
Abstract
Cyclodextrins (CDs) are of interest to synthetic chemists because of their chemical stability and the possibility to modify these molecules in a regioselective manner. For supramolecular chemistry CDs are of great importance, since they represent a homologous series of water-soluble and chiral host molecules which can be used as models for studying weak interactions. In addition, their low price provides motivation for the discovery of new applications. Thus, CDs are used for the solubilization and encapsulation of drugs, perfumes, and flavourings. These applications are due to their low toxicity and their biodegradablity. Finally, they are of general interest because they are obtained from a renewable resource, starch (Eggersdorfer, M., Warwel, S., Wulff, CT.: Nachwachsende Rohstoffe Perspektiven für die Chemie. VCH, Weinheim (1993) .
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
6.1 Cyclodextrins as Hydrophilic Hosts
A supramolecular structure results from defined non-covalent interactions between several individual molecules. [1–9]. Host–guest complexes [10–13] are important examples of this type of structure. The host molecule offers the guest molecule a suitable environment, generally a cavity (Scheme 1). The driving forces for complexation can arise from coulombic, dipole–dipole, van der Waals, hydrophobic, solvatophobic, or hydrogen bonding interactions between host and guest [14, 15]. The outer sphere of the host should be compatible with the required solvent in order to avoid aggregation or insolubility problems.

Schematic illustration of complexation and polymerization of β-CD-complexed monomers
The formation of the complexes leads to significant changes of the solubility and reactivity of the guest molecules without any chemical modification. Thus, water insoluble molecules may become completely water soluble simply by mixing with an aqueous solution of native CD and CD derivatives, e.g. methylated (Me) or hydroxypropylated CD. The water solubility of these inclusion compounds enables detection of complex formation in solution by spectroscopic methods, such as NMR [16], UV, fluorescence, or circular dichroism spectroscopy, as well as by thermodynamic methods, e.g. microcalorimetry [17] or density [18, 19], or by solubility measurements. Likewise, mass spectrometry was used [20].
A crystal of the complexes suitable for X-ray diffraction analysis shows an orthorhombic structure, in which the guest molecule is complexed by the CD cavity (Fig. 1) [21,22]. It is interesting to note that the complexes are packed in columns, and the CD toruses are tilted in an alternatively oriented way, wherein the orientation of the entire column can be either upwards or downwards. The guest molecules are located almost perpendicularly to the host.

2D–ROESY measurements can be evaluated along with the constitution of the complexes (Fig. 2) [23]. Cross peaks can be detected between the protons of the guest molecule and the inner protons of the Me-β-CD torus.

2D-ROESY-NMR-spectrum of N-methacryloyl-1-aminohexane/Me-β-CD [23]
A further important and easy method to prove the formation of a complex is thin layer chromatography. For example, the R f-values of methylated β-CD (Me-β-CD, R f = 0.69) and uncomplexed phenyl methacrylate (R f = 0.92) in methanol are significantly different from the values of the complex (R f = 0.54) [24].
The FT–IR spectra of the complexed and uncomplexed guest molecules 11-(methacryloylamino)-undecanoic acid showed that the carbonyl bonds of the complexed monomer are shifted to higher frequencies from 1,708 to 1,712 cm−1. This clearly indicates the influence of the host ring molecule on the carbonyl vibration [25].
6.2 Influences of CDs on Co- and Homopolymerization
We applied CDs to solubilize various types of vinyl monomers which are otherwise water insoluble. This unique behaviour of CD allows it to copolymerize hydrophobic monomers complexed in CDs with water soluble comonomers.
6.2.1 Polymerization of CD Complexed Monomers
In earlier works we reported on the preparation and structural analysis of 1:1 host–guest compounds of several types of CDs with pyrrole or 3,4-ethylenedioxythiophene (EDT), and their oxidative polymerization in water. These complexes were prepared simply by adding one equivalent of monomer to one equivalent of CD in water (Fig. 3) [26].

Pyrrole or 3,4-ethylenedioxythiophene (EDT)/CD complex formation in water and oxidative polymerization of CD complexes
From some of these complexes, i.e. pyrrole/α-CD, EDT/α-CD, and EDT/β-CD, we were able to obtain single crystals that were studied by X-ray analysis. These show the herringbone fashioned cage-type crystal structure of pyrrole/α-CD, proving that it is a 1:1 complex with the pyrrole molecule located within the CD-cavity. The solid host–guest complexes are very stable under ambient conditions [21, 22]. After several weeks, the crystals remain unchanged while the unmodified monomers themselves change colour after a short time under the same conditions due to partial oxidation. Surprisingly, in the case of the EDT/α-CD, X-ray analysis showed the formation of a 1:1 non-inclusion channel-type packed supramolecular complex based on multiple hydrogen bonds. These colourless, crystalline pyrrole/CD and EDT/CD complexes that are barely soluble at room temperature were polymerized in water at 60°C under oxidative conditions.
All experiments showed that the corresponding polymers precipitated from solution. After filtration and washing with hot water, polypyrrole and poly(EDT) were obtained as dark powders in their oxidized state. Conductivity measurements showed that these materials have the same electrical properties (10–100 S cm−1) as conventionally prepared polypyrrole or poly(EDT) [27, 28].
We also reported on the first examples of the free radical polymerization and copolymerization of a fluorinated 2-vinylcyclopropane monomer in aqueous solution via the host–guest complexation with Me-β-CD using a water-soluble initiator [29]. 2-Vinylcyclopropanes containing electron-withdrawing and radical-stabilizing groups are known to undergo radical ring-opening polymerization leading to polymers consisting predominantly, but not solely, of 1,5-linear olefin structures. The fluorinated vinylcyclopropane was homopolymerized in the presence of 7 mol equivalent of random Me-β-CD in aqueous medium at 60°C by the water-soluble azo initiator V50 within 2 h, in 50% yield (Fig. 4).

Complexation of fluorinated vinylcyclopropane monomer with Me-β-CD and its homopolymerization and copolymerization with hexyl substituted vinylcyclopropane in aqueous solution
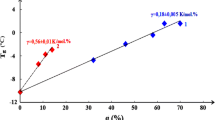
Fluorinated vinylcyclopropane was copolymerized with different ratios of hexyl substituted vinylcyclopropane via their respective complexes in Me-β-CD in aqueous solution in an analogous manner (Table 1). The prepared polymers exhibited liquid crystal behaviour. No crystallinity was detected by our DSC analyses of the fluorinated copolymers and the thermotropic mesophases occurred above the glass transition temperature. The fluorinated homopolymer is known to form smectic phases [30], and we assume that consistently the copolymers poly(1-co-2) also formed a smectic mesophase. This was generated by the self-assembly of the fluorinated side groups into an ordered registry of smectic layers. The degree of order of the mesophase could vary with copolymer composition according to the different isotropization enthalpies measured.
It is noteworthy that the formation of a mesophase was sustained in the copolymers even incorporating low proportions of mesogenic fluorinated side groups. Moreover, the introduction of repeat co-units with hexyl substituents lowered T g by internal plasticization and resulted in a somewhat broader mesophase range for the copolymers relative to the fluorinated homopoymer (e.g. T i − T g = 164°C for copolymer).
Furthermore, we have reported on the first examples of free radical homopolymerization and copolymerization of 2,3,4,5,6-pentafluorostyrene (3) with styrene (5) and its derivative 4-(N-adamantylamino)-2,3,5,6-tetrafluorostyrene (4) in aqueous solution via the host–guest complexation with Me-β-CD using water-soluble initiators (Fig. 5) [31]. The fluorinated monomers (3 and 4) and styrene (5) were complexed by Me-β-CD in water. The stoichiometries of the host-guest complexes were determined by NMR spectroscopy according to the Job method [32–34]. It was clearly shown that styrene (5) forms a defined 1:1 complex while the fluorinated monomers (3 and 4) are encapsulated by two Me-β-CD molecules. In the case of fluorinated monomer (3) this result was not expected since 3 and 5 have the same molecular scaffold and molecular modelling showed that the fluorinated styrene derivative 3 is only slightly bigger than styrene itself (Fig. 6) [35].

Job plots by considering the shifts of protons of three kinds of monomers in the presence of Me-β-CD

Homopolymerization of pentafluorostyrene in toluene and water by CD-complexation of the monomer respectively
The Me-β-CD/monomer complexes 3a and 4a were homopolymerized in water at room temperature using the redox initiator system K2S2O8/Na2S2O5 (Figs. 6 and 7). To compare our technique with traditional polymerization methods, all polymerizations were also carried out starting from the free monomers 3, 4, and 5 in organic solvents (toluene or benzene) at 80°C using AIBN as initiator (Table 2).

Homopolymerization of free monomer in toluene and with Me-β-CD in water followed by degradation of Me-β-CD with trifluoroacetic acid
In all cases almost quantitative yields of the homo and copolymers from complexed monomers were obtained. Poly(pentafluorostyrene) (P3a) and polymer P(3a-co-4a) could be isolated via simple filtration (Table 2). Polymer P4a was completely water-soluble due to the presence of non-covalently attached Me-β-CD. After treatment with trifluoroacetic acid to decompose the Me-β-CD ring, polymer P4 precipitated after some hours and could be isolated by filtration (Fig. 7).
As an example, myrcene, an isoprene dimer, is found in many natural oils. Most of the polymerizations of myrcene were affected by a metal catalyst in organic solvents [36–39]. We tried to synthesize copolymers containing myrcene, styrene and diethyl fumarate (DEF) by using Me-β-CD in water (Fig. 8) [40].

Copolymers containing myrcene, styrene and diethyl fumarate (DEF) after Me-β-CD mediated copolymerization using redox initiator in aqueous media
We have found that the polymerization in homogenous, aqueous CD solution is normally much faster and ends up with higher yields and molecular weights than polymerization under similar conditions in an organic solvent [41, 42]. The polydispersities (M w/M n) of polymers were similar in all cases. Only the M w/M n values of the polymer containing styrene monomer units showed higher values.
The copolymerization of CD-complexed styrene and DEF led to a precipitation of the corresponding water-insoluble copolymers [43]. The reactivity of the styrene complex is higher than that of complexed DEF. The conversions of both monomers were finished after about 6 h at room temperature. The lower reactivity of DEF in comparison to styrene can be explained by the fact that the double bond is sterically hindered by the two ester groups in the trans-1,2 position (Fig. 9).

Kinetic plots of the radical copolymerization of a 1:1 molar mixture of CD-complexed styrene (filled triangles) and diethyl fumarate (filled squares) in water at room temperature (using 2.5 mol% redox initiator K2S2O8/Na2S2O5)
Additionally, the double bond is preferentially covered by the CD ring. In contrast, the double bond of styrene is located more or less out of the CD centre, which allows easy access of the growing radicals. Furthermore, copolymerization parameters indicate that styrene is incorporated preferentially into the copolymer chain (r styrene = 1.95 ± 0.02 and r DEF = 0.97 ± 0.01). The reduced rate of copolymerization of DEF with styrene-rich compositions remains constant up to a fairly high conversion. It can be shown from the r styrene and r DEF values that the monomer composition changes during the progress of the reaction. Thus, the consumption of the DEF monomer is slower than that of styrene, leading to polymer compositions with an increased amount of homo segments. The strong influence of CD on the copolymerization parameters becomes obvious when comparing the values given in the literature for polymerization in an organic solvent (r styrene = 0.36 ± 0.04 and r DEF = 0.10 ± 0.02) giving rise to almost alternating copolymers [44]. In contrast to this, the CD-complexed monomers copolymerize in a completely different manner, as discussed above.
DEF (6) and diethyl maleate (DEM, 7) were complexed with equimolar amount of Me-β-CD and polymerized with redox initiator (K2S2O8/Na2S2O5) at room temperature and with VA44 at 50°C in aqueous media (Fig. 10) [22]. The homopolymerization of CD complexed monomers 6a and 7a with redox initiator led to a precipitation of the corresponding water-insoluble polymer P6, which was separated by centrifugation.

Preparation of poly(ethoxycarbonylmethylene) from 6 and 7 with/without Me β-CD
Normally, the maleic ester cis-isomers do to not give any homopolymer under similar condition as applied in the case of trans-isomers [45–47]. Surprisingly, in the presence of both Me-β-CD and redox initiator 7 undergoes homopolymerization via complexation with CD to give poly(ethoxycarbonylmethylene) P5. We evaluated the independent potential influence of CD and redox initiator on observed isomerization. It was shown that in the presence of only one of the components no isomerization takes place. Since 7 can be radically polymerized via a cis-trans-isomerization of the monomer, this reaction obviously needs both redox initiator and CD (Fig. 10).
The molecular weight of polymer was estimated by MALD-TOF MS (Fig. 11). The peaks show exactly the molecular masses of the polymers. The glass transition temperature (T g) of the polymers obtained from DEF 6 and DEM 7 are almost identical. The effective CD-mediated homopolymerization reactivity of 6a and 7a are visualized in Fig. 12. The uncomplexed monomers 6 and 7 did not polymerize under the same conditions.

MALDI-TOF spectrum of polymer P5

Kinetic plot of homopolymerization of methylated β-CD-complexed monomer 6a (filled triangles) and 7a (filled inverted triangles) and CD-uncomplexed monomer 6 (open squares) and 7 (open circles) using redox initiator in aqueous media at room temperature
The unreacted residual trans-monomer was found to consist of only fumarate after polymerization of the complex 6a by means of HPLC. In contrast, the residual 7a was detected to consist of cis- and trans-isomer during the polymerization. This means that the complexed cis-isomer 7a isomerized to trans configuration in the presence of radicals. Figure 13 shows the chromatograms of monomers obtained during the polymerization of complex 7a with redox initiator. It indicates that the peak of trans-monomer increased with increasing reaction time. Therefore, it was clear that the polymerization of cis-olefin monomers 7 in the presence of both CD and redox initiator takes place via an isomerization radical polymerization mechanism, i.e. maleates radicals isomerize first into fumarates and then these fumarates undergo radical polymerization.

HPLC Chromatograms of 7a showing cis/trans isomerization during the polymerization with redox initiator (R f(DEM) = 1.63 and R f(DEF) = 2.50)
Copolymerization of the CD-complexed macromonomer 8a with hydrophilic comonomers 2-acrylamido-2-methylpropansulfonate (AMPS) and N,N-dimethyl-acrylamide (DMAA) leads to formation of hydrophobically associative polymers (Fig. 14) [48].

Synthesis of hydrophobically associative polymers P6, P7 and P8
To study the aggregation of the synthesized polymers, 8-anilino-1-naphthalin-sulfonacid ammonium (ANS) was used as the fluorescence probe. ANS has a minimum solubility of 28 g L−1 in water but a higher solubility in hydrophobic media. With increasing polarity of the media its fluorescence intensity increases sensitively, and a simultaneous blue shift of the fluorescence band was also observed. Its fluorescence is independent on pH value for pH > 6. Therefore, ANS is a suitable fluorescence probe to study the aggregation of the polymers (P6–P8) [49]. According to Fig. 15 the fluorescence spectra of ANS in differently concentrated aqueous solutions of polymer P8 indicate the potential aggregation. It is interesting to note that the copolymer is a sidechain polyrotaxane. The content of CD was proved by GPC equipped with a chiral detector.

Fluorescence spectra of ANS in differently concentrated aqueous solution of polymers at RT (P8:contains 5 wt% of the macromonomer)
In Fig. 16 the maximum fluorescence intensity of ANS was plotted against the concentration of the polymers with different content of associative macromonomer (P6–P8). In diluted polymer solutions, the maximum fluorescence intensity increased gradually with increasing polymer concentration. At the same polymer concentration, ANS showed higher fluorescence intensity in a solution of polymer that contains more of the associative macromonomer 8.

Plots of the maximum fluorescence intensity as a function of polymer concentration at RT (P6: 1.0 wt% of 8; P7: 3.0 wt% of 8; P8: 4.9 wt% of 8)
The monomers N-methacryloyl-1-aminopropane (9), N-methacryloyl-1-aminobutane (10), N-methacryl-oyl-1-aminopentane (11), and N-methacryloyl-1-aminohexane (12) are synthesized directly from the corresponding amine and methacrylic acid by microwave irradiation [50], or classically by the treatment of amines with methacryloyl chloride (Fig. 17).

Structures of the complexed Me-β-CD monomers and subsequent polymerization
1H–NMR spectroscopic measurements were carried out to investigate the stoichiometry of the CD complexes [51]. D2O is used as the solvent at pH 7.3 and different mole fractions are considered ranging from 0.1 to 1.0 at increments of 0.1 with Me-β -CD as the host. The plot of −Δδ* • X CD vs the mole fraction X 4 for the Me-β -CD-complexed hexyl methacrylamide (12a) shows a maximum at X 4 = 0.50. This is a strong hint that a 1:1 complex was formed (Fig. 18).

Job plot of the complex of monomer 12a in D2O with Me-β-CD (pH 7.3), the maximum is at X 4 = 0.50
The hydrodynamic volumes of both Me-β-CD and the Me-β-CD complexes were measured by use of dynamic light scattering to prove the existence of molecularly dispersed complexes. Interestingly, results indicate that no aggregates are formed. However, because of guest monomer incorporation the hydrodynamic volume of the complex increases slightly in comparison to Me-β-CD (Fig. 19).

Particle size distribution by dynamic light scattering of free Me-β-CD (filled circles) and Me-β-CD complexes 9a (filled squares) and 12a (filled triangles) in water at 50°C
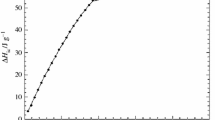
To evaluate the effect of Me-β-CD on polymerization kinetics, the free uncomplexed monomers 9–12 and the corresponding Me-β-CD complexes (9a–12a) are homopolymerized in water by a free radical mechanism under identical conditions. In the case of the aqueous polymerization of the free monomers 9–12 the initial polymerization rate increases with increasing water solubility. The opposite effect is observed in the case of the polymerization of the Me-β-CD complexed methacrylamide monomers 9a–12a. The polymerization rates are increased with increasing alkyl chain length of the complexed monomers 9a–12a and the decreasing water solubility of the free monomers 9–12 (Fig. 20).

Initial polymerization rate v0 of CD complexed monomers 9a–12a (open circles) vs water solubilities of monomers 9–12 (filled squares)
6.2.2 Polymerization of a CD Complexed Photoinitiator and a Water Soluble Monomer
For evaluating the influence of CD on the photoinitiated polymerization process, the polymerization of a water soluble monomer, N-isopropylacrylamide (NIPAAM), using complex of Me-β-CD and 2-hydroxy-2-methyl-1-phenylpropan-1-one (13) as the initiator, was performed in aqueous medium [52]. After the addition of 2 mol% of photoinitiator or CD-complexed photoinitiator the mixtures were irradiated simultaneously at constant temperature (20°C) with a high-pressure mercury lamp as the light source. In accordance with TLC measurements the photo initiator stays in the complexed form also in the presence of NIPAAM monomer, indicating that the equilibrium between free monomer and CD-monomer complex can be neglected.
UV spectroscopic measurements revealed that the polymerization reaction initiated with CD-complexed photo initiator is faster and, thus, ends up with higher yields than the polymerization reaction initiated with the same molar concentration of uncomplexed photo initiator. Radical photo polymerization is achieved by the homolytic fragmentation of carbon–carbon bonds of a photo-excited molecule as illustrated in Fig. 21.

Preparation of the CD-photoinitiator complex in water and proposed decomposition mechanism of the complex
6.2.3 Polymerization of CD Complexed Monomer and Water Soluble Monomer
Copolymerization of CD complexed myrcene (14) and NIPAAM was carried out (Fig. 22). The obtained polymer P13 was water soluble and little amounts of CD still remained in the polymers as detected by 1H NMR spectroscopy. The thermosensitive properties of aqueous solutions of copolymers were investigated by monitoring the changes of turbidity as a function of temperature. Surprisingly, P13 shows exceptional high LCST transition temperature of 80°C. Thus, the turbidity points of copolymers is significantly higher than of poly(NIPAAM) itself (32°C) because of the existence of hydrophilic CD complexes of the side groups.

Polymerization of CD-complexed myrcene with NIPAAM in H2O using redox initiator
The LCST value can be influenced, for example, by copolymerization of NIPAAM or by chemical modification of the acrylamide polymer itself. The slightly cross-linked LCST polymers, so-called hydrogels, find potential application in the medical and biochemical fields, for controlled drug delivery [53] and as materials for bioreactors [54]. One general explanation of the LCST effect is that strong hydrogen bonds exist between water molecules and the hydrophilic groups of the polymer at low temperature [55]. With increasing temperature, intramolecular interactions between hydrophobic chain components of the polymer increase.
For the investigation of the LCST behaviour further NIPAAM containing copolymers containing hydrophobic adamantine or cyclohexayl groups were synthesized (Fig. 23) [56–59].

Radical copolymerization of acrylamide with R = adamantyl (15) or cyclohexyl (16) and NIPAAM in DMF without any host compound; n/m = 1:20
Optical turbidity measurements showed a LCST for aqueous solutions of P14 up to 17°C. On the other hand, solutions of copolymer with cyclohexyl P15 have a transition at 31°C, which is 4°C lower than that of pure NIPAAM polymer P16 (Table 3). The lower LCST for P15 compared to P16 is attributed to the hydrophobic effect [60], which is the decrease of the LCST with increasing numbers of hydrophobic side-chain groups.
In addition, the thermosensitive properties of aqueous solutions of copolymers P17–P19 and of their supramolecular non-covalent cross-linked networks were investigated by monitoring the changes of turbidity as a function of temperature [61,62]. The N,N-dimethylacrylamide-containing copolymer P17 shows no turbidity point at temperatures between 10°C and 90°C in water. For copolymer P18 we obtained the LCST transition at 23°C; for copolymer P19 we found a cloud point of 21°C (Fig. 24). Both copolymers showed a sharp phase transition in response to a small temperature change. The fact that the turbidity points of copolymers P18 and P19 are significantly lower than that of poly(NIPAAM) itself (32°C) is caused by the hydrophobic adamantyl units in the copolymers; copolymers P18 and P19 are less soluble in water than a homopolymer of NIPAAM.

NIPAAM and N,N-dimethylacrylamide containing copolymers (n:m = 20:1)
To investigate the effect of adding monomeric and dimeric CD on the change of phase transition temperature of the polymers P18 and P19, we performed turbidity measurements at the same polymer concentration as above in the presence of a defined amount of Me-β-CD and CD dimer, respectively (Fig. 25 a,b) [61,62]. As reported previously [56–59], we found that addition of Me-β-CD led to cloud points of polymers P18 and P19 of 32°C, which correlates to the LCST of pure poly(NIPAAM). This increase of the cloud points relative to the cloud points of pure P18 and P19 results from the inclusion of the hydrophobic adamantyl units by Me-β-CD.

a,b Turbidity measurements of aqueous solutions of the polymers P18 a and P19 b without addition of host molecules and measurements of the supramolecular complexes of the polymers with CD dimer and Me-β-CD respectively. C P = 10 g L−1, C Me-β-CD = 10 g L−1, and C CD dimer = 5 g L−1, 10°C
When a CD dimer was added to the aqueous polymer solutions at half the molar ratio (relative to the amount of Me-β-CD), the cloud points decreased significantly. This observed effect again can be explained by the supramolecular cross-linkage of single polymer chains upon complexation of the CD dimer. The resulting supramolecular structures are restricted in their mobility and solubility, which is reflected in cloud points of 14.0°C for polymer P18 and 15.7°C for polymer P19. As a result of the LCST behaviour of the polymer/CD dimer mixtures the obtained hydrogels show different transparencies. The gel formed from copolymer P17 is transparent at room temperature, whereas those from polymers P18 and P19 are turbid at room temperature and transparent below their cloud points of 14.0°C and 15.7°C, respectively.
As another example, when the transparent solutions of the complexed polymers P20a were heated above 70°C, a sudden turbidity was observed [63, 64]. Surprisingly, when the solution was cooled, transparency was completely restored. Exact turbidity measurements were performed to evaluate the solubility as a function of temperature. We found that the complexed monomer 17a is completely soluble in the temperature range 10–85°C. This means that the monomer complex is stable enough to keep the monomer in solution independent of temperature. In contrast, the polymer complex P20a showed a definite clouding point at a temperature of 54°C for the heating run (Fig. 27 a, ba) and a clearing point of about 50°C for the cooling run (Fig. 27 a, bb).

a,b Transmittance of a solution of complexed polymer P20a vs temperature during heating a and cooling b
These temperatures were found to be a function of the molecular mass of the β-CD-free polymers. Figure 26 shows the reversible decomplexation–complexation process of polymer P20 in water. The transmittance change of a solution of P20a is plotted as a function of temperature in Fig. 27 a,b. The decrease in the transmittance of the complexed polymer to 0% occurs within a temperature interval of roughly 5°C. In the subsequent cooling step the polymer returns to solution by new formation of polymer/β-CD complex. In contrast, the β-CD free polymer is not soluble in water at any temperature in this range.

Temperature-dependent reversible unthreading of CD from the bulky side group of P20a during the heating procedure
The interesting polymer-solubility behaviour led us to compare this phenomenon with classical LCST effects. In our case, because of the reversible complex formation between the polymer P20 and CD, the optical effect is based on supramolecular interactions. This means that the discovered pseudo-LCST behaviour is a result of non-covalent interactions between the CD host and polymer guest. Furthermore, in this system competitive inhibition or control of the LCST is possible by addition of other suitable guest molecules of low molecular weight, for example, potassium 1-adamantylcarboxylate. CD complexes these molecules preferably and the polymer precipitates. This special effect cannot be observed in regular LCST systems.
A further important application of CD is the preparation of relatively low polydispersity polystyrenes (compared to radical CD polymerization) using the water-soluble RAFT reagent 3-benzylsulfanyl thiocarbonylsulfanylpropionic acid (TTC) complexed in aqueous Me-β-CD solution (Fig. 28) [65]. This method also allows the direct synthesis of amphiphilic block copolymers in aqueous solution without compatibility issues.

Schematic presentation of complexation and RAFT polymerization of Me-β-CD complexed styrene 18a with 3-benzylsulfanylthiocarbonylsulfanylpropionic acid (TTC) as RAFT agent
Controlled living radical polymerization of CD-complexed styrene in water can be conducted via the RAFT process, especially at low conversion (<20%). The molecular weight of PS can be controlled by variation of the RAFT agent concentration and the number-average molecular weight increases linearly with conversion (Fig. 29).

Evolution of the full molecular weight distributions in the CD-mediated styrene RAFT polymerization in aqueous solution at 70°C. RAFT agent/initiator to 10:1. Samples were taken after 75, 180, 300 and 420 min, respectively. The individual conversions to polystyrene are given within the figure
The polymers produced with the CD-RAFT system exhibited narrower polydispersities (1.23 < M w/M n < 2.36) than those without RAFT agent (5.24 < M w/M n < 9.21). However, the polydispersities in the CD-RAFT system are considerably higher (especially at increased conversions) than those in conventional (bulk or solution) RAFT systems and the molecular weight evolution deviates considerably from the theoretically expected one. We attribute these effects to the continuous precipitation of the polymer with increasing conversion, which effectively leads to a hybrid system between conventional and controlled living free radical polymerization.
Furthermore, we investigated temperature-dependent solution behaviour of Me-β-CD with poly(meth)acrylamides bearing bulky hydrophobic side groups (Fig. 30) [66].

Schematic illustration of the polymerization of 19a using 1 mol% water-soluble azo initiator (VA044) at different temperatures in water
The free radical polymerization of complexed monomer 19a was carried out at different temperatures in water in the presence of 1 mol% water-soluble azoinitiator. The temperature range of interest was 50–90°C, to determine v 0 below and above the T crit of 65°C of the given CD-polymer system (P21a).
In Fig. 31, the transparency τ of an aqueous solution of P21a is plotted against the temperature. While heating, it can be observed that the transparency of the solution of P21a decreases from almost 100% to 0% around 65°C (± 3°C). We would like to emphasize that the point of turbidity certainly depends on the sample concentration. With increasing concentration of CD, the LCST shifts to higher values [67,68]. During the cooling phase, the water insoluble polymer P21 remains insoluble. However, the reformation of the complex P21a takes place within 12 h of stirring at room temperature or below. We suspect that, once precipitated, the bulky adamantyl-side group of polymer P21 is hardly accessible by the Me-β-CD, so that reconstitution of the complex P21a takes hours (Figs. 30 and 31).

Transparency t of an aqueous solution of P21a (c = 30 g L−1) against the temperature during heating and cooling
The complexation–decomplexation equilibrium of the polymer is strongly entropy driven. The monomer complex 19a (Fig. 30) is stable up to 100°C, since it has a high mobility in solution. However, in contrast, a strong increase in total mobility of the polymer system takes place above T crit because the CD rings are released from the polymer.
The incorporation of a flexible spacer between the polymer backbone and the adamantyl groups strongly affects the thermosensitive properties of the supramolecular complex [67,68]. In Fig. 32, the turbidity measurement of a 100 g L−1 aqueous solution of polymer/Me-β-CD-complex P22a is presented. The heating run indicates the cloud point of P22a at 38.6°C, which is 6°C lower than that of P21a (Fig. 32). In the cooling run the transparency recovers from 0% to almost 100% at about the same temperature as in the heating run. Apparently recomplexation of the spacer-containing polymer P22 is significantly faster than that of polymer P21a, which contains directly attached adamantyl groups. These results correlate with the degree of mobility of the adamantyl groups attached to the polymer.

Rapid complexation of P22 by Me-β-CD. Transmittance as a function of temperature for an aqueous solution of polymer/Me-β-CD complex P22a at a heating/cooling rate of 1°C min−1. [P22a] = 100 g L−1 (13.75 g L−1 polymer, 86.25 g L−1 Me-β-CD)
In addition, we reported about the behaviour of an LCST copolymer P23 bearing a covalently attached solvatochromic 4-azastilbene dye and investigated the solvatochromism of this polymer in the presence of Me-β-CD [69].
A characteristic bathochromic shift from orange to dark red could be observed when the aqueous polymer solution at basic pH is heated above the LCST value of about 31°C (Figs. 33 and 34). This means that, during precipitation of the polymer due to heating above the LCST value, most of the polar water molecules are pushed out of the polymer coil. This causes a negative change of polarity next to the dye moiety, which results in a colour change. This temperature-dependent colour change is fully reversible. However, the protonated copolymer showed no colour change effect due to the solubility change at its LCST.

Transmittance (τ) of a solution of polymer P23 in water (5 mg mL−1) at pH 10 vs temperature during heating and cooling

Colour effects of copolymer P23 at pH 1 (no shift) and at pH 10 (bathochromic shift)
We clearly found that the bathochromic shift due to negative polarity change was also visible after excessive CD addition to the aqueous polymer solution below its LCST. This means that the complexation of the copolymer P23 by CD could also be recognized simply by the naked eye.
The unpolar cavity of CD creates an unpolar environment around the dye moiety. The colour change due to CD threading is nearly identical compared to the colour change caused by phase transition. This proves that the dye–dye interaction plays a minor role. As mentioned above, this effect influenced only the deprotonated polymer at high pH. Addition of CD to the polymer solution with the protonated dye (pH 1) showed only a small effect on the visible spectrum. UV measurements started directly after acid addition. Thus, decomposition of CD is not to be expected. This small effect might be due to weak interaction of CD with the protonated dye. In measurements of an aqueous solution of P23 without CD, glucose was excessively added to this solution to verify the host–guest interactions. As it was expected, no colour change could be detected (Fig. 35 a,b).

a,b Visible spectra of a 1.25 g L−1 aqueous solution of P23 with (filled squares) (addition of 60 mg of CD) or without (open squares) (addition of 60 mg of glucose) at pH 10 a and at pH 1 b
In conclusion, CDs constitute excellent hosts for homo- and copolymerizations. This important property has encouraged the use of CDs in a range of applications related to polymers, as described in this review. These investigations demonstrate the successful application of CDs in polymer synthesis in aqueous medium via free radical polymerization or via a oxidative recombination mechanism. CDs as reaction media are useful to alter both polymerization kinetics and copolymerization-parameters. New aqueous LCST systems can be designed with switchable physical properties.
As expected, within only a few decades an important revolution in the polymer chemistry through CD-mediated reactions might change the current production processes in chemical industry. Undoubtedly, the unique host properties of CDs should be taken into account when novel polymerization processes are created.
Abbreviations
- AIBN:
-
2,2′-Azobis(2-methylpropionitrile)
- AMPS:
-
2-Acrylamido-2-methylpropansulfonate
- ANS:
-
8-Anilino-1-naphthalinsulfonacid ammonium
- CD:
-
Cyclodextrin
- 2D-ROESY:
-
Two-dimensional rotating-frame overhauser spectroscopy
- DEF:
-
Diethyl fumarate
- DEM:
-
Diethyl maleate
- DMF:
-
N,N-Dimethyl formaldehyde
- DSC:
-
Differential scanning calorimetry
- MDAA:
-
N,N-Dimethylacrylamide
- EDT:
-
3,4-Ethylenedioxythiophene
- FT-IR:
-
Fourier transformations infra red
- GPC:
-
Gel Permeation Chromatography
- HPLC:
-
High pressure liquid chromatography
- LCST:
-
Lower critical solution temperature
- MALDI-TOF MS:
-
Matrix assisted laser desorption ionization time of flight mass spectroscopy
- Me:
-
Methylated
- M w/M n :
-
Polydispersities
- NIPAAM:
-
N-Isopropylacrylamide
- NMR:
-
Nuclear magnetic resonance spectroscopy
- r:
-
Copolymerization parameters
- RAFT:
-
Reversible addition–fragmentation chain transfer
- R f :
-
Retention factor
- T crit :
-
Critical solution temperature
- T g :
-
Glass transition temperature
- t:
-
Transparency
- TLC:
-
Thin layer chromatography
- TTC:
-
Thiocarbonylsulfanylpropionic acid
- UV:
-
Ultra violet spectroscopy
- V50:
-
2-Methylpropionamidine dihydrochloride
- VA44:
-
2,2′-Azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride
References
Lehn J-M (1985) Science 227:849
Lehn J-M (1988) Angew Chem 100:91
Lehn J-M (1988) Angew Chem Int Ed Engl 27:89
Lehn J-M (1990) Angew Chem Int Ed Engl 102:1347
Lehn J-M (1990) Angew Chem Int Ed Engl 29:1304
Rebek J Jr (1990) Angew Chem Int Ed Engl 102:245
Rebek J Jr (1990) Angew Chem Int Ed Engl 29:261
Vogtle F (1992) Supramolekulare Chemie. Teubner, Stuttgart
Vogtle F (1991) Supramolecular chemistry. Wiley, New York
Cram DJ, Cram JM (1974) Science 183:803
Cram DJ, Cram JM (1978) Arch Chrom Res 11:8
Ritter H, Tabatabi M (2002) Prog Polym Sci 27:1713
Ritter H, Tabatabi M (2003) Adv Macromol Supramol Mater Prog 41
Schneider HJ (1991) Angew Chem 103:1419
Schneider HJ (1991) Angew Chem Int Ed Engl 30:1417
Funasaki N, Ishikawa S, Neya SJ (2004) Phys Chem B 108:9593
Höfler T, Wenz G (1996) J Inclusion Phenom 25:81
Isaacs NS, Young DJ (1999) Tetrahedron Lett 40:3953
Abou-Hamdan A, Bugnon P, Saudan C, Lye PG, Merbach AE (2000) J Am Chem Soc 122:592
Hori K, Hamai S (1999) J Inclusion Phenom Macrocyclic Chem 34:245
Mejias L, Schollmeyer D, Sepulveda-Boza S, Ritter H (2003) Macromol Biosci 3(8):395
Choi SW, Frank W, Ritter H (2006) React Funct Polym 66:149–156
Steffens C, Choi SW, Ritter H (2006) Macromol Rapid Commun 27:542–547
Jeromin J, Ritter H (1998) Macromol Rapid Commun 19:377
Jeromin J, Ritter H (1999) Macromolecules 32:5236
torsberg J, Ritter H, Peilatzik H, Groenendaal L (2000) Adv Mater 12:567
Rodriguez J, Grande H-J, Otero TF (1997) Handbook of organic conductive molecules and polymers, vol. 2. Wiley, Chichester, pp 417
Heywang G, Jonas F (1992) Adv Mater 4:116
Choi SW, Kretschmann O, Ritter H, Ragnoli M, Galli G (2003) Macromol Chem Phys 204:1475
Galli G, Gasperetti S, Bertolucci M, Gallot B, Chiellini (2002) Macromol Rapid Commun 23:814
Cinar H, Kretschmann O, Ritter H (2005) Macromolecules 38:5078–5082
Blanda T, Horner JH, Newcomb M (1989) J Org Chem 54:4626–4636
Job P (1925) Compet Rend 180:928
Connors A (1987) Binding constants, the measurement of molecular complex stability. Wiley, New York
Wenz G, Han A (2006) Chem Rev 106:782
Frank RL, Adams CL, Blegen JR, Deanin R, Smith PV (1947) Ind Eng Chem 39:887
Marvel CS, Williams JLR, Baumgarten HE (1949) J Polym Sci 4:583
Clabaugh WS, Leslie RT, Gilchrist RJ (1955) Res Natl Bur Stand 55:261
Marvel CS, Hwa CCL (1960) J Polym Sci 28:25
Choi SW, Ritter H (2007) e-Polymers 045
Storsberg J, Ritter H (2000) Macromol Rapid Commun 21:236
Storsberg J, Aert H van, Roost C van, Ritter H (2003) Macromolecules 36:50
Choi SW, Ritter H (2004) Macromol Rapid Commun 25:716
Mayo FR, Lewis FM (1944) J Am Chem Soc 66:1594
Otsu T, Toyoda N (1981) Macromol Chem Rapid Commun 2:79
Toyoda N, Yoshida M, Otsu T (1983) Polym J 15:255
Otsu T, Shiraishi K (1985) Macromolecules 18:1795
Pang Y, Ritter (2006) Macromol Chem Phys 207:201
Yusa S-I, Shimada Y, Mitsukami Y, Yamamoto T, Morishima Y (2003) Macromolecules 36:4208
Goretzki C, Krlej A, Steffens C, Ritter H (2004) Macromol Rapid Commun 25:513
Steffens C, Choi SW, Ritter H (2006) Macromol Rapid Commun 27:542
Alupei IC, Alupei V, Ritter H (2002) Macromol Rapid Commun 23:55
Han JH, Krochta JM, Kurth MJ, Hsieh Y-L (2000) J Agric Food Chem 48:5278–5282
Heo J, Thomas KJ, Seong G, Crooks RM (2003) Anal Chem 75:22–26
Feil HY, Bae H, Feijen J, Kim SW (1993) Macromolecules 26:2496–2500
Ritter H, Sadowski O, Tepper E (2003) Angew Chem 115:3279
Ritter H, Sadowski O, Tepper E (2003) Angew Chem Int Ed 42:3171
Ritter H, Sadowski O, Tepper E (2005) Angew Chem 117:6253 (Corrigendum)
Ritter H, Sadowski O, Tepper E (2005) Angew Chem Int Ed 44:6099
Heskins M, Guilett JE (1969) J Macromol Sci Chem 2:1441
Kretschmann O, Choi SW, Miyauchi M, Tomatsu I, Harada A, Ritter H (2006) Angew Chem 118:4468–4472
Schmitz S, Ritter H (2006) Angew Chem Int Ed 45:4361–4365
Schmitz S, Ritter H (2005) Angew Chem 117:3000–3006
Schmitz S, Ritter H (2005) Angew Chem Int Ed 44:5658–5661
Köllisch H, Barner-Kowollik Ch, Ritter H (2006) Macromol. Rapid Commun 27:848
Steffens C, Kretschmann O, Ritter H (2007) Macromol. Rapid Commun 28:623
Kretschmann O, Steffens C, Ritter H (2007) Angew Chem 119:1–5
Kretschmann O, Steffens C, Ritter H (2007) Angew Chem Int Ed 46:2708
Koopmans C, Ritter H (2007) J Am Chem Soc 129(12):3502
Eggersdorfer M, Warwel S, Wulff (1993) Nachwachsende Rohstoffe Perspektiven für die Chemie. VCH, Weinheim
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer-Verlag London
About this chapter
Cite this chapter
Choi, S., Amajjahe, S., Ritter, H. (2009). Polymerization of Included Monomers and Behaviour of Resulting Polymers. In: Wenz, G. (eds) Inclusion Polymers. Advances in Polymer Science, vol 222. Springer, Berlin, Heidelberg. https://doi.org/10.1007/12_2008_6
Download citation
DOI: https://doi.org/10.1007/12_2008_6
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-01409-3
Online ISBN: 978-3-642-01410-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)