
Abstract
PEGylation refers to the covalent attachment of polyethylene glycol to proteins to reduce immunogenicity and extend their time in blood circulation. PEGylation is recognized as a promising method for increasing the therapeutic efficacy of medicines in clinical settings. The main advantages of PEGylation are (1) an increase in the size of drug molecule, resulting in reduced filtration by kidneys, (2) an increase in solubility, and (3) protection from enzymatic digestion and recognition by antibodies. A variety of molecules, such as small molecules, peptides, proteins, enzymes, antibodies and their fragments, and nanoparticles have been modified with PEG. Several PEGylated drugs have been approved by the US Food and Drug Administration (FDA) and several more are being tested in clinical settings. This review summarizes the methodologies and effects of PEGylation on drug delivery and highlights recent developments in PEGylated drugs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
In 1977, Abuchowski et al. reported that covalent attachment of polyethylene glycol (PEG) to albumin reduced the immunogenicity of albumin [1]. Subsequently, this group also found that PEGylated biomolecules had a longer blood circulation time than the corresponding normal biomolecules [2]. On the basis of this discovery, PEGylation has been widely recognized as one of the more promising methods for exploration of therapeutic drugs. This exploration includes developments in the methodologies of PEGylation [3]. In the first generation of PEGylated molecules, the target molecule was nonspecifically and irreversibly PEGylated with linear PEG chains (Fig. 1). In the second generation, the molecule was PEGylated with branched PEG chains at specific positions and covalently bound, so that PEG could be released by stimuli from the outside environment. A variety of molecules, including small molecules, peptides, proteins, enzymes, antibodies, antibody fragments, and nanoparticles have been modified with PEG. At present, 11 PEGylated drugs have been approved for clinical use by the US Food and Drug Administration (FDA) and several more trials in clinical settings are ongoing.

PEGylation technologies. Nonspecific and irreversible PEGylation is associated with several limitations, such as altered drug properties. To overcome this problem, a new generation of PEGylation technologies that enable highly specific and reversible PEGylation have been developed
2 PEGylation Chemistry
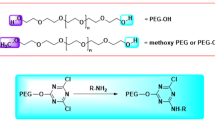
At present, the most frequently used methods for PEGylation are chemical conjugation between reactive groups in the drug, such as the primary amine of lysine in protein, and end-reactive PEG derivatives, such as the N-hydroxysuccinimide-terminated PEG derivative. These conventional methods are summarized in Fig. 2. Amine-specific and thiol-specific reagents are efficient and afford a good yield of PEGylated products. These simple methods can be applied to PEGylation of various molecules. Indeed, several PEGylated drugs have been approved for clinical use by the FDA. However, nonselective conjugations of the amino and/or thiol groups in protein molecules results in product heterogeneity, which often causes significant deactivation of the product. For example, 20–70% of native interferon-beta-1b (IFN-β-1b) antiviral activity was retained in mono-PEGylated IFN-β-1b, but the activity was greatly reduced or disappeared almost completely in multi-PEGylated IFN-β-1b [4]. Therefore, the development of site-specific PEGylation technology is quite important for developing more active and safer drugs.

Examples of activated PEG derivatives commonly used for PEGylation
2.1 Site-Specific PEGylation
PEGylation of drugs with an amine- or thiol-specific reagent is effective and thus the most popular method in current use. The thiol group of cysteine is often used for PEGylation of protein because PEGylation at cysteine is more specific than that of the amino group of lysine. Generally, cysteine forms dithiol linkages and the free thiol group is not available. To utilize the free thiol group for conjugation, it is necessary to engineer a new and free cysteine into the protein via recombination techniques. Although this approach works well, the genetic engineering involved in the process requires high skill, and protein misfolding and aggregation often occur during the purification process. To overcome this problem, Brocchini et al. developed site-specific PEGylation technology [5, 6]. An outline of their technology is shown in Fig. 3. Briefly, this technique involves the synthesis of a novel bis-thiol-specific PEG reagent (PEG monosulfone) containing a thiol-specific bis-alkylating group, which comprises an α, β-unsaturated carbonyl group possessing a sulfomethyl group at the α-position of the unsaturated double bond. After the reduction of the disulfide bond in the protein, both free reactive thiol groups react with the PEG reagent to produce disulfide-bridging PEGylation with a three-carbon bridge.

The N-terminal methionine residue of protein can also be employed for selective PEGylation using aldehyde-terminated PEG via a reductive amination reaction, because the N-terminal primary amine has a lower pKa of 7.8 than other amines such as lysines, whose pKa is 10.1 [7]. After reaction with aldehyde-terminated PEG at low pH, the resultant imine is reduced with sodium cyanoborohydrate to provide PEGylated protein (Fig. 4) [8, 9]. This technique was used for the production of Neulasta, which was approved for use by the FDA in 2002 [10].

PEGylation at the N-terminal methionine residue. The difference in pKa between the N-terminal amine and other amines in the protein enables site-specific PEGylation. After reaction with aldehyde-terminated PEG at low pH, reduction of the resultant imine produces PEGylated protein
2.2 Enzymatic PEGylation
Novel methods have been proposed by various researchers to achieve site-specific PEGylation using enzymatic PEGylation reactions. Sato et al. utilized transglutaminase (TGase; protein-glutamine γ-glutamyltransferase), an emerging enzyme [11]. This enzyme catalyzes an acyl transfer reaction between the γ-carboxyamide group of the glutamine residue (acyl donor) in a protein and a variety of primary amines (acyl acceptor) (Fig. 5) [12]. TGase is believed to require special sequential structures of the acyl donor for efficient modification; however, it is interesting to note that a variety of primary amines are accepted as acyl donors. Thus, TGase is a very useful reagent for protein modification [13]. An amino derivative of PEG, such as PEG-NH2, can be used as an acyl acceptor for PEGylation of proteins. Several clinically important proteins, including human growth hormone and interleukin-2, have been PEGylated using TGase [14].

Enzymatic site-specific PEGylation by transglutaminase (TGase). The alkylamine derivative of PEG can be introduced into proteins in a site-specific manner
2.3 Heterobifunctional PEG
Heterobifunctional PEG, which possesses different functional groups at the α- and ω-chain ends, is very useful in the field of drug delivery [15]. For example, heterobifunctional PEG can conjugate drug-containing nanoparticles with a targeting ligand (Fig. 6). In one study, a method for the synthesis of heterobifunctional PEG by direct ring-opening polymerization of ethylene oxide (EO) using a metal alkoxide initiator with a protected functional group was developed (Fig. 7) [16, 17]. This useful method was further developed by Akiyama et al., [18–21]. To date, a variety of heterobifunctional PEGs have been reported. Some popular derivatives are shown in Fig. 8. Several PEG derivatives are now commercially available from NOF, Japan.

Utility of the heterobifunctional PEG. Two different components such as drug and ligand can be introduced at the different ends of PEG

Synthesis of heterobifunctional PEG. (a) Nagasaki et al. developed a method for the polymerization of EO using an initiator containing defined functionalities [16, 17]. (b) Akiyama et al. further developed a synthetic route to prepare a series of heterobifunctional PEGs [18–21]. After the ring-opening polymerization of ethylene oxide, a second functional group was introduced at the ω-end of PEG

Examples of heterobifunctional PEGs popularly used for PEGylation
2.4 Linear and Branched PEGs
Early PEGylation technology utilized linear PEG chains for conjugation. As stated above, multiple and nonspecific conjugations often change the activity of native proteins significantly, and it has been reported that the large molecular weight of PEG causes a tendency to accumulate in the liver [22]. Branched PEG derivatives are effective candidates for solving these issues. The same PEGylation effect on a pharmaceutical can be obtained by introducing a smaller branched PEG with fewer conjugation points. The second generation of PEGylation technology often utilized branched PEGs because branched PEGylated products circulate longer in the blood than linear PEGylated products [23]. This effect is thought to be because of the steric hindrance of branched PEG [24]. PEGylation with branched-chain PEG has been adopted in the development of FDA-approved drugs, including PEGASYS [25], Macugen [26], and Cimzia [27].
Size-exclusion chromatography (SEC) showed no significant difference in size between branched and linear PEGylated proteins [28]. Therefore, the longer in vivo half-life of branched PEGylated drugs was not due to the size of the conjugate in solution, but probably to the more effective masking of the protein surface by branched PEGs.
Although the detailed mechanism of the longer circulation time of branched PEGylated protein is unclear, the architecture of PEG affects the release profile, the pharmacokinetics of the drug [29], and the behavior of the protein at the interface (e.g., protein absorption on hydrophobic surfaces [30]).
2.5 Releasable PEGylation
Although covalent attachments of PEG to drugs prolongs the lifetime of the drug in vivo, they often have the opposite effect on biological and pharmacological properties because the active site of drugs is inactivated due to shielding by massive PEG chains [31, 32]. Even optimized site-specific PEGylation often results in insufficient pharmacological properties. It also prevents internalization of the drug into the cell [33]. New emerging technologies, such as de-PEGylation from complex drugs, have been developed. Specific biodegradable linkages between the drug and PEG chains are introduced to allow de-PEGylation. After the release of PEG chains, drugs such as small molecules and proteins recover their original structures, activities, and cellular-uptake capacities.
Roberts et al. reported the synthesis of PEG–drug conjugates via an ester bond between the drug and PEG chain [34]. Despite its simplicity and efficacy, it is difficult to regulate the release specificity because numerous esterases exist in the cellular environment. In addition, many biologically active compounds often lack a hydroxyl or carboxyl group, which is required for ester formation. In this system, residues of linking reagent that connect PEG to the drug were left on the parent molecules, even after the cleavage of PEG chains [34]. These residues may affect the biological activities of the drug and might be a potential source of immunogenicity.
Shulman et al. synthesized bifunctional linking reagents containing 2-sulfo-9-fluorenyl-methoxycarbonyl (FMS) to produce a PEG conjugate that can be cleaved by spontaneous hydrolysis under physiological conditions, on the basis of the FMS cleavable system (Scheme 1) [35]. Amino groups in drugs can be utilized for conjugation to PEG in this case.

Releasable PEGylation based on the FMS principle
Zalipsky et al. reported a drug–PEG conjugate via a benzyl carbamate linkage (Scheme 2) [36]. This linkage is cleaved by a benzyl elimination reaction initiated by the thiolytic cleavage of disulfide in the para or ortho position. Filpula et al. developed a series of releasable PEG linkers that enable the controlled release of drugs [37]. In their system, the cleavage reaction is initiated by a trigger reaction, such as ester bond cleavage by esterase (Scheme 3). By controlling the rate-determining step with an optimized linker structure, the release rate of PEG chains can be controlled. For example, the introduction of steric hindrance, which slows the triggered hydrolysis reaction of esters, results in a diminishing release rate [37].

Drug–PEG conjugate via a benzyl carbamate linkage and its thiolytic cleavage

Releasable PEGylation based on 1,6-benzyl elimination prodrug strategy
ProLynx LLC has developed another type of releasable PEGylation technology based on the β-elimination reaction shown in Scheme 4. In this system, the release rate of the native drug is determined by the acidity of the proton adjacent to the modulator. Half-lives of molecules prepared using this approach range from several hours to several weeks.

Releasable PEGylation based on the β-elimination reaction
The concept of de-PEGylation can be applied to the development of nanoparticle-based drug delivery systems. PEG is used for the modification of liposomes to increase their blood circulation time [38]. However, it also prevents cellular uptake, resulting in a decrease in therapeutic efficiency; thus, modifications of the liposome surface with PEG interfere with membrane fusion to the cell membrane and liposome decomposition [39]. One of the possible strategies to solve this problem is to cleave the PEG chains after the nanoparticle reaches the target site (Fig. 9). This system of de-PEGylation of liposomes is also useful in avoiding the immune response called the accelerated blood clearance phenomenon (ABC phenomenon), in which the circulation time of a second dose of injected PEGylated liposome is substantially reduced [40, 41]. Harashima et al. developed a multifunctional envelope-type nanodevice (MEND) for a nonviral gene delivery system [39, 42]. In this system, multiple device functions are assembled into a single system. In particular, PEGs on the surface of liposomes were designed to be cleaved by enzymes such as matrix metalloproteinases (MMPs) that are specifically expressed at tumor sites. Removing PEGs resulted in an improvement of cellular uptake and subsequent endosomal escape of the liposome.

Utility of de-PEGylation technology in liposomes. (a) PEG derivative possessing a lipid moiety. The covalent bond between PEG and the lipid moiety can be cleaved by stimuli such as those within the acid environment of cancer and inflammation. (b) After binding the target cell via specific recognition of the receptor by the ligand, PEG molecules on the surface of the liposome are cleaved. The release of PEG facilitates membrane fusion of the liposome and liposome decomposition, resulting in efficient drug delivery
3 Effect of PEGylation on Pharmaceuticals
3.1 Reduction of Renal Clearance
One of the remarkable properties of PEGylation is to increase the hydrodynamic radius in order to decrease renal clearance. For example, Kubetzko et al. have reported that antibody fragments (theoretically 29 kDa) showed retention volumes corresponding to a size range of 200–300 kDa by SEC upon mono-PEGylation with PEG (20 kDa) [43]. This demonstrates the strong hydrodynamic properties of PEGylated molecules. The increase in hydrodynamic radius significantly decreases renal clearance. Although the threshold of the molecular weight cut-off of renal filtration of protein is about 65 kDa, the 30-kDa PEG demonstrates minimal renal permeability [44].
3.2 Molecular Recognition of PEGylated Molecules
Although PEGylated molecules in the blood stream have longer half-lives than the parent molecules, studies have reported conflicting conclusions about changes in the binding affinities of PEGylated molecules. Chapman et al. demonstrated that site-specific modification of antibody fragments at the termini of PEG diminishes the loss of activity of the antibody fragment [45]. In contrast, Kubetzko et al. reported a fivefold decrease in apparent affinity upon attachment of the 20-kDa PEG molecule at the C-terminus of the antibody fragment [43]. By analyzing the binding kinetics, they found that the reduction in affinity was mainly due to a slower association rate constant, whereas the dissociation rate constant was nearly unchanged. Using a mathematical model, intramolecular and/or intermolecular blocking by tethered PEG were proposed as the main factors behind the decrease in the observed association rate constant. The model suggested that more than 90% of PEGylated ligands are not capable of binding the target, indicating that accessibility to PEGylated molecules is significantly restricted [43]. This model can partially explain the lower immunogenicity and higher enzymatic resistance of PEGylated molecules.
Although the affinity was decreased fivefold upon PEGylation, PEGylated antibody fragments showed an 8.5-fold higher accumulation in tumors than unmodified antibody fragments, because of a longer serum half-life [46].
3.3 PEGylation on the Surface
PEGylation technology is also relevant for solid surfaces. Immobilization of proteins, antibody fragments, and whole antibodies has been widely used in biosensing and bioseparation systems [47]. There are several factors that affect the ability of these systems. These factors include the quantity, density, conformation, and orientation of the immobilized molecules. A common method of immobilization of a protein is based on the reaction between reactive residues in the protein, such as lysine, and the reactive surface. Yoshimoto et al. immobilized antibody fragments (Fab′) on a gold surface through S–Au linkage [48]. However, after the initial absorption of Fab′ onto the gold surface, reactions between the interactive residues of Fab′ and the gold surface changed the conformation and orientation of Fab′, resulting in the inactivation of the antigen-binding function of Fab′. To overcome this problem, they used a mixed-PEG layer formation in which different molecular weights of PEG (2 and 5 kDa) were used for the formation of a densely packed PEG layer. Formation of a mixed-PEG layer was originally developed by our group [49]. A highly dense mixed-PEG layer almost completely prevented nonspecific protein absorption and facilitated biospecific interactions (Fig. 10) [48]. This methodology can be applied to the construction of targeted drug delivery systems, in which a ligand is needed on the surface of nanoparticles to bind the target molecule with high specificity and efficiency.

PEGylation on the surface. A highly dense PEG layer composed of mixed-PEG prevents nonspecific protein interactions and inactivation of Fab′
4 PEGylated Drugs
PEGylation technology has been applied to the development of various kinds of drugs, including small molecules, peptides, proteins, antibody fragments, whole antibodies, oligonucleotides, and macromolecules such as polymer micelles and liposomes. Currently, there are ten PEGylated drugs, which utilize proteins, enzymes, antibody fragments, and oligonucleotides, and a PEGylated nanoparticle named Doxil available on the market (Table 1). PEGylated small drugs are currently under investigation in clinical tests; however, there are no approved drugs available on the market. Sections 4.1–4.4 describe various PEGylated drugs that have been approved or are in clinical trials.
4.1 Small Molecules
PEGylation of small drugs has several advantages. First, PEGylation improves pharmacokinetic properties due to the increased blood circulation time. Second, the immunogenicity of immunogenic small drugs is reduced. Third, PEGylation increases drug accumulation in tumors via the enhanced permeability and retention (EPR) effect. Finally, the toxicity of the drug is reduced by the massive PEG molecule.
Enzon pharmaceuticals have developed a PEGylated drug called SN-38 (7-ethyl-10-camptothecin) [50]. SN-38 is an active metabolite of irinotecan and is produced by hydrolysis of CPT-11 [51]. Several problems arise in the development of drugs using SN-38 or CPT-11. First, carboxylesterase-2 is thought to be the main esterase that hydrolyzes CPT-11; however, the expression of this enzyme in the blood is low. Accordingly, only 1–9% of injected CPT-11 is converted to the active form SN-38. The second problem associated with CPT-11 is the opening reaction of the lactone-E ring, which results in a form that is inactive against the target protein. Finally, SN-38 cannot be used for systemic applications because of poor solubility. This problem was solved by PEGylation of SN-38. PEGylation of SN-38 by acylation of the 20-hydroxyl functional group improved water solubility and preserved the active lactone form in the circulation. To increase the loading of the drug onto PEG, multi-arm PEG (a four-armed-PEG derivative) was used to enable the conjugation of four drugs to one molecule [52]. This conjugate will now be assessed in further preclinical development and clinical trials [53].
4.2 Peptides, Proteins, Antibodies, and Antibody Fragments
The majority of approved PEGylated drugs are categorized into this group. Eight PEGylated proteins, including antibody fragment conjugates, have been approved to date. Adagen is the first approved PEGylated product in which bovine adenosine deaminase is randomly conjugated with a 5-kDa mono-methoxy PEG [54]. This conjugate is synthesized using PEG succinimidyl succinate. This activated ester group can be reacted with nucleophilic amino acid units such as lysine. Since the approval of Adagen, seven other protein–PEG conjugates have been approved. Although nonspecific PEGylation has been reported to decrease the activity of protein in some cases, several approved drugs employ nonspecific PEGylation. Krystexxa, which was approved in 2010 for the management of treatment-resistant gout and hyperuricemia, was also prepared using nonspecific PEGylation. A conjugate containing six strands of 10-kDa PEG per subunit was found to have a significantly longer half-life in blood and dramatic urate-lowering potency [55].
In contrast to nonspecific PEGylation, several protein–PEG conjugates have adopted site-specific PEGylation. Cimzia is a PEGylated anti-tumor necrosis factor (TNF)-α antibody fragment used for the treatment of Crohn’s disease and rheumatoid arthritis [27]. Recent progress in biotechnology has enabled low-cost production of the recombinant antibody fragment by Escherichia coli expression [56]. However, such proteins, which are obtained and purified from E. coli, often possess immunogenicity. PEGylation on the protein reduces the immunogenicity of the recombinant non-human protein. To prepare Cimzia, the C-terminal cysteine is reacted with maleimide, which is introduced at the end of the 40-kDa branched PEG chain [27].
Another type of PEG–antibody fragment is PEGylated di-Fab, in which two antibody fragments are attached to PEG [57]. CDP791 is prepared using a bis-maleimide PEG and a humanized antibody fragment, resulting in a divalent PEGylated Fab fragment. This unique architecture enables high affinity for vascular endothelial growth factor receptor 2 (VEGFR-2), resulting in reduction of solid tumors [58].
4.3 Oligonucleotide–PEG Conjugates
Oligonucleotide-based drugs such as antisense drugs, aptamers, and small interfering RNA (siRNA) have attracted considerable attention as promising therapeutic agents for the treatment of various human diseases [59]. To develop therapeutic oligonucleotides, several issues must be addressed [60]. These issues include the instability of native oligonucleotides and their rapid clearance from the blood. To overcome these problems, PEGylation of the oligonucleotide is very useful. For example, Macugen, which is used for the treatment of the wet form of age-related macular degeneration (ARMD), is an aptamer drug modified with branched 40-kDa PEG at the 5′-terminus [26]. A number of PEGylated oligonucleotides are now at various stages of clinical trials.
4.4 PEGylated Nanoparticles
Precise design of the surface of nanoparticles is very important for the efficient and specific delivery of the drug by the nanocarrier. Surface modification of nanoparticles, such as micelles and liposomes, with PEG represents an essential strategy for reducing nonspecific interactions with serum proteins and endothelial cells in the blood stream, as well as avoiding recognition by immune system components such as the reticuloendothelial system (RES) [61]. Thus, stabilized nanocarriers tend to yield long blood circulation times and facilitate accumulation in the tumor tissue through the effects of EPR. Tamura et al. reported the influence of the surface PEG density on nanoparticles in the blood circulation [62] by using a nanogel composed of chemically crosslinked poly[2-N,N-(diethylamino)ethyl methacrylate] (PEAMA) gel cores surrounded by PEG palisade layers [63]. Because of their chemically crosslinked polyamine gel core, the PEGylated nanogels show higher stability against extremely dilute and high salt conditions than self-assembled nanocarriers such as liposomes and micelles. This stable nanoparticle is suitable for studying the influence of the physicochemical properties of nanoparticles on pharmacokinetics. The density of PEG on the surface of the nanogels was controlled by the post-PEGylation method. It was clearly demonstrated that the blood circulation time of the post-PEGylated nanogels was definitely prolonged as the PEG content was increased [62].
4.4.1 PEGylated Liposomes
Several liposome-based drugs have been approved for clinical application [64]. One of the clinically approved liposomes is Doxil, a PEGylated liposome containing doxorubicin (DOX), which is used for the treatment of a number of diseases [65]. As shown in this case, in the field of liposome drug development, PEG is widely used to protect the liposome from recognition by opsonins, thereby reducing liposome clearance.
A number of PEGylation reactions with liposomes have been developed [66]. One of the methods utilizes lipophilic compounds that possess reactive groups such as amino and carboxyl groups. By incorporating these components into the bilayer membrane, 500–2,000 functional groups can be introduced onto the liposome surface. This functionalized liposome can be used for the preparation of PEGylated liposomes (Fig. 11).

Methods for the construction of PEGylated liposomes. (a) Liposomes possessing reactive groups, such as amino and carboxyl groups, can be prepared by incorporating lipophilic components containing these functional groups into a bilayer membrane. Functionalized liposomes can be PEGylated by reaction with activated PEG derivatives. (b) Preparation of PEGylated liposomes using PEG derivatives possessing lipid moieties
Another method for the preparation of PEGylated liposomes utilizes PEG, which possesses a lipid moiety at one end, in conjunction with low molecular weight lipid molecules during the preparation of the liposome (Fig. 11). This method was originally reported by Kilbanov et al. in 1990 [67]. They conjugated phosphatidylethanolamine with PEG possessing activated carboxyl groups. For liposome preparation, 7.4 mol% of the PEG lipid was incubated. At this ratio of components, no increase in the leakage of the entrapped compound was observed. This methodology was adopted for the preparation of Doxil.
4.4.2 Micelles
Polymeric micelles containing anticancer drugs were originally developed by Kataoka and Kabanov, independently [68, 69]. Anticancer drugs are incorporated into micelles via physical entrapment or chemical conjugation. A number of micelles are being assessed in clinical trials [70], and progress on the polymer micelle system is emerging. Nishiyama et al. reported the development of a cisplatin-loaded micelle [71], in which platinum is coordinated by carboxylate groups in block copolymers consisting of PEG and polyaspartate (Fig. 12). DOX has been also loaded into micelles by chemical conjugation. Bae et al. developed a novel method for the conjugation of DOX with PEG-b-poly(aspartic acid) diblock copolymers [72], via a hydrazine linkage, which enables the release of DOX in acidic environments such as acidosis and endocytosis. This property facilitates the stimuli-responsive delivery of the drug (Fig. 13).

Cisplatin-loaded micelle developed by Nishiyama et al. [71]. Cisplatin is bonded to block polymers via coordination by carboxylate groups in the core of the micelle

The structure of DOX-conjugated PEG-b-poly(aspartic acid) diblock copolymer. DOX molecules are covalently bonded to a diblock copolymer via hydrazine linkage, which can be cleaved in acidic conditions, enabling the release of DOX in a site-specific manner
4.4.3 Inorganic Nanoparticles
Inorganic nanoparticles have also attracted interest in the field of drug delivery [73]. These inorganic nanoparticles include calcium phosphate, gold, silicon oxide, and iron oxide. They can be prepared easily with controllable size and can be readily functionalized. Inorganic nanoparticles, however, are generally unstable and may be toxic in biological systems. Accordingly, surface modification is needed to improve the biological stability and biocompatibility.
Surface modification of inorganic nanoparticles with PEG is a very useful way to overcome this problem. For instance, thiol groups are suitable anchors on gold nanoparticles. A variety of drugs, such as small compounds, oligonucleotides, and proteins, have been delivered by gold nanoparticles that carry drugs co-immobilized with thiol-PEG [74, 75]. For instance, recombinant human TNF-α was immobilized on PEGylated colloidal gold nanoparticles [76] to facilitate preferential accumulation of TNF-α in tumors and minimal uptake in healthy organs. This TNF-α-immobilized gold nanoparticle has been evaluated in a Phase I clinical trial [77]. Our group has investigated the stabilization of nanoparticles by block copolymers possessing PEG as one of the segments. For example, PEG-b-poly[2-(N,N-dimethylamino)ethyl methacrylate] significantly improves the stabilization of gold nanoparticles under physiological conditions [78, 79]. Multi-anchoring of amino groups in the poly[2-(N,N-dimethylamino)ethyl methacrylate] segment strongly improves adsorption efficiency. Luminescent nanoparticles have also been modified by several block copolymers [80, 81].
5 Conclusions and Future Prospects
Since PEGylation of proteins was first reported in the 1970s, extensive research on PEGylation technology and pharmaceutical development of PEGylated molecules has been conducted. A variety of molecules, including small organic molecules, proteins, antibody fragments, and nanoparticles have been modified with PEG. Currently, 11 PEGylated drugs have been marketed, and many other PEGylated drugs are in clinical trials. In recent years, the success rate for bringing new drugs to market has been decreasing [82]. One of the reasons for this is that the FDA is highly focused on the safety of new drugs. In this regard, PEGylation is very useful because PEG is categorized as “generally regarded as safe” (GRAS) by the FDA. Although there are potential concerns regarding non-degradability, product heterogeneity, and accumulation of large linear PEG chains in the liver [22], PEG provides substantial benefits, such as reduced immunogenicity and antigenicity of the drug. As in the cases of PEG-INTRON, Neulasta, and Doxil, emerging drugs can be developed by PEGylation of previously commercialized non-PEGylated drugs. By further development of cost-effective PEGylation technologies that enable more controlled release of PEG from the drug and site-specific modifications to deliver homogeneous products, the market of PEGylated drugs will continue to grow.
References
Abuchowski A, van Es T, Palczuk NC, Davis FF (1977) Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J Biol Chem 252:3578–3581
Abuchowski A, McCoy JR, Palczuk NC, van Es T, Davis FF (1977) Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J Biol Chem 252:3582–3586
Roberts MJ, Bentley MD, Harris JM (2002) Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev 54:459–476
Basu A, Yang K, Wang M, Liu S, Chintala R, Palm T, Zhao H, Peng P, Wu D, Zhang Z, Hua J, Hsieh MC, Zhou J, Petti G, Li X, Janjua A, Mendez M, Liu J, Longley C, Zhang Z, Mehlig M, Borowski V, Viswanathan M, Filpula D (2006) Structure-function engineering of interferon-beta-1b for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective mono-PEGylation. Bioconjug Chem 17:618–630
Balan S, Choi JW, Godwin A, Teo I, Laborde CM, Heidelberger S, Zloh M, Shaunak S, Brocchini S (2007) Site-specific PEGylation of protein disulfide bonds using a three-carbon bridge. Bioconjug Chem 18:61–76
Brocchini S, Godwin A, Balan S, Choi JW, Zloh M, Shaunak S (2008) Disulfide bridge based PEGylation of proteins. Adv Drug Deliv Rev 60:3–12
Wong SS (1991) Chemistry of Protein Conjugation and Cross- linking. CRC Press, Boston
Hu J, Sebald W (2011) N-terminal specificity of PEGylation of human bone morphogenetic protein-2 at acidic pH. Int J Pharm 413:140–146
Lee H, Jang IH, Ryu SH, Park TG (2003) N-terminal site-specific mono-PEGylation of epidermal growth factor. Pharm Res 20:818–825
Renwick W, Pettengell R, Green M (2009) Use of filgrastim and pegfilgrastim to support delivery of chemotherapy: twenty years of clinical experience. BioDrugs 23:175–186
Sato H (2002) Enzymatic procedure for site-specific pegylation of proteins. Adv Drug Deliv Rev 54:487–504
Lorand L, Parameswaran KN, Stenberg P, Tong YS, Velasco PT, Jönsson NA, Mikiver L, Moses P (1979) Specificity of guinea pig liver transglutaminase for amine substrates. Biochemistry 18:1756–1765
Griffin M, Casadio R, Bergamini CM (2002) Transglutaminases: nature’s biological glues. Biochem J 368:377–396
Fontana A, Spolaore B, Mero A, Veronese FM (2008) Site-specific modification and PEGylation of pharmaceutical proteins mediated by transglutaminase. Adv Drug Deliv Rev 60:13–28
Otsuka H, Nagasaki Y, Kataoka K (2003) PEGylated nanoparticles for biological and pharmaceutical applications. Adv Drug Deliv Rev 55:403–419
Nagasaki Y, Iijima M, Kato M, Kataoka K (1995) Primary amino-terminal heterobifunctional poly(ethylene oxide). Facile synthesis of poly(ethylene oxide) with a primary amino group at one end and a hydroxyl group at the other end. Bioconjug Chem 6:702–704
Nagasaki Y, Kutsuna T, Iijima M, Kato M, Kataoka K, Kitano S, Kadoma Y (1995) Formyl-ended heterobifunctional poly(ethylene oxide): synthesis of poly(ethylene oxide) with a formyl group at one end and a hydroxyl group at the other end. Bioconjug Chem 6:231–233
Akiyama Y, Nagasaki Y, Kataoka K (2004) Synthesis of heterotelechelic poly(ethylene glycol) derivatives having alpha-benzaldehyde and omega-pyridyl disulfide groups by ring opening polymerization of ethylene oxide using 4-(diethoxymethyl)benzyl alkoxide as a novel initiator. Bioconjug Chem 15:424–427
Akiyama Y, Otsuka H, Nagasaki Y, Kato M, Kataoka K (2000) Selective synthesis of heterobifunctional poly(ethylene glycol) derivatives containing both mercapto and acetal terminals. Bioconjug Chem 11:947–950
Hiki S, Kataoka K (2007) A facile synthesis of azido-terminated heterobifunctional poly(ethylene glycol)s for “click” conjugation. Bioconjug Chem 18:2191–2196
Hiki S, Kataoka K (2010) Versatile and selective synthesis of “click chemistry” compatible heterobifunctional poly(ethylene glycol)s possessing azide and alkyne functionalities. Bioconjug Chem 21:248–254
Pasut G, Veronese FM (2007) Polymer-drug conjugation, recent achievements and general strategies. Progr Polym Sci 32:933–961
Zhao H, Yang K, Martinez A, Basu A, Chintala R, Liu HC, Janjua A, Wang M, Filpula D (2006) Linear and branched bicin linkers for releasable PEGylation of macromolecules: controlled release in vivo and in vitro from mono- and multi-PEGylated proteins. Bioconjug Chem 17:341–351
Veronese FM, Caliceti P, Schiavon O (1997) Branched and linear poly(ethylene glycol): Influence of the polymer structure on enzymological, pharmacokinetic and immunological properties of proteinconjugates. J Bioact Compatible Polym 12:196–207
Aghemo A, Rumi MG, Colombo M (2010) Pegylated interferons alpha2a and alpha2b in the treatment of chronic hepatitis C. Nat Rev Gastroenterol Hepatol 7:485–494
Campa C, Harding SP (2011) Anti-VEGF compounds in the treatment of neovascular age related macular degeneration. Curr Drug Targets 12:173–181
Patel AM, Moreland LW (2010) Certolizumab pegol: a new biologic targeting rheumatoid arthritis. Expert Rev Clin Immunol 6:855–866
Fee CJ (2007) Size comparison between proteins PEGylated with branched and linear poly(ethylene glycol) molecules. Biotechnol Bioeng 98:725–731
Veronese FM, Schiavon O, Pasut G, Mendichi R, Andersson L, Tsirk A, Ford J, Wu G, Kneller S, Davies J, Duncan R (2005) PEG-doxorubicin conjugates: influence of polymer structure on drug release, in vitro cytotoxicity, biodistribution, and antitumor activity. Bioconjug Chem 16:775–784
Pinholt C, Bukrinsky JT, Hostrup S, Frokjaer S, Norde W, Jorgensen L (2011) Influence of PEGylation with linear and branched PEG chains on the adsorption of glucagon to hydrophobic surfaces. Eur J Pharm Biopharm 77:139–147
Somack R, Saifer MG, Williams LD (1991) Preparation of long-acting superoxide dismutase using high molecular weight polyethylene glycol (41,000–72,000 daltons). Free Radic Res Commun 12–13:553–562
Knauf MJ, Bell DP, Hirtzer P, Luo ZP, Young JD, Katre NV (1988) Relationship of effective molecular size to systemic clearance in rats of recombinant interleukin-2 chemically modified with water-soluble polymers. J Biol Chem 263:15064–15070
Brandenberger C, Mühlfeld C, Ali Z, Lenz AG, Schmid O, Parak WJ, Gehr P, Rothen-Rutishauser B (2010) Quantitative evaluation of cellular uptake and trafficking of plain and polyethylene glycol-coated gold nanoparticles. Small 6:1669–1678
Robers MJ, Harris JM (1998) Attachment of degradable Poly(ethylene glycol) to proteins has the potential to increase therapeutic efficacy. J Pharm sci 11:1440–1445
Peleg-Shulman T, Tsubery H, Mironchik M, Fridkin M, Schreiber G, Shechter Y (2004) Reversible PEGylation: a novel technology to release native interferon alpha2 over a prolonged time period. J Med Chem 47:4897–4904
Zalipsky S, Qazen M, Walker JA 2nd, Mullah N, Quinn YP, Huang SK (1999) New detachable poly(ethylene glycol) conjugates: cysteine-cleavable lipopolymers regenerating natural phospholipid, diacyl phosphatidylethanolamine. Bioconjug Chem 10:703–707
Filpula D, Zhao H (2008) Releasable PEGylation of proteins with customized linkers. Adv Drug Deliv Rev 60:29–49
Yatuv R, Robinson M, Dayan I, Baru M (2010) Enhancement of the efficacy of therapeutic proteins by formulation with PEGylated liposomes; a case of FVIII, FVIIa and G-CSF. Expert Opin Drug Deliv 7:187–201
Hatakeyama H, Akita H, Harashima H (2011) A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: a strategy for overcoming the PEG dilemma. Adv Drug Deliv Rev 63:152–160
Ishida T, Kiwada H (2008) Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int J Pharm 354:56–62
Xu H, Wang KQ, Deng YH, da Chen W (2010) Effects of cleavable PEG-cholesterol derivatives on the accelerated blood clearance of PEGylated liposomes. Biomaterials 31:4757–4763
Hatakeyama H, Akita H, Ito E, Hayashi Y, Oishi M, Nagasaki Y, Danev R, Nagayama K, Kaji N, Kikuchi H, Baba Y, Harashima H (2011) Systemic delivery of siRNA to tumors using a lipid nanoparticle containing a tumor-specific cleavable PEG-lipid. Biomaterials 32:4306–4016
Kubetzko S, Sarkar CA, Plückthun A (2005) Protein PEGylation decreases observed target association rates via a dual blocking mechanism. Mol Pharmacol 68:1439–1454
Yamaoka T, Tabata Y, Ikada Y (1994) Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice. J Pharm Sci 83:601–606
Chapman AP, Antoniw P, Spitali M, West S, Stephens S, King DJ (1999) Therapeutic antibody fragments with prolonged in vivo half-lives. Nat Biotechnol 17:780–783
Kubetzko S, Balic E, Waibel R, Zangemeister-Wittke U, Plückthun A (2006) PEGylation and multimerization of the anti-p185HER-2 single chain Fv fragment 4D5: effects on tumor targeting. J Biol Chem 281:35186–35201
Andresen H, Bier FF (2009) Peptide microarrays for serum antibody diagnostics. Methods Mol Biol 509:123–134
Yoshimoto K, Nishio M, Sugasawa H, Nagasaki Y (2010) Direct observation of adsorption-induced inactivation of antibody fragments surrounded by mixed-PEG layer on a gold surface. J Am Chem Soc 132:7982–7989
Uchida K, Otsuka H, Kaneko M, Kataoka K, Nagasaki Y (2005) A reactive poly(ethylene glycol) layer to achieve specific surface plasmon resonance sensing with a high S/N ratio: the substantial role of a short underbrushed PEG layer in minimizing nonspecific adsorption. Anal Chem 77:1075–1080
Yu D, Peng P, Dharap SS, Wang Y, Mehlig M, Chandna P, Zhao H, Filpula D, Yang K, Borowski V, Borchard G, Zhang Z, Minko T (2005) Antitumor activity of poly(ethylene glycol)-camptothecin conjugate: the inhibition of tumor growth in vivo. J Control Release 110:90–102
Chabot GG (1997) Clinical pharmacokinetics of irinotecan. Clin Pharmacokinet 33:245–259
Zhao H, Rubio B, Sapra P, Wu D, Reddy P, Sai P, Martinez A, Gao Y, Lozanguiez Y, Longley C, Greenberger LM, Horak ID (2008) Novel prodrugs of SN38 using multiarm poly(ethylene glycol) linkers. Bioconjug Chem 19:849–859
Pastorino F, Loi M, Sapra P, Becherini P, Cilli M, Emionite L, Ribatti D, Greenberger LM, Horak ID, Ponzoni M (2010) Tumor regression and curability of preclinical neuroblastoma models by PEGylated SN38 (EZN-2208), a novel topoisomerase I inhibitor. Clin Cancer Res 16:4809–4821
Hershfield MS (1995) PEG-ADA: an alternative to haploidentical bone marrow transplantation and an adjunct to gene therapy for adenosine deaminase deficiency. Hum Mutat 5:107–112
Burns CM, Wortmann RL (2011) Gout therapeutics: new drugs for an old disease. Lancet 377:165–177
Laden JC, Philibert P, Torreilles F, Pugnière M, Martineau P (2002) Expression and folding of an antibody fragment selected in vivo for high expression levels in Escherichia coli cytoplasm. Res Microbiol 153:469–474
Jayson GC, Parker GJ, Mullamitha S, Valle JW, Saunders M, Broughton L, Lawrance J, Carrington B, Roberts C, Issa B, Buckley DL, Cheung S, Davies K, Watson Y, Zinkewich-Péotti K, Rolfe L, Jackson A (2005) Blockade of platelet-derived growth factor receptor-beta by CDP860, a humanized, PEGylated di-Fab', leads to fluid accumulation and is associated with increased tumor vascularized volume. J Clin Oncol 23:973–981
Ton NC, Parker GJ, Jackson A, Mullamitha S, Buonaccorsi GA, Roberts C, Watson Y, Davies K, Cheung S, Hope L, Power F, Lawrance J, Valle J, Saunders M, Felix R, Soranson JA, Rolfe L, Zinkewich-Peotti K, Jayson GC (2007) Phase I evaluation of CDP791, a PEGylated di-Fab' conjugate that binds vascular endothelial growth factor receptor 2. Clin Cancer Res 13:7113–7118
Davidson BL, McCray PB Jr (2011) Current prospects for RNA interference-based therapies. Nat Rev Genet 12:329–340
Lares MR, Rossi JJ, Ouellet DL (2010) RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol 28:570–579
Joralemon MJ, McRae S, Emrick T (2010) PEGylated polymers for medicine: from conjugation to self-assembled systems. Chem Commun (Camb) 46:1377–1393
Tamura M, Ichinohe S, Tamura A, Ikeda Y, Nagasaki Y (2011) In vivo and in vitro characteristics of core-shell-type nanogel particles: optimization of core cross-linking density and surface PEG density in PEGylated nanogels. Acta Biomaterialia. doi:10.1016/j.actbio.2011.05.027
Oishi M, Nagasaki Y (2010) Stimuli-responsive smart nanogels for cancer diagnostics and therapy. Nanomedicine (Lond) 5:451–468
Elbayoumi TA, Torchilin VP (2008) Liposomes for targeted delivery of antithrombotic drugs. Expert Opin Drug Deliv 5:1185–1198
Jiang W, Lionberger R, Yu LX (2011) In vitro and in vivo characterizations of PEGylated liposomal doxorubicin. Bioanalysis 3:333–344
Torchilin VP (2005) Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov 4:145–160
Klibanov AL, Maruyama K, Torchilin VP, Huang L (1990) Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett 268:235–237
Yokoyama M, Okano T, Sakurai Y, Ekimoto H, Shibazaki C, Kataoka K (1991) Toxicity and antitumor activity against solid tumors of micelle-forming polymeric anticancer drug and its extremely long circulation in blood. Cancer Res 51:3229–3236
Alakhov V, Klinski E, Lemieux P, Pietrzynski G, Kabanov A (2001) Block copolymeric biotransport carriers as versatile vehicles for drug delivery. Expert Opin Biol Ther 1:583–602
Matsumura Y, Kataoka K (2009) Preclinical and clinical studies of anticancer agent-incorporating polymer micelles. Cancer Sci 100:572–579
Nishiyama N, Okazaki S, Cabral H, Miyamoto M, Kato Y, Sugiyama Y, Nishio K, Matsumura Y, Kataoka K (2003) Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res 63:8977–8983
Bae Y, Kataoka K (2009) Intelligent polymeric micelles from functional poly(ethylene glycol)-poly(amino acid) block copolymers. Adv Drug Deliv Rev 61:768–784
Karakoti AS, Das S, Thevuthasan S, Seal S (2011) PEGylated inorganic nanoparticles. Angew Chem Int Ed Engl 50:1980–1994
Brown SD, Nativo P, Smith JA, Stirling D, Edwards PR, Venugopal B, Flint DJ, Plumb JA, Graham D, Wheate NJ (2010) Gold nanoparticles for the improved anticancer drug delivery of the active component of oxaliplatin. J Am Chem Soc 132:4678–4684
Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS, Mirkin CA (2006) Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science 312:1027–1030
Visaria RK, Griffin RJ, Williams BW, Ebbini ES, Paciotti GF, Song CW, Bischof JC (2006) Enhancement of tumor thermal therapy using gold nanoparticle-assisted tumor necrosis factor-alpha delivery. Mol Cancer Ther 5:1014–1020
Libutti SK, Paciotti GF, Byrnes AA, Alexander HR Jr, Gannon WE, Walker M, Seidel GD, Yuldasheva N, Tamarkin L (2010) Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin Cancer Res 16:6139–149
Ishii T, Otsuka H, Kataoka K, Nagasaki Y (2004) Preparation of functionally Pegylated gold nanoparticles with narrow distribution through autoreduction of auric cation by alpha-biotinyl-PEG-block-[poly (2-(N, N-dimethylamino) ethyl methacrylate)]. Langmuir 20:561–564
Miyamoto D, Oishi M, Kojima K, Yoshimoto K, Nagasaki Y (2008) Completely dispersible PEGylated gold nanoparticles under physiological conditions: modification of gold nanoparticles with precisely controlled PEG-b-polyamine. Langmuir 24:5010–5017
Kamimura M, Miyamoto D, Saito Y, Soga K, Nagasaki Y (2008) Design of poly(ethylene glycol)/streptavidin co-immobilized upconversion nanophosphors and their application to fluorescence biolabeling. Langmuir 24:8864–8870
Kamimura M, Kanayama N, Tokuzen K, Soga K, Nagasaki Y (2011) Near-infrared (1550 nm) In vivo bioimaging based on rare-earth doped ceramic nanophosphors modified with PEG-b-poly(4-vinylbenzyl phosphonate). Nanoscale. doi:10.1039/C1NR10466G
Mullard A (2011) 2010 FDA drug approvals. Nat Rev Drug Discov 10:82–85
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Ikeda, Y., Nagasaki, Y. (2011). PEGylation Technology in Nanomedicine. In: Kunugi, S., Yamaoka, T. (eds) Polymers in Nanomedicine. Advances in Polymer Science, vol 247. Springer, Berlin, Heidelberg. https://doi.org/10.1007/12_2011_154
Download citation
DOI: https://doi.org/10.1007/12_2011_154
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-27855-6
Online ISBN: 978-3-642-27856-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)