
Abstract
This review details recent developments in the Pd-catalyzed C–H bond arylation and alkenylation of indoles and pyrroles, aromatic heterocycles that are frequently displayed in natural products and medicinal agents.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Chemical synthesis has been revolutionized by the development of metal-catalyzed cross-coupling reactions. Palladium-catalyzed reactions such as the Heck [1], Negishi [2], Stille [3], Suzuki [4] and Sonogoshira [5], to name but a few, have become crucial tools for synthetic organic chemists. It is now possible to couple aryl halides, triflates and tosylates with olefins, alkanes or other functionalized arenes in high yield with very low catalyst loading. These tactics rely on strategically installed metal-active groups and such starting materials require additional steps to prepare them from feedstock chemical sources. C–H bonds are ubiquitous in organic molecules but are often unreactive. Introduction of functionality through the direct transformation of C–H bonds would provide the opportunity for distinctly different assemblies of synthetic targets. While this is an attractive concept, achieving selectivity among different C–H bonds remains a major challenge; this means that developing new catalyst systems that display both high reactivity and predictable selectivity is essential in order to increase the efficiency with which complex molecular architectures can be assembled.
1.1 Conventional Palladium-Catalyzed Cross-Coupling
Biaryl C–C bond formation, via metal-catalyzed cross-coupling tactics, is a widely used transformation in the synthesis of both complex natural products and medicinal agents. Efficient catalytic methods have emerged for the union of complex molecular fragments together in a highly selective manner. Typically one reaction component bears a metal containing functionality (Y) (nucleophilic component) whilst the other contains a halide atom or pseudo-halogen (X) (electrophilic component) (Scheme 1).

Arylation via conventional cross-coupling tactics
Similarly, the Heck reaction has become one of the most fundamental metal-catalyzed C–C bond forming processes for the synthesis of complex molecules [6]. Heck type reactivity comes from the ability of Pd(0) species to undergo oxidative addition to various C–X bonds and the addition of the RPdX intermediates to unsaturated bonds (Scheme 2).

Alkenylation via conventional Heck cross-coupling
Metal-catalyzed cross-coupling has had a major impact in the union of complex fragments during natural product synthesis programmes. In many cases structurally and functionally complex molecules can be coupled, often as part of a synthesis endgame, to reveal the architecture of natural products with exquisite control and in high yield. Furthermore, in many of these cases, there would be no alternative method to complete the synthesis.
While it is difficult to find fault in complex examples of conventional cross couplings, when applied to smaller molecules there is room for potential improvement. First, the overall coupling of two fragments in this manner requires two or three discrete steps: (1) pre-functionalization of starting materials, i.e. formation of the aryl halide and/or metallated component, and (2) Pd(0)-catalyzed union of reaction partners. This unfortunately gives rise to loss of material and significant accumulation of waste products, although this is less of an issue when coupling larger fragments together.
1.2 Palladium-Catalyzed C–H Bond Functionalization
The concept of “atom economy” has frequently been used to emphasize the minimal number of reactants, as well as selection of “low energy” starting materials as substrates [7], for example, using substrates that contain a reactive C–H bond rather than a C–X bond. Ideally, a direct catalytic cross-coupling reaction that utilizes “traditionally inert” C–H bonds would underline this concept as only the loss of nontoxic hydrogen gas or water as by-products would be necessitated (Scheme 3).

Direct cross-coupling by C–H activation
The ability to activate a specific “inert” C–H bond and utilize it as a more versatile functional group is an emerging area in chemistry [8]. Activation of C–H bonds with metal catalysts and further functionalization of these intermediates to valuable products has the potential to replicate the utility of reactions such as the Suzuki or Heck whilst also satisfying the high atom and step efficiency demanded by environmental constraints. This would result in a more efficient transformation and also allow us to look at new approaches in the synthesis of complex molecules that are free from the restraints of functional group manipulation.
Despite many advantages, there are still significant challenges posed by this concept. First, the synthetic challenge arising from the high strength of C–H bonds in alkanes and arenes (e.g. CH3–H, 105 kcal mol−1; Ar–H, 110 kcal mol−1) relative to the C–X bonds activated in traditional cross-coupling (e.g. Ar–I, 65 kcal mol−1). Often C–H bond activation processes require harsh reaction conditions which severely impinge from their use in complex molecule synthesis. Second, whilst the ubiquitous nature of C–H bonds in organic molecules gives it the potential to become a highly general process, one of the greatest limitations of this type of chemistry is the ability to activate the desired C–H bond in a chemoselective manner.
Over the past 30 years there has been a massive effort to achieve selective C–H bond activation by transition metal catalysis and there now exists a variety of mechanisms for the activation of C–H bonds using a range of transition metal catalysts (Scheme 4) [8–18].

Various mechanism for the activation of C–H bonds
1.3 Activation Mechanisms of the C–H Bond
1.3.1 Electrophilic C–H Bond Activation at Palladium(II) Centres
The most common means of activating aromatic C–H bonds via palladium catalysis is by electrophilic C–H activation. This proceeds more like a Freidel-Craft type metallation mechanism, followed by rearomatization to form versatile aryl-metal intermediates (Scheme 5) [19]. It can occur with electrophilic palladium(II) catalysts such as Pd(OAc)2, PdCl2, Pd(TFA)2 (Scheme 5a) or on electrophilic aryl-palladium(II) complexes, that result from oxidative addition of palladium(0) into an aryl halide (Scheme 5b). The resultant aryl-palladium(II) complexes are analogous to those observed in conventional cross-coupling reactions and as such are versatile intermediates in the formation of new C–C bonds.

Electrophilic C–H bond activation at Pd(II) centres
1.3.2 Proton-Transfer Metallation Pathway
Mechanistically proton-transfer metallation is thought to proceed via a concerted arene-metallation and C–H bond cleaving process, which depends on the acidity of the C–H bond being cleaved (Scheme 6). The reaction shows complete inversion of reactivity relative to the electrophilic C–H activation pathway with electron deficient arenes reacting preferentially [20, 21].

Proton-transfer metallation
1.4 Regiocontrol in the C–H Bond Functionalization Event
As discussed previously, another major challenge relates to achieving selectivity in the C–H bond functionalization step. The prevalence of C–H bonds in organic molecules means that it is necessary to activate C–H bonds specifically, usually amongst similar C–H environments. In terms of reactions at Pd(II) centres, described above, there are four main factors used to control the regioselectivity of the C–H functionalization.
-
1.
Intramolecular reactions: tethered reacting groups are employed to limit the degrees of freedom in a system, thereby controlling the regioselectivity of the reaction.
-
2.
Directing groups: auxiliary groups usually containing Lewis basic heteroatoms, coordinate palladium and bring the metal centre into close proximity of a specific C–H bond allowing formation of palladacycles.
-
3.
Electronic properties of the substrate: often “activated” C–H bonds are targeted, for example, with electron rich heteroarenes; electrophilic palladation is favored at the most nucleophilic position and relies on the inherent reactivity of the system. Similarly if a proton-transfer pathway is under operation, activation is favoured on the most acidic C–H bond.
-
4.
Steric properties of the substrate: depending on substrate structure, certain C–H bonds may be more accessible to the catalyst than others.
2 Pd-Catalyzed C–H Bond Functionalization on the Indoles and Pyrrole Nucleus
This review will cover recent advances in the palladium-catalyzed C–H activation of the indole and pyrrole nucleus, specifically for the formation of new C–C bonds via arylation and alkenylation. Most commonly, activation is achieved through mechanisms based on electrophilic substitution of aromatic C–H bonds by a palladium(II) species. In particular, we will discuss the effects of mechanistic differences on substrate reactivity and how various levels of regioselectivity can be achieved within aromatic and heteroaromatic systems.
The indole and pyrrole nucleus are common structural motifs in a range of natural products and medicinal agents (Fig. 1). Therefore, methods for their selective and efficient functionalization are important targets for chemical synthesis. The inherent reactivity of these heteroarenes has attracted widespread interest as ideal substrates for direct metal-catalyzed C–H bond functionalization reactions. However, related to their intrinsic reactivity is their sensitivity to harsh aerobic reaction conditions, and so methods to enable direct transformations on these heteroarenes must take this into account.

Indole and pyrrole containing natural products and therapeutics
A common strategy employed to effect selectivity is exploitation of inherent substrate reactivity and utilization of “activated” C–H bonds. Heteroaromatic compounds represent common motifs in both natural products and medicinal agents and contain certain C–H bonds that are intrinsically more reactive than others. By using heteroaromatic motifs as the Ar–H unit, the inherent differences in reactivity of C–H bonds around the motif can be exploited to achieve selectivity.
3 Pd-Catalyzed C–H Bond Arylation on Indole and Pyrrole
3.1 Mechanisms of Pd-Catalyzed C–H Bond Arylation
In terms of palladium-catalyzed direct arylation reactions, there are three general mechanisms that are commonly applied (Scheme 7).

Proposed Pd(0)/Pd(II) catalytic cycle for direct arylation
The first case involves direct arylation of arenes with aryl halides via a Pd(0)/Pd(II) cycle (Scheme 8). After initial oxidative addition of Pd(0) into the aryl halide, the C–H bond activation event takes place at the Ar–Pd(II)–X species 4 and is followed by C–C bond forming reductive elimination and regeneration of Pd(0).

Proposed Pd(II)/Pd(IV) catalytic cycle for direct arylation
More recently, direct arylations have been developed that are proposed to proceed via a Pd(II)/Pd(IV) cycle (Scheme 9). In this case the C–H bond functionalization takes place first, at the highly electrophilic Pd(II) catalyst species, and is followed by oxidative addition of an Ar–X type compound, generating the corresponding Pd(IV) species II. Reductive elimination would then form a new C–C bond and release Pd(II) back into the cycle.

Proposed Pd(II)/Pd(IV) catalytic cycle for direct arylation
A final common arylation mechanism also involves C–H bond palladation with a Pd(II) catalyst, but then a transmetallation with an organometallic such as a boronic acid. Reductive elimination to form the desired product also releases Pd(0) and this species must be oxidized back to the active Pd(II) catalyst. A key aspect of this process is developing an oxidative system that does not result in homo-coupling of the aryl boronic acid (Scheme 9).
3.2 Regioselective Pd-Catalyzed C–H Bond Arylation on Indole and Pyrrole
Initial work in the area focussed on applying Pd(0)/Pd(II) manifolds to electron rich and nucleophilic heterocycles such as imadazoles, indoles and various other azoles. For an overview of this particular aspect of C–H bond functionalizations, the reader is directed to other excellent reviews of this field [17].
In the context of this review, the indole motif, in particular, has attracted a great deal of attention in the development of C–H functionalization processes. Some of the earliest studies were reported in 1989 by Ohta, who investigated palladium-catalyzed coupling of 2-chloro-3,6-dialkyl pyrazines with protected indoles [22]. Despite requiring highly elevated temperatures they were able to affect selective C2 or C3 heteroarylation of the indole core in moderate yields. Use of 1-tosyl-indole led, predominantly, to 3-heteroaryl indoles whilst 1-alkyl analogues were shown to undergo substitution at C2 (Scheme 10).

Ohta’s regioselective heteroarylation of indoles
Shortly after this, palladium-catalyzed intramolecular cyclizations on indole were also reported. Work by Grigg [23], Kozikowski [24] and Merour [25] showed that under analogous Pd(0) conditions, at high temperature, a variety of polycyclic indoles could be made (Scheme 11).

Intramolecular arylation of indoles
In 2004, Sames reported a practical method by which N-substituted indoles could be selectively arylated at the C-2 position with a range of aryl iodides [26]. The investigations found two competitive processes in operation (Scheme 12), namely the desired cross-coupling (Path A) and biphenyl formation (Path B), caused by palladium-catalyzed Ullmann type coupling. It was found that decreased catalyst loading favored production of the desired product due to requirement for bimolecular transmetallation of the aryl-palladium species I for biphenyl formation.

Sames’ strategy towards regioselective intermolecular arylation of indoles
A more robust process, suitable for the production of a greater scope of heterocycle, was later developed using SEM-protected azoles that could be readily converted to the N–H counterparts under a variety of conditions (Scheme 13a) [27]. Palladium complexes containing imidazyl carbene ligands were utilized to improve the stability of palladium throughout the catalytic cycle, modulate reactivity and disfavour biphenyl formation.

Sames’ intermolecular C2-arylation of indoles
Sames’ discovery that phosphine ligands inhibited the reaction has lead to the development of a new phosphine-free arylation for N–H indoles and pyrroles (Scheme 13b) [28]. Increased indole concentration makes possible the coupling of sterically demanding arene donors and indole motifs.
In general these methods, developed by Sames for C–H arylation of indoles, do not follow the expected “electrophilic” regiochemistry, instead showing high C2 selectivity (Scheme 14). Sames described how their mechanistic experiments point towards a mechanism involving electrophilic C–H activation [29]. First, the reaction was shown to be first order in both substrate and catalyst. Moreover, a Hammett plot indicated a positive charge accumulated at the 3-position of indole and a larger KIE was obtained for the 3-position, where the substitution does not occur. This evidence provides support for an electrophilic palladation mechanism at C3 suggesting, perhaps, that it is then followed by a 1,2-migration of palladium. It should be noted that Fagnou et al. recently reported that a number of reactions that were thought to proceed via electrophilic metalation could in fact be explained by a proton-transfer (concerted metalation deprotonation) mechanism. They reported that the free energy of activation for the direct arylation would be lower at the C2 position, hence explaining the regiochemical outcome of the reaction [30, 31].

Proposed catalytic cycle for Sames’ intermolecular C2-arylation of indoles
A related example from He and co-workers demonstrated that phosphinous ligands on Pd(0) are also able affect an indole arylation reaction. A small selection of C3 arylated indoles can be realized using this method. Of particular note is the C3 selectivity observed in this reaction, when compared to the work of Sames who observed C2 selectivity (Scheme 15).

He’s C3 selective arylation of indole
Sames and co-workers also demonstrated that rhodium(I) catalysts also facilitate a C–H arylation of indole at the C-2 position (Scheme 16). While not explicitly described in their paper, they infer that the pivolate ligand may be involved in an internal proton-transfer, and it seems likely that this is in line with a CMD type activation step reported by Fagnou and Echaverran [20, 21] (opinion of Gaunt and Beck). The arylation was demonstrated on a range of indole and pyrrole molecules in good yields. In all cases arylation was observed at the C2 position of the heteroarene [30].

Rh-catalyzed C2-arylation of indoles
An extension of the palladium(0) catalyzed direct arylation reactions was reported by Lautens et al. in 2005. Based on the Catellani reaction [32], a direct intramolecular arylation of indole (C2) followed ortho-alkylation, via a norbornene-mediated tandem aromatic alkylation/Heck reaction (Scheme 17) [33]. An analogous process was later developed for thiophenes and furans, allowing formation of a range of interesting hetero-aryl polycyclic products (Scheme 17) [34].

Lautens’ norbornene-mediated tandem alkylation/Heck reaction
This unusual strategy is thought to make use of the different reactivities of palladium(0), palladium(II) and palladium(IV) intermediates. Mechanistically, the reaction is proposed to occur via initial oxidative addition of palladium(0) to the aryl iodide, followed by carbopalladation with norbornene to afford alkyl palladium species II (Scheme 18).

Proposed mechanism for the norbornene-mediated alkylation/Heck reaction
Intermediate II is incapable of β-hydride elimination and therefore directed ortho-palladation on the arene takes place generating intermediate III. This Pd(II) complex (III) is proposed to add oxidatively to the alkyl halide tether containing indole moiety. The proposed palladium(IV) intermediate IV undergoes reductive elimination to form an alkyl–aryl bond (V). If the ortho position is blocked the norbornyl-palladium species V undergoes decarbopalladative expulsion of norbornene to regenerate an aryl-palladium species VI. This intermediate can then undergo intramolecular cyclization onto the indole to give a variety of 6- or 7-membered annulated indole products.
As well as those discussed, Pd(0)/Pd(II) catalyst systems have also been used to affect arylation on a range of other heterocycles including imidazopyrimidines, indolizines, furans, thiophenes and related benzo-analogs (Fig. 2) [18, 35–41]. In almost all cases regioselectivity and mechanistic studies support the suggestion that arylation proceeds via an electrophilic mechanism.

Observed regioselectivities for direct arylation of heterocycles
While synthetically useful, the Pd(0)/Pd(II) catalyst systems generally suffer from a couple of notable disadvantages. In particular, high temperatures and long reaction times (125–150 °C for 12–48 h) are often required to achieve reactivity that can results in moderate scope and functional group tolerance as well as high sensitivity to ambient air and moisture.
More recently, there has been an emphasis on designing direct arylation reactions that occur under milder conditions and lower temperatures. Since the initial findings concerning the use of Pd(II) catalysts in oxidative Heck type transformations and directed palladations, it has been shown that utilization of these highly electron deficient Pd(II) catalysts can enhance the rate of the key electrophilic palladation step, allowing arylation of nucleophilic arenes under much milder and tolerant conditions. Heteroaromatics such as indole and pyrrole are particularly suited to such transformations, due to their electron rich and highly nucleophilic character. The strategy most often applied in order to effect oxidative palladium(II) catalyzed C–H bond arylation with Ar–X type species is proposed to proceed via a Pd(II)/Pd(IV) catalytic cycle. An oxidant that simultaneously creates a carbon–palladium bond and oxidizes palladium(II) to palladium(IV) is used. This also benefits from avoiding generation of Pd(0), which often causes problems in terms of catalyst turnover due to precipitation of palladium black.
Using this concept the Sanford group found that direct C2-arylation of indoles and pyrroles could be effected under remarkably mild conditions with aryl iodonium salts and palladium(II) catalysts (Scheme 19) [42]. The high C2 selectivity of the functionalization is attributed to a mechanism involving initial palladation at C3 followed by fast palladium migration to C2 under acidic conditions as initially proposed by Gaunt [43] and Sames [29]. The reaction also works well for simple pyrroles, again with C2 selectivity.

Sanford’s oxidative C2-arylation of indoles and pyrroles
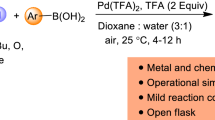
The mild reaction conditions and compatibility with ambient air/moisture are features attributed to be a consequence of the proposed Pd(II)/ Pd(IV) pathway operating in these systems. Whilst C2-phenylation was observed under extremely mild conditions in the presence of catalytic Pd(OAc)2 (5 min, room temperature), only moderate yields were obtained, presumably due to catalyst deactivation. Palladium(II) complexes containing stabilizing ancillary ligands (e.g. IMesPd(OAc)2) resulted in significantly improved performance, albeit slower rate caused, by the more electron rich palladium(II) catalyst). Phenylation could be extended to a variety of electronically diverse indole substrates as well as pyrroles and notably both N–H and N–Me indole were comparably reactive. As with the previous arylations a range of aryl groups could be installed via [Mes-I–Ar]BF4 salts; however superior yields were obtained with symmetrical iodine(III) compounds ([Ar–I–Ar]BF4). Having identified a potential limitation as being the requirement for independent synthesis of the iodonium salt arylating reagents, a one-pot approach to the transformation starting with ArI(OAc)2 and commercially available ArB(OH)2 reagents was developed. Combining PhB(OH)2 and PhI(OAc)2 in AcOH for 15 min at room temperature in the presence of 5 mol% Pd(OAc)2 followed by addition of the 1-methyl indole resulted in formation of the C2-phenylation product in excellent yield (Scheme 20).

One-pot strategy for C2-arylation of indole
Interestingly, a related copper catalyzed reaction enables controllable formation of either the C3 or C2 indole arylation products (Scheme 21). Gaunt et al. showed that when free (NH)-indole was treated with diaryliodonium triflates in the presence of copper(II)triflate at room temperature the C3 arylation product was observed in excellent selectivity and high yields [44]. However, if N–Ac indole is used then the product of the reaction is the C2 arylation product. The mechanism is proposed to proceed through a Cu(III)–aryl intermediate via an electrophilic metalation pathway. The origin of the C3/C2 selectivity is reasoned to arise form the ability of the metalated indole intermediate to undergo C3 to C2 migration, with the 1,2-shift favored on N–Ac indoles. A broad range of indole arylations has been demonstrated with this method. In comparison to the C2 selectivity displayed by palladium catalysts, it is interesting to note the complimentary selectivity displayed by copper catalysts.

Controllable and selective Cu-catalyzed C–H bond arylation of indole
Shi described a palladium(II) catalyzed cross-coupling of electron rich (hetero)arenes with aryl boronic acids (Scheme 22) [45]. A major strategic challenge was avoiding homo-coupling of the aryl boronic acids in the presence of palladium(II). However, it was found that acidic conditions helped to facilitate fast electrophilic attack and hindered the transmetallation of the aryl boronic acids to palladium(II).

Shi’s direct coupling of arenes and aryl boronic acids
Various aromatic rings show good selectivities without requiring directing groups and a range of substituted boronic acids are tolerated. Electron rich heterocycles such as indole and pyrrole are readily employed in the coupling reaction and selectivity appears to be controlled by the electronic properties of the arenes.
These preliminary studies suggest a catalytic cycle initiated by electrophilic attack of palladium(II) on the indole, followed by transmetallation with a boronic acid and reductive elimination to produce the desired arylated products (Scheme 23).

Proposed catalytic cycle for coupling of arenes and aryl boronic acids
Larrosa reported the direct C2-arylation of indoles with aryl iodides at room temperature (Scheme 24) [46]. The mild conditions allow a broad range of functionalities on both coupling partners and the method is particularly advantageous due to the large pool of commercially available aryl iodides.

Larrosa’s C2-arylation of indoles with aryl iodides
Interestingly, they propose a mechanism that proceeds via a Pd(0)/Pd(II) cycle (Scheme 36). Silver carboxylates, generated in situ from Ag2O and the corresponding carboxylic acid, are thought to have an important role in the reaction. The limiting step in the arylation of indoles via a Pd(0)/Pd(II) cycle is thought to be the necessary electrophilic palladation of the electron rich Pd(II) species I. It was proposed that after oxidative addition of Pd(0) into the aryl halide, the silver salt removed the iodide from the Pd(II) complex I forming a cationic Pd(II) species II which would be more electrophilic towards the indole unit thus increasing the rate of this palladation step. A poorly coordinating counter ion would allow dissociation to cationic species II, forming III, and also could act as a base in the palladation step (Scheme 25).

Proposed catalytic cycle for arylation of indoles with aryl iodides
However, it is not clear how the proposed Pd(0) is formed from Pd(OAc)2 under these reaction conditions and so a mechanism more similar to that proposed by Sanford [42] or Daugulis [47], involving a Pd(II)/Pd(IV) reaction manifold, cannot be ruled out (opinion of Gaunt and Beck). Rapid electrophilic attack from the Pd(II) catalyst would be expected with nucleophilic indole substrates, under the acidic conditions reported. The silver salt could then facilitate oxidative addition to the resulting palladium(II) species I by complexing the aryl iodide. Subsequent reductive elimination from the Pd(IV) centre II followed by ligand exchange with the silver carboxylate could then remove iodide from the reaction mixture and regenerate the active Pd(II) catalyst (Scheme 26).

Alternative proposed catalytic cycle for oxidative coupling of indoles and aryl iodides (Gaunt and Beck)
The most common mechanism of C–H bond cleavage in the arylation examples discussed above has been assumed to be electrophilic aromatic substitution involving reaction of an electrophilic palladium catalyst with an electron rich, nucleophilic aromatic ring. In order to effect direct arylation on simple, electron deficient arenes, a basic directing group or intramolecular reaction is usually necessary to enable formation of a metalocycle. As a brief introduction to the effect of this area on the functionalization of indoles and pyrroles, we provide an overview of the mechanistic rationale for this mode of activation. The reader is also directed to the seminal contributions [20, 21]. These studies investigated the effect of various electronically biased substituents on aromatic C–H donors, shown in Scheme 27. Notably, the results of the reactions were inconsistent with an electrophilic palladation mechanism. In conjunction with computational studies, a mechanism for Pd-catalyzed arylation was proposed, involving proton abstraction by a carbonate, or related ligand.

Alternative proposed catalytic cycle for oxidative coupling of indoles and aryl iodides
In 2006 Fagnou described the direct intermolecular arylation of perfluoro benzene derivatives via the proposed proton-transfer pathway (Scheme 6). The reaction shows complete inversion of reactivity relative to the electrophilic C–H activation pathway and is thought to proceed via a concerted arene-metallation and C–H bond cleaving process, which depends on the acidity of the C–H bond being cleaved.
They were able to extend the scope of the direct arylation to simple, completely unactivated arenes, such as benzenes by development of a palladium-pivalic acid co-catalyst system (Scheme 28) [48].

Proposed catalytic cycle for direct arylation via proton-transfer palladation
The pivalate anion is a key component in the C–H bond cleaving step, lowering the energy of the C–H bond cleavage (Step 3) and acting as a catalytic proton shuttle from benzene to the stoichiometric carbonate base. Competition experiments indicate that electron deficient arenes react preferentially (fluoro benzene > benzene > anisole) with C–H acidity influencing the regioselectivity and reactivity correlating with the proposed proton-transfer pathway.
The formation of a C–C bond resulting from the coupling of two C–H bonds is a particularly attractive target, since the only formal by-product would be hydrogen, or water in an oxidative system. However, substantial hurdles impede the conception of a catalytic arene cross-coupling process that does not involve any substrate pre-activation at all. Aside from issues of reactivity and regioselectivity, the prevention of homo-coupling is a key factor for the development of this important class of reaction. The catalyst must be able to react with one arene in the first step of the catalytic cycle and then invert its selectivity in the second step to react exclusively with a different arene (Scheme 29).

Direct arylation via double C–H activation
One of the first reports of unsymmetrical biaryl formation via a double C–H activation event was published in 2006 by Lu et al. (Scheme 30) [49].

Lu’s intermolecular cross-coupling of simple arenes by double C–H activation
In 2007, the Fagnou group achieved a much more practical and selective Ar–H/Ar–H cross-coupling [50]. Electron deficient palladium(II) complexes can react via an electrophilic C–H activation mechanism with good selectivity for electron rich arenes. In contrast, Fagnou [51] recently showed that complimentary reactivity to this is displayed by some ArPd(II) complexes that react through a proton-transfer-palladation mechanism, and that they depend on arene C–H acidity rather than arene nucleophilicity (Scheme 31).

Complimentary mechanisms for C–H bond activation
Fagnou et al. were able to exploit these two complimentary reactivity modes within a single catalytic cycle to achieve a highly selective Ar–H/Ar–H cross-coupling (Scheme 32).

Fagnou’s proposed catalytic cycle for direct coupling of indoles and arenes
In the first step, it was proposed that the highly electrophilic PdII(TFA)2 catalyst affected selective electrophilic C–H bond activation exclusively on the electron rich indole. This generated an indole–Pd(II) complex I, which was able to selectively activate the benzene via a transfer-palladation pathway, which is controlled by C–H acidity. Reductive elimination afforded biaryl C–C bond formation and released Pd(0) which required oxidation to regenerate the active Pd(II) catalyst.
The palladium-catalyzed oxidative cross-coupling process allows access to both C3 and C2 arylindoles as well as displaying a degree of regioselectivity at the benzene component (Scheme 33) [51, 52]. C3 selectivity was achieved on N–Ac indoles using a stoichiometric copper(II) oxidant and catalytic Pd(TFA)2. Optimal catalytic activity was observed with 3-nitropyridine and cesium pivolate as additives. It is presumed the pyridine additive may stabilize the palladium(0) prior to reoxidation preventing formation of palladium black that precipitates out of the reaction. Switching to AgOAc as oxidant produced an inversion in selectivity, favouring C2-arylation. These conditions were optimized by switching to N–pivalyl indole and removing the additives and resulted in 100% conversion and a 1:25 C3:C2 ratio. Selectivity studies have indicated it is most likely the acetate base, not the metal counter ion, that imparts C2 selectivity; perhaps due to carboxylate-induced cleavage of higher order Pd clusters and the formation of monomeric Pd species. Pd–Cu complexes, analogous to trinuclear Pd carboxylate clusters seen at high [Pd], are thought to cause the pronounced C3 selectivity.

Regioselective coupling of indoles and arenes via double C–H activation
DeBoef and co-workers have reported a similar reaction, wherein direct C–H to C–H indole-arene cross-coupling can be controlled through the use of a particular oxidant (Scheme 34) [53, 54]. The basis of their selectivity concept is the formation of different polyvalent clusters between the Pd(OAc)2 and the AgOAc or Cu(OAc)2 oxidants respectively, and the subsequent reactivity of these complex in the arylation reaction. The same group also demonstrated the utility of an intermolecular C–H to C–H coupling reaction.

Regioselective coupling of indoles and arenes via double C–H activation
Although not directly relevant to this review, it should be noted that Sanford, Shi and Buchwald have also reported a palladium-catalyzed reaction for the chemo- and regioselective oxidative cross-coupling between ligand coordinated L–CAr–H substrates and simple arenes (Ar–H) [55–57].
Although not a palladium-catalyzed reaction, the Ir(I)-catalyzed C–H borylation reaction developed independently by Smith and Malezcka [58] and Hartwig and Miyaura [59] deserves some mention in the context of indole and pyrrole functionalization. Based on the original studies, indoles and pyrroles can be borylated (and hence cross coupled under Suzuki conditions) to form either the C2 or C3 functionalised products (Scheme 35) [60, 61]. Free (NH)-indoles and pyrroles react exclusively at the C2, whereas N-TIPS indole and pyrroles are borylated at the C3 positions. Interestingly, Smith, Maleczka and co-workers also demonstrated that when the C2 position of indole is blocked, then the borylation reaction takes place at the C7-position of the indole nucleus [62]. They propose that an N-chelation to Ir (or B) is responsible for the observed selectivity.

Regioselective Ir(I)-catalyzed C–H borylation of indoles and pyrroles
4 Pd-Catalyzed C–H Alkenylation of Indole and Pyrrole
4.1 Mechanisms of Pd-Catalyzed C–H Bond Alkenylation
Indoles and pyrroles have also been shown to undergo C–H alkenylation reactions. In these cases the process involves the coupling of the unfunctionalized (hetero)arene with an olefin (Scheme 36).

C–H bond alkenylation of arenes
There are two typical mechanisms generally applied for palladium-catalyzed C–H alkenylation. First, the oxidative Heck mechanism, initially proposed by Fujiwara and Moritoni [63].
σ-Aryl–Pd complexes such as I, formed via electrophilic substitution of aromatic C–H bonds by Pd(II) species, are thought to be key intermediates in the catalytic cycle (Scheme 37). It is then proposed that a standard Heck arylation pathway is followed. The olefin coordinates to the unstable σ-aryl–palladium complex I and subsequently undergoes 1,2-migratory insertion into the aryl–palladium bond to form III. This species then rapidly decomposes, via β-hydride elimination, to the arylated olefin and palladium hydride. The latter subsequently reductively eliminates to generate HX and palladium(0), which can be reoxidized to the active palladium(II) source. Second, C–H alkenylation of arenes can occur through initial olefin activation, via coordination to palladium(II) (I), followed by subsequent nucleophilic attack from a electron rich (hetero)arene (Scheme 38).

Proposed catalytic cycle for Pd(II) catalyzed oxidative Heck reaction

Proposed catalytic cycle for Pd(II) catalyzed alkenylation via alkene activation
Fujiwara and Moritoni carried out seminal work in the area of C–H alkenylation; they reported that palladium(II) complexes could mediate the coupling of unfunctionalized arenes with olefins in refluxing acetic acid [64]. The initial reactions used stoichiometric quantities of palladium salts; however the reaction was subsequently made catalytic using oxidants such as Ag(I) or Cu(II) in the presence of oxygen, t-BuOOH, t-BuOOBz or benzoquinone [65]. Analogous to the limitations described for direct arylations, C–H alkenylation was restricted in its utility due to the inability to effect single and selective functionalization of simple substituted arenes. The initial work with simple benzene derivatives indicated that electron donating substituents (e.g. Me, Et, OMe) directed ortho/para, where as electron withdrawing groups (e.g. NO2) tended to give meta products. However, reactions were never completely selective on the arene coupling partner and typically gave mixtures of regioisomers.
4.2 Regioselective C–H Bond Alkenylation of Indole and Pyrrole
Utilization of “activated” C–H bonds in heteroaromatic compounds is particularly suited to these transformations, where the key C–H activation step involves electrophilic palladation at an electron deficient Pd(II) catalyst. Fujiwara et al. investigated the application of the oxidative Heck chemistry to various heterocycles (furan, thiophene, benzofuran, indole) (Scheme 39) [66, 67].

Fujiwara’s application of the oxidative Heck reaction to various heterocycles
The reaction gave mono- and bis-alkenylated products on furan and thiophene, regioselective for the 2 and 5 positions, although limiting reaction to a single alkenylation was not addressed. In the reaction of 2-substituted furans bearing electron withdrawing or donating groups, the reaction selectively occurred at the 5-position. In an isolated example, reaction with indole occurred at the naturally more nucleophilic C3 position. As with the benzenoid examples, for heteroaromatics Fujiwara et al. and Tsuji et al. both reported more efficient catalytic systems using small amounts of palladium acetate and benzoquinone in the presence of tert-butyl peroxybenzoate as an inexpensive reoxidant [66, 67]. Various heteroaromatics and olefins undergo coupling reaction with high TON (up to 280) albeit with limited regioselectivity and substrate scope.
In 2003, Beccalli et al. reported an intramolecular variant, allowing chemoselective cyclization of indole carboxamide derivatives into β-carbolinones (path A) or pyrazino[1,2-a]indoles (path B) (Scheme 40) [68]. The chemoselectivity of the cyclization could be controlled by the reaction conditions. PdCl2(MeCN)2 and benzoquinone in DMF/THF made possible C–H functionalization at the C3 position of indole, whilst Pd(OAc)2 in Na2CO3 and n-Bu4NCl in DMF resulted in intramolecular amination of the double bond (N–H functionalization).

Beccalli’s chemoselective cyclization of indole carboxamide derivatives
Stoltz et al. also reported an intramolecular reaction where C–H bond cyclization of indoles onto unactivated olefins was possible using palladium(II) and molecular oxygen as the sole stoichiometric oxidant (Scheme 41) [69]. They found the C2 position on indole could be activated if the more nucleophilic C3 position was blocked.

Stoltz’s intramolecular annulation of indoles
Following work from Uemura [70], research focused on the use of palladium-pyridine systems, which had the potential for modification with chiral ligands to catalyze enantioselective processes. A correlation between the electronic nature of the pyridine ligand and its ability to facilitate the cyclization was observed. More electron withdrawing ligands resulted in a more electrophilic, and therefore more reactive, palladium catalyst; however, if too electron deficient, they were unable to ligate palladium sufficiently and as a result hampered both reactivity and Pd(0) oxidation. The mechanism is consistent with the previously discussed oxidative Heck process involving initial electrophilic palladation followed by olefin insertion and β-hydride elimination). This was indicated by subjecting a designed indole to the annulation conditions (Scheme 42). The observed stereochemistry of the product supports the mechanism if the requirements for syn migratory insertion and syn β-hydrogen elimination are operative.

Proposed mechanism for intramolecular alkenylation of indoles
The group have also developed an analogous process for direct C–H functionalization of electron rich aromatic rings and cyclization with unactivated alkenes to access substituted benzofuran and dihydrobenzofuran derivatives (Scheme 43) [71].

Intramolecular alkenylation of aromatic rings
Interestingly, Widenhoefer reported a similar palladium(II) catalyzed cyclization of indoles onto alkenes (Scheme 58) [72]. This mild protocol for cyclization/carboxylation of 2-alkenyl indoles makes possible catalytic addition of a carbon-nucleophile and carbonyl group across a C=C bond. The mechanism, however, is thought to involve outer-sphere attack of indole onto a palladium–olefin complex rather than the electrophilic C–H activation of the indole C(3)–H bond, exhibited by the Stoltz carbocyclization.
Copper(II) chloride was found to be the best oxidant for the system and a range of esters could be formed if ten equivalents of the corresponding alcohol was added to a THF solution. A range of substituted indoles were subjected to the reaction conditions and furnished both 6- and 7-membered annulation products, in good to excellent yields, 58–91%. 3-Alkenyl indoles were also effective substrates, under going cyclization onto the less nucleophilic C2 position in good yield albeit requiring extended reaction time (Scheme 44).

Widenhoefer’s cyclization/carboxylation of alkenyl indoles
It was also possible to carry out an analogous intermolecular version of the alkylation/carboxylation process between 2-substituted indoles and styrenes (Scheme 45) [73]. While sterically and electronically diverse styrene derivatives reacted with moderate to good yields (40–78%), indoles without substitution at C2 failed to undergo efficient reaction. The reactions were always selective for the indole C3-position (as C2 was blocked) and produced products substituted to the phenyl ring.

Intermolecular alkylation/carboxylation of indoles with styrene derivatives
The stereochemistry of the reaction was investigated for both (Z) and (E) 3-(4-deuterio-3-butenyl)indoles. Treatment of (Z)-deuterated alkene with a catalytic quantity of PdCl2(CH3CN)2 in the presence of CuCl2 gave the cis-product as a single diastereomer whilst treatment of corresponding (E)-isomer with the same conditions gave trans-product also as a single diastereomer. This indicated the palladium-catalyzed cyclization/carboalkoxylation was stereospecific and that the indole and carbomethoxy group add in an anti fashion across the C=C bond of the olefin (Scheme 46).

Mechanistic investigations of the cyclization/carboxylation reaction
The stereochemical outcome was in agreement with a mechanism for the palladium-catalyzed cyclization/carboalkoxylation of a substituted alkene (Scheme 47) that involves outer-sphere attack of the indole on the palladium-olefin complex I which, coupled with loss of HCl, would form the alkylpalladium intermediate II. 1,1-Migratory insertion of CO into the Pd–C bond of II with retention of stereochemistry would form the acyl–palladium complex III, which could undergo methanolysis to release cis-product and form a palladium(0) complex. Oxidation with Cu(II) would then regenerate the active Pd(II) catalyst.

Proposed catalytic cycle for the cyclization/carboxylation of alkenyl indoles
Widenhoefer has also reported two isolated examples of C2-cyclization/carboxylation on N-alkenyl pyrrole substrates (Scheme 48) [73]. In order to achieve moderate to good yields, addition of molecular sieves and dropwise addition of a Brønsted base was required to counteract the competitive acid promoted pyrrole polymerization.

C2 C–H cyclization/carboxylation on N-alkenyl pyrroles
Although much has been learnt about the reactivity and regioselectivity in direct functionalization of heteroarenes, the ability to manipulate and controllably switch the selectivity is extremely rare. In 2005, a method for direct and selective C2 or C3 elaboration of free-(NH) indoles using palladium-catalyzed C–H functionalization was developed by Gaunt and co-workers (Scheme 49) [43].

Gaunt’s regioselective intermolecular C–H alkenylation of indoles
This relatively mild and highly regioselective process allowed C–H alkenylation with both electron deficient acrylates and more unactivated alkenes. Switchable regioselectivity was achieved by varying the reaction media and chemical oxidant.
It is proposed that, for both pathways, indole initially undergoes electrophilic palladation at the more nucleophilic C3 position via intermediate I (Scheme 50). Under neutral conditions the acetate ion, formed from the attack of indole on Pd(OAc)2, will readily remove a proton from intermediate I to form palladated species II (Scheme 50 , retention pathway). This species undergoes 1,2-migratory insertion with the olefin, and subsequent β-hydride elimination then affords C3-alkenylated products. In contrast, under acidic conditions (t-BuOOBz, AcOH/dioxane) it is thought that rearomatization is slowed, allowing a migration pathway to dominate (Scheme 50 , migration pathway). The C3-PdX bond in I is proposed to shift to the highly activated C2 position of the iminium intermediate to give species Ia and ultimately IIa. Subsequent 1,2-migratory olefin insertion and β-hydride elimination would then generate the corresponding C2-alkenylated products.

Proposed catalytic cycle for regioselective C–H alkenylation of indoles
Concurrent with these studies, Brown and co-workers reported a regioselective C–H functionalization of indoles via directing group control (Scheme 51) [74]. When a benzyl group was attached to the indole nitrogen, alkenylation took place selectively at the more nucleophilic C3 position (in agreement with Fujiwara’s observations). Alternatively N-pyridyl protected indoles directed alkenylation to the more unusual C2 position via coordination to the protecting group.

Brown’s directing group controlled intermolecular alkenylation of indoles
Gaunt and co-workers have also developed an efficient palladium(II) oxidation system for C–H functionalization of pyrroles under ambient conditions (Scheme 52) [75]. It is possible to control the position of reaction via simple steric and electronically tuned N-pyrrole protecting groups to form products with either C-2 or C-3 elaboration. N-Boc pyrroles react at the C2 position, exploiting the inherent reactivity of this heteroarene. In contrast, N-TIPS pyrrole is shielded from reacting at the C2 position by the sterically demanding silyl group and so instead undergoes the C–H bond alkenylation at the C3 position. This method can be used to generate a range of alkenylated products. The regioselectivity concept can also be applied in an intramolecular sense forming different pyrrole architectures depending on the group on the pyrrole nitrogen. Despite the high propensity of pyrroles to oxidation and polymerization, this mild C–H functionalization method affords excellent yields of either C2 or C3 derived products.

Gaunt’s regioselective inter- and intramolecular C–H bond functionalization of pyrroles
In terms of the mechanism, both reactions are thought to proceed via an electrophilic palladation step followed by Heck type coupling with the olefin (oxidative Heck mechanism) (Scheme 53). For C2 palladation, a slightly electron withdrawing group on nitrogen deactivates the ring sufficiently that the natural C2 selectivity controls the electrophilic palladation. In the case of C3 elaboration, the TIPS group is extremely bulky and slightly electron donating. Presumably, the steric bulk deters reaction at the adjacent C2 position whilst the highly activated nature of the pyrrole allows palladation at the less nucleophilic C3 even under these mild catalytic conditions. After regioselective palladation on pyrrole it is thought that the respective aryl–palladium intermediates (IIa and IIb) undergo a 1,2-migratory insertion with the alkene, which after β-hydride elimination gives the alkenylated products and palladium(0).

Proposed regioselectivity concept and catalytic cycle for C–H alkenylation
5 Pd-Catalyzed C–H Bond Functionalization of Indoles and Pyrroles in Complex Molecule Synthesis
In terms of making natural products, synthetic strategies have been revolutionized by the development of cross-coupling reactions, which are particularly useful for uniting complex molecular fragments. The successful assembly of complex molecules via cross-coupling tactics exploits the reactivity of strategically installed “metal-active” functional groups in the key bond forming events. In recent years the advance of metal-catalyzed C–H bond activation technology has suggested that similar transformations are possible without the need to pre-functionalize the parent molecules. There has been a wealth of methodological advances that have highlighted the potential efficacy of these processes in synthesis. However, there are few examples of the application of such catalytic tactics in total synthesis. This is possibly due to complications with the harsh reaction conditions often required and the selectivity and stability issues associated with the more complex substrates inevitably needed for these purposes. In spite of this, direct C–H functionalization tactics could have a huge impact in streamlining natural product synthesis and so further development in this area represents an important goal for synthetic chemists.
The following section looks at how palladium-catalyzed C–H functionalization has been successfully applied in synthetic strategies enabling rapid and elegant routes to complex natural products containing the indole and pyrrole nucleus. There are a number of metal-mediated examples where stoichiometric quantities of transition metals are employed to affect the desired transformation; however, there are very few cases of catalytic functionalization with in the context of complex molecule synthesis.
An early example of a natural product synthesis featuring an indole C–H bond alkenylation was report by Trost and co-workers (Scheme 54). Using stoichiometric Pd(II)-salts in combination with silver(I) tetrafluoroborate, they were able to mediate a oxidative Heck process onto the bicyclic amino-alkene. Using a reductive work-up to reduce the Pd–C bond, they were able to complete a synthesis of ibogamine [76].

Trost’s synthesis of ibogamine featuring a Pd(II)-mediated carbocyclization
In 2002 Corey and Baran employed a similar palladium-mediated indole-dihydroindoloazocine cyclization to construct the core 8-membered ring in their synthesis of (+)-austamide, (Scheme 55) [77, 78]. Treatment with Pd(OAc)2 in 1:1:1 THF–H2O–AcOH at 23 °C under 1 atm of O2 for 36 h allowed conversion of an N-prenylated tryptophan derivative to the dihydroindoloazocine tricyclic architecture in one step (29%) making possible a impressively short route to the natural product. The proposed carbocyclization mechanism involved initial C2 indole palladation followed by intramolecular 7-exo-trig Heck type cyclization. It was thought that the aqueous acetic acid solvent system subsequently caused C–PdX bond heterolysis and Pd(0) which produced a cationic intermediate that by migration of the electron rich β-[2-indolyl] group resulted in ring enlargement and formation of the dihydroindoloazocine product.

Corey’s synthesis of (+)-austamide featuring a Pd(II)-mediated carbocyclization
Similarly, in 2004, the Stoltz group employed a heteroarene/olefin oxidative C–C coupling transformation as a key step in their synthesis of pyrrole alkaloid (+)-dragmacidin F (Scheme 56) [79, 80]. They found that exposure of the key pyrrole fragment to Pd(OAc)2 under a variety of conditions led to carbocyclization and after optimization they were able to isolate the desired fused pyrrole-bicyclic structure as a single stereo- and regio-isomer in 74% yield. The transformation is noteworthy since it results in functionalization of the electronically deactivated C3 position. They were, however, unable to effect catalytic turnover of palladium with a stoichiometric oxidant, which they presumed was due to extensive oxidative decomposition of both starting material and product. This Pd(II)-mediated strategy did, however, provide the desired cyclized product in around twice the yield of the conventional Heck route, using equivalent amounts of palladium, which highlights the positive influence of C–H functionalization processes.

Stoltz’s synthesis of dragmacidin F featuring a Pd(II)-mediated carbocyclization
Trauner applied a C–H bond activation approach to the synthesis of rhazinilam [81]. The synthetic power of direct C–H arylation on nucleophilic heteroarenes was demonstrated through formation of the strained 9-membered ring using intramolecular coupling to an unactivated pyrrole (Scheme 57).

Trauner’s application of direct C–H arylation in the synthesis of (±)-rhazinilam
Conditions first described by Fagnou were used to affect the C–H to C–H bond cyclization, which proceeded in 47% yield. Mechanistically the direct coupling reaction is thought to proceed via intramolecular nucleophilic attack of the pyrrole moiety onto the Pd(II) centre. It was postulated that the electron rich “DavePhos” ligand facilitates both oxidative addition and forms a more reactive cationic Pd(II) species by dissociation of the halide. Following a deprotonation step, reductive elimination of Pd(0) then resulted in formation of the biaryl bond, completing the core framework. Application of this “direct” palladium-catalyzed biaryl coupling facilitates a very efficient and concise synthesis of rhazinilam as a racemate.
Gaunt and co-workers have also completed the first total synthesis of rhazinicine, a related natural product to rhazinilam, achieved in 11 steps from commercial materials using a short synthetic strategy that demonstrates how iterative metal-catalyzed C–H bond functionalizations can positively influence complex molecule assembly (Scheme 58) [82].

Iterative C–H bond functionalization strategy for the synthesis of (±)-rhazinicine
They found that combination of the Ir(I) catalyzed C–H borylation and Suzuki coupling sequence led to a two-step, one-pot “C–H Suzuki” arylation that enabled direct transformation of the N-Boc pyrrole to the C3 arylated intermediate in 78% yield. Following installation of the required acyl group, an application of their oxidative Pd-catalyzed C–H alkenylation reaction enabled formation of the key structural architecture of the natural deliver the natural product. The orthogonal selectivity characteristics displayed by these C–H functionalization processes makes possible iterative functionalization of the heteroaromatic pyrrole core. Utilization of the highly versatile C–H borylation – Suzuki coupling to install the aromatic functionality opens up possibilities of facile analogue synthesis via this route.
C–H functionalization is an area of rapid growth that pushes the limits of chemical reactivity. Even within the subsection of palladium-catalyzed C–H bond functionalization of indole and pyrrole, many new modes of reactivity and strategies for controlling selectivity have emerged over the past 5–10 years. Discovery of new catalytic species and reaction conditions with novel selectivity modes will be crucial to the future utility of C–H activation within the context of complex molecule synthesis. Achieving selectivity among different C–H bonds remains a major challenge and so the development of new catalyst systems that are both highly reactive and predictably selective is essential to significantly increase the efficiency with which carbon frameworks can be constructed. Combining various mechanisms of activation should allow us to selectively functionalize at a variety of different positions around an aromatic system. Such an ability to transform a variety of C–H bonds with high levels of selectivity would unlock an entirely new perspective in complex molecule synthesis and should lead to major simplifications of synthetic sequences.
Abbreviations
- Ac:
-
Acyl
- Am:
-
Amyl
- Ar:
-
Aryl
- atm.:
-
Atmospheres
- Bn:
-
Benzyl
- Boc:
-
tert-Butyloxycarbonyl
- BQ:
-
Benzoquinone
- Bu:
-
Butyl
- Bz:
-
Benzoyl
- °C:
-
Degrees centigrade
- cat.:
-
Catalytic
- cod:
-
Cycloocta-1,5-diene
- DavePhos:
-
2-(Dicyclohexylphosphino)-2′-(dimethylamino)biphenyl
- dba:
-
Dibenzylideneacetone
- DCE:
-
1,2-Dichloroethane
- DMA:
-
N,N-Dimethylacetamide
- DMF:
-
N,N-Dimethylformylamide
- DMSO:
-
Dimethylsulfoxide
- DTBP:
-
2,6-Di-tert-butylpyridine
- dtbpy:
-
4,4′-Di-tert-butyl 2,2′-bipyridine
- ee:
-
Enantiomeric excess
- eq:
-
Equivalent
- Et:
-
Ethyl
- FG:
-
Functional group
- Fmoc:
-
Fluorenylmethyloxycarbonyl chloride
- h:
-
Hour
- HATU:
-
(O-(7-Azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HetAr:
-
Heteroaromatic
- IMes:
-
1,2-bis(2,4,6-trimethyl phenyl)imidazol-2-ylidene
- Kcal:
-
Kilocalories
- L:
-
Ligand
- M:
-
Metal
- Mbs:
-
p-Methoxy benzene-sulfonyl
- Me:
-
Methyl
- Mes:
-
Mesityl
- MOM:
-
Methoxymethyl
- NHC:
-
N-Heterocyclic carbene
- [O]:
-
Oxidant
- Ph:
-
Phenyl
- PIDA:
-
Phenyliodo(III)diacetate
- pin:
-
Pinacolato
- Piv:
-
Pivaloyl
- i-Pr:
-
iso-Propyl
- Py:
-
Pyridine
- SEM:
-
2-Trimethylsilyl ethoxymethoxy
- Sol:
-
Solvent
- S-Phos:
-
2-Dicyclohexylphosphino-2′,6′-dimethoxybiphenyl
- TIPS:
-
Triisopropylsilyl
- TFA:
-
Trifluoroacetic acid
- Tf:
-
Triflate
- THF:
-
Tetrahydrofuran
- TMS:
-
Trimethylsilyl
- TON:
-
Turnover number
- Ts:
-
Tosyl
- TSE:
-
Trimethylsilylethyl
- μw:
-
Microwave
- X-Phos:
-
2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl
References
Heck RF (1968) J Am Chem Soc 90:5518–5526
Negishi E, Baba S (1976) JCS Chem Commun 596–597
Milstein D, Stille JK (1978) J Am Chem Soc 100:3636–3638
Suzuki A, Miyaura N, Yanagi T (1981) Synth Commun 11:513–520
Sonogoshira K, Tohda Y, Hagihara N (1975) Tetrahedron Lett. 50:4467–4470
Beletskaya IP, Cheprakov AV (2000) Chem Rev 100:3009–3066
Trost BM (1991) Science 254:1471–1477
Labinger JA, Bercaw JE (2002) Nature 417:507–514
Ritleng V, Sirlin C, Pfeffer M (2002) Chem Rev 102:1731–1769
Kakiuchi F, Murai S (2002) Acc Chem Res 35:826–834
Doye S (2001) Angew Chem Int Ed 40:3351–3353
Guari Y, Sabo-Etienne S, Chaudret B (1999) Eur J Inorg Chem 7:1047–1055
Dyker G (1999) Angew Chem Int Ed 38:1699–1712
Jia C, Kitamura T, Fujiwara Y (2001) Acc Chem Res 132:633–639
Godula K, Sames D (2006) Science 312:67–72
Alberico D, Scott ME, Lautens M (2007) Chem Rev 107:174–238
Seregin IV, Gevorgyan V (2007) Chem Soc Rev 36:1173–1193
Synlett (2006) Duagulis review, 3382
Jia C, Kitamura T, Fujiwara Y (2001) Acc Chem Res 34:633
Garcia-Cuadrado D, Braga AAC, Maseras F, Echavarren AM (2006) J Am Chem Soc 128:1066–1067
Lafrance M, Rowley CN, Woo TK, Fagnou K (2006) J Am Chem Soc 128:8754–8756
Akita Y, Itagaki Y, Takizawa S, Ohta A (1989) Chem Pharm Bull 37:1477
Grigg R, Shridharan V, Stevenson P, Sukirthalingam S, Worakun T (1990) Tetrahedron 46:4003–4108
Kozikowski AP, Ma D (1991) Tetrahedron Lett 32:3317–3320
Desarbre E, Merour J-Y (1995) Heterocycles 41:1987–1998
Lane BS, Sames D (2004) Org Lett 6:2897–2900
Toure BB, Lane BS, Sames D (2006) Org Lett 8:1979–1982
Wang X, Gribkov DV, Sames D (2007) J Org Chem 72:1476–1479
Lane BS, Brown MA, Sames D (2005) J Am Chem Soc 127:8050–8057
Wang X, Lane BS, Sames D (2005) J Am Chem Soc 127:4996
Gorelsky SI, Lapointe D, Fagnou KJ (2008) Am Chem Soc 130:10848
Catellani M, Frignani F, Rangoni A (1997) Angew Chem Int Ed 36:119
Bressy C, Alberico D, Lautens M (2005) J Am Chem Soc 127:13148–13149
Martins A, Alberico D, Lautens M (2006) Org Lett 8:4827–4829
Li W, Nelson DP, Jensen MS, Hoerrner RS, Javadi GJ, Cai D, Larsen RD (2003) Org Lett 5:4835–4837
Park C-H, Ryabova V, Seregin IV, Sromek AW, Gevorgyan V (2004) Org Lett 6:1159–1162
McClure MS, Glover B, McSorely E, Millar A, Osterhout MH, Roschangar F (2001) Org Lett 3:1677–1680
Ohta A, Akita Y, Ohkuwa T, Chiba M, Fukunaga R, Miyajuji A, Nakata T, Tani N, Aoyagi Y (1990) Heterocycles 31:1951–1958
Lavenot L, Gozzi C, Ilg K, Orlova I, Penalva V, Lemaire M (1998) Organomet Chem 567:49–56
Gozzi C, Lavenot L, Ilg K, Penalva V, Lemaire M (1997) Tetrahedron Lett 38:8867–8870
Chabert JFD, Joucla L, David E, Lemaire M (2004) Tetrahedron 60:3221–3230
Deprez NR, Kalyani D, Krause A, Sanford MS (2006) J Am Chem Soc 128:4972–4973
Grimster NP, Gauntlet C, Godfrey CRA, Gaunt MJ (2005) Angew Chem Int Ed 44:3125–3129
Phipps RJ, Grimster NP, Gaunt MJ (2008) J Am Chem Soc 130:8172
Yang S-D, Sun C-L, Fang S, Li B-J, Li Y-Z, Shi Z-J (2008) Angew Chem Int Ed 47:1473–1476
Lebrasseur N, Larrosa I (2008) J Am Chem Soc 130:2926–2927
Daugulis O, Zaitsev VG, Shabashov D, Pham Q-N, Lazarev A (2006) Synlett 20:3382
Lafrance M, Fagnou K (2006) J Am Chem Soc 128:16496–16497
Li R, Jiang L, Lu W (2006) Organometallics 25:5973–5975
Stuart DR, Fagnou K (2007) Science 316:1172–1175
Stuart DR, Villemure E, Fagnou K (2007) J Am Chem Soc 129:12072–12073
Sloan OD, Thorton P (1986) Inorganica Chim Acta 120:173–175
Potavathri S, Dumas AS, Dwight TA, Naumiec GR, Hammann JM, DeBoef B (2008) Tetrahedron Lett 49:4050
Dwight TA, Rue NR, Charyk D, Josselyn R, DeBoef B (2007) Org Lett 9:3137
Hull KL, Sanford MS (2007) J Am Chem Soc 129:11904–11905
Li B-J, Tian S-L, Fang Z, Shi Z-J (2008) Angew Chem Int Ed 47:1115
Brasche G, García-Fortanet J, Buchwald SL (2008) Org Lett 10:2207
Cho J-Y, Tse MK, Holmes D, Maleczka RE Jr, Smith MR III (2002) Science 295:305
Murphy JM, Liao X, Hartwig JF (2007) J Am Chem Soc 129:15434
Tse MK, Cho J-Y, Smith MR III (2001) Org Lett 3:2831
Takagi J, Sato K, Hartwig JF, Ishiyama T, Miyaura N (2002) Tetrahedron Lett 43:5649
Paul S, Chotana GA, Holmes D, Reichle RC, Maleczka RE Jr, Smith MR III (2006) J Am Chem Soc 128:15552
Fujiwara Y, Moritani I, Danno S, Teranishi S (1969) J Am Chem Soc 91:7166–7169
Moritani I, Fujiwara Y (1967) Tetrahedron Lett 8:1119–1122
Fujiwara Y, Maruyama O, Yoshidomi M, Taniguchi H (1981) J Org Chem 46:851–855
Tsuji J, Nagashima H (1984) Tetrahedron 40:2699–2702
Fujiwara Y, Jia C, Kitamura Y (1999) Org Lett 1:2097–2100
Abbiati G, Beccalli EM, Broggini G, Zoni C (2003) J Org Chem 68:7625–7628
Ferreira EM, Stoltz BM (2003) J Am Chem Soc 125:9578–9579
Nishimura T, Onoue T, Ohe K, Uemura S (2003) J Am Chem Soc 125:9578–9579
Zhang H, Ferreira EM, Stoltz BM (2004) Angew Chem Int Ed 43:6144–6148
Liu C, Widenhoefer RA (2004) J Am Chem Soc 126:10250–10251
Liu C, Widenhoefer RA (2006) Chem Eur J 12:1371–2382
Capito E, Brown JM, Ricci A (2005) Chem Commun 1854–1856
Beck EM, Grimster NP, Hatley R, Gaunt MJJ (2006) Am Chem Soc 128:2528
Trost BM, Godleski SA, Genet JPJ (1978) Am Chem Soc 100:3930
Baran PS, Corey EJ (2002) J Am Chem Soc 124:7904–7905
Baran PS, Guerrero CA, Corey EJJ (2003) Am Chem Soc 125:5628
Garg NK, Caspi DD, Stoltz BM (2004) J Am Chem Soc 126:5970–5971
Garg NK, Caspi DD, Stoltz BM (2005) J Am Chem Soc 127:5970–5978
Bowie AL, Hughes CC, Trauner D (2005) Org Lett 7:5207–5209
Beck EM, Hatley R, Gaunt MJ (2008) Angew Chem Int Ed 47:3004
Acknowledgment
We are grateful to the EPSRC and GlaxoSmithKline for funding (E.M.B.) and the Royal Society and Philip and Patricia Brown (for University Research Fellowship and Next Generation Fellowship to M. J. G).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Beck, E.M., Gaunt, M.J. (2009). Pd-Catalyzed C–H Bond Functionalization on the Indole and Pyrrole Nucleus. In: Yu, JQ., Shi, Z. (eds) C-H Activation. Topics in Current Chemistry, vol 292. Springer, Berlin, Heidelberg. https://doi.org/10.1007/128_2009_15
Download citation
DOI: https://doi.org/10.1007/128_2009_15
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-12355-9
Online ISBN: 978-3-642-12356-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)