations of ionic groups of the polymeric components. For nonstoichiometric mixtures, one takes as C 0 the initial concentration of ionic units of the minority polyelectrolyte.
From the above consideration, it follows that from the experimental point of view equilibrium (1) can be easily investigated for polyelectrolytes whose charged groups possess a strong specific affinity to counterions. Such systems comprising weak polymeric acids or weak polymeric bases have been studied in detail due to the simplicity and availability of experimental techniques for study of interpolyelectrolyte reactions by means of potentiometric titration. As an illustration, one can consider equilibria (4) and (5):


where (4) describes a reversible charging of a weak polyacid in the presence of polycations and (5) represents a reversible charging of a weak polybase in the presence of polyanions. Both charging reactions lead to the formation of IPECs. A detailed analysis of these equilibria in terms of Θ against the pH has been published [27]. A number of experimental Θ versus pH dependences for various polyelectrolyte systems are shown in Fig. 1.

Plots of Θ versus pH for various polyelectrolyte systems: (1) poly(acrylic acid) (PAA) – poly(l-lysine); (2) PAA – poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA); (3) poly(l-glutamic acid) – PDMAEMA; (4) PAA – poly(N-ethyl-4-vinylpyridinium bromide) (P4VPQ); (5) poly(4-vinylpyridine) – poly(sodium styrene sulfonate) (PSSNa); (6) PDMAEMA – PSSNa. Reprinted from [27] with kind permission from MAIK Nauka/Interperiodica Copyright 1999
The obtained findings point to a high stability of the formed IPECs, which in turn results from a high cooperativity of the coupling reactions (1), (4), and (5) between oppositely charged polyelectrolytes (also referred to as polyion addition reactions) [7, 8].
2.2 Water-Soluble Nonstoichiometric IPECs
IPECs have attracted significant attention mainly as novel amphiphilic materials, which are promising for applications in medicine, biology, and ecology [10, 28, 29]. A breakthrough and further progress in the domain of interpolyelectrolyte reactions and IPECs were achieved in the middle of the 1970s. They were associated with the discovery of so-called water-soluble nonstoichiometric IPECs, which contain charged groups of the polymeric components in a nonequivalent ratio. For the first time, such macromolecular co-assemblies were reported [30]. Extensive studies of the water-soluble nonstoichiometric IPECs considerably advanced knowledge about the structure of these extremely interesting and promising macromolecular co-assemblies and their self-organization. It was found that the solution behavior of such IPECs decisively depends on their charge-to-charge stoichiometry. In particular, it was shown that IPEC species become compact if the ratio of oppositely charged groups of their polymeric components approaches 1:1 (i.e., stoichiometric).
These and other results allow one to consider water-soluble nonstoichiometric IPECs as peculiar amphiphilic block copolymers comprising a number of complex blocks assembled from the electrostatically coupled polymeric components. Such complex blocks are rather hydrophobic, thereby manifesting a tendency to a mutual segregation in aqueous media. Free (uncomplexed) segments of the excess polyelectrolyte are obviously hydrophilic and therefore grant IPEC species a solubility in aqueous media. This excess polyelectrolyte is typically referred to as a “host” polyelectrolyte (HPE). Its polymeric counterpart bearing opposite charge, which is incorporated into IPEC as a minority component, is generally referred to as a “guest” polyelectrolyte (GPE). The structure of water-soluble nonstoichiometric IPECs proposed by Kabanov and Zezin [31, 32] is schematically depicted in Fig. 2.

The unique behavior of water-soluble nonstoichiometric IPECs manifests itself, first of all, by polyion exchange reactions, which proceed between IPEC species and lead to transfer of a chain (or chains) of GPE from one HPE chain to another. These polyion exchange reactions cause such systems to undergo so-called disproportionation, which is known to result in phase separation of solutions of such macromolecular co-assemblies. The IPECs contained in coexisting phases considerably differ in their charge-to-charge stoichiometries: the insoluble phase is typically composed of a stoichiometric IPEC while a nonstoichiometric IPEC (or even a pure HPE) remains in the solution. Such phase separations can be caused by various factors, for example, by varying charge-to-charge stoichiometry of the mixture, by adding low molecular weight salts, by changing the temperature, etc.
These and other findings show that charge-to-charge stoichiometry of IPEC species can be remarkably different to the ratio between the amounts of monomer units of oppositely charged polymeric components in their mixture, i.e., the charge-to-charge stoichiometry of the mixture. To overcome this, one should introduce two independent parameters to characterize the charge-to-charge stoichiometry of the mixture: first, Z = [GPE]/[HPE] (here, the basemolar concentrations of the polymeric components in a reaction mixture are given in the brackets) and second, the stoichiometry of the formed IPEC, φ = N GPE/N HPE (here, the amounts of monomer units of the polymeric components incorporated into an IPEC are given). These parameters are defined already in early works [31, 32] and will be used throughout this review.
Figure 3 illustrates transformations of the water-soluble nonstoichiometric IPECs upon increasing Z. Here, three regions (A, B, and C) are marked on the Z-axis, where Zcr and Zlim correspond to the boundaries between the regions A and B and B and C, respectively, with the stoichiometry of the IPEC changing differently in each region. This scheme describes the behavior of mixtures of oppositely charged polyelectrolytes with considerably different degrees of polymerization, where \( {\mathrm{ D}}{{\mathrm{ P}}_{\mathrm{ HPE}}} \gg {\mathrm{ D}}{{\mathrm{ P}}_{\mathrm{ GPE}}} \) (here, DP is the degree of polymerization of the corresponding polyion) in the range 0 < Z ≤ 1.

Behavior of mixtures of oppositely charged linear polyelectrolytes with DPHPE >> DPGPE upon an increasing content of GPE. Z=[GPE]/[HPE]. For descriptions of regions A, B, and C, see text
In region A, 0 < Z ≤ Z cr and free (uncomplexed) macromolecules of HPE and IPEC species with stoichiometry being constant and equal to φ cr = DPGPE/DPHPE coexist in the solution. Numerous experimental results provide evidence that each of the IPEC species contains only one HPE chain. Disproportionation observed in region A results from an insufficient amount of GPE chains, which cannot occupy all HPE chains in the mixture. With increasing Z, the fraction of free HPE chains in the reaction mixture linearly decreases, with all HPE chains being incorporated into IPEC species at Z = Z cr.
In region B, Z cr < Z ≤ Z lim and all HPE chains are occupied but they are able to host further GPE. Experimental results indicate that GPE chains, which are additionally incorporated into the IPEC species of stoichiometry φ cr, are uniformly distributed among the macromolecular co-assemblies. However, a change of charge-to-charge stoichiometry of the IPEC species upon increasing Z in the region B has not been investigated in detail so far. It was only found that IPEC species with φ > φ lim lose their solubility in aqueous media. This happens at Z = Z lim = φ lim.
On further addition of GPE to a mixture of oppositely charged polymeric components (Z lim < Z ≤ 1), the system becomes heterogeneous (the region C). As previously described [26, 33], IPEC species considerably differing in their charge-to-charge stoichiometries coexist in the mixture of oppositely charged polyelectrolytes. Particles of an insoluble IPEC in region C are present in a colloidally dispersed state and can be easily separated from the reaction mixture, for example, by centrifugation. A direct determination of their charge-to-charge stoichiometry by means of elemental analysis indicates that it remains constant and corresponds to the equimolar ratio between charged groups of HPE and GPE (i.e., φ = 1). At the same time, a water-soluble nonstoichiometric IPEC with stoichiometry φ = φ lim remains in the solution. At Z = 1, only a precipitate of a stoichiometric IPEC (φ = 1) is formed and the supernatant contains no polyions.
These insoluble stoichiometric IPECs have found their application as binders of various disperse systems, including soils and grounds, thus acting as effective agents for preventing wind and/or water erosion. Specifically, they have been used to suppress spreading of radioactive contamination in soils and grounds resulting from accidents in atomic power stations [10, 14–16].
Addition of low molecular weight salts into mixtures of oppositely charged polyelectrolytes has a pronounced influence on the behavior of water-soluble nonstoichiometric IPECs. In particular, this manifests itself in a shift of the boundary between the regions A and B, that is, Z cr, to lower values. The low molecular weight salts induce conformational transformations of particles of nonstoichiometric IPECs,(i.e., a coil–globule transition) followed by phase separation of the solution and macroscopic precipitation of a stoichiometric IPEC [31, 34].
The above phenomena, which are to be investigated for understanding of self-organization in multicomponent systems containing oppositely charged polyions, imply that IPEC species can exchange their polymeric components with each other. Turbidimetric titrations of aqueous solutions of various HPEs with aqueous solutions of different GPEs (DPHPE > DPGPE) provide evidence for such polyion exchange. The reaction mixture remains transparent until Z = Z lim. When Z exceeds Z lim, turbidity appears and then linearly increases with increasing Z (in the range Z lim < Z < 1). This is schematically illustrated by the turbidimetric titration curves given in Fig. 4.

Turbidimetric titration curves of (1) an aqueous solution of sodium polyphosphate (PPNa) acting as HPE with an aqueous solution of 5,6-ionene bromide acting as GPE at pH 7.0 and 0.01 M NaBr; and (2) an aqueous solution of PDMAEMA·HCl (HPE) with an aqueous solution of PPNa (GPE) at pH 4.5 and 0.1 M NaCl
These results undoubtedly point to the formation of a water-soluble nostoichiometric IPEC at low Z-values (Z < Z lim), followed by the formation and the subsequent concentration of an insoluble stoichiometric IPEC at Z > Z lim. This completely corresponds to the transformations of IPECs, which are described above (Fig. 3).
2.3 Polyion Transfer in IPEC Systems
The dynamic properties of IPECs have been considered in more detail by Kabanov and colleagues [7, 8, 35], who reported on the kinetics of interpolyelectrolyte exchange reactions. These reactions comprise a transfer of GPEs from some HPEs to other HPEs. The reaction represented by (6) illustrates this process:

Here, HPE1 and HPE2 are chemically equal polyions with the same macromolecular characteristics. They exchange with each other by GPE. The reaction described by (6) represents a simple exchange process between particles of a nonstoichiometric IPEC (each containing one GPE chain) and free (uncomplexed) HPE chains. Such IPECs with \( \varphi = {N_{\mathrm{ GPE}}}/{N_{\mathrm{ HPE}}} = {\mathrm{ D}}{{\mathrm{ P}}_{\mathrm{ GPE}}}/{\mathrm{ D}}{{\mathrm{ P}}_{\mathrm{ HPE}}} \) are formed in region A (see Fig. 3). It is obvious that the polyion exchange reaction proceeds for any Z-value and even not necessarily in homogeneous media. In the heterogeneous region (C), the reaction is matched to an interphase transfer of GPE chains.
From the basic point of view, the reaction represented by (6) is most suitable for fundamental investigations on the kinetics and mechanism of interpolyelectrolyte exchange reaction. Such studies provide key understanding of processes proceeding in self-organizing multicomponent systems comprising oppositely charged polyions. They are also necessary for deep insight into the structure and properties of a novel generation of amphiphilic polymeric materials based on IPECs.
The experimental investigation of interpolyelectrolyte exchange reactions described by (6) requires that HPE1 and HPE2, which participate in the process, are distinguishable. It is also necessary to be able to detect a transfer of GPE chains. The first was achieved by fluorescent labeling of HPE2 or HPE1 [8, 36]. As fluorescent labels, antracenyl or pyrenyl ones, were used and poly(methacrylate) (PMA−) anions were exploited. The second problem was overcome by choosing GPEs, which are effective fluorescence quenchers. Typically, these are polymeric aromatic quaternary ammonium salts. Mostly, salts of poly(N-ethyl-4-vinylpyridinium) (P4VPQ+) cations were used. Thus, a transfer of GPE-quenchers results either in fluorescence enhancing or fluorescence quenching in the system, depending on which HPE (i.e., HPE2 or HPE1) is fluorescently labeled.
The kinetics of a single chain transfer, which is represented by (6), was studied more completely. In the simplest case, equal amounts of IPEC species, each containing only one HPE chain and one GPE chain, and free fluorescently labeled HPE (HPE*) are taken so that [HPE] = [HPE*]. Conversion (q) of the transfer was defined as the fraction of HPE* chains complexed with GPE chains. It was found that the kinetics of the transfer are well described in terms of a second-order reaction with respect to the concentrations of macromolecular reagents, i.e., IPEC {HPE-GPE} and HPE*. Accordingly, experimentally obtained kinetic curves were fitted as linear functions using Eq. (7):
Here, k 2 is the rate constant for the transfer of a GPE chain from one HPE chain onto another, [HPE*]0 = [HPE]0 is the initial basemolar concentration of the reacting species in the system, and t is time. It turned out that the measured values of k 2 are three to five orders of magnitude less than the calculated values for the rate constant, which is determined by diffusion-driven collisions [36]. This finding implies that indeed only one transfer of a GPE chain from an IPEC onto a HPE* chain takes place per 103–105 collisions. The Scheme (8) below illustrates this process:

According to this scheme, transfer of a GPE chain from an IPEC onto a HPE* chain proceeds in a united macromolecular coil comprising three polyions (i.e., HPE, HPE*, and GPE) through redistribution of interpolymer contacts (interpolymer salt bonds). It was shown that the probability of the chain transfer strongly increases with increasing ionic strength of the solution [36]. This is explained by an enhanced segmental mobility of GPE (due to breakup of some interpolymer salt bonds) in the united macromolecular coil (an intermediate state).
Thus, the kinetics of polyion exchange reactions can be governed by varying the concentrations of a low molecular weight salt in the system. If the ionic strength of solutions is low, then macromolecular dissociation of IPECs to their polymeric components does not happen under such conditions. The rate of polyion exchange reaction also strongly increases with the decreasing linear charge density of GPE as well as with the decreasing degree of polymerization of the latter [36].
The considered processes play an extremely important role during preparation of IPECs. Thus, mixing aqueous solutions of oppositely charged polyelectrolytes at charge-to-charge ratios at which water-soluble nonstoichiometric IPECs are to be finally formed results, at first, in appearance of turbidity in the system. However, the turbidity gradually disappears with time, depending on the mixing conditions, and particles of a nonstoichiometric IPEC with φ = Z can be detected in the system [36]. The first, rapid stage of the reaction proceeds with a diffusion-controlled rate. It is accompanied by appearance of rather large aggregates, whose charge-to-charge stoichiometry differs from the charge-to-charge stoichiometry of the reaction mixture, i.e., φ ≠ Z. The next, relatively slow stage is a polyion exchange between these aggregates and free HPE, leading to the formation of thermodynamically stable water-soluble nonstoichiometric IPECs.
The processes described above are also of importance for insoluble stoichiometric IPECs as well as for IPEC-based materials, including composites. This manifests itself in the fact that the swollen-in-water IPECs behave as viscous liquids and can creep. When incorporated into complex disperse systems (e.g., soils), they quickly recognize complementary sites on the surface of soil particles and strongly stick them together. In such systems, oppositely charged polyions constantly migrate relative to each other due to Brownian motion, remaining at the same time incorporated into the IPECs [8]. This behavior explains the fact that composites based on such macromolecular co-assemblies in a wet state exhibit pronounced self-healing behavior.
The results of studies on the kinetics of interpolyelectrolyte exchange reactions provide evidence that fluorescent labels of HPE*, which were taken to be hydrophobic, act as “anchors” for GPE-quenchers. It was found that an antracenyl label is a weak “anchor” compared to a pyrenyl label and that an increasing amount of labels in a HPE* chain leads to its more selective binding with GPE-quenchers [36, 37]. This is because of an additional gain of free energy upon formation of a contact between a label and a monomer unit of the polymeric quencher due to the donor–acceptor interaction and also because of incorporation of the label into a rather hydrophobic domain consisting of hydrophobic moieties of the coupled polyions. Such “recognition” plays an important role in biological systems.
Analysis of the equilibrium of interpolyelectrolyte coupling and kinetics of interpolyelectrolyte exchange led to the conclusion that IPECs are stable macromolecular co-assemblies. In aqueous media, IPECs do not dissociate to their polymeric components at concentrations of low molecular weight salts typically below about 0.5 M and, at the same time, retain high dynamics. At such salt concentrations, polyions building up macromolecular co-assemblies at ambient temperatures are able to easily migrate with respect to each other. In aqueous solutions of nonstoichiometric IPECs, GPE chains easily change their hosts through interpolyelectrolyte exchange reactions. These processes provide remarkable self-organization of IPECs. Being of a pronounced amphiphilic character, IPECs quickly find their optimal location in a complex environment. Thus, they “recognize” complementary sites on surfaces of particles in natural dispersions and anchor onto them, as depicted in Fig. 5.

A fragment of an IPEC and a fragment of an IPEC in a soil pore
Nowadays, IPECs are typically considered to be self-organizing and ordered macromolecular co-assemblies. This concept is based not only on semi-intuitive understanding of such systems as products of the cooperative interpolyelectrolyte interaction but also on some supporting experimental findings.
Self-adjustment phenomena in IPECs manifest themselves on different scales: both within single particles in aqueous solutions of nonstoichiometric IPECs and in bulk insoluble stoichiometric IPECs. A transfer of GPE chains from some HPE chains to other identical chains has been considered above. It has been assumed that this transfer is possible because a GPE chain permanently changes its location in a HPE coil due to Brownian motion. In a united {HPE1-GPE-HPE2} coil, it can “choose” another host within the lifetime of the coil and, therefore, migrate from the original HPE1 to the added HPE2. Besides, one assumes that all locations of the GPE chain in the HPE coil are equal because there is no available information about their preferable locations. At the same time, when the GPE chain is rather short it remains relatively strained on the oppositely charged longer HPE chain. This implies a certain order in mutual locations of the polymeric components in an IPEC particle. Such a consideration follows from numerous results on the conformational behavior of particles of water-soluble nonstoichiometric IPECs. Thus, the migration of GPE chains resembles a “worm”-like motion along a stretched thread, this motion being permanent and chaotic in nature.
The Brownian motion of GPE-quenchers becomes vectorized even if a minor amount of pyrenyl labels is attached to HPE chains; as little as about one label per 350 monomer units of HPE* is sufficient. This manifests itself in the fact that GPE-quenchers, as described above, predominantly occupy HPE* chains bearing the fluorescent labels. Considering a single particle of a water-soluble nonstoichiometric IPEC*, this also means that a random walk of a GPE chain in a HPE* coil is directed, with the GPE chain spending more time on sites of HPE*, which contain the anchoring label.
In the context of self-organization of IPECs, the so-called interpolyelectrolyte substitution reactions are of particular interest. In these reactions, HPE1 chains coupled to GPE (i.e., forming IPEC1) are substituted by HPE2 chains, which are different in nature, thereby generating IPEC2 as shown in (9)
Such reactions are described in terms similar to those that have been applied for consideration of polyion exchange reactions (6). A reaction with a participation of water-soluble nonstoichiometric IPECs comprising fluorescently labeled PMA− anions as HPE* and P4VPQ+ cations as GPE-quenchers was studied. On addition of polyanions containing sulfo- or sulfonate groups to these systems, a full transfer of GPE chains from IPEC* particles onto polysulfo/sulfonate anions is observed, as illustrated by Scheme (10):

This process is accompanied by enhancing fluorescence, which is a measure of the shift of the equilibrium of the reaction shown in (10) from left to the right. Experimentally observed [7, 8] extremely high selectivity of interpolyelectrolyte interaction is explained by additional donor–acceptor interactions between the electron-donating sulfo/sulfonate groups of HPE2 and the electron-accepting pyridinium groups of GPE. Even at rather low values of the energy of the non-Coulomb interaction between monomer units of the oppositely charged polymeric components, the total energy of the additional interaction of GPE chains with a HPE2 chain, which is summed over all GPE monomer units, is sufficiently high to provide an error-free recognition and an almost complete selectivity of the interpolyelectrolyte coupling.
Published works [7, 38, 39] convincingly demonstrated that in a complex biological environment (e.g., in blood plasma) polycations find highly sulfated polysaccharides (e.g., heparin) in the system in an error-free manner. Polycations form stable IPECs with heparin, thereby suppressing its activity as a blood anticoagulant. Further detailed investigation on the kinetics and equilibrium of interpolyelectrolyte substitution reactions have provided a basis for the development of complex heparin antagonists with low toxicity and an improvement in systems for immunodiagnostics [7].
Thus, self-organization processes are widely represented in complex interpolyelectrolyte systems. The kinetics of such processes can be finely tuned by varying environmental conditions, in particular, by changing the concentration of low molecular weight salts. The course of the process can be controlled by incorporating “anchor” groups into the polymeric components of IPECs as well as by a directed choice of polyion competitors that differ in the chemical nature of their ionic groups. These phenomena have been considered on the nanometer scale, which corresponds to the typical size of charged macromolecules.
Self-organization processes proceeding in IPECs are not limited by the nanometer scale, pronouncedly manifesting themselves on the scale corresponding to mesostructures and in bulk. The self-organization can lead to the selective formation of highly ordered structures. An example of such a structure is stoichiometric IPEC formed by PA− anions and protonated linear poly(ethylene imine) (PEI) cations. The study of chemical transformations in such macromolecular co-assemblies provided evidence that more than about 80% (mol) of interpolymer salt bonds can be converted to covalent amide bonds, as illustrated by Scheme (11):

The reaction (11) proceeds at a temperature of 170–270°C in solid glassy specimens. The high yields are due to high ordering of both the original IPEC and the reaction product [8, 40, 41]. From an application point of view, these processes are extremely useful for chemical modification of such macromolecular co-assemblies with the aim of enhancing their stability in aggressive media and improving their mechanical properties. The structured IPECs {PAA-PEI} are found to be suitable macromolecular templates for the design of novel metallo-containing IPECs, which contain transition and/or heavy metal ions sandwiched between the polymeric components [42].
During the last decade, the potential for synthesis and construction of novel complex multicomponent self-organized IPEC-based structures has considerably increased as polyelectrolytes and polyionic species with nonlinear topology have become available. Among those are polymeric micelles with polyelectrolyte coronas, star-shaped polyelectrolytes, and cylindrical polyelectrolyte brushes. Polyionic species of such types themselves possess often a pronounced capability for self-organization, which is expected to be enhanced when they are incorporated into complex macromolecular structures such as IPECs. The advanced IPECs based on polyionic species with nonlinear topologies are considered in Sect. 3 of this review.
3 Advanced Interpolyelectrolyte Complexes
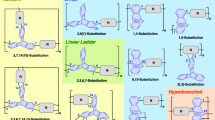
Further progress in the field of IPECs has been associated with involvement of more complex polyionic architectures, such as branched ionic (co)polymers (polyelectrolyte stars and cylindrical polyelectrolyte brushes) as well as self-assemblies of linear ionic diblock copolymers (polymeric micelles) (Fig. 6a–c), into interpolyelectrolyte complexation. Synthesis of well-defined polymeric architectures with nonlinear topology has become possible only recently due to considerable developments in living and controlled polymerizations. In this section, we briefly consider and update the main achievements regarding the preparation and investigation of such advanced IPECs, which are also reported in a recent review [43].

A polyelectrolyte star (a), a micelle with a polyelectrolyte corona (b), and a cylindrical polyelectrolyte brush (c)
The increasing interest to IPECs based on polyionic species of nonlinear topology is mainly due to the possibility of using them as a basis for building (optionally, to template) easily available and multifunctional polymeric architectures, which are of high potential for applications in rapidly developing nanotechnologies connected with such important areas as nonviral gene delivery, targeted/prolonged drug delivery, preparation of nanosized catalytic systems, etc.
3.1 IPECs Based on Star-Shaped (Co)Polymers
Star-shaped polymers (also referred to as polymer stars or star polymers) represent isotropic centrosymmetric macromolecules, each containing a single branching point in a center of a macromolecule (Fig. 6a). They can be prepared via a “core-first” or an “arm-first” approaches. The branching point represents a small core remaining from an oligofunctional initiator (“core-first” approach), which is used to polymerize arms, or from a multifunctional cross-linker (“arm-first” approach), which is used to cross-link arms. The core determines the number of arms while the arms determine the overall size of the synthesized polymer star. Macromolecules of star-shaped polymers are characterized by a well-defined size (in the nanometer range) and a spherical morphology, both resulting from their inherent structuring.
Successful synthesis of well-defined star-shaped ionic (co)polymers [44] is a prerequisite for preparation of their IPECs. In connection with this, we consider papers [45, 46] that report on water-soluble IPECs with a star-shaped polyion, e.g., poly(sodium acrylate) (PANa) stars, acting as HPE. Water-soluble IPECs are formed when charged groups of the star-shaped HPE are in certain excess compared to charged groups of its polymeric counterpart (P4VPQ). Otherwise, macroscopic phase separation (precipitation) is observed in mixtures of the oppositely charged polymeric components. It was found that the number of arms of the star-shaped HPE determines a window of Z-values, which correspond to the formation of water-soluble nonstoichiometric IPECs, this effect becoming more pronounced with increasing ionic strength of the surrounding solution [45].
A structural picture of complex species based on PANa stars was ascertained for the star-shaped HPE having a large number of arms (21 arms) coupled with a short linear polycation, quaternized poly(2-vinylpyridine) (P2VPQ), acting as a GPE [46]. The authors suggested that each complex particle formed has a compartmentalized structure of micellar (core–corona) type. A hydrophobic complex core is composed of oppositely charged segments of the polymeric counterparts while a hydrophilic corona, which grants to the whole macromolecular co-assembly solubility in aqueous media, is built up from excess segments of HPE (Fig. 7). If the total number of charged groups of the star-shaped HPE exceeds the degree of polymerization of the linear GPE, then typically one polyelectrolyte star, whose charge is partially compensated by chains of its polymeric counterpart, is incorporated into each complex particle formed [45, 46].

Core–corona structure of the water-soluble IPECs formed by the star-shaped PANa (HPE), having a large number of arms, with the linear P2VPQ (GPE). Reprinted with kind permission from Springer Science + Business Media from [46] Copyright 2009, MAIK Nauka/Interperiodica
An examination of such systems by means of molecular dynamic simulations (Fig. 8) supported a proposed core–corona structure of nonstoichiometric IPECs based on star-shaped polyions and further refined their structural picture [47]. It was shown that some of the arms of the star-shaped HPE are completely charge-compensated by chains of the linear GPE and embedded into the core while the remaining arms are free and build up an ionic corona (Fig. 8b). This pronounced partition of arms between two populations explains an experimentally observed hydrodynamic size invariance (or a slight change in a hydrodynamic size) of the star-shaped HPE upon its loading with the oppositely charged linear GPE [45].

Snapshots of the typical conformations of (a) a bare (uncomplexed) polyelectrolyte star and (b) its IPEC with the linear GPE. Reprinted with permission from [47] Copyright 2009 American Chemical Society
IPECs of very complex morphologies, such as long fibers forming fiber bundles or double wall vesicles tending to aggregate, were observed upon the interaction of oppositely charged cationic and anionic polyelectrolyte stars in extremely dilute aqueous solutions, e.g., quaternized poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMAQ) stars and poly(methacrylic acid) (PMAA) stars, both having six arms [48]. By adjusting the molecular weights of the star-shaped polyelectrolytes, their concentrations, the matching degree of oppositely charged polymeric components, and preparation conditions, it is possible to tune different morphologies of the resulting IPECs.
To broaden the window of Z-values corresponding to the formation of water-soluble IPECs, star-shaped polyions can be complexed with double hydrophilic (bis-hydrophilic) diblock copolymers comprising an ionic block and a nonionic hydrophilic block. In this case, the formed macromolecular co-assemblies remain water-soluble even if oppositely charged groups of the polymeric counterparts are taken in 1:1 ratio (Z = 1) [49], provided that the length of hydrophilic nonionic block is sufficiently long. Apart from the enhanced water-solubility of the formed complex species, one can additionally impart new properties and desired functionalities, such as biocompatibility, thermosensitivity, etc., through the hydrophilic nonionic block of the copolymer.
It was found that each complex particle formed in mixtures of PANa stars and P2VPQ-block-poly(ethylene oxide) (P2VPQ-b-PEO) at Z = 1 is of a distinct micellar (core–corona) structure. Here, a hydrophobic complex core is composed of oppositely charged segments of the polymeric counterparts while a hydrophilic corona, which provides solubility in aqueous media to the whole macromolecular co-assembly, is built up from nonionic blocks of the bis-hydrophilic diblock copolymer (Fig. 9). In this case, several polyelectrolyte stars, whose charge is fully compensated by the ionic blocks of its polymeric counterpart, are typically (though not always) incorporated into each complex particle [49], their number being determined by the number of arms of the star-shaped polyion and degree of polymerization of the ionic block of the bis-hydrophilic diblock copolymer.

(a) Core–corona structure of IPECs formed by the star-shaped PANa fully charge-compensated with P2VPQ-b-PEO. (b) Cryogenic transmission electron microscopy (cryo-TEM) image of the IPEC cores. Reprinted with kind permission from Springer Science + Business Media from [49] Copyright 2011, Pleiades Publishing, Ltd.
Similar micelle-like macromolecular co-assemblies were reported [50] for an anionic star-shaped bis-hydrophilic heteroarm copolymer PMAA-PEO coupled with a cationic bis-hydrophilic diblock copolymer, PDMAEMAQ-block-PEO (PDMAEMAQ-b-PEO), in alkaline media. In this case, however, the PEO blocks of both polymeric counterparts form a hydrophilic corona of each of the complex species formed.
During recent years, IPECs based on star-shaped (co)polymers with nucleic acids (also referred to as polyplexes) have attracted considerable attention in the context of nonviral gene delivery [51–56]. In particular, polyplexes of star-shaped PDMAEMA, having three and five arms, with DNA were shown to combine acceptable transfection efficiency with lower cytotoxicity [56].
3.2 IPECs Based on Star-Like Micelles of Diblock Copolymers
Ionic amphiphilic diblock copolymers are well known to self-assemble into core–corona aggregates (micelles) in aqueous media. The micelle comprises a hydrophobic core formed by nonpolar blocks and a hydrophilic corona built up from polyelectrolyte blocks. The properties of such macromolecular self-assemblies are reviewed in detail elsewhere [57, 58]. In many cases, the micelles are characterized by a spherical morphology. When the radius of the hydrophobic core is considerably smaller than the thickness of the polyelectrolyte corona, such macromolecular self-assemblies are regarded as star-like micelles (Fig. 6b). They resemble star-shaped polyelectrolytes with a large number of arms, though the number of arms in such macromolecular self-assemblies might change if the micelles are of “dynamic” nature, that is, if they are able to change their aggregation numbers with variations in the environmental conditions. Historically, the micelles of ionic amphiphilic diblock copolymers were the first star-like polyionic species involved in interpolyelectrolyte complexation and their IPECs have attracted considerable attention during the recent years.
Because the micelles generated by ionic amphiphilic diblock copolymers in aqueous media possess polyelectrolyte coronas, they are naturally expected to form IPECs with oppositely charged polyions. To the best of our knowledge, the first attempt to study interpolyelectrolyte complexation in such systems was performed by Talingting et al. [59], who reported a study on the interaction of protonated polystyrene-block-poly(2-vinylpyridine) (PS-b-P2VPH+) micelles with linear PSSNa of different molecular weights. Under a considerable excess of PSSNa to avoid any bridging or aggregation by linear polyions, the formation of macromolecular co-assemblies with a large mass excess (by a factor of ca. 5–6) of the charged groups of the PSSNa over charged groups of P2VPH+ was found, thereby leading to charge inversion of the PS-b-P2VPH+ micelles. This concomitant massive charge overcompensation resulting from the considerable molar excess (ca. 4.7–5.5) of sulfonate groups over pyridinium ones causes the formed IPECs to be colloidally stable.
Water-soluble (or colloidally stable) IPECs formed by oppositely charged micelles with polyelectrolyte coronas and linear polyions were subsequently found for a number of other systems. In particular, a detailed characterization of such macromolecular co-assemblies was performed for polyisobutylene-block-poly(sodium methacrylate) (PIB-b-PMANa) micelles complexed with a linear P4VPQ [60–62]. It was found that the formed IPECs remain water-soluble only when loading of the original micelles (acting as a HPE) by the linear polyion (acting as a GPE) does not exceed a certain threshold value. Such nonstoichiometric IPECs were thoroughly examined by means of various techniques, which provided evidence on their peculiar core–shell–corona (also referred to as “onion-like”) structure (Fig. 10a). Specifically, each of the complex species comprises a hydrophobic core from nonpolar blocks, which is surrounded by a layer (inner shell) assembled from oppositely charged segments of the polymeric counterparts, and a hydrophilic corona (outer shell) from excess segments of ionic blocks, which do not form interpolymer salt bonds.

(a) “Onion-like” core–shell–corona structure of IPECs formed by the star-like PIB-b-PMACs micelles (HPE) with the linear P4VPQ (GPE). Reprinted from [61] Copyright 2004 with permission from Elsevier. (b) Cryo-TEM image of the micellar IPECs. Reprinted with permission from [62] Copyright 2008 American Chemical Society
A similar multilayer structure with an inner complex shell was also proposed for IPECs formed by PS-b-P4VPQ, PS-b-PANa, and PS-b-PMANa micelles complexed with oppositely charged linear polyions [63–65]. In these cases, a polyelectrolyte corona is formed either by excess segments of ionic blocks of the copolymer (no overcharging of the original micelles by the linear polymeric counterpart) or by excess segments of the linear polyion (overcharging of the original micelles by the linear polymeric counterpart), depending on the actual ratio between molar concentrations of charged groups of the polymeric components in the system.
As in the case of star-shaped polyelectrolytes described in Sect. 3.1, the micelles acting as HPE demonstrate only minor changes in their hydrodynamic size upon interaction with oppositely charged linear polyions [61–64]. This finding strongly suggests that, similarly to IPECs based on polyelectrolyte stars, the ionic blocks forming a polyelectrolyte corona of such complex species split into two populations: a certain number of such blocks are fully embedded into the complex inner shell, while the rest of the coronal blocks remain nearly free, thereby being responsible for solubility of the whole macromolecular co-assembly in aqueous media.
It was found that the aggregation numbers of the original micelles hardly changes upon their interaction with the oppositely charged linear polyions [61–64]. This remarkable invariance of the aggregation numbers was observed not only for “frozen” micelles such as PS-b-P4VPQ [63, 64] with a glassy PS block but also for “dynamic” micelles such as the PIB-b-PMANa [61, 62] with a soft PIB block, which can change their aggregation numbers with variations in environmental conditions [66]. This implies that the micelles act as peculiar macromolecular templates or “nucleating” particles for a buildup of core–shell–corona architectures. At the same time, it was shown that interpolyelectrolyte complexation does not render the dynamic PIB-b-PMANa micelles frozen as their aggregation numbers remain nevertheless sensitive to variations in the conditions of the surrounding solution (e.g., pH) [62].
Finally, we should mention that IPECs based on micelles of ionic amphiphilic diblock copolymers can participate in so-called polyion interchange (exchange and substitution) reactions, accompanied by a transfer of GPE chains from one to another HPE micelle. Such reactions were previously thoroughly investigated for aqueous mixtures of oppositely charged linear polyelectrolytes [8, 31, 36, 67]. It was found that the aggregation state of the polymeric component(s) involved in such polyion interchange reactions has a remarkable effect on the reaction rate [68]. Specifically, the rate of the polyion interchange reaction decreased in the following order: coil–coil (seconds) > coil–micelle (tens of seconds) > micelle–micelle (thousands of seconds). A similar tendency was also observed for polyion coupling (polyion addition) reactions, which result in the formation of IPECs, though in this case the complexation between oppositely charged micelles (a micelle–micelle system) was not examined [68].
3.3 IPECs Based on Cylindrical (Co)Polymer Brushes
Cylindrical polymer brushes (also referred to as molecular polymer brushes or “bottle-brushes”) represent anisotropic macromolecules with cylindrical symmetry, each containing a long linear backbone and a large number of rather short linear side chains, which are densely attached to the backbone (Fig. 6c). Therefore, they have a multitude of branching points located along the backbone. Cylindrical polymer brushes can be synthesized via “grafting-from”, “grafting-to”, or “grafting-through” approaches. Macromolecules of cylindrical polymer brushes exhibit a distinct worm-like morphology, which results from their pronounced anisotropic nature.
Recent advances in the synthesis of well-defined cylindrical ionic (co)polymer brushes, which represent another type of branched ionic polymers [44], offer the possibility of preparing their IPECs. An early attempt to obtain such macromolecular co-assemblies was made [69, 70] and describes formation of large complex aggregates with a rod-like cylindrical morphology resulting from the interaction of a PEO/PSSNa copolymer brush (so-called anionic prototype copolymer brush) with P4VPQ. These large rod-like co-assemblies are considered to be highly anisotropic supermicelles.
IPECs formed between cylindrical polyelectrolyte brushes with P2VPQ or PEI side chains and DNA were examined [71]. It was found that, at a large excess of either of the polymeric counterparts, nanosized complex species coexist with the uncomplexed macromolecules of the excess polymeric component, thereby demonstrating an inhomogeneous distribution of macromolecules of the minority component among macromolecules of the excess one. This phenomenon was qualitatively explained in terms of the kinetically controlled formation of IPECs in such systems. The large charge density mismatch between the P2VPQ or PEI brushes and DNA was supposed to result in the formation of strongly positively charged complex species at an excess of the charged groups of the brushes and a slightly negatively charged complex species at an excess of the charged groups of DNA (Fig. 11).

Charge mismatch for IPECs of cylindrical polyelectrolyte brushes with DNA: (a) excess of DNA and (b) excess of the cylindrical polyelectrolyte brush. Reprinted with permission from [71] Copyright 2007 American Chemical Society
It has been reported that IPECs based on cylindrical ionic (co)polymer brushes formed when macromolecular co-assemblies were prepared in organic solvents [72, 73]. To dissolve polymeric components to be involved in interpolyelectrolyte complexation in such solvents, a PSS brush with dodecylammonium counterions (PSS DDA) and a PEI/PEO copolymer brush were used. In particular, it was shown [72] that in dimethylformamide one can obtain both kinetically controlled and topologically controlled IPECs, depending on the “charge” of the PEI/PEO brush created by adding HCl. The interaction of the PSS DDA brush with the protonated PEI/PEO brush resulted in kinetically controlled IPECs whereas the interaction with the non-protonated PEI/PEO brush led to topologically controlled IPECs of a cylindrical morphology. The formation of similar topologically controlled IPECs was also observed for the PSS DDA brush interacting with a polyamidoamine dendrimer of the fifth generation in such organic solvents as dimethylformamide or N-methylformamide, whereas kinetically controlled macromolecular co-assemblies were formed in methanol [73].
Water-soluble nonstoichiometric IPECs formed by a cylindrical PAA brush (acting as a HPE) and linear P4VPQ (acting as a GPE) are described [74]. Similarly to the water-soluble IPECs based on PAA stars considered in Sect. 3.1, they are formed only when charged groups of the HPE brush are in a certain excess compared to charged groups of the linear GPE. Under this condition, IPECs based on the PAA brush exhibit a hydrodynamic size that is very close to that of the original brush, and P4VPQ chains are uniformly distributed among its macromolecules. Solubility of such macromolecular co-assemblies in aqueous media points to their core–corona structure. Their structural organization was further examined by means of molecular dynamics simulations [74]. The snapshots resulting from the simulations (Fig. 12) clearly demonstrate the formation of a necklace of core–corona pearls, each consisting of a hydrophobic complex core made from oppositely charged segments of the polymeric counterparts, which is decorated by a hydrophilic corona built up from excess side chains of the cylindrical polyelectrolyte brush. This leads to a nanopatterned structure with longitudinal undulations. It is remarkable that such longitudinal undulations were experimentally observed for IPECs based on a PDMAEMAQ brush complexed with a short linear PSSNa [74, 75] (Fig. 13). Similarly to the IPECs based on PAA stars described in Sect. 3.1, side chains of the cylindrical polyelectrolyte brush demonstrate distinct repartitioning between the corona and the complex core domains; that is, some of the side chains of the HPE brush are nearly completely charge-compensated by chains of the linear GPE and embedded into the cores while the remaining side chains are free and build up coronas of the pearls (Fig. 12).

Snapshots of the typical conformation of (a) the bare (uncomplexed) cylindrical polyelectrolyte brush, and (b,c) its IPECs with the linear GPE at the degrees of charge compensation of 0.25 (b) and 0.5 (c). Reprinted from [74] with permission from the Royal Society of Chemistry Copyright 2009

(a) AFM image of IPEC formed by the PDMAEMAQ brush (HPE) with linear PSSNa (GPE). (b) Section analysis of the upper species. Reprinted with permission from [75] Copyright 2010 American Chemical Society
A possibility for manipulating the morphologies of cylindrical polyelectrolyte brushes via their complexation with oppositely charged linear polyions has been described [75]. In particular, it was found that increased loading of a PDMAEMAQ brush with a short linear PSSNa induces morphological changes of the original brush from a worm-like structure through an intermediate pear-necklace structure to fully collapsed spheres. However, an extremely long linear PSSNa resulted in a transition of the PDMAEMAQ brush to fully collapsed spheres, without intermediate states, even at very low loading by the linear GPE. As described previously [71], a pronounced inhomogeneous distribution of PSSNa chains (the minority component) among macromolecules of the PDMAEMAQ brush (the excess component) was observed.
Cylindrically shaped dendronized polymers resemble cylindrical polyelectrolyte brushes to a certain extent. The interaction of highly charged cationic dendronized polymers with DNA was examined [76]. It was shown that DNA wraps around such polymers, the overall charge of the formed IPECs and pitch size of the wrapped DNA being determined by dendron generation. These macromolecular co-assemblies might be used for development of novel nonviral gene delivery systems.
4 Metallo-Containing Interpolyelectrolyte Complexes
Polymer–metal hybrids based on polyelectrolyte systems have attracted growing interest during recent decades [77–79]. Metallo-containing compounds can provide polymer materials with special optical, electrical, magnetic, and mechanical properties as well as catalytic activity [77–81]. The capability of functional groups on polyelectrolytes to bind metal ions offers a possibility for their application as sorbing agents, ion-exchange materials, components of selective membranes [81–83], or as precursors for preparation of polymer–inorganic hybrids via reduction or precipitation of metal ions [81–85]. Polymer–inorganic nanocomposites are important candidates for construction of photonic devices, band-pass filters, components of nonlinear optical systems, optical limiters, elements of microcircuit chips, etc. [78, 79, 86]. Polyelectrolyte-based materials, including ultrafine particles of silver and noble metals, exhibit antibacterial properties and are therefore promising for application in medicine [87–90].
Metal ions coordinated with functional groups of polyelectrolytes may be used as a motif for assembling supramolecular and colloid systems [80, 91–93]. Polymer colloids and nanocomposites are very important as heavy metal carriers [80]. The dynamic nature of the coordination bonds between functional groups of polyelectrolytes and metal ions provides switchability to the system, mimicking the behavior of natural suprastructures [94]. The application of ionic amphiphilic block copolymers and terpolymers as templates for nanostructured systems or as precursors for synthesis of polymer–metal hybrids with different architectures [95–97] might be advantageous for design of prototypes for advanced catalytic, medicine-relevant, and electronic systems.
Sequences of interpolymer salt bonds built up by monomer units of oppositely charged polyelectrolytes form nanosized, structured domains from coupled functional groups. In some cases [8, 40, 41], even a perfect ladder-like arrangement of interpolymer salt bonds has been demonstrated. Such nanosized structured domains may act as scaffolds for sandwiching metal ions, which are incorporated (e.g., via the formation of coordination bonds with functional groups of the polymeric components) into the macromolecular co-assemblies with high selectivity [81]. The great variety of structures realized by IPEC-based systems, combined with the possibility of controlling the interaction of functional groups of the polymeric components with metal ions, has provoked a great interest in development of such novel functional materials.
This section describes, first, IPEC-based systems that contain metal ions. Then, a preparation of polymer–inorganic hybrids comprising metal nanoparticles (NPs) embedded into IPEC matrices is considered. Finally, advanced structures based on IPECs containing metal ions and IPEC-based hybrids containing metal NPs are reviewed.
4.1 IPECs Containing Metal Ions
The high dialysis permeability of IPECs allows transport of metal ions through the complex polymer matrix. An attractive possibility for an application of IPECs as sorbents or ion-exchange materials has stimulated considerable interest in the process of embedding metal ions into such macromolecular co-assemblies, distribution of metal ions throughout the complex polymer matrices, and their complexation with functional groups on the polymeric components. The preparation of nanocomposites in polyelectrolyte systems is another important stimulus for preparation of IPECs with a controlled content and regulated distribution of metal ions [81].
To study sorption of metal ions by IPECs and the formation of metallo-containing macromolecular co-assemblies based on them, both stoichiometric and nonstoichiometric IPECs {PAA-PEI} were used [81, 98, 99]. The interaction of metal ions with the IPECs implies sandwiching of these ions between PAA and PEI, being coordinated by their functional groups, which results in the formation of triple (tricomponent) macromolecular co-assemblies containing metal ions, as shown in (12):

The scheme shown in (12) demonstrates that the sorption of metal ions by IPECs causes the pH of the environment to decrease. To shift the equilibrium, it is necessary to add alkali, which leads to structures with a high content of incorporated (sandwiched) metal ions.
Generally, the ligand environment of metal ions includes functional groups of both PAA and PEI. The formation of the triple macromolecular co-assemblies containing metal ions results in coloration of initially colorless IPECs. For example, IPEC films become deeply dark blue in the case of Cu2+ whereas incorporation of Ni2+ leads to a green coloration. Therefore, the coloration of the IPEC {PAA-PEI} films is a qualitative indicator of transformation of the IPEC into metallo-containing macromolecular co-assemblies. The structure of {PAA-Cu2+-PEI} was revealed by means of electron paramagnetic resonance (EPR) [99]. The application of EPR provides direct information about coordination of paramagnetic metal ions with functional groups on the polymeric components. Figure 14 shows the characteristic EPR signals of Cu2+ incorporated into IPEC {PAA-PEI} films with different stoichiometries in different ligand environments.

EPR spectra of the triple metallo-containing macromolecular co-assembly {PAA-Cu2+-PEI} with Cu2+ content of 6% (wt) where [PAA]:[PEI] = 1:9 (a), 1:1 (b), and 9:1 (c). Arrows point to different ligand environments: (1) 2 NH and 2 COO−, (2) 4 NH, and (3) 2 COO−
In the stoichiometric IPECs, Cu2+ may exist in two ligand environments (Fig. 14b), i.e., 4 NH (see (13), structure A) or 2 NH and 2 COO− (see (13), structure B). However, the ligand environment with 2 NH and 2 COO− groups was shown to be preferable for Cu2+. The ratio between structures A and B in the triple macromolecular co-assembly depends on the proportions of the reagents and the pH of the solution [81, 99]. The EPR spectra point to a gradual increase in the linewidth observed with rising of Cu2+ content in IPECs, thereby suggesting a relatively uniform distribution of metal ions throughout the complex polymer matrices.

Table 1 demonstrates a high sorption capacity of the IPEC {PAA-PEI} films with respect to various metal ions. The largest capacity was found for Cu2+ because the geometry of the functional groups of the polymeric counterparts in the IPEC fits suitably to that required for the ligand environment of the metal ion. The ion-exchange capacity in this case is as high as 8.6 mg-equivalent Cu2+ per gram of the dry IPEC, that is, very close to that expected for structure B (13) of the triple macromolecular co-assembly {PAA-Cu2+-PEI} with fully occupied ligand sites. Thus, IPECs may act as precursors for the further templated formation of polymer–inorganic hybrid structures containing metal ions. Such precursors are characterized by high regularity of an array of the metal-mediated coordination bonds between the polymeric components.
A transformation of IPECs into triple macromolecular co-assemblies containing metal ions leads to a considerable decrease in their degree of swelling in water (Table 2) and, accordingly, to a substantial increase in their durability. It was found that transition metal and silver ions sorbed by IPECs {PAA-PEI} are bound extremely strongly [81, 98, 99] due to the formation of coordination bonds with ligands of both polymeric counterparts and a chelating effect. For example, the IPEC {PAA-PEI} films were able to sorb Cu2+ from aqueous solutions of Сu(NO3)2 at concentrations as low as 10−5 M. It is also the case for other metal ions, for example, Co2+, Ni2+, Fe2+. This makes it possible to use the IPECs for effective extraction of metal ions from dilute aqueous solutions and for ion-exchange concentrating of metal ions for analytical purposes. The high sorption capacity of IPECs in combination with their stability both in alkali and acidic media (especially for thermally crosslinked IPECs {PAA-PEI}) provide the prerequisites for development of novel highly effective and easy-to-prepare sorbents or ion-exchange materials.
The method of a preparation of multilayer polyelectrolyte films by exposing a surface to solutions of a polyanion and a polycation in a cyclic (alternating) fashion was reported by Decher [100]. This became known as the “layer-by-layer” (LbL) technique, which fabricates free-standing multilayer polyelectrolyte films and films on solid substrates [101–103]. These polyelectrolyte-based systems arise from the cooperative ionic interaction between functional groups of polyanions and polycations at the interface. They may be considered as IPEC films with a heterogeneous distribution of the polymeric components. The incorporation of metal ions into such polyelectrolyte multilayer systems is a general approach for preparation of macromolecular co-assemblies containing metal ions. It was found that polyelectrolyte films prepared via the LbL technique are able to bind metal ions [82, 83, 103] as they typically contain polyanions (such as PSSNa and PAA) and polycations [such as PEI, poly(allylamine hydrochloride) (PAH), and poly(N,N-diallyldimethylammonium chloride (PDADMAC)].
The functional groups of polyions can coordinate metal ions, thereby acting as ligands. The sorption of metal ions from solutions is a common way of embedding metal ions into multilayer polyelectrolyte films. For example, Ag+ ions were incorporated into PAA/PAH multilayers from solutions of silver acetate [104]. A variation in the pH made it possible to change the content of metal ions in the films [105]. Also, palladium ions [104] were successfully included into PAA/PAH multilayers from aqueous solutions containing [Pd(NH3)4]2+ ions. It should be mentioned that polyelectrolyte multilayers containing metal ions can be further used as precursors for preparation of polymer–metal nanohybrids [82, 83, 103–105].
Control of the assembly of multilayer polyelectrolyte films and the incorporation of metal ions provide the possibility for development of polymer matrices characterized by a varying content and distribution of the metal ions. The spatial localization of Ag+ within multilayer polyelectrolyte films was realized using two different types of bilayer building blocks [104]. By adding fully ion-paired oppositely charged polyelectrolyte, a series of PAH/PAA bilayers, which do not include metal ions, can be inserted between the bilayers, which contain Ag+. This technique allows one to prepare a sandwich-like metallo-containing film structure.
An alternative technique is based on a directed precoordination of metal ions with functional groups on polymers, which transforms them into polyions. This method was developed [106] for precursors of silver NPs synthesized in PEI/PAA films. Specifically, the PEI-Ag+ polycation was used as a polymeric component for construction of polyelectrolyte multilayers with PAA.
Another approach for preparation of similar macromolecular co-assemblies containing metal ions, which is discussed in the literature [80, 92, 93, 107, 108], uses so-called “coordination polymers”. Use of bifunctional compounds with two terminal ligand groups, differing in a spacer length, demonstrates that chains and rings can be formed in aqueous solutions via coordinating metal ions [109]. An increasing solution concentration favors generation of chains (Fig. 15). This makes preparation of reversible coordination polymers possible [80, 109]. IPECs may include such coordination polymers as one of the polymeric components, though cationic molecules have been used so far. For example, LbL assemblies were obtained on planar substrates and surfaces of colloid particles using Fe2+-metallo-supramolecular coordination polyelectrolytes (Fig. 16a) as polycations [108]. In this strategy, multilayer fabrication is achieved by repeated immersion of the substrates in solutions containing Fe2+-metallo-supramolecular coordination polyelectrolytes and negatively charged polyions (Fig. 16b,c).

(a) Structure of water-soluble bifunctional ligands. (b) Schematic representation of the formation of coordination polymers and rings. Reprinted with permission from [109] Copyright 2003 American Chemical Society

(a) Formation of Fe2+-metallo-supramolecular coordination polyelectrolytes and a LbL assembly on planar (b) and spherical (c) surfaces derived from Fe2+-metallo-supramolecular coordination polyelectrolytes and PSSNa. Reprinted from [80] Copyright 2010 with permission from Elsevier
4.2 IPEC Hybrids Containing Metal Nanoparticles
A development of methods for preparation of polymer composites containing inorganic NPs is one of the hottest topics in nanoscience because of the unique electrophysical behavior, antibacterial and catalytic activity of such composites. During recent years, attention has been focused on elaboration of a simple single-stage method for production of polymer–metal hybrid materials directly in swollen polymer matrices via a reduction of metal ions incorporated into hydrogels [110–112] or bound to polyelectrolytes [81–85, 103–105, 113–117].
Due to simplicity, the chemical reduction of metal ions in a liquid phase is the most commonly applied approach for the preparation of NPs in aqueous and nonaqueous media. A number of compounds (aluminum hydrides, borohydride, hypophosphites, formaldehyde, salts of oxalic and tartaric acids, hydroquinone, hydrogen, hydrazine, etc. [77, 78]) can be used as reducing agents. A complete reduction usually requires multiple excess of a reducing agent. Generally, this approach is applicable for the synthesis of NPs of silver and noble metals. The NPs obtained through chemical reduction often contain impurities [78, 79]. A typical drawback of this approach is a broad size distribution of the generated NPs. However, chemical reduction in the presence of polymers may yield NPs of controlled sizes [78, 82–85, 103–105, 115, 116]. In this context, a specific technique [97, 112, 118, 119], whereby the polymers are used both as reducing agents of metal ions and for effective stabilization of the prepared NPs, should be mentioned.
Radiation–chemical approaches for the preparation of NPs were also found to be exceptionally useful, both for studying the processes that underlie the formation of NPs and for preparation of nanocomposites [81, 89, 90, 111, 114, 115, 120]. The main advantages of ionizing radiation for the preparation of NPs are control over the formation of reducing radiolysis products and controlled changes in the rate of reduction of metal ions in a broad interval [111, 120]. This method does not require any specific chemicals (reducing agents), thereby leading to NPs with high purity [78, 79, 111, 120]. Ionizing irradiation allows one to obtain NPs with a desired average size and a narrow size distribution [111, 120]. Due to high reduction potentials [121, 122] of radiolysis products, the radiation-induced reduction makes it possible to prepare NPs from many metal ions (not only those of noble metals). The synthesis of copper NPs having a metal lattice is an illustrative example of the advantages of the radiation–chemical method, whereas chemical reduction, as a rule, leads to formation of copper protoxide [121].
Reduction or precipitation of metal ions is commonly applied for preparation of inorganic NPs by so-called intermatrix synthesis [81–85, 103–106, 113–117, 123–127] in polyelectrolyte matrices. Stabilization of inorganic NPs in polymeric matrices prevents their aggregation and, in addition, allows control of their growth rate and size. Polyelectrolytes or ion-exchange resins, which are filled by metal ions, are widely used precursors for fabrication of nanocomposites. The intermatrix synthesis technique has proved to be applicable for preparation of catalytically, electrocatalytically, and magnetically active composites with zero-valent metals (e.g., Cu, Pd, Ag, and others) via a chemical reduction of metal ions in matrices of cation-exchange resins. Due to the fact that functional groups of the polymeric component appear to be regenerated after each cycle (converted back into their initial ionic form), undertaking consecutive cycles with other metals will result in the formation of NPs with advanced structures (e.g., bimetallic core–shell, tri-metallic core–sandwich, etc.) [116, 117]. A precipitation of metal ions opens the possibility to obtain nanocomposites on the basis of metal sulfide NPs [104, 116].

Intermatrix synthesis of palladium–nickel bimetal NPs in the multilayer film PAA/PAH. Reprinted with permission from [123] Copyright 2003 American Chemical Society
The advantages of synthesis of metal NPs in multilayer systems and their development are discussed in [103]. A number of studies [82–84, 104–106, 125–127] demonstrate that the LbL deposition of polyelectrolytes containing metal ions and subsequent reduction of these metal ions provides a straightforward technique for obtaining encapsulated NPs with a controlled size. Encapsulation into polymers appears to be advantageous because, apart from stabilization and protection of NPs, polymers offer unique possibilities for both a modification of the environment around catalytic sites and a change of access to these sites [103]. Various polyelectrolytes and metal ions with different binding activities can be used for assembling LbL-based hybrids, which are precursors for polymer–inorganic composites. Control of the precursor structure through conditions for a buildup of the LbL films and regimes of metal ion reduction provide prerequisites for the development of composites containing NPs with various sizes and even with different spatial distributions of NPs [105, 106]. The PAA/PEI films with silver NPs exhibit electrocatalytic behavior and antibacterial activity [106]. Palladium–nickel bimetallic core–shell NPs with magnetic properties were obtained by chemical reduction in the PAA/PAH films [123]. Through a chemical reduction, palladium NPs with a diameter of 2 nm were obtained as seeds for further growth of Ni2+ shells, with control of their thicknesses (Fig. 17).
The LbL technique is widely applied for assembly of preformed NPs with oppositely charged polyelectrolytes [128–131]. The metal or semiconductor NPs of appropriate sizes in stabilizing media should be prepared before assembly of the LbL films. However, it appears to be quite difficult to realize an effective control over the concentration of NPs [103].
During the last few years, progress has been made in preparation of polymer–inorganic composites based on the LbL films, mainly related to membrane catalytic systems (Fig. 18). The LbL adsorption in porous polymeric membranes provides a simple way to create membrane catalysts. A universal approach for obtaining catalytic systems is a reduction of metal ions associated with charged groups of polyions, which constitute a film on the membrane, to obtain metal NPs [85, 125, 126] (Fig. 18c). On the other hand, the LbL adsorption of polyelectrolytes and a subsequent binding of preprepared gold or platinum NPs with charged groups on the polymers is an effective way of modifying the pores of hollow fibers and of developing catalytic membrane reactors [85, 125] (Fig. 18b).

Membrane catalytic systems: (a) TEM images of pores of an alumina membrane modified with a PAA/PAH/(platinum NP) film. Modification of membrane pore surfaces using (b) the LbL deposition of PAA/PAH/(platinum NP) films, or (c) the LbL deposition of PAA/PEI-Pt2+ films followed by a reduction. Reprinted from [125] Copyright 2009 with permission from Elsevier
IPECs containing metal ions swell in water and in water–organic media. Similarly to the precursor IPECs, they possess high permeability for polar low molecular weight substances and salts. These properties make it possible to reduce or precipitate metal ions directly in/into a polymeric matrix for a further preparation of inorganic NPs. Chemical and radiation–chemical approaches lead to composite materials with different structures [81, 114, 115, 132–134]. They contain NPs of metals or their oxides in matrices of IPEC {PAA-PEI}. The behavior of metallo-containing complex polymer materials is controlled by a size and a spatial distribution of the embedded NPs. An investigation of factors that regulate the formation of the metal NPs in the polymer matrices is of a key importance for a development of composites with required characteristics. Controlled variations in the content of the metal ions and their distribution in polymer samples [81, 115, 133] offer a unique opportunity to use IPECs to reveal such factors. The IPEC {PAA-PEI} loaded with metal ions has provided information about the main features of reduction of the metal ions, processes of nucleation and growth of the generated NPs as well as about the environmental conditions that control a spatial distribution of the NPs in the polymer matrices.
NPs of magnetic oxides show a pronounced superparamagnetic behavior. A polymer–inorganic hybrid material including particles of iron oxide was synthesized with the use of an IPEC and its magnetic properties were studied [134]. Alternative approaches were used for a preparation of metal nanoclusters. When IPEC films containing Fe3+ are kept in an alkaline solution, iron hydroxide nanoclusters are formed. According to the low-temperature Mössbauer spectra, subsequent drying of the IPEC films at 60–70°C results in the formation of iron oxide NPs in the IPEC {PAA-PEI}:
The structure and properties of such polymer–inorganic hybrid materials substantially depend on the method of reduction of the metal ions [81, 114, 115, 132, 133]. The chemical reduction of Cu2+ in IPEC matrices leads to the formation of copper protoxide NPs [81]. Micrographs of the irradiated film demonstrate that their size is of about 10 nm. The electric conductivity of the prepared hybrids is very low (Table 3).
The process of the reduction of Cu2+ in IPECs {PAA-PEI} may be schematically described by the reaction:
It is worth noting that the redox potential of NaBH4 is −1.24 V [79]. The redox potential for Cu2+/Cu+ is −0.15 V, and that of a reduction of Cu+ to metal atoms is −2.9 V [121]. For this reason, a chemical reduction of Cu2+ leads to Cu+ and, therefore, to the formation of copper protoxide NPs in an alkaline environment. Nanocomposites including Cu2О possess about the fivefold lower ion-exchange capacity of Cu2+ in comparison to the initial IPEC films. The dramatic decrease in the ion-exchange capacity shows that considerable fractions of functional groups of PAA and PEI are blocked because of their interaction with copper prototoxide NPs, acting as active filler.
The chemical reduction of Ni2+ in IPECs {PAA-PEI} results in metal NPs. The magnetization curves of dry films of the obtained nanocomposites qualitatively resemble those of metallic nickel [81], suggesting that the reaction proceeds as follows:
Table 3 demonstrates that this polymer–metal hybrid material shows a relatively high electric conductivity. The ion-exchange capacity of the obtained nanocomposite is several times lower than that of the IPEC precursor. This large effect indicates that nickel NPs strongly interact with the IPEC matrix as effectively as in the case of the IPEC-Cu2О nanocomposite. The swelling coefficient of PAA-PEI-Ni hybrids [10% (wt) Ni] obtained in matrices of noncrosslinked IPECs is about 100%. It considerably exceeds that for the triple metallo-containing IPEC {PAA-Ni2+-PEI} (Table 2) but is lower than the value for the noncrosslinked IPEC {PAA-PEI}. This result proves the interaction between NPs and the IPEC matrix and shows that, similarly to copper protoxide NPs, ultrafine nickel NPs act as active filler.
The irradiation source and irradiation conditions influence the structure of the resulting hybrid materials [114, 115, 132, 133]. Microdifractograms (Fig. 19) and X-ray images show that copper, silver, nickel, and palladium NPs can be successfully obtained via reduction of the corresponding metal ions in the IPEC {PAA-PEI} films, using electron accelerators as well as X-ray and γ-radiation sources [81, 114, 115, 132, 133].

Microdifractograms of irradiated IPEC {PAA-PEI} film loaded with (a) Cu2+ (initial content of 6% (wt), dose of γ-irradiation 320 kGy) Reprinted from [81] Copyright 2010 with permission from Elsevier; (b) Ni2+ (initial content of 6% (wt), dose of γ-irradiation 400 kGy) Reprinted from [81] Copyright 2010 with permission from Elsevier; (c) Cu2+ (initial content of 4% (wt), dose of e-beam irradiation 200 kGy) Reprinted with kind permission from Springer Science + Business Media from [115] Copyright 2011, Pleiades Publishing, Ltd.; and (d) Ag+ (initial content of 16% (wt), dose of X-ray irradiation 800 kGy) Reprinted with kind permission from Springer Science + Business Media from [115] Copyright 2011, Pleiades Publishing Ltd. The numbers 1–5 signify reflexes
In the IPEC films irradiated in water–organic media, the species involved in the reduction of metal ions and the formation of NPs were primarily produced by the radiolysis of water [81, 114, 132, 133]:
The hydrated electrons, H atoms, and hydrogen can act as reducing agents. The OH radicals may oxidize metal atoms and ions in intermediate oxidation states. To increase the efficiency of the reduction processes, it is common to use scavengers of OH radicals (e.g., aliphatic alcohols):
Both active species and stable radiolysis products can act as reducing species. The penetrating ability and dose rate are important parameters of ionizing irradiation. They influence the localization of the formation of reducing species as well as determine the contributions of processes related to generation and growth of NPs [111, 120].
The high penetrating ability of γ-irradiation and the EU-4 accelerator (Skobeltsyn Institute of Nuclear Physics, M.V. Lomonosov Moscow State University) [115, 133] provide a uniform generation of reducing species in the irradiated polymer system. The use of isotopic 60Co sources for a reduction of Cu2+ and Ni2+ embedded into the IPEC {PAA-PEI} allows one preparation of a material containing rather small NPs (2–5 nm) [81, 115] that are uniformly distributed throughout the IPEC films (Fig. 20a). This accelerator offers an exceptionally high dose rate (more than 800 Gy/s). Under these conditions, the high rate of formation of metal clusters inside an IPEC film proceeds mainly via active particles formed in the swollen polymer matrix. The probability of cluster formation is high, and, in this case, the ratio between the rate of NP nucleation and the rate of NP growth determines the appearance of ultrafine NPs (less than 1.5 nm in size).

Transmission electron microscopy (TEM) micrographs of irradiated IPEC {PAA-PEI} film loaded with (a) Cu2+ (initial content of 6% (wt), dose of γ-irradiation 320 kGy) Reprinted from [81] Copyright 2010 with permission from Elsevier; (b) Cu2+ (initial content of 4% (wt), dose of e-beam EU-4 irradiation 200 kGy) Reprinted with kind permission from Springer Science + Business Media from [115] Copyright 2011, Pleiades Publishing, Ltd.; (c, d, e) Cu2+ (initial content of 4% (wt), dose of X-ray irradiation 35, 70, and 140 kGy, correspondingly) Reprinted with kind permission from Springer Science + Business Media from [133] Copyright 2011, Pleiades Publishing, Ltd.; and (f) Ag+ (initial content of 16% (wt), dose of X-ray irradiation 800 kGy) Reprinted with kind permission from Springer Science + Business Media from [115] Copyright 2011, Pleiades Publishing, Ltd.
Use of the EU-4 accelerator (with electron energy of 250 keV) and the X-ray irradiator provide different regimes for NP nucleation and NP growth (Fig. 20b) on the surface and in bulk of the polymer matrix [115]. In the case of the EU-4 accelerator, particles of 2–10 nm in size are primarily formed on the film surface whereas the particles generated inside the film are larger (~30–50 nm). In the case of X-ray irradiation, the nucleation of NPs is similarly controlled by the formation and transport of radiolysis products in the IPEC film and in the external water–alcohol medium. However, in this case, some other regimes of reduction come into play, being determined by specific features of the energy transfer from X-ray radiation to the substance [81, 132, 133]. The structure of the material depends on the nature of the reduced metal ions and their initial content in the sample. The unique ability of the IPEC matrices to stabilize NPs allows one to obtain information about peculiarities of their formation at different stages of X-ray irradiation (Fig. 20c–e) [115, 133]. A study of nanostructures obtained in IPECs {PAA-PEI} under the radiation–chemical reduction of Cu2+ using X-ray irradiation demonstrated that the NPs are selectively formed in the subsurface layer of the IPEC films. The observed effect is due to favorable conditions for reduction of metal ions in the surface region because of the effective diffusion of radiolysis products from the outer water–alcohol medium and transport of Cu2+ through the IPEC matrix to the ligand vacancies near the interface boundary. The specific feature of the interaction of X-rays with matter at the physical stage results in significant heterogeneity of energy absorption and increases the rate of formation of reducing radiolysis products near the surface of the IPEC films. The reduction of Cu2+ is a slow, two-stage process [121]. The duration of the reduction of Cu2+ and the formation of metal clusters provides an almost complete localization of NPs near the surfaces of the samples. In contrast to the formation of copper nanoclusters, single-stage radiation-induced chemical reduction of Ag+ to silver atoms proceeds with a relatively high rate [122]. In this case, the efficient formation of NPs of 10–30 nm in size occurs not only at the surface of the IPEC film but also inside the IPEC film (Fig. 20f) [115].
Generation and transport of radiolysis products and the migration of metal ions across the polymer matrix control the formation of NPs in the irradiated IPEC films [115]. When irradiation is performed in a water–alcohol medium, metal clusters form via local processes, which proceed with participation of active species having high reduction potentials. Here, growth of NPs is provided by interfacial processes, in which the stable radiolysis products take part.
The ratio between the rates of NP nucleation and growth is determined by the dose rate and by the mechanisms of reduction of metal ions and formation of metal nanoclusters. The character of the formation of NPs is strongly controlled by the mechanisms of energy transfer from the ionizing radiation to the substance that determine the spatial distribution of radiolysis products. Variations in radiation parameters provide the different regimes for reduction on the surface of the IPEC film and inside the IPEC film. They make it possible to prepare composites both with NPs that are uniformly distributed throughout the polymer matrix and with a regular spatial distribution of NPs across the film thickness, including their localization in the subsurface layers.
Microdifractograms show a size effect on the packing of metal atoms in the prepared NPs. In the case of ultrasmall NPs with a mean size of 2–3 nm, the wide reflexes correspond to the interplane distances of ca. 1.20 Å and 2.00 Å for copper NPs (Fig. 19a) and 1.16 Å and 2.00 Å for nickel NPs (Fig. 19b) [81]. This suggests only the short order of metal atoms and demonstrates the amorphous character of the NPs. In the case of larger NPs, distinct reflections of metal lattices (Fig. 19c,d) (copper, nickel, or silver) are clearly observed [115, 132, 133].
Since metal NPs act as active filler in the IPEC {PAA-PEI} matrix, their formation leads to the immobilization of carboxylate groups on their positively charged surfaces [115]. A high radiation–chemical yield for the reduction of metal ions [81, 114, 115, 132, 133] embedded into the IPEC matrices can be used for the development of single-stage methods for preparation of nanocomposites, applying various types of ionization radiation.
In general, one may conclude that both chemical and radiation–chemical approaches provide effective reduction of metal ions directly in IPEC matrices. This leads to the formation of stabilized NPs. However, it was found that the chemical method is applicable only for a preparation of NPs in thermally crosslinked IPECs. Apparently, a poor regularity of the chemical processes causes noncrosslinked matrices to disintegrate. In contrast, the irradiation technique allows one to derive nanocomposites from both crosslinked and noncrosslinked IPEC films due to specific control over the radiation-induced processes.
4.3 Advanced Structures Based on Metallo-Containing IPECs
A controlled macromolecular assembly in solution is a universally exploited way for preparation of materials for nanotechnology. In particular, the LbL technology has been applied to the fabrication of coated core–shell particles and hollow capsules [103, 135, 136]. The fabricated core–shell and hollow particles may be used for various applications in catalysis, optics, drug delivery, and biosensing. They can serve as precursors for polymer–metal nanohybrids [103]. Reduction reactions are usually used for preparation of metal NPs inside of such multilayered polyelectrolyte systems. Specifically, polyelectrolytes whose charged groups are coordinated with metal ions may act as one of the components in the LbL systems. Alternatively, the sorption of metal ions from a solution leads to incorporation of metal ions into core–shell particles and hollow capsules [137]. Hollow capsules composed of PSS-doped polyaniline and PAH with bound Ag+ were used for a preparation of capsules containing silver [138]. In this case, the PSS-doped polyaniline acts as reducing agent for Ag+. Laser scanning during confocal microscope imaging can accelerate the reduction (Fig. 21a).

(a) Photoreduction of Ag+ on the surface of the (polyaniline-PSS/PAH)4 shells. Inset: (polyaniline-PSS/PAH)4 capsules after the addition of AgNO3 and photo-irradiation of the lower capsule (arrow). Scale bar: 10 mm. Reprinted with permission from [137] Copyright 2002 American Chemical Society (b) Formation of palladium NPs in a multilayered polyelectrolyte film on colloids. Reprinted with permission from [138] Copyright 2004 American Chemical Society
Micrometer-sized hollow spheres with metal NPs (10–30 nm) were obtained by photoreduction of Ag+ in polyelectrolyte multilayers comprising Ag+-PSS layers immobilized onto submicrometer-sized PS particles [138]. Hollow capsules with metal NPs can be formed either via a reduction of Ag+ followed by a core dissolution or by a core dissolution with a subsequent reduction of Ag+. The formed spherical nanocomposites with silver NPs were stable for long time (over 3 months). The silver-based core–shell particles and hollow spheres may find interesting applications in catalysis and molecular photoprinting. The PEI-Pd2+ metallo-containing polycations were assembled by PAA interlayers onto alumina particles of 150 μm diameter [138]. Then, metal NPs (1–3 nm) with catalytic activity were obtained by a reduction of Pd2+ with NaBH4 in a multilayered polyelectrolyte matrix (Fig. 21b).
Micellar IPECs (also referred to as complex coacervate core micelles) [139, 140] are formed from bis-hydrophilic diblock copolymers comprising a charged and a nonionic hydrophilic block, which are complexed with oppositely charged macromolecules. One important application of such IPECs is their potential for mimicking biological phenomena or gene delivery using complexes of DNA with cationic bis-hydrophilic diblock copolymers. The incorporation of metal ions via their coordination with functional groups on the polymers is a particular approach for the assembly of micellar IPECs [80, 92]. In aqueous media, a bis-hydrophilic diblock copolymer P2VPQ-b-PEO forms on its chains a reversible supramolecular polyelectrolyte through coordination of metal ions to functional groups of the P2VPQ block (Fig. 22a). The reversible supramolecular polyelectrolyte acts as a homopolyelectrolyte in this system. It was mentioned in Sect. 4.1 that an increase in solution concentration results in the formation of chains, in the case of bifunctional ligands, with an extended spacer (Fig. 15). The negatively charged coordination compound consisting of Zn2+ connected by ditopic ligands based on terdendate ligand groups [92] (Fig. 22a) induces assembly of the cationic blocks. The formation of complex micelles in the system containing the coordination polymer involves two simultaneous synergistic processes [80]: the formation of micelles and polymerization of coordination supramolecules due to a sharp local increase in the concentration of metallo-containing units. The cryo-TEM images prove the formation of micelles with cores, which are highly contrasted by metal ions (Fig. 22b).

(a) Formation of IPEC micelles in Zn-L2EO4/P2VPQ-b-PEO. (b) Cryo-TEM image of the aggregates in the mixture of P2VPQ-b-PEO/Zn-L2EO4 (0.02% P2VPQ-b-PEO with 1:1 molar ratio between P2VPQ-b-PEO and Zn-L2EO4). Scale bar: 200 nm. Reprinted with permission from [92] with Copyright 2007 American Chemical Society
The morphology of micellar IPECs is under the control of mixture stoichiometry. By direct mixing of the components at a 1:1 charge-to-charge ratio, spherical micelles (Fig. 22) with a radius of ca. 25 nm are formed [80, 92]. Worm-like micelles (Fig. 23) with a hydrodynamic radius of over 150 nm were found in a mixture with excess positive charge [80, 92].

(a) Illustration of the bridging effect of Zn-L2EO4 coordination oligomers (small rings) between positively charged premicelles. (b) Cryo-TEM image of aggregates in the mixture of P2VPQ-b-PEO/Zn-L2EO4 (0.04% P2VPQ-b-PEO with 3:1 molar ratio between P2VPQ-b-PEO and Zn-L2EO4). Scale bar: 200 nm. Reprinted with permission from [92] Copyright 2007 American Chemical Society
Design of advanced medicines and delivery drug systems has stimulated interest in microcapsulation of magnetic or antibacterial NPs incorporated into polymers [141, 142]. Fabrication of superfine catalytic or electronic devices is another reason for development of hierarchically organized metallo-containing polymer systems [95, 96, 143–145]. The nanostructured polymer systems give a fine tool for control of NP growth through variation of the interface interactions [143]. Immobilization of metal NPs into polymeric matrices such as micelles [146], microemulsions and microgels [147, 148], dendrimers [149, 150], block co-/terpolymer self-/co-assemblies [95, 96, 143, 145, 151], and spherical polyelectrolyte brushes [152, 153] provides a convenient approach for the fabrication of composites with various spatial orders of the NPs.
A method for preparation of a composite based on the three-dimensional nanosized copolymer template has been discussed [118]. The IPEC of the metallo-containing PEI-Ag+ polycation with the ionic amphiphilic diblock copolymer PS-b-PAA was obtained for further synthesis of encapsulated metal NPs. The silver NPs with diameter 20–40 nm were successfully synthesized in coronas of micelles (Fig. 24). In this case, PEI was used as both reducing and stabilizing agent. The cryo-TEM images suggest that the Ag+ content determines the size and spatial distribution of silver NPs.

Synthesis of silver NPs in the triple metallo-containing macromolecular co-assembly {PS-b-PAA-Ag+-PEI}. (a) Procedure for preparation of Ag/PS-b-PAA composite. (b, c) Typical TEM images of Ag/PS-b-PAA composites with different Ag content: (b) 0.012% (wt) PS-b-PAA, 0.02 mol/L AgNO3; and (c) 0.012.% (wt) PS-b-PAA, 0.05 mol/L AgNO3. Reprinted from [118] Copyright 2008 with permission from Elsevier
Polymer-assisted synthesis and environment-sensitive stabilization of metal NPs can be achieved through the formation of IPECs based on diblock copolymers in the presence of Ag+ [97]. Silver NPs were obtained in micellar IPECs consisting of P2VPQ-b-PEO and PAA-block-poly(N-isopropyl acrylamide) (PAA-b-PNIPAAm). Temperature was used to trigger the structural transition of a core–shell structure. The P2VPQ and PAA segments acted as containers for Ag+ ions within micellar cores (25°C) or shells (60°C). PEO is supposed to ensure a spontaneous reduction of Ag+ to Ag through oxidation of the oxyethylene groups [119] in the micellar IPECs. Control was demonstrated over the size of the formed silver NPs, over the size and shape of the micelles containing the metal NPs, and over the location of the silver NPs within the micellar IPECs. Spherical and elongated micelles with the metal NPs were observed. The authors suggested a potential application of such nanocomposites as environment-sensitive silver quantum dots and as antimicrobial agents in antifouling surface coatings that can be prepared upon exposure of hydrophilic surfaces to a solution of micellar IPECs containing the silver NPs.
The IPECs based on triblock terpolymers containing polybutadiene (PB), P2VPQ, and PMAA blocks acted as scaffolds for the preparation of polymer–inorganic nanohybrids [96]. The IPEC shell on a hydrophobic PB core (Fig. 25a) was assembled from the P2VPQ and PMAA blocks while the excess segments of the PMAA blocks formed a hydrophilic corona. The PMAA blocks and the discontinuous (patchy) intramicellar IPEC {PMAA-P2VPQ} shell were loaded with AuCl4 − (Fig. 25a). The polymer micelles with coordinated AuCl4 − were exposed to UV irradiation to generate gold NPs with a narrow size distribution (3–4 nm in size). Interestingly, the formation of gold NPs takes place predominantly at the interface between the intramicellar IPEC and the PMAA corona as well as within the PMAA corona (Fig. 25b). Further addition of a bis-hydrophilic diblock copolymer P2VPQ-b-PEO to the PB-b-P2VPQ-b-PMANa micelles generated multilayered micellar IPECs (Fig. 25c). In this case, gold NPs obtained via reduction of AuCl4 − incorporated into such structures were located exclusively within the IPEC shell surrounded by the PEO corona (Fig. 25d).

(a) Formation of gold NPs in PB-b-P2VPQ-b-PMAA triblock terpolymer micelles. (b) TEM micrograph of PB-b-P2VPQ-b-PMAA triblock terpolymer micelles loaded with gold NPs. (c) The IPEC from PB-b-P2VPQ-b-PMAA and P2VPQ-b-PEO. (d) Cryo-TEM micrograph of the IPEC from PB-b-P2VPQ-b-PMAA and P2VPQ-b-PEO, which is loaded with gold NPs. Reprinted with permission from [96] Copyright 2009 American Chemical Society
The gold, platinum, and palladium NPs were generated within cylindrical micelles [95] of a PB-b-P2VPQ-b-PMANa triblock terpolymer. The PB cylindrical cores were covered by an intramicellar IPEC patchy shell assembled from the P2VPQ blocks complexed with PMAA segments while the excess PMAA segments formed a hydrophilic corona (Fig. 26c). The metal NPs were obtained using UV irradiation (gold) or reduction with NaBH4 (platinum, palladium). TEM and cryo-TEM images demonstrate a difference in the localization of NPs within the polymer scaffold. The platinum and gold NPs (Fig. 26a,b) were randomly distributed all over the cylinders, indicating a location within both the PMAA corona and the intramicellar IPEC domains (Fig. 26b). At the same time, the situation is different for palladium NPs in the PB-b-P2VPQ-b-PMANa micelles (Fig. 26d–f). Here, the palladium NPs are located in the IPEC patches distributed along the cylindrical PB core. Thus, the experiments with templates based on micelles of ionic amphiphilic triblock terpolymers have demonstrated that both hydrophilic (PMAA corona) and rather hydrophobic (IPEC domains) compartments present in such self-assembled structures can be used for deposition of NPs.

TEM micrographs of PB-b-P2VPQ-b-PMAA cylinders/metal NP hybrids deposited from aqueous solution: (a) gold , (b) platinum, and (d) palladium. (c) The proposed solution structure. (e, f) Cryo-TEM images from palladium-containing PB-b-P2VPQ-b-PMAA. Insets show a higher magnification. Reprinted from [95] with permission from the Royal Chemical Society Copyright 2011
5 Conclusions and Perspective
Recent advances in controlled synthesis of macromolecules, in particular in controlled radical polymerization, have allowed the construction of polymers with various topologies and compositions. Successful design of star-shaped polymers, molecular polymer brushes, and amphiphilic block copolymers and terpolymers has stimulated extensive studies of self-organization phenomena in multicomponent macromolecular systems. Electrostatically driven co-assembly in such systems provides a simple route towards micelle-like polymeric structures with spatially separated domains (compartments) having different functionalities. An outer domain grants solubility in solution to the macromolecular co-assemblies while an inner domain (or domains) can accumulate from or release into the environment various compounds in response to variations in environmental conditions. Variations in the pH or in the ionic strength are an effective way to change the structure and properties of the formed IPECs. During the last few years, various advanced functional IPEC structures based on polyions (or polyionic species) with nonlinear (branched) topologies have been successfully prepared and thoroughly examined.
IPECs are proved to effectively bind metal ions. Such hybrid macromolecular co-assemblies containing noble and transition metal ions can be considered, therefore, as universal matrices for further preparation of polymer–metal nanocomposites with controlled and variable NP size as well as various spatial distributions of NPs in such polymer systems. In this context, the use of polyions (or polyionic species) with nonlinear (branched) topologies can be, beyond all doubt, advantageous and very attractive. The IPECs based on such polyions (or polyionic species) act as autonomous nanoreactors and provide the prerequisites for successful directed design of promising hybrids for applications related to medicine, biotechnology, and catalysis. During the next few years, a breakthrough in this field is anticipated, resulting in the formation of novel families of nanosized advanced functional materials, which can be further applied for the needs of rapidly developing nanotechnologies.
Abbreviations
- Cryo-TEM:
-
Cryogenic transmission electron microscopy
- EPR:
-
Electron paramagnetic resonance
- GPE:
-
“Guest” polyelectrolyte
- HPE:
-
“Host” polyelectrolyte
- IPEC:
-
Interpolyelectrolyte complex
- LbL:
-
Layer-by-layer
- NP:
-
Nanoparticle
- P2VPH+ :
-
Protonated poly(2-vinylpyridine)
- P2VPQ:
-
Quaternized poly(2-vinylpyridine)
- P4VPQ:
-
Quaternized poly(4-vinylpyridine)
- PAA:
-
Poly(acrylic acid)
- PAH:
-
Poly(allylamine hydrochloride)
- PANa:
-
Poly(sodium acrylate)
- PB:
-
Polybutadiene
- PDADMAC:
-
Poly(N,N-diallyldimethylammonium chloride)
- PDMAEMA:
-
Poly[2-(dimethylamino)ethyl methacrylate]
- PDMAEMAQ:
-
Quaternized poly[2-(dimethylamino)ethyl methacrylate]
- PEI:
-
Poly(ethylene imine)
- PEO:
-
Poly(ethylene oxide)
- PIB:
-
Polyisobutylene
- PMAA:
-
Poly(methacrylic acid)
- PMANa:
-
Poly(sodium methacrylate)
- PPNa:
-
Poly(sodium phosphate)
- PS:
-
Polystyrene
- PSSNa:
-
Poly(sodium styrene sulfonate)
- TEM:
-
Transmission electron microscopy
- PNIPAAm:
-
Poly(N-isopropylacrylamide)
- DDA:
-
Dodecylammonium
References
Kabanov VA (1997) Journal of Journals 1:35
Manning GS (2001) Macromolecules 34:4650
Sergeyev VG, Pyshkina OA, Gallyamov MO, Yaminsky IA, Zezin AB, Kabanov VA (1997) Prog Polym Sci 107:198
Smid J, Fish D (1988) In: Mark HF, Kroschwitz JI (eds) Encyclopedia of polymer science and engineering, vol 11. Wiley, New York, p 720
Tsuchida E, Abe K (1982) Adv Polym Sci 45:1
Philipp B, Dautzenberg H, Linow K-J, Kötz J, Dawydoff W (1989) Prog Polym Sci 14:91
Kabanov VA (1994) Polym Sci 36:143
Kabanov VA (2005) Russ Chem Rev 74:3
Thünemann B, Müller M, Dautzenberg H, Joanny JF, Löwen H (2004) Adv Polym Sci 166:113
Kabanov VA, Zezin AB, Kasaikin VA, Yaroslavov AA, Topchiev DA (1991) Russ Chem Rev 60:288
Petzold G, Nebel A, Buchhammer HM, Lunkwitz K (1998) Colloid Polym Sci 276:125
Buchhammer HM, Kramer G, Lunkwitz K (1995) Colloids Surf A 95:299
Buchhammer HM, Petzold G, Lunkwitz K (1999) Langmuir 15:4306
Zezin AB (1999) Chemistry Today 6:30
Zezin AB, Rogacheva VB, Yaroslavov AA, Mikheikin SV (2012) Proceedings of international science symposium on combating radionuclide contamination in agro-soil environment, Korijama, Japan, 8–10 March 2012, p 438
Zezin AB, Rogacheva VB, Yaroslavov AA, Mikheikin SV (2012) Proceedings of symposium on the application and R & D of the technologies of decontamination, remediation, and restoration of environment, Tokyo, Japan, 3 February 2012, p 93
Kabanov AV, Kabanov VA (1994) Polym Sci 36:157
Kabanov AV, Kabanov VA (1998) Adv Drug Deliv Rev 30:49
Matyjaszewski K, Davis TP (2002) Handbook of radical polymerization. Wiley-Interscience, Weinheim
Müller AHE, Matyjaszewski K (2009) Controlled and living polymerizations. Wiley-VCH, Weinheim
Prud’homme RE (1989) In: Saegusa T, Higashimura T, Abe A (eds) Proceedings of the IUPAC 32nd international symposium on macromolecules “Frontiers of macromolecular science”, Kyoto, Japan, 1–5 August 1988. Blackwell, Oxford, p 55
Kabanov VA, Papisov IM (1979) Vysokomol Soed Ser A 21:243
Baranovsky VY, Litmanovich AA, Papisov IM, Kabanov VA (1981) Eur Polym J 17:969
Michaels A (1965) Ind Eng Chem 57:32
Kalyuzhnaya RI, Volynskii AL, Rudman AR, Vengerova NA, Razvodovskii EF, Eltsefon BS, Zezin AB (1976) Vysokomol Soed Ser A 18:71
Gulyaeva ZG, Aldoshina IV, Zansokhova MF, Rogacheva VB, Zezin AB, Kabanov VA (1990) Vysokomol Soed Ser A 32:776
Zezin AB, Lutsenko VV, Rogacheva VB, Aleksina OA, Kalyuzhnaya RI, Kabanov VA, Kargin VA (1999) Polym Sci Ser A 41:1250
Zezin AB, El’tsefon BS, Rudman AR, Vengerova NA, Kalyuzhnaya RI, Valueva SP, Kopylova EM, Chepurov AK, Efimov VS, Kabanov VA (1987) Pharm Chem J 21:464
Zezin AB, Izumrudov VA, Kabanov VA (1989) Makromol Chem Macromol Symp 26:249
Tsuchida E, Osada Y, Sanada K (1972) J Polym Sci Polym Chem Ed 10:3399
Kabanov VA, Zezin AB (1984) Pure Appl Chem 56:343
Kabanov VA, Zezin AB (1982) In: Vol’pin ME (ed) Soviet Sci Rev Sect B Chem Rev, vol 4. Harwood Academic Publishing, p 207
Rogacheva VB, Ryzhikov SV, Zezin AB, Kabanov VA (1984) Vysokomol Soed Ser A 26:1674
Rogacheva VB, Ryzhikov SV, Shchors TV, Zezin AB, Kabanov VA (1984) Vysokomol Soed Ser A 26:2417
Kabanov VA, Zezin AB, Izumrudov VA, Bronich TK, Bakeev KN (1985) Makromol Chem Suppl 13:137
Bakeev KN, Izumrudov VA, Kuchanov SI, Zezin AB, Kabanov VA (1992) Macromolecules 25:4249
Izumrudov VA, Zezin AV, Kabanov VA (1991) Russ Chem Rev 60:792
Skorodinskaya AM, Kemenova VA, Chernova OV, Efimov VS, Lakin KM, Zezin AB, Kabanov VA (1983) Khim-Farm Zh 17:1463
Skorodinskaya AM, Kemenova VA, Efimov VS, Mustafaev MI, Kasaikin VA, Zezin AB, Kabanov VA (1984) Pharm Chem J 18:146
Zezin AB, Rogacheva VB, Komarov VS, Razvodovskii EF (1975) Vysokomol Soed Ser A 17:2637
Komarov VS, Rogacheva VB, Bezzubov BB, Zezin AB (1976) Vysokomol Soed Ser B 18:784
Zezin AB, Rogacheva VB, Valueva SP, Nikonorova NI, Zansokhova MF, Zezin AA (2006) Nanotechnol Russ 1:191
Pergushov DV, Borisov OV, Zezin AB, Müller AHE (2011) Adv Polym Sci 241:131
Xu Y, Plamper FA, Ballauff M, Müller AHE (2010) Adv Polym Sci 228:1
Pergushov DV, Babin IA, Plamper FA, Zezin AB, Müller AHE (2008) Langmuir 24:6414
Pergushov DV, Babin IA, Plamper FA, Schmalz H, Müller AHE, Zezin AB (2009) Dokl Phys Chem 425:57
Larin SV, Darinskii AA, Zhulina EB, Borisov OV (2009) Langmuir 25:1915
Ding J, Wang L, Yu H, Huo J, Liu Q, Xiao A (2009) J Phys Chem C 113:5126
Babin IA, Pergushov DV, Wolf A, Plamper FA, Schmalz H, Müller AHE, Zezin AB (2011) Dokl Phys Chem 441:219
Ge Z, Xu J, Wu D, Narain R, Liu S (2008) Macromol Chem Phys 209:754
Yin M, Ding K, Gropeanu RA, Shen J, Berger R, Weil T, Müllen K (2008) Biomacromolecules 9:3231
Xu FJ, Zhang ZX, Ping Y, Li J, Kang ET, Neoh KG (2009) Biomacromolecules 10:285
Nemoto Y, Borovkov A, Zhou Y-M, Talewa Y, Tatsumi E, Nakayama Y (2009) Bioconjug Chem 20:2293
Schallon A, Jérôme V, Walther A, Synatschke CV, Müller AHE, Freitag R (2010) React Funct Polym 70:1
Li D, Ping Y, Xu F, Yu H, Pan H, Huang H, Wang Q, Tang G, Li J (2010) Biomacromolecules 11:2221
Synatschke CV, Schallon A, Jérôme V, Freitag R, Müller AHE (2011) Biomacromolecules 12:4247
Förster S, Abetz V, Müller AHE (2004) Adv Polym Sci 166:173
Borisov OV, Zhulina EB, Leermakers FAM, Müller AHE (2011) Adv Polym Sci 241:57
Talingting MR, Voigt U, Munk P, Webber SE (2000) Macromolecules 33:9612
Pergushov DV, Remizova EV, Feldthusen J, Zezin AB, Müller AHE, Kabanov VA (2003) J Phys Chem B 107:8093
Pergushov DV, Remizova EV, Gradzielski M, Lindner P, Feldthusen J, Zezin AB, Müller AHE, Kabanov VA (2004) Polymer 45:367
Burkhardt M, Ruppel M, Tea S, Drechsler M, Schweins R, Pergushov DV, Gradzielski M, Zezin AB, Müller AHE (2008) Langmuir 24:1769
Chelushkin PS, Lysenko EA, Bronich TK, Eisenberg A, Kabanov AV, Kabanov VA (2004) Dokl Phys Chem 395:72
Lysenko EA, Chelushkin PS, Bronich TK, Eisenberg A, Kabanov VA, Kabanov AV (2004) J Phys Chem B 108:12352
Chelushkin PS, Lysenko EA, Bronich TK, Eisenberg A, Kabanov VA, Kabanov AV (2007) J Phys Chem B 111:8419
Burkhardt M, Martinez-Castro N, Tea S, Drechsler M, Babin IA, Grishagin IV, Schweins R, Pergushov DV, Gradzielski M, Zezin AB, Müller AHE (2007) Langmuir 23:12864
Kabanov VA (1994) In: Dubin P, Bock J, Davis R, Schulz DN, Thies C (eds) Macromolecular complexes in chemistry and biology. Springer, Berlin, p 151
Chelushkin PS, Lysenko EA, Bronich TK, Eisenberg A, Kabanov VA, Kabanov AV (2008) J Phys Chem B 112:7732
Ishizu K, Toyoda K, Furukawa T, Sogabe A (2004) Macromolecules 37:3954
Ishizu K (2005) Polym Degrad Stab 90:386
Störkle D, Duschner S, Heimann N, Maskos M, Schmidt M (2007) Macromolecules 40:7998
Duschner S, Störkle D, Schmidt M, Maskos M (2008) Macromolecules 41:9067
Krohne K, Duschner S, Störkle D, Schmidt M, Maskos M (2010) Macromolecules 43:8645
Larin SV, Pergushov DV, Xu Y, Darinskii AA, Zezin AB, Müller AHE, Borisov OV (2009) Soft Matter 5:4938
Xu Y, Borisov OV, Ballauff M, Müller AHE (2010) Langmuir 26:6919
Gössl I, Shu L, Schlüter D, Rabe J (2002) J Am Chem Soc 124:6860
Schubert US, Newkome GR, Manners I (eds) (2006) Metal-containing and metallo-supramolecular polymers and materials. ACS, Washington
Wohrle D, Pomogailo AD (2003) Metal complexes and metals in macromolecules: synthesis, structure and properties. Wiley-VCH, Weinheim
Pomogailo AD, Kestelman VN (2005) Metallopolymer nanocomposites. Springer, New York
Yan Y, Huang J (2010) Coord Chem Rev 254:1072
Zezin AB, Rogacheva VB, Feldman VI, Afanasiev P, Zezin AA (2010) Adv Colloid Interface Sci 158:84
Bruening ML, Dotzauer DM, Jain P, Ouyang L, Baker GL (2008) Langmuir 24:7663
Liu G, Dotzauer DM, Bruening ML (2010) J Membr Sci 354:198
Ruiz P, Macanás J, Muñoz M, Muraviev DN (2011) Nanoscale Res Lett 6:343
Macanás J, Ouyang L, Bruening ML, Muñoz M, Remigya J-C, Lahittea J-F (2010) Catal Today 156:181
Nicolais L, Carotenuto G (eds) (2005) Metal-polymer nanocomposites. Wiley-VCH, Weinheim
Shih C-M, Shieh Y-T, Twu Y-K (2009) Carbohydr Polym 78:309
Geng B, Jin Z, Li T, Qi X (2009) Sci Total Environ 407:4994
Yoksan R, Chirachanchai S (2009) Mater Chem Phys 115:296
Long D, Wu G, Chen S (2007) Radiat Phys Chem 76:1126
Landsmann S, Winter A, Chiper M, Fustin C-A, Hoeppener S, Wouters D, Gohy J-F, Schubert US (2008) Macromol Chem Phys 209:1666
Yan Y, Besseling NAM, de Keizer A, Drechsler M, Fokkink R, Cohen Stuart MA (2007) J Phys Chem B 111:11662
van der Gucht J, Spruijt E, Lemmers M, Cohen Stuart MA (2011) J Colloid Interface Sci 361:407
Lehn J-M (2007) Chem Soc Rev 36:151
Schacher FH, Rudolph T, Drechsler M, Müller AHE (2011) Nanoscale 3:288
Schacher F, Betthausen E, Walther A, Schmalz H, Pergushov DV, Müller AHE (2009) ACS Nano 3:2095
Voets IK, de Keizer A, Frederik PM, Jellema R, Cohen Stuart MA (2009) J Colloid Interface Sci 339:317
Zezin AB, Kabanov VA (1982) Sov Sci Rev B 4:207
Kabanov NM, Kokorin AI, Rogacheva VB, Zezin AB (1979) Vysokomol Soed Ser A 21:209
Decher G (1997) Science 277:1232
Sukhorukov GB, Donath E, Davis S, Lichtenfeld H, Caruso F, Popov VI, Möhwald H (1998) Polym Adv Technol 9:759
Lavalle P, Boulmedais F, Ball V, Mutterer J, Schaaf P, Voegel JC (2005) J Membr Sci 253:49
Shi X, Shen M, Möhwald H (2004) Prog Polym Sci 29:987
Joly S, Kane R, Radzilowski L, Wang T, Wu A, Cohen RE, Thomas LE, Rubner MF (2000) Langmuir 16:1354
Wang TC, Rubner MF, Cohen RE (2003) Chem Matter 15:299
Dai J, Bruening ML (2002) Nano Lett 2:497
Schutte M, Kurth DG, Linford MR, Cölfen H, Möhwald H (1998) Angew Chem Int Ed 37:2891
Yan Y, Harnau L, Besseling NAM, de Keizer A, Ballauf M, Rosenfeld S, Cohen Stuart MA (2008) Soft Matter 4:2207
Vermonden T, van der Gucht J, de Waard P, Marcelis ATM, Besseling NAM, Sudhölter EJR, Fleer GJ, Cohen Stuart MA (2003) Macromolecules 36:7035
Corain B, Burato C, Centomo P, Lora S, Meyer-Zaika W, Shmid G (2005) J Mol Catal A Chem 225:189
Chmielewski AG, Chmielewska DK, Michalik J, Sampa MH (2007) Nucl Instrum Methods Phys Res B 265:339
Porel S, Singh S, Harsha SS, Rao DN, Radhakrishnan TP (2005) Chem Mater 1:9
Ziolo RF, Giannelis EP, Weinstein BA, O'Horo MP, Ganguly BN, Mehrotra V, Russell MW, Huffman DR (1992) Science 257:219
Zezin AA, Feldman VI, Shmakova NA, Valueva SP, Ivanchenko VK, Nikanorova NI (2007) Nucl Instrum Methods Phys Res B 265:334
Zezin AA, Feldman VI, Abramchuk SS, Ivanchenko VK, Zezina EA, Shmakova NA, Shvedunov VI (2011) Polym Sci Ser C 53:61
Muraviev DN, Ruiz P, Muñoz M (2008) Phys Status Solidi A 205:1460
Ruiz P, Muñoz M, Macanás J, Turta C, Prodius D, Muraviev DN (2010) Dalton Trans 39:1751
Lei Z, Wei X, Zhang L, Bi S (2008) Colloids Surf A 317:705
Zhang DB, Qi LM, Ma JM, Cheng HM (2001) Chem Mater 13:753
Belloni J (2006) Catal Today 113:141
Ershov BG (1994) Russ Chem Bull 43:25
Ershov BG (2001) Russ Chem J 45:20
Wang TC, Rubner MF, Cohen RE (2003) Chem Mater 15:299
Ji T, Shi H, Zhao J, Zhao Y (2000) J Magn Magn Mater 212:189
Dotzauer DM, Abusaloua A, Miachon S, Dalmon J-A, Bruening ML (2009) Appl Catal B 91:180
Bhattacharjee S, Dotzauer DM, Bruening ML (2009) J Am Chem Soc 131:3602
Ouyang L, Dotzauer DM, Hogg SR, Macanás J, Lahitte J-F, Bruening ML (2010) Catal Today 156:100
Kotov NA, Dekany I, Fendler JH (1995) J Phys Chem 99:13065
Kleinfeld ER, Ferguson GS (1994) Science 265:370
Lvov Y, Ariga K, Onda M, Ichinose I, Kamitake T (1997) Langmuir 13:6195
Shmitt J, Decher G, Dressick WJ, Brandow SL, Geer RE, Shashidhar R, Culver JM (1997) Adv Mater 9:61
Zezin AA, Feldman VI, Dudnikov AV, Zezin SB, Abramchuk SS, Belopushkin SI (2009) High Energy Chem 43:100
Zezin AA, Feldman VI, Zezina EA, Belopushkin SI, Tsybina EV, Abramchuk SS, Zezin SB (2011) High Energy Chem 45:99
Suzdalev IP, Maksimov UV, Prusakov VE, Matveev VV, Imshennik VK, Novochihin SV, Zezin AB, Rogacheva VB, Valueva SP (2008) Nanotechnol Russ 3:729
Caruso F (2001) Adv Mater 13:11
Caruso F, Caruso RA, Möhwald H (1999) Science 282:1111
Antipov AA, Sukhorukov GB, Federik YA, Hartman J, Giersing M, Möhwald H (2002) Langmuir 18:6687
Kidami S, Dai J, Li J, Bruening ML (2004) J Am Chem Soc 126:2658
Cohen Stuart MA, Besseling NAM, Fokkink RG (1998) Langmuir 14:6846
Gohy JF, Varshney SK, Antoun S, Jérôme R (2000) Macromolecules 33:9298
Gubin SP (ed) (2009) Magnetic nanoparticles. Wiley-VCH, Berlin
De M, Ghost PS, Rotello VM (2008) Adv Mater 20:4225
Bronstein LM, Sidorov SN, Valetsky PM (2004) Russ Chem Rev 73:501
Saito H, Okamura S, Isuzu K (1992) Polymer 33:1099
Zhang J, Xu S, Kumacheva E (2004) J Am Chem Soc 26:7908
Ingert D, Pileni MP (2001) Adv Funct Mater 11:136
Manziek L, Langenmayr E, Lamola A, Gallagher M, Brese N, Annan N (1998) Chem Mater 10:3101
Antonietti M, Grohn F, Hartmann J, Bronstein L (1997) Angew Chem Int Ed 36:2080
Zhao M, Sun L, Crooks RM (1998) J Am Chem Soc 120:4877
Esumi K (2003) Top Curr Chem 227:31
Whilton NT, Berton B, Bronstein L, Hernze H, Antonietti M (1999) Adv Mater 11:1014
Sharma G, Mei Y, Lu Y, Ballauff M, Irrgang T, Proch S, Kempe R (2007) J Catal 246:10
Ballauff M, Lu Y (2007) Polymer 48:1815
Acknowledgments
The support from the Deutsche Forschungsgemeinschaft within the framework of the Sonderforschungsbereich 840 “Von partikulären Nanosystemen zur Mesotechnologie” and from the Russian Foundation for Basic Research (project no. 12-03-00762) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Pergushov, D.V., Zezin, A.A., Zezin, A.B., Müller, A.H.E. (2013). Advanced Functional Structures Based on Interpolyelectrolyte Complexes. In: Müller, M. (eds) Polyelectrolyte Complexes in the Dispersed and Solid State I. Advances in Polymer Science, vol 255. Springer, Berlin, Heidelberg. https://doi.org/10.1007/12_2012_182
Download citation
DOI: https://doi.org/10.1007/12_2012_182
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-40733-8
Online ISBN: 978-3-642-40734-5
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)