
Abstract
Metabolomics is an essential technology for functional genomics and systems biology. It plays a key role in functional annotation of genes and understanding towards cellular and molecular, biotic and abiotic stress responses. Different analytical techniques are used to extend the coverage of a full metabolome. The commonly used techniques are NMR, CE-MS, LC-MS, and GC-MS. The choice of a suitable technique depends on the speed, sensitivity, and accuracy. This chapter provides insight into plant metabolomic techniques, databases used in the analysis, data mining and processing, compound identification, and limitations in metabolomics. It also describes the workflow of measuring metabolites in plants. Metabolomic studies in plant responses to stress are a key research topic in many laboratories worldwide. We summarize different approaches and provide a generic overview of stress responsive metabolite markers and processes compiled from a broad range of different studies.

Graphical Abstract
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
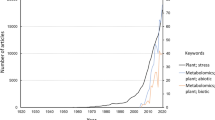


Keywords
- Abiotic stress
- Analytical platforms
- Biotic stress
- Data mining
- Functional genomics
- Mass spectrometry
- Metabolomics
- Systems biology
1 Introduction
Recent advances in technology have revolutionized the approach in which biological systems are visualized and questioned. Progressive developments in the field of genetics and automated nucleotide sequencing have supported the large-scale mapping and sequencing of many genomes, which include Arabidopsis thaliana [1], rice [2, 3], tomato [4], humans [5], etc. Technologies like expressed sequence tag (EST), mRNA profiling using microarrays [6], or serial analysis of gene expression (SAGE) [7] have allowed comprehensive analysis of the transcriptome. Advancements in mass spectrometry have enabled the analysis of cellular proteins and metabolites on a large scale, which was previously not possible [8,9,10,11,12,13,14,15,16,17,18]. The cumulative application of these technologies in various fields has led to advancement in the research of functional genomics and systems biology [19,20,21,22,23]. The foundations of both functional genomics and systems biology rely on comprehensive genome-scale molecular analysis [16, 18]. These approaches are commonly referred to as genomics, transcriptomics, proteomics, and metabolomics (Fig. 1).

Flow of biological information from genome to metabolome of a given cellular state in order to generate desired phenotype
Metabolomics is a complementary tool for functional genomics and systems biology, together with well established “omics” technologies for high-throughput data acquiring [16, 24,25,26]. The components of the metabolome can be viewed as the end-product of gene expression that defines the biochemical phenotype of a cell or a tissue. Quantitative and qualitative measurement of cellular metabolites thus provides a broad view of the biochemical status of an organism that can be used to monitor or assess gene function [16, 24]. The transcriptome represents mRNA changes in cellular machinery required for protein synthesis, but an increase in the levels of mRNA does not always correlate with protein levels [27]. Furthermore, translated proteins may or may not be enzymatically active. Thus, due to these reasons, changes in the transcriptome or proteome level do not necessarily correspond to the alteration in biochemical phenotypes. Moreover, transcriptome and proteome profiling includes the identification of mRNA and proteins through sequence similarity or database search (i.e., it depends on organism-specific genome information). Absence of these information/databases often limits the outcome of the analysis. Considering the above limitations, metabolite profiling provides essential functional information that has to be integrated with transcriptome and proteome analysis in order to increase the understanding of a given cellular state/biological sample [16,17,18, 28]. The qualitative and quantitative metabolomic profiling of a cell, tissue, or organism is very crucial because metabolites are structurally very small (less than 2,000 Da) and diverse molecules that are chemically transformed during cellular metabolism and hence pose a great challenge for analytical technologies [16, 29, 30]. Many stress responses lead to altered gene expression, particularly in plants, which results in qualitative changes in the metabolite pool, therefore identification of metabolites becomes even more critical [30].
One of the earliest metabolic profiling methods originated from Baylor College of Medicine in the early 1970s [31,32,33], which includes multicomponent analyses of steroids, acids, and neutral and acidic urinary drug metabolites using GC/MS. Thereafter, the concept of metabolite profiling was widely used for diagnostics and to assess health regimes [34, 35]. Gradually, there was an increase in research on automation [36] and expansion of GC-based methods to a wide range of chemical classes [37], followed by the use of high performance liquid chromatography (HPLC) and nuclear magnetic resonance (NMR) techniques for metabolite profiling [38, 39]. In the early 1990s, Sauter and colleagues used GC-MS metabolic profiling as the diagnostic technique in order to determine the mode of action of herbicides on barley plants [40]. Based on this and other studies, the concept of metabolic profiling and metabolomics in the context of functional genomics was introduced [24, 25, 41,42,43]. A first introduction of metabolomics as an integral technique for systems biology, linking metabolite profiling and metabolomics with genome-scale metabolic modelling, was described in 2003 [16]. There are many research institutes and commercial entities that are growing exponentially and working towards the development and improvisation of metabolomics science to broaden the range of its application.
2 Analytical Platforms in Metabolomics
A single analytical technique is not sufficient for detection and quantification of the metabolome and, therefore, multiple technologies are needed for a comprehensive view [16, 22, 29, 44]. Analytical technologies used in metabolomics include thin layer chromatography (TLC), HPLC with ultraviolet and photodiode array detection (LC/UV/PDA), gas chromatography–mass spectrometry (GC-MS), capillary electrophoresis–mass spectrometry (CE-MS), liquid chromatography–mass spectrometry (LC-MS), liquid chromatography–electrochemistry–mass spectrometry (LC-EC-MS), NMR, LC-NMR, direct infusion mass spectrometry (DIMS), and Fourier–transform infrared (FT-IR), etc. [16, 25, 43, 45, 46]. Of the above-mentioned techniques, NMR, GC-MS, LC-MS, and CE-MS are the most widely used technologies today [24, 45, 47,48,49,50]. Selection of the most suitable technology is based on speed, selectivity, sensitivity, and accuracy. NMR is rapid and selective, whereas mass spectrometry methods (GC-MS, LC-MS, and CE-MS) offer good selectivity and sensitivity but with longer analysis time [40, 51].
2.1 Nuclear Magnetic Resonance (NMR)
The use of NMR in metabolomics has opened the areas of biochemistry and phytochemical analysis [47, 52]. It is an unbiased, rapid, non-destructive technique that requires little sample preparation [48]. NMR analysis is not based on the analyte separation (as in the case of chromatographic analysis); rather it provides selectivity without separation and is also independent of the analyte polarity and does not require sample derivatization prior to the analysis. When the samples are placed in a strong magnetic field and irradiated with radio frequency, the absorption of energy promotes nuclei from a low-energy to a high-energy state. The subsequent emission of radiation generates resonance or signals that are recorded on the NMR spectrum as “chemical shifts,” representing frequencies from all NMR-visible nuclei in the sample, is relative to the reference proton present in a reference compound [53]. Hence, NMR analyses generally provide a global view of all the metabolites (primary and secondary) in a sample, provided they are in the detectable range [30, 49]. The disadvantage of low sensitivity and resolution has been addressed by using cryogenic probes, higher strength of superconducting magnets, miniaturized radio frequency coils, and multidimensional techniques (for example 2D-J-resolved and heteronuclear single quantum coherence) [48, 49]. 2D-NMR facilitates the identification of the compounds, which also includes the observation of minor compounds along with structural elucidation. Further, NMR has been applied in the areas of plant metabolism [54, 55], Duchenne muscular dystrophy [56], bioavailability, and metabolic responses of rats to epicatechin, hypertension, and acetaminophen toxicity [57, 58].
2.2 Mass Spectrometry (MS)
Flow-injection mass spectral analysis has been widely used in metabolic fingerprinting. Vaidyanathan et al. [59] demonstrated the use of flow injection ESI/MS for metabolic fingerprinting of cell-free extracts used for bacterial identification [59]. Similarly, multiple ionization techniques coupled to Fourier transform mass spectrometry (FT-MS) were used to identify metabolites associated with development and ripening of strawberry fruit [60]. FT-MS has a high resolution and high mass accuracy capacity, which allows separation and differentiation of very complex samples with the calculation of elemental composition, which facilitates structural differentiation and characterization. Unfortunately, this technology cannot differentiate chemical isomers that have an exact mass, e.g., hexoses [51].
Column chromatographic techniques (GC/LC) have a medium to high sensitivity that provides separation based on the physiochemical properties of an analyte [16, 50]. For complex samples, chromatographic separations include factors like column chemistry, an elution method for LC (gradient or isocratic), and a program–temperature method for GC. Multidimensional separation systems such as two-dimensional gas chromatography (GC × GC) and two-dimensional liquid chromatography (LC × LC) are used to enhance chromatographic separation of complex mixtures. Coupled to mass spectrometry (MS) for detection, these techniques are sensitive and capable of detecting low abundance metabolites [61,62,63,64,65]. GC-MS is a robust, technically reproducible, and sensitive approach. It is a well-suited platform for non-targeted metabolite profiling of volatile and thermally stable non-polar or derivatized polar metabolites [16, 26, 28, 66,67,68,69]. This technique is also used for targeted analysis of derivatized primary metabolites [70]. Electron impact (EI) is most commonly used in GC-MS, which results in fragmentation patterns that are highly reproducible. A mass analyzer like time-of-flight (TOF)-MS is widely used for detection because it has a faster “scan rate,” which improves deconvolution, high mass accuracy, and reduces the run time for complex mixtures [28, 66, 67, 69, 71, 72]. This technology is widely used for metabolite profiling, and thus contains several stable protocols for machine setup and maintenance, along with chromatogram evaluation and interpretation. Additionally, it has a short running time and a relatively low running cost. The use of the GC-MS platform is limited for thermally stable volatile compounds, thus making the analysis difficult for high molecular weight compounds (larger than 1 kDa) [16, 50, 73,74,75]. GC-MS facilitates the identification and quantification of hundreds of metabolites in plant samples, which include sugars, amino acids, organic acids, and polyamines, leading to the comprehensive coverage of primary metabolites in the central pathways. Quantitative metabolite profiling of potato tuber (Solanum tuberosum) using GC-MS leads to the identification of sugars, sugar alcohols, amino acids, and organic acids [69, 72, 76, 77]. The non-biased approach in the metabolite profiling of Arabidopsis leaf extract led to the identification of up to 652 metabolites and metabolite features [25, 28, 78]. Several studies were performed using other GC-MS methods, such as the metabolite profiling of tomato (Lycopersicon esculentum) and Lotus japonicus, which led to the identification of 200 and 87 metabolites respectively [79, 80]. LC-MS does not require prior sample treatment and separates the components in a liquid phase [16, 50]. The choice of columns includes reversed phase ion exchange and hydrophobic interaction columns that separate metabolites based on the different chemical properties. In plant metabolomics, LC-MS is frequently used in the profiling of secondary metabolites [71, 81,82,83,84,85]. Hydrophilic interaction liquid chromatography coupled to mass spectrometry (HILIC-MS) is also widely used to analyze highly polar plant extracts [86, 87].
There are three main components in all types of MS instruments: (1) an ionization source such as electron impact (EI), electrospray (ESI), and atmospheric pressure chemical ionization (API); (2) a mass analyzer such as time of flight (TOF), quadrupole mass filters, and quadruple ion trap; and (3) a detector such as an electron multiplier-based detector or micro-channel plate linked to a time-to-digital converter. The detected ions are recorded as pairs of m/z and abundance value, processed and displayed in a mass spectral format that allows us to identify and quantify a large variety of metabolites even with high molecular mass, high polarity, and low thermostability [45, 46].
2.3 Capillary Electrophoresis (CE) MS
Capillary electrophoresis (CE) uses charge-to-mass ratio to separate polar and charged compounds. CE is a powerful technique that can separate a diverse partially complementary range of chemical compounds compared to liquid chromatography [88,89,90]. In many CE-MS-based metabolomics studies, ESI is used for ionization in combination with TOF-MS, which provides high mass accuracy and high resolution. A small amount of a sample is required for the analysis (nanoliters of samples in the capillary). It can be used for volume-restricted sample analysis. One of the major drawbacks of this technique is less sensitivity, poor migration time, reproducibility, and lack of reference libraries. CE and LC can both separate a large variety of metabolites based on fundamentally different mechanisms, they can often be used together to provide a wider coverage of metabolites [91].
Wantanabe et al. quantified carbohydrates, amino acids, and primary metabolites using CE-MS in response to elevated CO2 [92]. Several studies have also been performed in rice using CE and CE-MS for the identification of primary metabolites. Maruyama and co-workers identified cold and dehydration-responsive metabolites, phytohormones, and gene transcription in rice. This analysis led to the identification of several genes that were up-regulated and involved in starch degradation, sucrose metabolism, and the glyoxylate cycle. It has also demonstrated the accumulation of glucose (Glc), fructose, and sucrose. Additionally, it was also observed that regulation of the glyoxylate cycle is correlated with glucose accumulation in rice, which was not observed in Arabidopsis [93].
2.4 Data Mining and Data Processing
All the “omics” analysis generates a large volume of data. In order to handle these large data sets, automated software is needed that can identify peaks from raw data, align the peaks among different samples, and replicate to identify and quantify each metabolite [81, 94,95,96,97,98]. Therefore, informatics and statistics are essential tools for processing metabolomics datasets. Data mining consists of data pre-processing, data pre-treatment, and statistical interpretation of the primary data [96]. The statistical interpretation is essential for central data analysis [68, 99, 100].
Metabolomics studies generate high-dimensional complex datasets that are difficult to analyze and interpret using univariate statistical analysis. Therefore, multivariate data analysis (MVDA) and mathematical modelling approaches are used to obtain meaningful information. These methods provide models that are well suited for a covariance pattern (both within and between variables) analyses [16, 17, 24, 28, 68, 98, 101,102,103]. Most commonly used methods for MVDA are principal component analysis (PCA), ANOVA, partial least square (PLS), and SIMCA (Soft Independent Modelling of Class Analogy). Web-based applications like MetaGeneAlyse implements standard normalization/clustering methods like k-means and independent component analysis (ICA) [104]. This software also provides other statistical analysis like the t-test, PLSDA (partial least square discriminant analysis), pathway enrichment analysis, etc. Other web-based applications are MetaboAnalyst [105], MetaMapp [106], metaP-Server [107], MeltDB [108], MetiTree [109], etc. These applications cover multiple steps from data pre-processing to biological interpretation. Many different multivariate statistical tools, metabolic modelling, and structural elucidation of unknown metabolites were integrated into a toolbox for metabolomics called COVAIN [96, 97]. The name stems from “covariance inverse,” implying that a covariance pattern is indeed used for functional interpretation of metabolite dynamics. This concept was recently developed and provides a fundamental novel approach to link causal relationships of biochemical networks with metabolite dynamics measured with metabolomics technology such as GC-MS, LC-MS, or any other technology [16, 18, 50, 68, 96, 97]. This approach goes beyond the classical MVDA analysis or correlation network analysis because it is able to identify causal biochemical perturbation points. It was recently applied to the analysis of the interface of primary and secondary metabolism in plants in Arabidopsis and Theobroma [81, 84, 85] and for the analysis of energy starvation and subsequent biochemical regulation [110, 111].
The high throughput metabolic data can be analyzed in supervised and unsupervised strategies [68]. The unsupervised method focuses on the intrinsic structure, relation, and interconnection of the data, which are sometimes referred to as descriptive and explorative models. Supervised methods seek to transform multivariate data from metabolite profiling into representations of biological interest under “supervision” and are often referred to as predictive models [29].
Metabolite data contain information such as metabolite name, change in levels, and their relationships, which are very useful for the interpretation of the biological significance. Most commonly, the identified metabolite is described in pathways or networks to understand its biological context. Some of the well curated databases for metabolic pathways in plants are KEGG (which is based on resources like GenBank/EMBL/DDJB) [112], ArcCyc [113], MetaCrop [114], UniPathway [115], SMPDB [116], and MapMan [117] etc. Table 1 shows the details of other metabolomic databases that are widely used for the analysis.
2.5 Compound Identification
Compound identification is the conclusive step in metabolite analysis; it is one of the critical steps because the biochemical interpretation of metabolomic data is based on the availability of a well-structured database for identification of metabolites [118, 119]. Putative compound identification is based on molecular properties like the mass spectral pattern and accurate mass to define molecular and/or empirical formulae from which the metabolite can be derived or identified by the comparative search [82]. Definitive compound identification is based on retention time (Rt), retention index (RI), mass spectral fragmentation, and NMR spectral shift. Confirmation of the identified compound is done by a comparative library search, authentic chemical standards, and by using in vivo labelling methods. Sometimes analytically detected entities with biological significance are reported as “unknowns” with no structural identification [46, 118]. Recently, we have introduced a novel algorithm for structural elucidation of unknown compounds and even full pathways from untargeted metabolomics data [82]. This algorithm is especially suited for stress-related secondary metabolites such as flavonoids as an antioxidative response to cold and light stress [82]. In this study we also demonstrated how cold and light stress change the oxygen-to carbon-ratio in secondary metabolites systematically using so-called van Krevelen plots [82]. For compound identification at different levels of accuracy, minimum reporting standards need to be described. The metabolomics community has developed a nomenclature for publication of metabolomics data [120].
2.6 Limitations of Metabolomics
Metabolomic platforms lack the ability to comprehensively profile all the metabolites of a given cell/tissue [98]. This limitation is directly linked to the chemical complexity of the metabolites, the biological variance that is inherent in living organisms, and the dynamic range of the instruments. The genome and transcriptome consist of linear polymers of nucleotides with high chemical similarity; this structure facilitates high-throughput analytical approaches. The proteome is substantially more complex, but it is still based on a limited set of amino acids. The chemistry of these biopolymers is well defined and analytical technologies like 2DE gel electrophoresis and shotgun proteomics can readily identify and differentiate a large number of proteins in a single analysis and even post-translational modifications such as phosphorylation or methylation [14, 121, 122]. In the case of the metabolome, the chemical complexities are significantly greater and range from an ionic inorganic moiety to hydrophilic carbohydrates, hydrophobic lipids, and complex natural products. Hence, the chemical diversity and complexity make metabolome profiling extremely difficult. This obstacle can be circumvented by using selective extraction protocols and combinations of technologies for the analysis to obtain a more comprehensive coverage of the metabolome [16, 28, 71, 81].
Analytical variation can be defined as the coefficient of variation or relative standard deviation that is directly related to the experimental approach; this variance differs depending on the technology platform being employed. Biological variance arises from the quantitative variation in metabolite levels between plants of the same species that are grown under identical conditions [28, 123]. Biological variance is the major limitation of “resolution” in metabolomics. Pooling of the samples tends to avoid or reduce biological variance. This strategy helps to minimize random variation by using statistical knowledge, but also leads to dilution of the samples, which results in the dilution of sites or tissues that are important for specific regulations (up/down) of the metabolite. Therefore, more targeted analysis can help to minimize the variation. In the case of plants, parameters like the synthesis of natural products, growth stage, environmental cues, etc. make the sampling critical and strategies are required to minimize the variations [44, 51].
The major analytical challenge encountered in metabolomics is dynamic range. Dynamic range can be defined as the concentration boundaries for analytical determination. The dynamic range can be critically limited by a sample matrix or by the presence of interfering and competing compounds. Most of the mass spectrometers have a dynamic range of 104–106 for individual components; however, this range is reduced significantly due to the presence of other chemical components (i.e., the presence of excessive metabolites that can cause significant interference that limits the range for the identification of other metabolites) [44, 51]. For example, high levels of sugars (primary metabolites) often interfere with the identification of secondary metabolites such as flavonoids. However, many of the highly expressed metabolites are often unique and provide a basis for the differentiation of the cellular states, organs, tissues, and organisms. These exclusive compounds are often referred as “biomarkers.” Selective profiling of these biomarkers is very useful for high-throughput diagnosis of a specific disorder, for example diabetes (i.e., glucose monitoring) or cancer. This detection should not be regarded or classified as metabolomics due to the highly targeted nature of profiling [24]. Another problem is salts; low levels of these ionic species reduce the ionization efficiency in ESI/MS and significantly interfere with the profiling of all other species [124]. Therefore, different analytical approaches have been developed to improve the dynamic range and to increase the level of identifications [51].
3 Plant Metabolomics
Metabolomic platforms can be used for unbiased identification of the metabolite levels in different genotypes that may or may not produce visible phenotypes [16, 72, 125]. The total number of metabolites found in plants are currently estimated to be ~200,000, with ~7,000–15,000 found in any individual species [73], of which 3,000–5,000 were exclusively determined in leaves [48]. So far metabolite profiling has been performed on a wide range of plant species, which includes Arabidopsis, tomato, potato, rice, wheat, strawberry, Medicago, cucumber, lettuce, tobacco, poplar, and Eucalyptus.
Selective metabolite profiling has been used in many studies to provide biological information beyond simple identification of the plant constituents. These include: (1) fingerprinting of species, genotypes, and ecotypes for taxonomic or biochemical information [29, 44, 126]; (2) monitoring the behavior of a specific class of metabolites in response to exogenous chemicals or physical stimuli [127, 128]; (3) understanding the developmental process and symbiotic associations [129]; and (4) comparison of the metabolite content of mutant or transgenic plants with the wild type [44, 51]. In each of these studies, metabolite profiling can be coupled with other “omics” technologies to provide an integrated picture of all aspects of information from genome to metabolome and the resulting phenotype [18]. For example, metabolite profiling can be combined with marker-assisted selection providing integral information and understanding about the chemical composition of crop species [130]. Apart from the wide application of metabolomics in plant research, there has been considerable progress in the metabolomic analysis of single cells from different plant species, which includes tissues like pollen, trichrome, root hairs, guard cells, etc.; these studies have been well reviewed [131].
The phenotype of a plant depends on the synthesis and accumulation of a series of metabolites in specific organs, at specific developmental stages [132]. It also depends on the environmental signals. Therefore, there are various kinds of metabolites in plants that have organ-/tissue-specific characteristics. For example, sphingolipids, a class of lipids that are critical in the development of the male gametophyte, are significantly different in the pollen and leaf tissues of Arabidopsis [133]. Anthocyanins accumulate in hypocotyls of young tomato seedlings, whereas several flavonols and phenolic compounds have been identified in cotyledons and some alkaloidal compounds are found in the root [132]. There are a lot of variations in biochemical pathways at cellular and sub-cellular levels in plants; therefore, the use of metabolomic platforms with different methods has significantly increased.
4 Workflow for Plant Metabolomic Analysis
Plant metabolomes are very complex and diverse in their chemical structures. Comprehensive identification and a broad range of metabolic pictures can be achieved by the combination of two or more metabolomic strategies and analytical systems, including variation in the extraction protocols [16, 28, 49, 71, 81, 134,135,136].
Metabolomic analyses consist of three main experimental strategies: (1) sample preparation; (2) acquisition of the data using analytical methods; and (3) compound identification and data mining. These steps are crucial and inter-related, as is illustrated in Fig. 2, with each step consisting of a series of sub-steps with various experimental phases to form a meaningful biochemical interpretation [118, 136].

Flowchart detailing the steps involved in plant metabolomics. There are three main steps in the metabolomic analysis, which include sample preparation, data acquisition, and data analysis, which lead to the biochemical interpretation of the identified metabolite
Sample preparation is one of the critical steps as it contributes to the identification of the wide array of metabolites. This step consists of selection and harvesting of samples, drying or quenching procedure, and extraction of metabolites for analysis (derivatization). The selection of plant material depends on the researcher and the experimental design. Throughout this step, care must be taken to avoid the introduction of unwanted variability, which could significantly affect the outcome of the analysis. Sample degradation (oxidative or enzymological) and contamination are the major factors. Various enzyme quenching methods like drying, use of enzyme inhibitors, use of acids, or high concentrations of organic solvents can also affect the analyses/identification [24, 136].
Plant metabolites are structurally diverse with high complexities like different size, solubility, volatility, polarity, quantity, and stability [51]. There are several metabolite extraction protocols; the choice of method depends on a variety of factors such as physiochemical properties of the targeted metabolites, biochemical composition, and solvent used. Some of the common extraction protocols include solvent extraction, supercritical fluid extraction, solid phase extraction, and sonication [30, 52, 136]. However, no comprehensive extraction technique exists that can lead to the identification of all classes of metabolites with high reproducibility and robustness.
Sample analysis requires an advanced analytical platform (separately/combination) to measure the ultra-complex metabolite samples [51]. The ranges of analytical platforms are well discussed above, and each platform has its own limitations, either in sensitivity or selectivity. The choice of the platform depends on the study undertaken, consideration of the class of compounds, and their chemical and physical properties along with their concentration levels [45, 137].
Recently we have developed an integrative protocol that combines comprehensive metabolite extraction and analysis with proteomics and RNA analysis from one sample [28]. This protocol is adapted to allow the simultaneous analysis of all molecular levels and investigate their inter-relation and covariance structure [28, 66, 67, 138, 139]. This covariance structure of molecular dynamics of a cellular system is a result of biochemical regulation [139]. Therefore, it is possible to read biochemical regulation from the molecular association networks or in other words the corresponding covariance data [16,17,18, 66, 68, 72, 84, 110, 139].
5 Metabolomic Studies in Plant Stress Responses
Metabolomic studies have become increasingly common in plant physiology and biochemistry. In the following section, we review applications of metabolomics to study plant responses to environmental stress (like abiotic and biotic factors) (Table 2). Abiotic factors include drought, temperature, salt, oxidative stress, flooding, nutrient deficiency, heavy metals, and effects of combinations of stress. We describe the nature and symptoms of various stresses and introduce several metabolomic studies with major metabolite changes (Table 2).
5.1 Drought Stress
Drought is one of the major threats to crop production worldwide and the situation is projected to get worse in the near future [193, 194]. The physiological response of plants under drought stress includes reduction of leaf area, leaf abscission, and increase in root growth to enhance the uptake of nutrients. Additionally, closure of leaf stomata takes place, which reduces water loss through transpiration. These physiological changes improve the water use efficiency (WUE) of the plant in the short term, but have a negative effect on photosynthesis, for example, closure of stomata lowers the intercellular CO2 concentration, which adversely affects photosynthesis [193, 194]. Fine metabolomic adjustment is another strategy of plants to cope with drought stress. These metabolomic adjustments include net accumulation of osmolytes in the cell to retain or promote the uptake of water into the cell via osmosis in order to maintain turgor pressure [193].
Compatible solutes are highly soluble and small molecular-weight osmolytes that do not inhibit cellular metabolism even at high concentrations [195, 196]. Common osmolytes include soluble sugars (e.g., glucose, sucrose, and trehalose), the RFOs (raffinose, stachyose, and verbascose), polyols (e.g., mannitol, sorbitol), amino acids (e.g., proline), quaternary ammonium compounds (e.g., glycine betaine), and other polyamines (e.g., putrescine, spermidine, and spermine). Accumulation of these osmolytes in plant cells is important for sustaining cell turgor by osmotic adjustment, and stabilization of enzymes that reduce the levels of reactive oxygen species (ROS) in order to maintain the cellular redox balance.
The targeted metabolite approach using LC-MS was established by Antonio and co-workers in 2008 to investigate the effect of drought in Lupinus albus stem tissues [140]. In this analysis, 12 water-soluble organic osmolytes – like mono- and disaccharides, raffinose, stachyose, and verbascose – and sugar alcohols were identified by chromatographic analysis using PGC stationary phase. Although the role of RFOs is not completely understood, there are several reports that show evidence for the strong correlation between accumulation of RFOs and development of desiccation tolerance [140].
Urano et al. [146] demonstrated metabolomic changes in A. thaliana (ecotype Col-0 (WT) and the NCED3 knockout mutant) under drought stress. Many metabolites were identified including amino acids such as proline, raffinose family oligosaccharides, and γ-amino butyrate (GABA). In this study an nc3-2 mutant that lacks the NCED 3 gene is involved in the dehydration-inducible biosynthesis of abscisic acid (ABA), to determine the effect of ABA under drought stress. In combination with the transcriptome analysis, it was clearly demonstrated that ABA-dependent transcriptional regulation is important to activate metabolomic pathways like branched amino acids, polyamine and proline biosynthesis, GABA shunt, etc., but regulation of a raffinose biosynthetic pathway still remains unknown [146].
Polyamines such as putrescine, spermidine, and spermine are ubiquitous in nature and also provide protection to plants under drought stress [145, 197, 198]. GC-TOF-MS analysis by Do et al. [144] determined the level of selected metabolites related to polyamine metabolism in rice cultivar (Oryza sativa L. Ecotype indica and japonica) under moderate long-term drought stress. The combination of gene expression and GC-TOF-MS metabolite data showed coordinated adjustment of polyamine biosynthesis in order to facilitate the accumulation of spermine under drought stress conditions [144].
Gechev and collaborators combined transcriptomics and metabolomics to investigate desiccation tolerances in Haberla rhodopensis in four different conditions (i.e., well-watered, partially dehydrated, desiccated, and rehydrated) [141]. Transcripts of proteins involved in carbohydrate metabolism (e.g., genes that encode galactinol synthase and stachyose synthase), sucrose synthase, and sucrose-6-phosphate synthase showed increased levels. These finding showed the importance of carbohydrate metabolism for protecting cells during desiccation. Genes that mainly encode proteins to prevent cellular damage and participate in antioxidant defense (e.g., LEA) were found to be most abundant in response to dehydration. In this study, the GC-TOF-MS approach was used to determine metabolite profiling combined with two different LC-MS approaches, which allowed the broad range of metabolome identification. This study also revealed that sucrose, maltose, and RFOs such as stachyose and verbascose accumulate in H. rhodopensis in significantly high levels upon dehydration. Additionally, other metabolites like amino acids, phenylalanine, and tyrosine were also observed in the dehydrated state, which suggests the activation of the shikimate pathway that results in the synthesis of antioxidants.
Skirycz and co-workers performed metabolite profiling of Arabidopsis leaves under mild drought stress. The response to stress in the growing and mature leaves was significantly distinct. Metabolites such as proline, erythritol, and putrescine were identified in mature leaves. Comparing the data with other studies revealed that decreases in the level of aspartate and increases in the level of proline are two common responses shared between mild and severely desiccated leaves [147, 199].
Metabolite profiling has been additionally carried out in other crop plants (maize, wheat, tomato, and soybean) under drought stress and it was observed that changes in the metabolite levels including branched chain amino acids are one of the common factors [143, 148, 149, 151].
A metabolic adjustment also depends on the severity of the stress. Maize was subjected to a drought stress for 17 days. GC-MS metabolomic analysis showed changes in the concentrations of 28 identified metabolites. Further, accumulation of carbohydrates, proline, amino acids, shikimate, serine, glycine, and aconitase were identified. Additionally, decreased levels of leaf starch, malate, fumarate, and 2-oxoglutarate were also observed in the drought-treatment course. However, between the 8th and 10th days, some metabolites were changed drastically, hence showing their dependency on stress severity [150].
The GC-MS metabolomic analysis was also performed on the moss Physcomitrella patens under drought stress. In this analysis, 2 weeks of physiological drought stress was applied, which showed that 26 metabolites were differentially affected in gametophores, including altrose, maltitol, L-proline, maltose, isomaltose, and butyric acid. More interestingly, a new compound, annotated as EITTMS_N12C_ATHR_2988.6_1135EC44, was also accumulated specifically in response to drought stress in this moss [142].
5.2 Temperature Stress
A freezing environment leads to the formation of ice, which can seriously damage plant cells and cellular membranes. Many plant species develop freezing tolerance during their exposure to non-freezing low temperatures, and this process is known as “cold acclimatization” [200]. The molecular basis of this process has been widely studied. The first metabolomic studies of cold acclimatization were performed by two groups in 2004. Cook et al. [152] compared the metabolomic changes during cold acclimatization in A. thaliana (ecotype Wassilewskija – 2 (Ws-2) and Cape Verde islands-1 (CVi-1)). A metabolome of the Ws-2 plant was significantly changed in response to low-temperature stress. Seventy-five percent of metabolites monitored were found to increase under cold-acclimated plants, which include amino acids such as proline and sugars (glucose, fructose, inositol, galactinol, raffinose, and sucrose). Additional changes were also identified with increased levels of trehalose, ascorbate, putrescine, citrulline, and some TCA-cycle intermediates. However, there were considerable overlaps in the metabolite changes between the two ecotypes in response to low temperature [152].
Time-course metabolomic analysis (from cold to heat conditions) showed an increase in the pool size of amino acids derived from pyruvate, oxaloacetate, polyamine precursors, and other compatible solutes [154]. The study concluded that the majority of the heat shock metabolite responses were shared with cold stress, while heat shock had a less pronounced effect on metabolism. Transcriptomics analysis revealed the regulation of GABA shunt and the accumulation of proline under a cold condition that was obtained by transcriptional and post-transcriptional processes [155]. Espinoza et al. [153] studied the effect of diurnal gene/metabolite regulation during cold acclimatization by using metabolomics and transcriptomics. Approximately 30 % of identified/analyzed metabolites showed the circadian rhythm in their pool size and low temperatures affected the cyclical pattern of metabolite abundance [153].
The number of important traits in plants, such as stress resistance, post-harvesting etc., largely dependents upon the metabolic content, which can be used for the manipulation of the metabolic phenotype via a classical breeding method. A study performed by Korn et al. [156] combined GC-MS metabolite profiling and statistical methods to decode or identify the combination of metabolites that can predict the freezing tolerance in Arabidopsis. One of the identified candidates was raffinose, which can also be considered as a good marker for freezing tolerance [156].
Metabolomics was also used for functional characterization of candidate genes involved in cold acclimatization. One of the well-characterized genes is a C-repeat binding factor (CBF) that increases freezing tolerance by multiple mechanisms. Cook et al. [152] investigated the regulation of CBF and its effect on the metabolome of the plant under low temperatures using GC-MS analysis. Metabolite profiling of the non-acclimated plants that over-expressed the CBF 3 was similar to that of the cold acclimated Ws-2 ecotype. Hence these data indicate that the CBF pathway plays a prominent role in determining metabolite regulation at low temperatures. Further, the analysis reveals the accumulation of raffinose and galactinol, which are synthesized through the action of CBF-targeted genes AtGolS3.
In another study, Wienkoop et al. investigated the dynamics of metabolite-protein covariance networks and the relation of starch metabolism during cold acclimation [139]. This study revealed that raffinose accumulation belongs to general cold and heat temperature stress responses and that starch metabolism can be compensated by increased sucrose synthesis for cold adaptation processes. A follow-up study revealed the essential role of starch metabolism during cold adaptation in different Arabidopsis ecotypes, thus demonstrating that different ecotypes developed different biochemical strategies to cope with cold acclimation [157]. In another study, Doerfler et al. investigated the interface of primary and secondary metabolism as a response to cold and light stress [81, 82]. Arabidopsis accumulated huge amounts of flavonoid structures due to the combined cold and light stress and a novel algorithm for metabolite identification in non-targeted LC-MS metabolomics data was able to identify new potential flavonoid structures as anti-oxidative response factors [82]. Several other metabolomics analyses have been performed in crop and grass species (tomato, wheat, maize, and miscanthus) [158,159,160, 201] that determined the diverse range of metabolites in plants to confers stress tolerance (Table 2).
5.3 Salt Stress
Increasing salt concentration in soil damages plants in various ways: (1) it hampers the uptake of water and nutrients from the environment, which in turn reduces the water potential of the soil and leads to osmotic stress. Salt stress reduces plant growth and damages cells/tissues. (2) The steady accumulation of sodium ions in plant tissues inhibits essential cellular processes [202, 203]. Plants have adapted strategies to cope with salt stress, which includes adjustment of metabolic status [204].
Gong et al. [165] conducted metabolite profiling on Thellungiella halophila, a distant relative of A. thaliana, under salt stress. This plant shows “extremophile” characteristics manifested by extreme tolerance to a variety of abiotic stresses like low humidity, freezing, and high salinity (it can even grow and reproduce in a 500 nM NaCl concentration) [205]. A comparative metabolomics study between T. halophila and Arabidopsis (under controlled conditions) showed increased levels of proline in both the species along with inositols, hexoses, and complex sugars. The concentration of these metabolites was higher in T. halophila. Transcriptome analysis showed similar results suggesting that T. halophila is primed for acclimatization under stress conditions [165]. Kim et al. [166] showed the effect of high salt concentrations on primary metabolism in a cell culture of A. thaliana (T87 cultured cells). The obtained results showed that the methylation cycle and phenylpropanoid pathways are synergistically induced over the short term in response to salt stress. Long-term responses include co-induction of glycolysis and sucrose metabolism as well as co-reduction of the methylation cycle [166].
There have been several metabolomic studies performed to assess the metabolic effect of salinity in various crops and other plant species, which include tomato [206, 207], grapevine [162], poplar [161], sea lavender (Limonium latifolium), [163] and rice [167]. A study performed by Sanchez and co-workers extensively used the integrated approach of genomic, transcriptomic, and metabolomic analysis on L. japonicus and other lotus species in long-term regimes of non-lethal salt stress. The metabolomic changes were characterized by steady-state increased levels of amino acids, sugars, and polyols with decreases in most organic acids [169]. A comparative metabolomic study between extremophile (Lotus crticus) and glycophyte (Lotus corniculatus) generated similar metabolomic responses under salt stress conditions [170].
Barley (Hordeum vulgare) cultivars that differ in their salt tolerance capacity were subjected to metabolite profiling. In this study long-term salt stress was applied to the barley plants [171]. The more tolerant cultivar (Sahara) demonstrated increased levels of hexose phosphates and intermediates of the TCA cycle, which include citrate, aconitate, isocitrate, α-ketoglutarate, succinate, and malate. The levels of metabolites remain unchanged in the less tolerant cultivar (Clipper) during salt stress. From this study it was proposed that accumulation of proline, γ-amino butyric acid (GABA), and other amino acids in Clipper showed growth or induction of leaf necrosis under salt stress; however, accumulation of these metabolites does not indicate the phenomenon of acclimatization [171]. Another comparative study of barley (cultivated vs. wild) demonstrated that the wild type of barley is more salt tolerant than cultivated barley and possesses an improved ability to regulate osmotic stress through the accumulation of more carbohydrates and proline in its roots. GC-MS-based metabolite profiling led to the identification of 82 metabolites, which include water-soluble carbohydrates (sucrose, trehalose, and raffinose) and proline, which were predominant and contribute potentially to salt tolerance in roots [172]. A change in the amino acid metabolism in leaves seems critical to develop a salt tolerance mechanism.
Rice is one of the most sensitive crops to elevated salt concentration because its roots are highly permeable to sodium ions present in the soil [203]. A GC-TOF-MS metabolomic analysis revealed the lower levels of TCA cycle intermediates and organic acid in the roots of tolerant rice cultivars in comparison to the sensitive cultivar. In addition, accumulation of amino acids was also observed in the tolerant cultivar [167]. Liu et al. [168] conducted metabolite profiling of rice cell culture focusing on the initial phase of salt stress that was exclusively characterized by osmotic stress. Further, glucose, fructose, galactose, hexose phosphates, glucose-6-phosphate, and fructose-6-phosphate accumulated in rice suspension culture subjected to 100 mM NaCl for 1–24 h. Several studies on different rice cultivars showed a decrease in the TCA cycle-dependent organic acids and accumulation of various amino acids [168].
Maize plants exposed to salt stress (50–150 mM NaCl saline solution) showed an increase in the levels of sucrose and alanine, but the levels of glucose were decreased in roots and shoots. Other osmoprotectants like GABA, malic acid, and succinate showed increased levels in roots, whereas acetoacetate showed decreased levels. Simultaneously glutamate, asparagine, and glycine betaine showed increased levels in shoots, whereas malic acid and trans-aconitic acid showed decreased levels. Increased metabolic response was more evident in shoots than in roots [164].
5.4 Oxidative Stress
Oxidative stress is one the major limiting factors for plant growth. It occurs by overproduction of ROS, for example hydrogen peroxide (H2O2), superoxide (O2−), and singlet oxygen (1O2.). A wide range of abiotic stresses like high light, low temperature, drought, and salt stress can cause oxidative stress [208]. ROS are produced constantly in the cell as a by-product of aerobic metabolism even under non-stress conditions, particularly during photosynthesis, in mitochondria via the mitochondrial electron transport chain, and in peroxisomes upon photorespiration and β-oxidation of fatty acids. However, the cellular antioxidant system can detoxify ROS via the ascorbate glutathione (GSH) cycle; a balanced cellular redox-status is maintained. Although low levels of ROS are important for signaling and response for certain stress signals, elevated ROS levels can contribute to a plants defense program by initiating programmed cell death, especially during pathogen attack [209].
In a study performed by Baxter et al. [173], heterotrophic Arabidopsis cells were treated with menadione, which enhances ROS production via the electron transport chain and hence changes metabolite abundance. Metabolomic abundance was quantified using 13C-labelling kinetics. It was observed that sugar phosphates related to glycolysis and oxidative pentose phosphate pathways (OPPPs) were accumulated, which directs the flow of the glycolytic carbon into the OPPP to provide NADPH for antioxidant activity. In addition, levels of ascorbate decreased and the accumulation of its degraded products like threonate was observed, which indicated activation of the antioxidative pathway in menadione-treated cells. Reduction in glycolytic activity probably leads to decreased levels of amino acids derived from glycolytic intermediates. Further inhibition of the TCA cycle intermediates was also observed and confirmed by 13C redistribution analysis [173].
Lehmann et al. [174] also performed a similar kind of study considering metabolite profiling and 13C-redistribution analysis on menadione-treated Arabidopsis roots. The results obtained were distinct from the heterotrophic cell study. In this analysis, roots showed pronounced accumulation of GABA, O-acetylserine (OAS), pyruvate, glucosinolates, and other amino acids. It is likely that cellular oxidation inhibits sulfur assimilation and leads to the accumulation of OAS [174]. Similarly, rice cell cultures treated with menadione redirected the carbon flux from glycolysis through the OPP pathway and subsequently increased the levels of NADPH. CE-MS analysis of these rice cultures showed depletion of sugar phosphates like pyruvate, 3-phosphoglyceric acid, dihydroxyacetone phosphate, fructose-6-phosphate, glucose-1-phosphate (G1P), G6P, G3P, phosphoenolpyruvate (glycolysis intermediates), and TCA intermediates like 2-oxoglutarate, aconitate, citrate, fumarate, isocitrate, malate, and succinate; followed by the increase in OPP pathway intermediates like 6-phosphogluconate, ribose 5-phosphate, and ribulose 5-phosphate. Additionally, an increase in the biosynthesis of GSH and its intermediates (O-acetyl-L-serine, cysteine, and γ-glutamyl-L-cysteine) was also observed in the menadione-treated rice cell cultures [175]. Down-regulated expression of manganese superoxide dismutase (MnSOD) levels, which cause oxidative stress on the metabolome, was also observed. GC-MS metabolite profiling in Arabidopsis revealed a redox shift in mitochondria, followed by a specific decrease in TCA cycle flux, due to the inhibition of aconitase and isocitrate dehydrogenase [210].
5.5 Flooding
Flooding imposes severe stress on plants. This is principally because excess water in the surroundings can deprive them of oxygen and CO2, which in turn hampers the process of photosynthesis and reduces growth and grain yield [211]. Mitochondrial fractions from the roots and hypocotyls of 4-day-old soybean seedlings that had been in flooding stress for 2 days were subjected to proteomics and metabolomics analysis. Proteins and metabolites related to the TCA cycle (like citrate, succinate, and aconitate), GABA shunt, and amounts of NADH and NAD were up-regulated under stress conditions, but ATP was significantly decreased by flooding stress [176]. Similarly, Komatsu et al. [177] investigated the root tips of soybean under flooding stress – in total 73 flood responsive metabolites were identified using capillary electrophoresis-mass spectrometry. The levels of gamma-aminobutyric acid, glycine, NADH2, and phosphoenol pyruvate were increased under flooding stress conditions [177].
5.6 Nutrient Deficiency
Nutrients are essential for plant growth and development. They can affect and regulate fundamental processes of plant physiology like photosynthesis and respiration. Depending upon the growth requirement of the plants, nutrients are referred to as either macronutrients (e.g., nitrogen, phosphorus, potassium, sulfur, and magnesium) or micronutrients (e.g., iron, zinc, etc.). Nutrient starvation or limitation of macronutrients has direct effects on the metabolism of the plant since most organic molecules are made up of a combination of these elements.
5.6.1 Nitrogen (N)
Nitrogen is the most important nutrient required by plants and its metabolism is highly coordinated with carbon metabolism in the fundamental process of plant growth. Paddy fields of rice plants preferentially use ammonium as a source of inorganic nitrogen. The conversion of ammonium to glutamine is catalyzed by glutamine synthetase (GS). Kusano et al. [178] performed comparative metabolomic analysis between the rice mutant lacking the OsGS1;1 gene and the wild type. The results revealed that mutant lines showed retardation in shoot growth in the presence of ammonium compared to the wild type. Overaccumulation of free ammonium in the leaf sheath and roots of the mutant lines demonstrated the importance of the OsGs1;1 gene for ammonium assimilation. The metabolomic profile of the mutant line revealed decreased levels of sugars, amino acids, and intermediates of the TCA cycle. In contrast, overaccumulation of secondary metabolites was observed particularly in roots under a continuous supply of ammonium [178]. Further, the effect of nitrogen deficiency at the metabolite levels in tomato leaves was investigated by Urbanczyk-Wochniak and Fernie [179]. Based on the analysis, the decreased levels of 2-oxoglutarate, as well as other TCA-cycle intermediates including citrate, isocitrate, succinate, fumarate, and malate, were observed under nitrogen stress [179]. Similarly, Tschoep et al. [180] analyzed the effect of mild nitrogen limitation in Arabidopsis, where the levels of malate and fumarate showed significant decreased levels [180]. These findings were in agreement with a previous study performed in tomato leaves [179].
5.6.2 Sulfur (S)
Sulphur is another macronutrient essential for the synthesis of sulfur-containing amino acids like cysteine and methionine as well as a wide range of sulfur-containing metabolites like glutathione. Sulfur stress has been well studied using metabolomics by several groups, and has been well elaborated by Hoefgen and Nikiforova [212]. Nikiforova et al. used GC-MS and LC-MS profiling methods to monitor the response of 134 metabolites and 6,023 unknown peaks of non-redundant ion trances under sulfur stress. Based on the profiling data, the coordinated network of metabolic regulation was successfully reconstructed under sulfur stress [213]. These data were also analyzed together with transcriptomic data in order to generate a gene-metabolite correlation network in Arabidopsis under sulfur stress [181].
5.6.3 Phosphorus (P)
Morcuende et al. [182] analyzed Arabidopsis seedlings grown in liquid culture under phosphorus starvation. The metabolite profile revealed the levels of sugar phosphates were decreased but other metabolites like glycolysis, glycerate-3- phosphate, glycerate-2-phosphate, and phosphoenolpyruvate were increased in P-deficient seedlings. P-deficient seedlings also showed accumulation of starch, sucrose, and reduced sugars as well as a general increase of organic acids including citrate, fumarate, malate, and oxoglutarate. The levels of aromatic amino acids like histidine, arginine, and threonine did not alter or increased slightly. Together with transcriptomic data, analysis of metabolites revealed that phosphorus deprivation leads to a shift towards the accumulation of carbohydrates, organic acids, and amino acids [182]. Hernández et al. performed metabolite profiling to understand the effect of phosphorus deficiency in the roots [184] and nodules [183] of the common bean. Huang et al. [185] performed metabolomic analysis in both shoots and roots of phosphorus-deficient barley. Severe phosphorus deficiency decreased the levels of phosphorylated intermediates (glucose-6-P, fructose-6-P, inositol-1-P, and glycerol-3-P) and organic acids (2-oxoglutarate, succinate, fumarate, and malate). It was also identified that phosphorous-deficient plants reconstruct carbohydrate metabolism initially in order to reduce phosphorus consumption, which consequently reduces the levels of organic acid in the TCA cycle [185].
5.6.4 Potassium (K)
Potassium (K) plays essential roles as a major cation in plants and as a cofactor of enzymes. Armengaud et al. [186] performed metabolite profiling in order to identify metabolic targets of potassium stress. Metabolite profiles of Arabidopsis plants (roots and shoots) under low-K concentration revealed increases in the levels of soluble sugars (sucrose, fructose, and glucose) and a slight net increase of total protein content and the overall amino acid level. Additionally, a strong decrease in pyruvate and organic acids was observed only in the roots but not in the shoots [186].
5.6.5 Heavy Metals
Heavy metals such as cadmium (Cd), cesium (Cs), lead (Pb), zinc (Zn), nickel (Ni), and chromium (Cr) are major soil pollutants (from the beginning of the industrial revolution) causing stress on plants. Heavy metals induce enzyme inhibition, cellular oxidation, and metabolic perturbation, resulting in growth retardation and plant death in extreme instances [214]. Sometimes essential nutrients including copper (Cu), iron (Fe), and manganese (Mn) can cause heavy metal stress if present in an inappropriate concentration. Jahangir et al. [187] analyzed the effects of Cu, Fe, and Mn on the metabolite levels of Brassica rapa (leaves and roots). The levels of metabolites such as glucosinolates and hydroxycinnamic acids as well as primary metabolites such as carbohydrates and amino acids were affected constitutively [187]. Similarly, Arabidopsis plants treated with Cd showed increased levels of alanine, ß-alanine, proline, serine, putrescine, sucrose, and other metabolites with compatible solute-like properties, notably GABA, raffinose, and trehalose. This study also indicated a significant increase in the concentrations of α-tocopherol, campesterol, ß-sitosterol, and isoflavone. When taken together these data indicate an important role of antioxidant defense in the mechanisms of resistance to cadmium stress [188].
5.7 Biotic Stress
In order to combat the stress from pathogens and pests, plants use complex chemical machinery as a major defense mechanism. The metabolic response of plants to biotic stresses depends highly on tissues, species, and plant-pathogen or pest interactions. A number of metabolites have been identified as metabolic biomarkers for biotic stress in diverse plant species [215]. For example, metabolic profiling of host tissues from the three rice varieties TN1, Kavya, and RP2068 exposed to gall midge biotype 1 (GMB1) was performed using GC-MS. In total, 16 fatty acids (such as unsaturated linoleic acid) together with two amino acids (glutamine and phenylalanine) were identified as the major components of the resistance features of gall midge (Orseolia oryzae Wood-Mason)-resistant rice varieties [189]. Similarly, metabolite profiling of sensitive and tolerant rice cultivars subjected to BLB (bacterial leaf blight) caused by Xanthomonas oryzae pv. oryzae (Xoo) led to the identification of several specific metabolites, such as acetophenone, xanthophylls, alkaloids, carbohydrates, and lipids [190]. Metabolomic analysis of barley, rice, and purple false brome grass, revealed identical changes in the metabolic patterns in which malate, polyamines, quinate, and non-polymerized lignin precursors accumulate during infection by Magnaporthe oryzae [191]. Phenylpropanoids are the precursors of lignin and constitute an important component of the plant stress defense mechanism by modulating cell wall composition and stiffness in roots. The thickened cell wall may help to defend against pathogen infection in the plant. Accumulation of phenylpropanoid and phenolic compounds was reported in response to Fusarium graminearum in wheat [192].
5.8 Stress Combination
In the natural habitat, plants are actually subjected to combinations of abiotic stress. Some of these stress conditions are already in combination, for example, high salt concentration leads to ionic and osmotic stresses. Although the metabolic response of plants under single stress conditions has been studied extensively as detailed in the previous section [216], Rizhsky et al. [217] applied the combination of drought and heat stress in Arabidopsis. The metabolite profiling showed high-level accumulation of sucrose and other sugars. Interestingly, proline was not accumulated in stress combination and hence it was concluded that sucrose replaces proline as an osmoprotectant in plants in order to avoid combined stress conditions because proline shows high-level toxic effects under heat stress conditions [217]. Wulff-Zottele et al. [218] also analyzed the effect of high light irradiance and sulfur depletion. In this study, it was observed that proline accumulated in a differential time course and other metabolites like raffinose and putrescine replaced proline during its delayed accumulation under stress [218].
6 Metabolite Accumulation: Adjustment in Response to Stress Conditions
Under abiotic stress conditions, a common defensive mechanism is activated in plants that leads to the production and accumulation of compatible solutes (metabolites). These are small molecular weight osmoprotectants that are diverse in nature, and include amino acids like asparagine, proline, and serine, and amines like polyamines, glycine betaine, and GABA (γ-amino-N-butyric acid). Additionally, they also include carbohydrates (e.g., fructose, sucrose, and trehalose), raffinose, and polyols (myo-inositol, D-pinitol), and antioxidants such as glutathione (GSH) and ascorbate [219]. These osmolytes have high levels of solubility/permeability in the cell and lack enzyme inhibition activities even at high concentrations. Accumulation of the solutes in response to stress is not only observed in plants, it is also a defense mechanism triggered by animal cells, bacteria, and marine algae, which indicate an evolutionarily conserved trait [196, 220, 221].
Several studies have been performed to understand the beneficial effects of these metabolites in plant tolerance against environmental stimuli. Proline is one of the best examples, and the correlation between proline accumulation and stress tolerance has been well described in Bermuda grass during water stress conditions [222]. In the follow-up studies, several research works were conducted to prove and correlate the importance of proline accumulation in plants under stress conditions. Based on the findings it can be concluded that proline can act as a protein stabilizer, a signaling molecule, a ROS scavenger (against several abiotic stresses), and a cryoprotectant/osmoprotectant in plants under stress conditions [223, 224]. Figure 3 represents the biosynthesis of the key metabolites that accumulate in plants in response to the stress condition.

Biosynthetic pathway of the metabolites that are accumulated under stress conditions (e.g. drought, temperature, salt, etc.). (1) proline, (2) GABA, (3) glycine betaine, (4) trehalose, (5) raffinose
Proline metabolism and its regulations are well characterized in plants (Fig. 3(1)). Proline is synthesized in cytoplasm or chloroplast from glutamate, which reduces to glutamate semialdehyde (GSA) by Δ-1-pyrroline-5-carboxylate synthetase (P5CS). GSA can spontaneously convert into pyrroline-5-carboxylate (P5C), which is further reduced by P5C reductase (P5CR) to proline. Proline is degraded within the mitochondrial matrix by proline dehydrogenase (ProDH) and P5C dehydrogenase (P5CDH) to glutamate [225]. Under stress conditions proline synthesis is stimulated, whereas during recovery from stress catabolism of proline is enhanced. In tobacco and petunia, over-expression of P5CS led to increasing accumulation of proline and showed enhanced tolerance to salt and drought [226]. Arabidopsis P5CS1 knock-out plants were impaired in proline synthesis and showed hypersensitivity to salt stress [227]. ProDH antisense Arabidopsis accumulated more proline, which shows enhanced tolerance against low temperature and high salinity [228]. An alternative pathway, mitochondrial P5C, can be produced by δ-aminotransferase (δ-OAT) from ornithine. Over-expression of Arabidopsis δ-OAT enhances proline levels, which in turn increases stress tolerance in rice and tobacco [229]. The core enzymes P5CS, P5C, P5CR, ProDH, and OAT are responsible for maintaining the balance between biosynthesis and catabolism of proline.
Extensive research work has been performed for other metabolites such as γ-aminobutyric acid (GABA), glycine betaine, trehalose, raffinose, and polyamines (like putrescine, spermidine, and spermine) etc.; these metabolites have also been proven to be efficient protectors against some abiotic stresses. GABA is a non-protein amino acid that accumulates rapidly under high-stress levels [230,231,232]. GABA is mainly synthesized from glutamate in cytosol and then transported into mitochondria. GABA-T (GABA transaminase) and SSADH (succinic semialdehyde dehydrogenase) convert GABA into succinate, which enters into the TCA cycle (Fig. 3(2)) [233]. GABA metabolism plays a major role in the carbon-nitrogen balance and ROS scavenging activity [234, 235]. GABA shunt also plays an important role in stress tolerance. Enzymes involved in GABA metabolism showed enhanced enzyme activity under salt stress [232]. GABA-T deficient Arabidopsis mutants showed hypersensitivity against ionic stress – increased levels of amino acids were accumulated while carbohydrate levels were diminished [232]. Similarly, disruption of the SSADH gene showed ROS accumulation and hypersensitivity against UV-B and heat stress [236].
Glycine betaine (GB) is a quaternary ammonium compound that occurs widely in the plant kingdom [237]. GB is synthesized from choline and glycine [238]. Arabidopsis and many other crops species do not accumulate GB. Plants with natural production of GB under stress conditions (cold, drought, and salt) showed enhanced accumulation of GB (Fig. 3(3)) [238]. In salt-tolerant species, a significant level of GB accumulation was determined. GB protects photosystem II, stabilizes the membrane, acts as a molecular chaperone, maintains water balance, and provides protection from oxidative stress [239,240,241].
Trehalose is a non-reducing disaccharide that accumulates in higher concentrations under some desiccation-tolerant plants like Myrothamnus flabellifolius [242]. At high levels, trehalose can act as an osmolyte to stabilize proteins [243]. In plants, trehalose is present in a small amount, but under high-stress conditions trehalose increases moderately [154, 244]. Biosynthesis of trehalose takes place in two steps: trehalose-6-phosphate synthase (TPS) produces trehalose-6-phosphate (T6P) from UDP-glucose and glucose-6-phosphate followed by dephosphorylation of trehalose by trehalose-6-phosphate phosphatase (TPP) [243] (Fig. 3(4)). Several research studies demonstrated the role of trehalose in stress tolerance; transgenic expression of the genes involved in trehalose biosynthesis enhances the tolerance of plants against abiotic stress. Heterologous expression of genes involved in the trehalose pathway from E. coli or Saccharomyces cerevisiae showed enhanced tolerance to abiotic stresses in several plants [245]. Further, over-expression of the TPS isoform conferred enhanced resistance in rice against salt, cold, and drought stress [246]. Loss of TPS5 (TPS with TPP domain) function lowered the basal thermotolerance in Arabidopsis [247]. Panikulangara et al. [248] demonstrated that levels of trehalose and activity of TPP increase under cold stress in rice, and overexpression OsTPP1 in rice showed enhanced tolerance against cold and salinity, although trehalose content was not observed to be increased [248].
Raffinose family oligosaccharides (RFOs) include raffinose, stachyose, and verbascose, which accumulate in various plant species (in desiccated seeds and leaves) under environmental stress like cold, heat, drought, or salinity [249]. RFO biosynthesis is initiated by formation of galactinol from myo-inositol and UDP-galactose by galactinol synthase (GolS). Sequential addition of galactose units by galactinol to sucrose leads to the formation of raffinose, and a higher class of RFO (Fig. 3(5)) [249]. The complete biosynthetic pathway of RFOs under stress conditions is not completely known. Research work performed by Taji et al. [250] and Nishizawa et al. [251] demonstrated that over-expression of Arabidopsis GolS1 and GolS2 accumulates high levels of galactinol and sucrose in Arabidopsis, which showed enhanced tolerance under drought and salt stress [250, 251].
Metabolomic adjustments play a very important role in plant survival; therefore, regulation of the metabolic pathways (biosynthesis and catabolism) is very critical in order to improve tolerance mechanisms against environmental stimuli in plants.
7 Conclusion and Outlook
Metabolomics is the comprehensive platform that can identify and quantify small molecules present in plants that lead to the ultimate expression of its genotype in response to environmental stimuli (abiotic and biotic stresses). Information obtained from the metabolomic studies in plants against different abiotic and biotic stresses have provided relevant information about the specific metabolites that are directly involved in physiological and biochemical changes. In an applied context, metabolomic approaches provide a broader, deeper, and integral perspective of the metabolic profiles in the acclimatization of plants against environmental stress. The obtained information is also transferable to the sensitive and economically important crops, to improve their adaptation strategies towards adverse conditions. The application of more advanced metabolomics tools will accelerate and improve plant breeding approaches, which will surely lead to the next generation of crops that are more tolerant to abiotic and biotic stresses worldwide.
References
Kaul S, Koo HL, Jenkins J, Rizzo M, Rooney T, Tallon LJ, Feldblyum T, Nierman W, Benito MI, Lin XY, Town CD, Venter JC, Fraser CM, Tabata S, Nakamura Y, Kaneko T, Sato S, Asamizu E, Kato T, Kotani H, Sasamoto S, Ecker JR, Theologis A, Federspiel NA, Palm CJ, Osborne BI, Shinn P, Conway AB, Vysotskaia VS, Dewar K, Conn L, Lenz CA, Kim CJ, Hansen NF, Liu SX, Buehler E, Altafi H, Sakano H, Dunn P, Lam B, Pham PK, Chao Q, Nguyen M, Yu GX, Chen HM, Southwick A, Lee JM, Miranda M, Toriumi MJ, Davis RW, Wambutt R, Murphy G, Dusterhoft A, Stiekema W, Pohl T, Entian KD, Terryn N, Volckaert G, Salanoubat M, Choisne N, Rieger M, Ansorge W, Unseld M, Fartmann B, Valle G, Artiguenave F, Weissenbach J, Quetier F, Wilson RK, De La Bastide M, Sekhon M, Huang E, Spiegel L, Gnoj L, Pepin K, Murray J, Johnson D, Habermann K, Dedhia N, Parnell L, Preston R, Hillier L, Chen E, Marra M, Martienssen R, Mccombie WR, Mayer K, White O, Bevan M, Lemcke K, Creasy TH, Bielke C, Haas B, Haase D, Maiti R, Rudd S, Peterson J, Schoof H, Frishman D, Morgenstern B et al (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815
Goff SA, Ricke D, Lan TH, Presting G, Wang RL, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchinson D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong JP, Miguel T, Paszkowski U, Zhang SP, Colbert M, Sun WL, Chen LL, Cooper B, Park S, Wood TC, Mao L, Quail P, Wing R, Dean R, Yu YS, Zharkikh A, Shen R, Sahasrabudhe S, Thomas A, Cannings R, Gutin A, Pruss D, Reid J, Tavtigian S, Mitchell J, Eldredge G, Scholl T, Miller RM, Bhatnagar S, Adey N, Rubano T, Tusneem N, Robinson R, Feldhaus J, Macalma T, Oliphant A, Briggs S (2002) A draft sequence of the rice genome (Oryza sativa L. ssp japonica). Science 296:92–100
Yu J, Hu SN, Wang J, Wong GKS, Li SG, Liu B, Deng YJ, Dai L, Zhou Y, Zhang XQ, Cao ML, Liu J, Sun JD, Tang JB, Chen YJ, Huang XB, Lin W, Ye C, Tong W, Cong LJ, Geng JN, Han YJ, Li L, Li W, Hu GQ, Huang XG, Li WJ, Li J, Liu ZW, Li L, Liu JP, Qi QH, Liu JS, Li L, Li T, Wang XG, Lu H, Wu TT, Zhu M, Ni PX, Han H, Dong W, Ren XY, Feng XL, Cui P, Li XR, Wang H, Xu X, Zhai WX, Xu Z, Zhang JS, He SJ, Zhang JG, Xu JC, Zhang KL, Zheng XW, Dong JH, Zeng WY, Tao L, Ye J, Tan J, Ren XD, Chen XW, He J, Liu DF, Tian W, Tian CG, Xia HG, Bao QY, Li G, Gao H, Cao T, Wang J, Zhao WM, Li P, Chen W, Wang XD, Zhang Y, Hu JF, Wang J, Liu S, Yang J, Zhang GY, Xiong YQ, Li ZJ, Mao L, Zhou CS, Zhu Z, Chen RS, Hao BL, Zheng WM, Chen SY, Guo W, Li GJ, Liu SQ, Tao M, Wang J, Zhu LH, Yuan LP, Yang HM (2002) A draft sequence of the rice genome (Oryza sativa L. ssp indica). Science 296:79–92
Sato S, Tabata S, Hirakawa H, Asamizu E, Shirasawa K, Isobe S, Kaneko T, Nakamura Y, Shibata D, Aoki K, Egholm M, Knight J, Bogden R, Li CB, Shuang Y, Xu X, Pan SK, Cheng SF, Liu X, Ren YY, Wang J, Albiero A, Dal Pero F, Todesco S, Van Eck J, Buels RM, Bombarely A, Gosselin JR, Huang MY, Leto JA, Menda N, Strickler S, Mao LY, Gao S, Tecle IY, York T, Zheng Y, Vrebalov JT, Lee J, Zhong SL, Mueller LA, Stiekema WJ, Ribeca P, Alioto T, Yang WC, Huang SW, Du YC, Zhang ZH, Gao JC, Guo YM, Wang XX, Li Y, He J, Li CY, Cheng ZK, Zuo JR, Ren JF, Zhao JH, Yan LH, Jiang HL, Wang B, Li HS, Li ZJ, Fu FY, Chen BT, Han B, Feng Q, Fan DL, Wang Y, Ling HQ, Xue YBA, Ware D, Mccombie WR, Lippman ZB, Chia JM, Jiang K, Pasternak S, Gelley L, Kramer M, Anderson LK, Chang SB, Royer SM, Shearer LA, Stack SM, Rose JKC, Xu YM, Eannetta N, Matas AJ, Mcquinn R, Tanksley SD, Camara F, Guigo R, Rombauts S, Fawcett J, Van De Peer Y, Zamir D, Liang CB, Spannagl M, Gundlach H, Bruggmann R et al (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485:635–641
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XQH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang JH, Miklos GLG, Nelson C, Broder S, Clark AG, Nadeau C, Mckusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng ZM, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge WM, Gong FC, Gu ZP, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke ZX, Ketchum KA, Lai ZW, Lei YD, Li ZY, Li JY, Liang Y, Lin XY, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue BX, Sun JT, Wang ZY, Wang AH, Wang X, Wang J, Wei MH, Wides R, Xiao CL, Yan CH et al (2001) The sequence of the human genome. Science 291:1304
Kehoe DM, Villand P, Somerville S (1999) DNA microarrays for studies of higher plants and other photosynthetic organisms. Trends Plant Sci 4:38–41
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW (1995) Serial analysis of gene-expression. Science 270:484–487
Chaturvedi P, Doerfler H, Jegadeesan S, Ghatak A, Pressman E, Castillejo MA, Wienkoop S, Egelhofer V, Firon N, Weckwerth W (2015) Heat-treatment-responsive proteins in different developmental stages of tomato pollen detected by targeted mass accuracy precursor alignment (tMAPA). J Proteome Res 14:4463–4471
Chaturvedi P, Ghatak A, Weckwerth W (2016) Pollen proteomics: from stress physiology to developmental priming. Plant Reprod 29:119–132
Chaturvedi P, Ischebeck T, Egelhofer V, Lichtscheidl I, Weckwerth W (2013) Cell-specific analysis of the tomato pollen proteome from pollen mother cell to mature pollen provides evidence for developmental priming. J Proteome Res 12:4892–4903
Ghatak A, Chaturvedi P, Nagler M, Roustan V, Lyon D, Bachmann G, Postl W, Schrofl A, Desai N, Varshney RK, Weckwerth W (2016) Comprehensive tissue-specific proteome analysis of drought stress responses in Pennisetum glaucum (L.) R. Br. (Pearl millet). J Proteome 143:122–135
Ghatak A, Chaturvedi P, Paul P, Agrawal GK, Rakwal R, Kim ST, Weckwerth W, Gupta R (2017) Proteomics survey of Solanaceae family: current status and challenges ahead. J Proteome 169:41–57
Ghatak A, Chaturvedi P, Weckwerth W (2017) Cereal crop proteomics: systemic analysis of crop drought stress responses towards marker-assisted selection breeding. Front Plant Sci 8:757
Glinski M, Weckwerth W (2006) The role of mass spectrometry in plant systems biology. Mass Spectrom Rev 25:173–214
Paul P, Chaturvedi P, Selymesi M, Ghatak A, Mesihovic A, Scharf KD, Weckwerth W, Simm S, Schleiff E (2016) The membrane proteome of male gametophyte in Solanum lycopersicum. J Proteome 131:48–60
Weckwerth W (2003) Metabolomics in systems biology. Annu Rev Plant Biol 54:669–689
Weckwerth W (2008) Integration of metabolomics and proteomics in molecular plant physiology--coping with the complexity by data-dimensionality reduction. Physiol Plant 132:176–189
Weckwerth W (2011) Green systems biology – from single genomes, proteomes and metabolomes to ecosystems research and biotechnology. J Proteome 75:284–305
Holtorf H, Guitton MC, Reski R (2002) Plant functional genomics. Naturwissenschaften 89:235–249
Ideker T, Galitski T, Hood L (2001) A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet 2:343–372
Kitano H (2000) Perspectives on systems biology. N Gener Comput 18:199–216
Oliver DJ, Nikolau B, Wurtele ES (2002) Functional genomics: high-throughput mRNA, protein, and metabolite analyses. Metab Eng 4:98–106
Somerville C, Somerville S (1999) Plant functional genomics. Science 285:380–383
Fiehn O (2002) Metabolomics – the link between genotypes and phenotypes. Plant Mol Biol 48:155–171
Fiehn O, Kopka J, Dormann P, Altmann T, Trethewey RN, Willmitzer L (2000) Metabolite profiling for plant functional genomics. Nat Biotechnol 18:1157–1161
Weckwerth W, Fiehn O (2002) Can we discover novel pathways using metabolomic analysis? Curr Opin Biotechnol 13:156–160
Gygi SP, Rochon Y, Franza BR, Aebersold R (1999) Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 19:1720–1730
Weckwerth W, Wenzel K, Fiehn O (2004) Process for the integrated extraction, identification and quantification of metabolites, proteins and RNA to reveal their co-regulation in biochemical networks. Proteomics 4:78–83
Goodacre R, Vaidyanathan S, Dunn WB, Harrigan GG, Kell DB (2004) Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol 22:245–252
Hall RD (2006) Plant metabolomics: from holistic hope, to hype, to hot topic. New Phytol 169:453–468
Devaux PG, Horning MG, Hill RM, Horning EC (1971) O-benzyloximes: derivatives for the study of ketosteroids by gas chromatography. Application to urinary steroids of the newborn human. Anal Biochem 41:70–82
Horning EC, Horning MG (1970) Metabolic profiles: chromatographic methods for isolation and characterization of a variety of metabolites in man. Methods Med Res 12:369–371
Horning EC, Horning MG (1971) Metabolic profiles: gas-phase methods for analysis of metabolites. Clin Chem 17:802–809
Cunnick WR, Cromie JB, Cortell R, Wright B, Beach E, Seltzer F, Miller S (1972) Value of biochemical profiling in a periodic health examination program: analysis of 1,000 cases. Bull N Y Acad Med 48:5–22
Mroczek WJ (1972) Biochemical profiling and the natural history of hypertensive diseases. Circulation 45:1332–1333
Vrbanac JJ, Braselton Jr WE, Holland JF, Sweeley CC (1982) Automated qualitative and quantitative metabolic profiling analysis of urinary steroids by a gas chromatography-mass spectrometry-data system. J Chromatogr 239:265–276
Niwa T (1986) Metabolic profiling with gas chromatography-mass spectrometry and its application to clinical medicine. J Chromatogr 379:313–345
Bales JR, Bell JD, Nicholson JK, Sadler PJ, Timbrell JA, Hughes RD, Bennett PN, Williams R (1988) Metabolic profiling of body fluids by proton NMR: self-poisoning episodes with paracetamol (acetaminophen). Magn Reson Med 6:300–306
Bales JR, Higham DP, Howe I, Nicholson JK, Sadler PJ (1984) Use of high-resolution proton nuclear magnetic resonance spectroscopy for rapid multi-component analysis of urine. Clin Chem 30:426–432
Sauter H, Lauer M, Fritsch H (1991) Metabolic profiling of plants – a new diagnostic-technique. ACS Symp Ser 443:288–299
Oliver SG, Winson MK, Kell DB, Baganz F (1998) Systematic functional analysis of the yeast genome. Trends Biotechnol 16:373–378
Trethewey RN, Krotzky AJ, Willmitzer L (1999) Metabolic profiling: a Rosetta stone for genomics? Curr Opin Plant Biol 2:83–85
Tweeddale H, Notley-Mcrobb L, Ferenci T (1998) Effect of slow growth on metabolism of Escherichia coli, as revealed by global metabolite pool (“metabolome”) analysis. J Bacteriol 180:5109–5116
Sumner LW, Duran AL, Huhman DV, Smith JT (2002) Metabolomics: a developing and integral component in functional genomic studies of Medicago truncatula. Phytochem Genom Post-Genom Era 36(31–61):258
Allwood JW, Goodacre R (2010) An introduction to liquid chromatography-mass spectrometry instrumentation applied in plant metabolomic analyses. Phytochem Anal 21:33–47
Moco S, Bino RJ, De Vos RCH, Vervoort J (2007) Metabolomics technologies and metabolite identification. TrAC-Trends Analyt Chem 26:855–866
Kikuchi J, Hirayama T (2007) Practical aspects of uniform stable isotope labeling of higher plants for heteronuclear NMR-based metabolomics. Methods Mol Biol 358:273–286
Kim HK, Choi YH, Verpoorte R (2010) NMR-based metabolomic analysis of plants. Nat Protoc 5:536–549
Kim HK, Choi YH, Verpoorte R (2011) NMR-based plant metabolomics: where do we stand, where do we go? Trends Biotechnol 29:267–275
Weckwerth W (2010) Metabolomics: an integral technique in systems biology. Bioanalysis 2:829–836
Sumner LW, Mendes P, Dixon RA (2003) Plant metabolomics: large-scale phytochemistry in the functional genomics era. Phytochemistry 62:817–836
Dunn WB, Ellis DI (2005) Metabolomics: current analytical platforms and methodologies. TrAC-Trends Analyt Chem 24:285–294
Tugizimana F, Piater L, Dubery I (2013) Plant metabolomics: a new frontier in phytochemical analysis. S Afr J Sci 109:11
Bligny R, Douce R (2001) NMR and plant metabolism. Curr Opin Plant Biol 4:191–196
Ratcliffe RG, Roscher A, Shachar-Hill Y (2001) Plant NMR spectroscopy. Prog Nucl Magn Reson Spectrosc 39:267–300
Griffin JL, Williams HJ, Sang E, Clarke K, Rae C, Nicholson JK (2001) Metabolic profiling of genetic disorders: a multitissue H-1 nuclear magnetic resonance spectroscopic and pattern recognition study into dystrophic tissue. Anal Biochem 293:16–21
Brindle JT, Nicholson JK, Schofield PM, Grainger DJ, Holmes E (2003) Application of Chemometrics to H-1 NMR spectroscopic data to investigate a relationship between human serum metabolic profiles and hypertension. Analyst 128:32–36
Solanky KS, Bailey NJC, Holmes E, Lindon JC, Davis AL, Mulder TPJ, Van Duynhoven JPM, Nicholson JK (2003) NMR-based metabonomic studies on the biochemical effects of epicatechin in the rat. J Agric Food Chem 51:4139–4145
Vaidyanathan S, Kell DB, Goodacre R (2002) Flow-injection electrospray ionization mass spectrometry of crude cell extracts for high-throughput bacterial identification. J Am Soc Mass Spectrom 13:118–128
Aharoni A, Ric De Vos CH, Verhoeven HA, Maliepaard CA, Kruppa G, Bino R, Goodenowe DB (2002) Nontargeted metabolome analysis by use of fourier transform ion cyclotron mass spectrometry. OMICS 6:217–234
Gu HW, Huang YA, Carr PW (2011) Peak capacity optimization in comprehensive two dimensional liquid chromatography: a practical approach. J Chromatogr A 1218:64–73
Guiochon G, Marchetti N, Mriziq K, Shalliker RA (2008) Implementations of two-dimensional liquid chromatography. J Chromatogr A 1189:109–168
Jandera P (2012) Programmed elution in comprehensive two-dimensional liquid chromatography. J Chromatogr A 1255:112–129
Kempa S, Hummel J, Schwemmer T, Pietzke M, Strehmel N, Wienkoop S, Kopka J, Weckwerth W (2009) An automated GCxGC-TOF-MS protocol for batch-wise extraction and alignment of mass isotopomer matrixes from differential 13C-labelling experiments: a case study for photoautotrophic-mixotrophic grown Chlamydomonas reinhardtii cells. J Basic Microbiol 49:82–91
Ralston-Hooper K, Hopf A, Oh C, Zhang X, Adamec J, Sepulveda MS (2008) Development of GCxGC/TOF-MS metabolomics for use in ecotoxicological studies with invertebrates. Aquat Toxicol 88:48–52
Morgenthal K, Wienkoop S, Scholz M, Selbig J, Weckwerth W (2005) Correlative GC-TOF-MS-based metabolite profiling and LC-MS-based protein profiling reveal time-related systemic regulation of metabolite-protein networks and improve pattern recognition for multiple biomarker selection. Metabolomics 1:109–121
Morgenthal K, Wienkoop S, Wolschin F, Weckwerth W (2007) Integrative profiling of metabolites and proteins: improving pattern recognition and biomarker selection for systems level approaches. Methods Mol Biol 358:57–75
Weckwerth W, Morgenthal K (2005) Metabolomics: from pattern recognition to biological interpretation. Drug Discov Today 10:1551–1558
Weckwerth W, Tolstikov V, Fiehn O (2001) Metabolomic characterization of transgenic potato plants using GC/TOF and LC/MS analysis reveals silent metabolic phenotypes, In: Proceedings of the 49th ASMS conference on mass spectrometry and allied topics: American Society Of Mass Spectrometry Chicago, pp 1–2
Dunn WB (2008) Current trends and future requirements for the mass spectrometric investigation of microbial, mammalian and plant metabolomes. Phys Biol 5:011001
Scherling C, Roscher C, Giavalisco P, Schulze ED, Weckwerth W (2010) Metabolomics unravel contrasting effects of biodiversity on the performance of individual plant species. PLoS One 5:e12569
Weckwerth W, Loureiro ME, Wenzel K, Fiehn O (2004) Differential metabolic networks unravel the effects of silent plant phenotypes. Proc Natl Acad Sci U S A 101:7809–7814
Fernie AR, Trethewey RN, Krotzky AJ, Willmitzer L (2004) Innovation – metabolite profiling: from diagnostics to systems biology. Nat Rev Mol Cell Biol 5:763–769
Halket JM, Waterman D, Przyborowska AM, Patel RKP, Fraser PD, Bramley PM (2005) Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J Exp Bot 56:219–243
Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR (2006) Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat Protoc 1:387–396
Farre EM, Tiessen A, Roessner U, Geigenberger P, Trethewey RN, Willmitzer L (2001) Analysis of the compartmentation of glycolytic intermediates, nucleotides, sugars, organic acids, amino acids, and sugar alcohols in potato tubers using a nonaqueous fractionation method. Plant Physiol 127:685–700
Roessner U, Willmitzer L, Fernie AR (2001) High-resolution metabolic phenotyping of genetically and environmentally diverse potato tuber systems. Identification of phenocopies. Plant Physiol 127:749–764
Fiehn O, Kopka J, Trethewey RN, Willmitzer L (2000) Identification of uncommon plant metabolites based on calculation of elemental compositions using gas chromatography and quadrupole mass spectrometry. Anal Chem 72:3573–3580
Desbrosses GG, Kopka J, Udvardi MK (2005) Lotus japonicus metabolic profiling. Development of gas chromatography-mass spectrometry resources for the study of plant-microbe interactions. Plant Physiol 137:1302–1318
Roessner-Tunali U, Hegemann B, Lytovchenko A, Carrari F, Bruedigam C, Granot D, Fernie AR (2003) Metabolic profiling of transgenic tomato plants overexpressing hexokinase reveals that the influence of hexose phosphorylation diminishes during fruit development. Plant Physiol 133:84–99
Doerfler H, Lyon D, Nagele T, Sun X, Fragner L, Hadacek F, Egelhofer V, Weckwerth W (2013) Granger causality in integrated GC-MS and LC-MS metabolomics data reveals the interface of primary and secondary metabolism. Metabolomics 9:564–574
Doerfler H, Sun X, Wang L, Engelmeier D, Lyon D, Weckwerth W (2014) Mzgroupanalyzer--predicting pathways and novel chemical structures from untargeted high-throughput metabolomics data. PLoS One 9:e96188
Meijon M, Feito I, Oravec M, Delatorre C, Weckwerth W, Majada J, Valledor L (2016) Exploring natural variation of Pinus pinaster Aiton using metabolomics: is it possible to identify the region of origin of a pine from its metabolites? Mol Ecol 25:959–976
Wang L, Nagele T, Doerfler H, Fragner L, Chaturvedi P, Nukarinen E, Bellaire A, Huber W, Weiszmann J, Engelmeier D, Ramsak Z, Gruden K, Weckwerth W (2016) System level analysis of cacao seed ripening reveals a sequential interplay of primary and secondary metabolism leading to polyphenol accumulation and preparation of stress resistance. Plant J 87:318–332
Wang L, Sun X, Weiszmann J, Weckwerth W (2017) System-level and granger network analysis of integrated proteomic and metabolomic dynamics identifies key points of grape berry development at the interface of primary and secondary metabolism. Front Plant Sci 8:1066
Tolstikov VV, Fiehn O (2002) Analysis of highly polar compounds of plant origin: combination of hydrophilic interaction chromatography and electrospray ion trap mass spectrometry. Anal Biochem 301:298–307
Tolstikov VV, Fiehn O, Tanaka N (2007) Application of liquid chromatography-mass spectrometry analysis in metabolomics: reversed-phase monolithic capillary chromatography and hydrophilic chromatography coupled to electrospray ionization-mass spectrometry. Methods Mol Biol 358:141–155
Ramautar R, Mayboroda OA, Somsen GW, De Jong GJ (2011) CE-MS for metabolomics: developments and applications in the period 2008–2010. Electrophoresis 32:52–65
Ramautar R, Somsen GW, De Jong GJ (2009) CE-MS in metabolomics. Electrophoresis 30:276–291
Soga T (2007) Capillary electrophoresis-mass spectrometry for metabolomics. Methods Mol Biol 358:129–137
Monton MR, Soga T (2007) Metabolome analysis by capillary electrophoresis-mass spectrometry. J Chromatogr A 1168:237–246. Discussion 236
Watanabe CK, Sato S, Yanagisawa S, Uesono Y, Terashima I, Noguchi K (2014) Effects of elevated CO2 on levels of primary metabolites and transcripts of genes encoding respiratory enzymes and their diurnal patterns in Arabidopsis thaliana: possible relationships with respiratory rates. Plant Cell Physiol 55:341–357
Maruyama K, Urano K, Yoshiwara K, Morishita Y, Sakurai N, Suzuki H, Kojima M, Sakakibara H, Shibata D, Saito K, Shinozaki K, Yamaguchi-Shinozaki K (2014) Integrated analysis of the effects of cold and dehydration on rice metabolites, phytohormones, and gene transcripts. Plant Physiol 164:1759–1771
Hoehenwarter W, Larhlimi A, Hummel J, Egelhofer V, Selbig J, Van Dongen JT, Wienkoop S, Weckwerth W (2011) Mapa distinguishes genotype-specific variability of highly similar regulatory protein isoforms in potato tuber. J Proteome Res 10:2979–2991
Hoehenwarter W, Van Dongen JT, Wienkoop S, Steinfath M, Hummel J, Erban A, Sulpice R, Regierer B, Kopka J, Geigenberger P, Weckwerth W (2008) A rapid approach for phenotype-screening and database independent detection of cSNP/protein polymorphism using mass accuracy precursor alignment. Proteomics 8:4214–4225
Sun X, Weckwerth W (2012) COVAIN: a toolbox for uni-and multivariate statistics, time-series and correlation network analysis and inverse estimation of the differential Jacobian from metabolomics covariance data. Metabolomics 8:1–13
Sun X, Weckwerth W (2013) Using COVAIN to analyze metabolomics data. In: Weckwerth W, Kahl G (eds) The handbook of plant metabolomics. Wiley-Blackwell, Weinheim, pp 305–320. https://doi.org/10.1002/9783527669882.ch17
Weckwerth W (2011) Unpredictability of metabolism-the key role of metabolomics science in combination with next-generation genome sequencing. Anal Bioanal Chem 400:1967–1978
Boccard J, Grata E, Thiocone A, Gauvrit JY, Lanteri P, Carrupt PA, Wolfender JL, Rudaz S (2007) Multivariate data analysis of rapid LC-TOF/MS experiments from Arabidopsis thaliana stressed by wounding. Chemom Intell Lab Syst 86:189–197
Liland KH (2011) Multivariate methods in metabolomics – from pre-processing to dimension reduction and statistical analysis. TrAC-Trends Analyt Chem 30:827–841
Jansen JJ, Smit S, Hoefsloot HCJ, Smilde AK (2010) The photographer and the greenhouse: how to analyse plant metabolomics data. Phytochem Anal 21:48–60
Van Den Berg RA, Rubingh CM, Westerhuis JA, Van Der Werf MJ, Smilde AK (2009) Metabolomics data exploration guided by prior knowledge. Anal Chim Acta 651:173–181
Vichi M, Saporta G (2009) Clustering and disjoint principal component analysis. Comput Stat Data Anal 53:3194–3208
Daub CO, Kloska S, Selbig J (2003) Metagenealyse: analysis of integrated transcriptional and metabolite data. Bioinformatics 19:2332–2333
Xia JG, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS (2012) Metaboanalyst 2.0-A comprehensive server for metabolomic data analysis. Nucleic Acids Res 40:W127–W133
Barupal DK, Haldiya PK, Wohlgemuth G, Kind T, Kothari SL, Pinkerton KE, Fiehn O (2012) MetaMapp: mapping and visualizing metabolomic data by integrating information from biochemical pathways and chemical and mass spectral similarity. BMC Bioinformatics 13:99
Kastenmuller G, Romisch-Margl W, Wagele B, Altmaier E, Suhre K (2011) metaP-server: a web-based metabolomics data analysis tool. J Biomed Biotechnol 2011:7
Neuweger H, Albaum SP, Dondrup M, Persicke M, Watt T, Niehaus K, Stoye J, Goesmann A (2008) MeltDB: a software platform for the analysis and integration of metabolomics experiment data. Bioinformatics 24:2726–2732
Rojas-Cherto M, Van Vliet M, Peironcely JE, Van Doorn R, Kooyman M, Te Beek T, Van Driel MA, Hankemeier T, Reijmers T (2012) MetiTree: a web application to organize and process high-resolution multi-stage mass spectrometry metabolomics data. Bioinformatics 28:2707–2709
Nagele T, Mair A, Sun X, Fragner L, Teige M, Weckwerth W (2014) Solving the differential biochemical Jacobian from metabolomics covariance data. PLoS One 9:e92299
Nagele T, Weckwerth W (2013) A workflow for mathematical modeling of subcellular metabolic pathways in leaf metabolism of Arabidopsis thaliana. Front Plant Sci 4:541
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40:D109–D114
Zhang PF, Foerster H, Tissier CP, Mueller L, Paley S, Karp PD, Rhee SY (2005) MetaCyc and AraCyc. Metabolic pathway databases for plant research. Plant Physiol 138:27–37
Schreiber F, Colmsee C, Czauderna T, Grafahrend-Belau E, Hartmann A, Junker A, Junker BH, Klapperstuck M, Scholz U, Weise S (2012) MetaCrop 2.0: managing and exploring information about crop plant metabolism. Nucleic Acids Res 40:D1173–D1177
Morgat A, Coissac E, Coudert E, Axelsen KB, Keller G, Bairoch A, Bridge A, Bougueleret L, Xenarios I, Viari A (2012) UniPathway: a resource for the exploration and annotation of metabolic pathways. Nucleic Acids Res 40:D761–D769
Frolkis A, Knox C, Lim E, Jewison T, Law V, Hau DD, Liu P, Gautam B, Ly S, Guo AC, Xia JG, Liang YJ, Shrivastava S, Wishart DS (2010) SMPDB: the small molecule pathway database. Nucleic Acids Res 38:D480–D487
Usadel B, Poree F, Nagel A, Lohse M, Czedik-Eysenberg A, Stitt M (2009) A guide to using MapMan to visualize and compare Omics data in plants: a case study in the crop species, Maize. Plant Cell Environ 32:1211–1229
Brown M, Dunn WB, Dobson P, Patel Y, Winder CL, Francis-Mcintyre S, Begley P, Carroll K, Broadhurst D, Tseng A, Swainston N, Spasic I, Goodacre R, Kell DB (2009) Mass spectrometry tools and metabolite-specific databases for molecular identification in metabolomics. Analyst 134:1322–1332
Okazaki Y, Saito K (2012) Recent advances of metabolomics in plant biotechnology. Plant Biotechnol Rep 6:1–15
Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TWM, Fiehn O, Goodacre R, Griffin JL, Hankemeier T, Hardy N, Harnly J, Higashi R, Kopka J, Lane AN, Lindon JC, Marriott P, Nicholls AW, Reily MD, Thaden JJ, Viant MR (2007) Proposed minimum reporting standards for chemical analysis. Metabolomics 3:211–221
Hoehenwarter W, Chen Y, Recuenco-Munoz L, Wienkoop S, Weckwerth W (2011) Functional analysis of proteins and protein species using shotgun proteomics and linear mathematics. Amino Acids 41:329–341
Klose J, Kobalz U (1995) 2-dimensional electrophoresis of proteins – an updated protocol and implications for a functional-analysis of the genome. Electrophoresis 16:1034–1059
Morgenthal K, Weckwerth W, Steuer R (2006) Metabolomic networks in plants: transitions from pattern recognition to biological interpretation. Biosystems 83:108–117
Smith RD, Loo JA, Loo RRO, Busman M, Udseth HR (1991) Principles and practice of electrospray ionization – mass-spectrometry for large polypeptides and proteins. Mass Spectrom Rev 10:359–451
Weckwerth W, Kahl G (2013) The handbook of plant metabolomics. Wiley, Hoboken
Gorinstein S, Zemser M, Vargasalbores F, Ochoa JL (1995) Classification of 7 species of Cactaceae based on their chemical and biochemical-properties. Biosci Biotechnol Biochem 59:2022–2027
Bednarek P, Franski R, Kerhoas L, Einhorn J, Wojtaszek P, Stobiecki M (2001) Profiling changes in metabolism of isoflavonoids and their conjugates in Lupinus albus treated with biotic elicitor. Phytochemistry 56:77–85
Lois R (1994) Accumulation of Uv-absorbing flavonoids induced by Uv-B radiation in Arabidopsis-thaliana L.1. Mechanisms of Uv-resistance in Arabidopsis. Planta 194:498–503
Harrison MJ, Dixon RA (1993) Isoflavonoid accumulation and expression of defense gene transcripts during the establishment of vesicular-Arbuscular mycorrhizal associations in roots of Medicago-truncatula. Mol Plant-Microbe Interact 6:643–654
Schauer N, Semel Y, Roessner U, Gur A, Balbo I, Carrari F, Pleban T, Perez-Melis A, Bruedigam C, Kopka J, Willmitzer L, Zamir D, Fernie AR (2006) Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat Biotechnol 24:447–454
Nägele T, Fragner L, Chaturvedi P, Ghatak A, Weckwerth W (2017) Pollen metabolome dynamics: biochemistry, regulation and analysis. Springer, Cham
Roldan MVG, Engel B, De Vos RCH, Vereijken P, Astola L, Groenenboom M, Van De Geest H, Bovy A, Molenaar J, Van Eeuwijk F, Hall RD (2014) Metabolomics reveals organ-specific metabolic rearrangements during early tomato seedling development. Metabolomics 10:958–974
Dietrich CR, Han G, Chen M, Berg RH, Dunn TM, Cahoon EB (2008) Loss-of-function mutations and inducible RNAi suppression of Arabidopsis LCB2 genes reveal the critical role of sphingolipids in gametophytic and sporophytic cell viability. Plant J 54:284–298
Allwood JW, Ellis DI, Goodacre R (2008) Biomarker metabolites capturing the metabolite variance present in a rice plant developmental period. Physiol Plant 132:117–135
Bino RJ, Hall RD, Fiehn O, Kopka J, Saito K, Draper J, Nikolau BJ, Mendes P, Roessner-Tunali U, Beale MH, Trethewey RN, Lange BM, Wurtele ES, Sumner LW (2004) Potential of metabolomics as a functional genomics tool. Trends Plant Sci 9:418–425
Kim HK, Verpoorte R (2010) Sample preparation for plant metabolomics. Phytochem Anal 21:4–13
Allwood JW, Ellis DI, Goodacre R (2008) Metabolomic technologies and their application to the study of plants and plant-host interactions. Physiol Plant 132:117–135
Valledor L, Escandon M, Meijon M, Nukarinen E, Canal MJ, Weckwerth W (2014) A universal protocol for the combined isolation of metabolites, DNA, long RNAs, small RNAs, and proteins from plants and microorganisms. Plant J 79:173–180
Wienkoop S, Morgenthal K, Wolschin F, Scholz M, Selbig J, Weckwerth W (2008) Integration of metabolomic and proteomic phenotypes: analysis of data covariance dissects starch and RFO metabolism from low and high temperature compensation response in Arabidopsis thaliana. Mol Cell Proteomics 7:1725–1736
Antonio C, Pinheiro C, Chaves MM, Ricardo CP, Ortuno MF, Thomas-Oates J (2008) Analysis of carbohydrates in Lupinus albus stems on imposition of water deficit, using porous graphitic carbon liquid chromatography-electrospray ionization mass spectrometry (Vol 1187, Pg 111, 2008). J Chromatogr A 1201:132–132
Gechev TS, Benina M, Obata T, Tohge T, Sujeeth N, Minkov I, Hille J, Temanni MR, Marriott AS, Bergstrom E, Thomas-Oates J, Antonio C, Mueller-Roeber B, Schippers JH, Fernie AR, Toneva V (2013) Molecular mechanisms of desiccation tolerance in the resurrection glacial relic Haberlea rhodopensis. Cell Mol Life Sci 70:689–709
Erxleben A, Gessler A, Vervliet-Scheebaum M, Reski R (2012) Metabolite profiling of the moss Physcomitrella patens reveals evolutionary conservation of osmoprotective substances. Plant Cell Rep 31:427–436
Bowne JB, Erwin TA, Juttner J, Schnurbusch T, Langridge P, Bacic A, Roessner U (2012) Drought responses of leaf tissues from wheat cultivars of differing drought tolerance at the metabolite level. Mol Plant 5:418–429
Do PT, Degenkolbe T, Erban A, Heyer AG, Kopka J, Kohl KI, Hincha DK, Zuther E (2013) Dissecting rice polyamine metabolism under controlled long-term drought stress. PLoS One 8:e60325
Yang JC, Zhang JH, Liu K, Wang ZQ, Liu LJ (2007) Involvement of polyamines in the drought resistance of rice. J Exp Bot 58:1545–1555
Urano K, Maruyama K, Ogata Y, Morishita Y, Takeda M, Sakurai N, Suzuki H, Saito K, Shibata D, Kobayashi M, Yamaguchi-Shinozaki K, Shinozaki K (2009) Characterization of the ABA-regulated global responses to dehydration in Arabidopsis by metabolomics. Plant J 57:1065–1078
Skirycz A, De Bodt S, Obata T, De Clercq I, Claeys H, De Rycke R, Andriankaja M, Van Aken O, Van Breusegem F, Fernie AR, Inze D (2010) Developmental stage specificity and the role of mitochondrial metabolism in the response of Arabidopsis leaves to prolonged mild osmotic stress. Plant Physiol 152:226–244
Semel Y, Schauer N, Roessner U, Zamir D, Fernie AR (2007) Metabolite analysis for the comparison of irrigated and non-irrigated field grown tomato of varying genotype. Metabolomics 3:289–295
Witt S, Galicia L, Lisec J, Cairns J, Tiessen A, Araus JL, Palacios-Rojas N, Fernie AR (2012) Metabolic and phenotypic responses of greenhouse-grown maize hybrids to experimentally controlled drought stress. Mol Plant 5:401–417
Sicher RC, Barnaby JY (2012) Impact of carbon dioxide enrichment on the responses of maize leaf transcripts and metabolites to water stress. Physiol Plant 144:238–253
Silvente S, Sobolev AP, Lara M (2012) Metabolite adjustments in drought tolerant and sensitive soybean genotypes in response to water stress. PLoS One 7:e38554
Cook D, Fowler S, Fiehn O, Thomashow MF (2004) A prominent role for the CBF cold response pathway in configuring the low-temperature metabolome of Arabidopsis. Proc Natl Acad Sci U S A 101:15243–15248
Espinoza C, Degenkolbe T, Caldana C, Zuther E, Leisse A, Willmitzer L, Hincha DK, Hannah MA (2010) Interaction with diurnal and circadian regulation results in dynamic metabolic and transcriptional changes during cold acclimation in Arabidopsis. PLoS One 5:e14101
Kaplan F, Kopka J, Haskell DW, Zhao W, Schiller KC, Gatzke N, Sung DY, Guy CL (2004) Exploring the temperature-stress metabolome of Arabidopsis. Plant Physiol 136:4159–4168
Kaplan F, Kopka J, Sung DY, Zhao W, Popp M, Porat R, Guy CL (2007) Transcript and metabolite profiling during cold acclimation of Arabidopsis reveals an intricate relationship of cold-regulated gene expression with modifications in metabolite content. Plant J 50:967–981
Korn M, Gartner T, Erban A, Kopka J, Selbig J, Hincha DK (2010) Predicting Arabidopsis freezing tolerance and heterosis in freezing tolerance from metabolite composition. Mol Plant 3:224–235
Nagler M, Nukarinen E, Weckwerth W, Nagele T (2015) Integrative molecular profiling indicates a central role of transitory starch breakdown in establishing a stable C/N homeostasis during cold acclimation in two natural accessions of Arabidopsis Thaliana. BMC Plant Biol 15:284
Paupiere MJ, Muller F, Li HJ, Rieu I, Tikunov YM, Visser RGF, Bovy AG (2017) Untargeted metabolomic analysis of tomato pollen development and heat stress response. Plant Reprod 30:81–94
Qi XL, Xu WG, Zhang JZ, Guo R, Zhao MZ, Hu L, Wang HW, Dong HB, Li Y (2017) Physiological characteristics and metabolomics of transgenic wheat containing the maize C-4 phosphoenolpyruvate carboxylase (PEPC) gene under high temperature stress. Protoplasma 254:1017–1030
Sun CX, Gao XX, Li MQ, Fu JQ, Zhang YL (2016) Plastic responses in the metabolome and functional traits of maize plants to temperature variations. Plant Biol 18:249–261
Brosche M, Vinocur B, Alatalo ER, Lamminmaki A, Teichmann T, Ottow EA, Djilianov D, Afif D, Bogeat-Triboulot MB, Altman A, Polle A, Dreyer E, Rudd S, Lars P, Auvinen P, Kangasjarvi J (2005) Gene expression and metabolite profiling of Populus euphratica growing in the Negev desert. Genome Biol 6:R101
Cramer GR, Ergul A, Grimplet J, Tillett RL, Tattersall EAR, Bohlman MC, Vincent D, Sonderegger J, Evans J, Osborne C, Quilici D, Schlauch KA, Schooley DA, Cushman JC (2007) Water and salinity stress in grapevines: early and late changes in transcript and metabolite profiles. Funct Integr Genomics 7:111–134
Gagneul D, Ainouche A, Duhaze C, Lugan R, Larher FR, Bouchereau A (2007) A reassessment of the function of the so-called compatible solutes in the halophytic Plumbaginaceae Limonium latifolium. Plant Physiol 144:1598–1611
Gavaghan CL, Li JV, Hadfield ST, Hole S, Nicholson JK, Wilson ID, Howe PWA, Stanley PD, Holmes E (2011) Application of NMR-based metabolomics to the investigation of salt stress in maize (Zea mays). Phytochem Anal 22:214–224
Gong QQ, Li PH, Ma SS, Rupassara SI, Bohnert HJ (2005) Salinity stress adaptation competence in the extremophile Thellungiella halophila in comparison with its relative Arabidopsis thaliana. Plant J 44:826–839
Kim JK, Bamba T, Harada K, Fukusaki E, Kobayashi A (2007) Time-course metabolic profiling in Arabidopsis thaliana cell cultures after salt stress treatment. J Exp Bot 58:415–424
Gupta P, De B (2017) Metabolomics analysis of rice responses to salinity stress revealed elevation of serotonin, and gentisic acid levels in leaves of tolerant varieties. Plant Signal Behav 12(7):e1335845
Liu D, Ford KL, Roessner U, Natera S, Cassin AM, Patterson JH, Bacic A (2013) Rice suspension cultured cells are evaluated as a model system to study salt responsive networks in plants using a combined proteomic and metabolomic profiling approach. Proteomics 13:2046–2062
Sanchez DH, Lippold F, Redestig H, Hannah MA, Erban A, Kramer U, Kopka J, Udvardi MK (2008) Integrative functional genomics of salt acclimatization in the model legume Lotus japonicus. Plant J 53:973–987
Sanchez DH, Pieckenstain FL, Escaray F, Erban A, Kraemer U, Udvardi MK, Kopka J (2011) Comparative ionomics and metabolomics in extremophile and glycophytic Lotus species under salt stress challenge the metabolic pre-adaptation hypothesis. Plant Cell Environ 34:605–617
Widodo, Patterson JH, Newbigin E, Tester M, Bacic A, Roessner U (2009) Metabolic responses to salt stress of barley (Hordeum vulgare L.) cultivars, Sahara and Clipper, which differ in salinity tolerance. J Exp Bot 60:4089–4103
Wu D, Cai S, Chen M, Ye L, Chen Z, Zhang H, Dai F, Wu F, Zhang G (2013) Tissue metabolic responses to salt stress in wild and cultivated barley. PLoS One 8:e55431
Baxter CJ, Redestig H, Schauer N, Repsilber D, Patil KR, Nielsen J, Selbig J, Liu JL, Fernie AR, Sweetlove LJ (2007) The metabolic response of heterotrophic Arabidopsis cells to oxidative stress. Plant Physiol 143:312–325
Lehmann M, Schwarzlander M, Obata T, Sirikantaramas S, Burow M, Olsen CE, Tohge T, Fricker MD, Moller BL, Fernie AR, Sweetlove LJ, Laxa M (2009) The metabolic response of Arabidopsis roots to oxidative stress is distinct from that of heterotrophic cells in culture and highlights a complex relationship between the levels of transcripts, metabolites, and flux. Mol Plant 2:390–406
Ishikawa T, Takahara K, Hirabayashi T, Matsumura H, Fujisawa S, Terauchi R, Uchimiya H, Kawai-Yamada M (2010) Metabolome analysis of response to oxidative stress in Rice suspension cells overexpressing cell death suppressor Bax inhibitor-1. Plant Cell Physiol 51:9–20
Komatsu S, Yamamoto A, Nakamura T, Nouri MZ, Nanjo Y, Nishizawa K, Furukawa K (2011) Comprehensive analysis of mitochondria in roots and hypocotyls of soybean under flooding stress using proteomics and metabolomics techniques. J Proteome Res 10:3993–4004
Komatsu S, Nakamura T, Sugimoto Y, Sakamoto K (2014) Proteomic and metabolomic analyses of soybean root tips under flooding stress. Protein Pept Lett 21:865–884
Kusano M, Tabuchi M, Fukushima A, Funayama K, Diaz C, Kobayashi M, Hayashi N, Tsuchiya YN, Takahashi H, Kamata A, Yamaya T, Saito K (2011) Metabolomics data reveal a crucial role of cytosolic glutamine Synthetase 1;1 in coordinating metabolic balance in rice. Plant J 66:456–466
Urbanczyk-Wochniak E, Fernie AR (2005) Metabolic profiling reveals altered nitrogen nutrient regimes have diverse effects on the metabolism of hydroponically-grown tomato (Solanum lycopersicum) plants. J Exp Bot 56:309–321
Tschoep H, Gibon Y, Carillo P, Armengaud P, Szecowka M, Nunes-Nesi A, Fernie AR, Koehl K, Stitt M (2009) Adjustment of growth and central metabolism to a mild but sustained nitrogen-limitation in Arabidopsis. Plant Cell Environ 32:300–318
Nikiforova VJ, Daub CO, Hesse H, Willmitzer L, Hoefgen R (2005) Integrative gene-metabolite network with implemented causality deciphers informational fluxes of Sulphur stress response. J Exp Bot 56:1887–1896
Morcuende R, Bari R, Gibon Y, Zheng WM, Pant BD, Blasing O, Usadel B, Czechowski T, Udvardi MK, Stitt M, Scheible WR (2007) Genome-wide reprogramming of metabolism and regulatory networks of Arabidopsis in response to phosphorus. Plant Cell Environ 30:85–112
Hernandez G, Valdes-Lopez O, Ramirez M, Goffard N, Weiller G, Aparicio-Fabre R, Fuentes SI, Erban A, Kopka J, Udvardi MK, Vance CP (2009) Global changes in the transcript and metabolic profiles during symbiotic nitrogen fixation in phosphorus-stressed common bean plants. Plant Physiol 151:1221–1238
Hernandez G, Ramirez M, Valdes-Lopez O, Tesfaye M, Graham MA, Czechowski T, Schlereth A, Wandrey M, Erban A, Cheung F, Wu HC, Lara M, Town CD, Kopka J, Udvardi MK, Vance CP (2007) Phosphorus stress in common bean: root transcript and metabolic responses. Plant Physiol 144:752–767
Huang CY, Roessner U, Eickmeier I, Genc Y, Callahan DL, Shirley N, Langridge P, Bacic A (2008) Metabolite profiling reveals distinct changes in carbon and nitrogen metabolism in phosphate-deficient barley plants (Hordeum vulgare L.) Plant Cell Physiol 49:691–703
Armengaud P, Sulpice R, Miller AJ, Stitt M, Amtmann A, Gibon Y (2009) Multilevel analysis of primary metabolism provides new insights into the role of potassium nutrition for glycolysis and nitrogen assimilation in Arabidopsis roots. Plant Physiol 150:772–785
Jahangir M, Abdel-Farid IB, Choi YH, Verpoorte R (2008) Metal ion-inducing metabolite accumulation in Brassica rapa. J Plant Physiol 165:1429–1437
Sun XM, Zhang JX, Zhang HJ, Ni YW, Zhang Q, Chen JP, Guan YF (2010) The responses of Arabidopsis thaliana to cadmium exposure explored via metabolite profiling. Chemosphere 78:840–845
Agarrwal R, Bentur JS, Nair S (2014) Gas chromatography mass spectrometry based metabolic profiling reveals biomarkers involved in rice-gall midge interactions. J Integr Plant Biol 56:837–848
Sana TR, Fischer S, Wohlgemuth G, Katrekar A, Jung KH, Ronald PC, Fiehn O (2010) Metabolomic and transcriptomic analysis of the rice response to the bacterial blight pathogen Xanthomonas oryzae pv. oryzae. Metabolomics 6:451–465
Parker D, Beckmann M, Zubair H, Enot DP, Caracuel-Rios Z, Overy DP, Snowdon S, Talbot NJ, Draper J (2009) Metabolomic analysis reveals a common pattern of metabolic re-programming during invasion of three host plant species by Magnaporthe grisea. Plant J 59:723–737
Gunnaiah R, Kushalappa AC, Duggavathi R, Fox S, Somers DJ (2012) Integrated metabolo-proteomic approach to decipher the mechanisms by which wheat QTL (Fhb1) contributes to resistance against Fusarium graminearum. PLoS One 7:e40695
Chaves MM, Maroco JP, Pereira JS (2003) Understanding plant responses to drought – from genes to the whole plant. Funct Plant Biol 30:239–264
Chaves MM, Oliveira MM (2004) Mechanisms underlying plant resilience to water deficits: prospects for water-saving agriculture. J Exp Bot 55:2365–2384
Hare PD, Cress WA, Van Staden J (1998) Dissecting the roles of osmolyte accumulation during stress. Plant Cell Environ 21:535–553
Yancey PH (2005) Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J Exp Biol 208:2819–2830
Bitrian M, Zarza X, Altabella T, Tiburcio AF, Alcazar R (2012) Polyamines under abiotic stress: metabolic crossroads and hormonal crosstalks in plants. Metabolites 2:516–528
Gill SS, Tuteja N (2010) Polyamines and abiotic stress tolerance in plants. Plant Signal Behav 5:26–33
Skirycz A, Inze D (2010) More from less: plant growth under limited water. Curr Opin Biotechnol 21:197–203
Guy CL (1990) Cold-acclimation and freezing stress tolerance – role of protein-metabolism. Annu Rev Plant Physiol Plant Mol Biol 41:187–223
Le Gall H, Fontaine JX, Molinie R, Pelloux J, Mesnard F, Gillet F, Fliniaux O (2017) NMR-based metabolomics to study the cold-acclimation strategy of two Miscanthus genotypes. Phytochem Anal 28:58–67
Hauser F, Horie T (2010) A conserved primary salt tolerance mechanism mediated by HKT transporters: a mechanism for sodium exclusion and maintenance of high K+/Na+ ratio in leaves during salinity stress. Plant Cell Environ 33:552–565
Munns R, Tester M (2008) Mechanisms of salinity tolerance. Annu Rev Plant Biol 65(59):651–681
Sanchez DH, Siahpoosh MR, Roessner U, Udvardi M, Kopka J (2008) Plant metabolomics reveals conserved and divergent metabolic responses to salinity. Physiol Plant 132:209–219
Inan G, Zhang Q, Li PH, Wang ZL, Cao ZY, Zhang H, Zhang CQ, Quist TM, Goodwin SM, Zhu JH, Shi HH, Damsz B, Charbaji T, Gong QQ, Ma SS, Fredricksen M, Galbraith DW, Jenks MA, Rhodes D, Hasegawa PM, Bohnert HJ, Joly RJ, Bressan RA, Zhu JK (2004) Salt cress. A halophyte and cryophyte Arabidopsis relative model system and its applicability to molecular genetic analyses of growth and development of extremophiles. Plant Physiol 135:1718–1737
Johnson HE, Broadhurst D, Goodacre R, Smith AR (2003) Metabolic fingerprinting of salt-stressed tomatoes. Phytochemistry 62:919–928
Shulaev V, Cortes D, Miller G, Mittler R (2008) Metabolomics for plant stress response. Physiol Plant 132:199–208
Mittler R (2002) Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci 7:405–410
Suzuki N, Koussevitzky S, Mittler R, Miller G (2012) ROS and redox signalling in the response of plants to abiotic stress. Plant Cell Environ 35:259–270
Morgan MJ, Lehmann M, Schwarzlander M, Baxter CJ, Sienkiewicz-Porzucek A, Williams TCR, Schauer N, Fernie AR, Fricker MD, Ratcliffe RG, Sweetlove LJ, Finkemeier I (2008) Decrease in manganese superoxide dismutase leads to reduced root growth and affects Tricarboxylic acid cycle flux and mitochondrial redox homeostasis. Plant Physiol 147:101–114
Jackson MB, Ishizawa K, Ito O (2009) Evolution and mechanisms of plant tolerance to flooding stress. Ann Bot 103:137–142
Hoefgen R, Nikiforova VJ (2008) Metabolomics integrated with transcriptomics: assessing systems response to sulfur-deficiency stress. Physiol Plant 132:190–198
Nikiforova VJ, Kopka J, Tolstikov V, Fiehn O, Hopkins L, Hawkesford MJ, Hesse H, Hoefgen R (2005) Systems rebalancing of metabolism in response to Sulfur deprivation, as revealed by metabolome analysis of Arabidopsis plants. Plant Physiol 138:304–318
Sharma SS, Dietz KJ (2009) The relationship between metal toxicity and cellular redox imbalance. Trends Plant Sci 14:43–50
Balmerl D, Flors V, Glauser G, Mauch-Mani B (2013) Metabolomics of cereals under biotic stress: current knowledge and techniques. Front Plant Sci 4:82
Obata T, Fernie AR (2012) The use of metabolomics to dissect plant responses to abiotic stresses. Cell Mol Life Sci 69:3225–3243
Rizhsky L, Liang HJ, Shuman J, Shulaev V, Davletova S, Mittler R (2004) When defense pathways collide. The response of Arabidopsis to a combination of drought and heat stress. Plant Physiol 134:1683–1696
Wulff-Zottele C, Gatzke N, Kopka J, Orellana A, Hoefgen R, Fisahn J, Hesse H (2010) Photosynthesis and metabolism interact during acclimation of Arabidopsis thaliana to high irradiance and sulphur depletion. Plant Cell Environ 33:1974–1988
Krasensky J, Jonak C (2012) Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J Exp Bot 63:1593–1608
Empadinhas N, Da Costa MS (2008) Osmoadaptation mechanisms in prokaryotes: distribution of compatible solutes. Int Microbiol 11:151–161
Rathinasabapathi B (2000) Metabolic engineering for stress tolerance: installing osmoprotectant synthesis pathways. Ann Bot 86:709–716
Barnett NM, Naylor AW (1966) Amino acid and protein metabolism in Bermuda grass during water stress. Plant Physiol 41:1222
Verbruggen N, Hermans C (2008) Proline accumulation in plants: a review. Amino Acids 35:753–759
Verslues PE, Agarwal M, Katiyar-Agarwal S, Zhu J, Zhu JK (2006) Methods and concepts in quantifying resistance to drought, salt and freezing, abiotic stresses that affect plant water status. (Vol 45, Pg 523, 2006). Plant J 46:1092–1092
Verslues PE, Sharma S (2010) Proline metabolism and its implications for plant-environment interaction. Arabidopsis Book 8:e0140
Kishor PBK, Hong ZL, Miao GH, Hu CAA, Verma DPS (1995) Overexpression of delta-pyrroline-5-carboxylate synthetase increases proline production and confers osmotolerance in transgenic plants. Plant Physiol 108:1387–1394
Szekely G, Abraham E, Cselo A, Rigo G, Zsigmond L, Csiszar J, Ayaydin F, Strizhov N, Jasik J, Schmelzer E, Koncz C, Szabados L (2008) Duplicated P5cs genes of Arabidopsis play distinct roles in stress regulation and developmental control of proline biosynthesis. Plant J 53:11–28
Nanjo T, Kobayashi M, Yoshiba Y, Sanada Y, Wada K, Tsukaya H, Kakubari Y, Yamaguchi-Shinozaki K, Shinozaki K (1999) Biological functions of proline in morphogenesis and osmotolerance revealed in antisense transgenic Arabidopsis thaliana. Plant J 18:185–193
Roosens NH, Al Bitar F, Loenders K, Angenon G, Jacobs M (2002) Overexpression of ornithine-delta-aminotransferase increases proline biosynthesis and confers osmotolerance in transgenic plants. Mol Breed 9:73–80
Kaplan F, Guy CL (2004) Beta-amylase induction and the protective role of maltose during temperature shock. Plant Physiol 135:1674–1684
Kempa S, Krasensky J, Dal Santo S, Kopka J, Jonak C (2008) A central role of abscisic acid in stress-regulated carbohydrate metabolism. PLoS One 3:e3935
Renault H, Roussel V, El Amrani A, Arzel M, Renault D, Bouchereau A, Deleu C (2010) The Arabidopsis pop2-1 mutant reveals the involvement of GABA transaminase in salt stress tolerance. BMC Plant Biol 10:20
Fait A, Fromm H, Walter D, Galili G, Fernie AR (2008) Highway or byway: the metabolic role of the GABA shunt in plants. Trends Plant Sci 13:14–19
Liu CL, Zhao L, Yu GH (2011) The dominant glutamic acid metabolic flux to produce gamma-amino butyric acid over proline in Nicotiana tabacum leaves under water stress relates to its significant role in antioxidant activity. J Integr Plant Biol 53:608–618
Song HM, Xu XB, Wang H, Wang HZ, Tao YZ (2010) Exogenous gamma-aminobutyric acid alleviates oxidative damage caused by aluminium and proton stresses on barley seedlings. J Sci Food Agric 90:1410–1416
Bouche N, Fait A, Bouchez D, Moller SG, Fromm H (2003) Mitochondrial succinic-semialdehyde dehydrogenase of the gamma-aminobutyrate shunt is required to restrict levels of reactive oxygen intermediates in plants. Proc Natl Acad Sci U S A 100:6843–6848
Cromwell BT, Rennie SD (1953) The biosynthesis and metabolism of betaines in plants.1. The estimation and distribution of Glycinebetaine (betaine) in Beta-vulgaris L and other plants. Biochem J 55:189–192
Chen TH, Murata N (2011) Glycinebetaine protects plants against abiotic stress: mechanisms and biotechnological applications. Plant Cell Environ 34:1–20
Goel D, Singh AK, Yadav V, Babbar SB, Murata N, Bansal KC (2011) Transformation of tomato with a bacterial coda gene enhances tolerance to salt and water stresses. J Plant Physiol 168:1286–1294
Park EJ, Jeknic Z, Sakamoto A, Denoma J, Yuwansiri R, Murata N, Chen TH (2004) Genetic engineering of glycinebetaine synthesis in tomato protects seeds, plants, and flowers from chilling damage. Plant J 40:474–487
Waditee R, Bhuiyan MN, Rai V, Aoki K, Tanaka Y, Hibino T, Suzuki S, Takano J, Jagendorf AT, Takabe T, Takabe T (2005) Genes for direct methylation of glycine provide high levels of glycinebetaine and abiotic-stress tolerance in Synechococcus and Arabidopsis. Proc Natl Acad Sci U S A 102:1318–1323
Bianchi G, Gamba A, Limiroli R, Pozzi N, Elster R, Salamini F, Bartels D (1993) The unusual sugar composition in leaves of the resurrection plant Myrothamnus-flabellifolia. Physiol Plant 87:223–226
Paul MJ, Primavesi LF, Jhurreea D, Zhang YH (2008) Trehalose metabolism and signaling. Annu Rev Plant Biol 59:417–441
Guy C, Kaplan F, Kopka J, Selbig J, Hincha DK (2008) Metabolomics of temperature stress. Physiol Plant 132:220–235
Iordachescu M, Imai R (2008) Trehalose biosynthesis in response to abiotic stresses. J Integr Plant Biol 50:1223–1229
Li HW, Zang BS, Deng XW, Wang XP (2011) Overexpression of the trehalose-6-phosphate synthase gene OsTPS1 enhances abiotic stress tolerance in rice. Planta 234:1007–1018
Suzuki N, Bajad S, Shuman J, Shulaev V, Mittler R (2008) The transcriptional co-activator MBF1c is a key regulator of thermotolerance in Arabidopsis thaliana. J Biol Chem 283:9269–9275
Panikulangara TJ, Eggers-Schumacher G, Wunderlich M, Stransky H, Schoffl F (2004) Galactinol synthase1. A novel heat shock factor target gene responsible for heat-induced synthesis of raffinose family oligosaccharides in Arabidopsis. Plant Physiol 136:3148–3158
Peterbauer T, Richter A (2001) Biochemistry and physiology of raffinose family oligosaccharides and galactosyl cyclitols in seeds. Seed Sci Res 11:185–197
Taji T, Ohsumi C, Iuchi S, Seki M, Kasuga M, Kobayashi M, Yamaguchi-Shinozaki K, Shinozaki K (2002) Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J 29:417–426
Nishizawa A, Yabuta Y, Yoshida E, Maruta T, Yoshimura K, Shigeoka S (2006) Arabidopsis heat shock transcription factor A2 as a key regulator in response to several types of environmental stress. Plant J 48:535–547
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Ghatak, A., Chaturvedi, P., Weckwerth, W. (2018). Metabolomics in Plant Stress Physiology. In: Varshney, R., Pandey, M., Chitikineni, A. (eds) Plant Genetics and Molecular Biology. Advances in Biochemical Engineering/Biotechnology, vol 164. Springer, Cham. https://doi.org/10.1007/10_2017_55
Download citation
DOI: https://doi.org/10.1007/10_2017_55
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-91312-4
Online ISBN: 978-3-319-91313-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)