
Abstract
Background
There is growing evidence for a relationship between gut microbiota and hepatic encephalopathy (HE). However, the causal nature of the relationship between gut microbiota and HE has not been thoroughly investigated.
Method
This study utilized the large-scale genome-wide association studies (GWAS) summary statistics to evaluate the causal association between gut microbiota and HE risk. Specifically, two-sample Mendelian randomization (MR) approach was used to identify the causal microbial taxa for HE. The inverse variance weighted (IVW) method was used as the primary MR analysis. Sensitive analyses were performed to validate the robustness of the results.
Results
The IVW method revealed that the genus Bifidobacterium (OR = 0.363, 95% CI: 0.139-0.943, P = 0.037), the family Bifidobacteriaceae (OR = 0.359, 95% CI: 0.133-0.950, P = 0.039), and the order Bifidobacteriales (OR = 0.359, 95% CI: 0.133-0.950, P = 0.039) were negatively associated with HE. However, no causal relationship was observed among them after the Bonferroni correction test. Neither heterogeneity nor horizontal pleiotropy was found in the sensitivity analysis.
Conclusion
Our MR study demonstrated a potential causal association between Bifidobacterium, Bifidobacteriaceae, and Bifidobacteriales and HE. This finding may provide new therapeutic targets for patients at risk of HE in the future.
Similar content being viewed by others


Introduction
Hepatic encephalopathy (HE) is a reversible neurocognitive impairment that commonly occurs in patients with liver cirrhosis and other advanced liver diseases [1]. HE, with its broad range of neurological symptoms from mild cognitive changes to severe confusion and coma, significantly impairs patients' quality of life and contributes substantially to increased hospitalizations, morbidity rates, and mortality risks [2, 3]. The pathogenesis of HE is complex and multifactorial, involving liver failure, hyperammonemia, systemic inflammation, and infection [4, 5]. In recent years, a mounting body of research findings has supported alterations of the gut microbiota play an important role in HE pathogenesis [6,7,8,9]. In particular, the perturbation of the crosstalk among gut, liver, and brain, often referred to as the “gut-liver-brain” axis, has been suggested to be a determinant factor in the development and severity of HE in patients with liver cirrhosis. A previous study has indicated that elderly patients, aged over 65 years, with cirrhosis tend to exhibit an altered gut–liver–brain axis, characterized by impaired cognitive performance, particularly affecting memory, in comparison to age-matched noncirrhotic controls [10]. In a randomized clinical trial (RCT), Bajaj et al. found that fecal microbiota transplantation from a rationally selected donor for cirrhotic patients with recurrent HE was safe, and associated with lower hospitalizations, improved cognition compared with whose received standard of care treatment [8]. Although changes in the gut microbiota composition and function have critical relationship with liver health and brain function, previous observation studies may encounter several obstacles in establishing a causal relationship between gut microbiota and HE due to limitations in sample size, population heterogeneity, and the presence of potential confounding factors.
Alternative methodologies aimed at reinforcing the causal association between the gut microbiota and HE will serve to determine the potential necessity of conducting RCTs and offer valuable insights for the advancement of targeted therapeutic interventions. Mendelian randomization (MR) offers an opportunity to infer the potential causal relationship between an exposure and an outcome through the utilization of genetic variants as instrumental variables (IVs) [11]. This method can help to address confounding and minimize the possibility of reverse causality, as genetic variants that are randomly assorted at conception and not influenced by the development or progression of the disease. Herein, we conducted a two-sample MR analysis of gut microbiota in relation to the risk of HE using the large genome-wide association studies (GWAS).
Methods
Study design and data sources
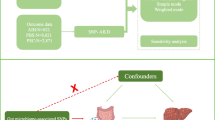
To investigate the potential causal relationship of the gut microbiome with the risk of HE, we applied a two-sample MR analysis. An overview of the study design is illustrated in Fig. 1.

Flow chart of the present MR study showing the causal association between gut microbiota and hepatic encephalopathy. SNPs, single-nucleotide polymorphism; MR, Mendelian randomization; IVW, inverse variance-weighted; MR-RAPS, MR-Robust adjusted profile score
Genetic variants for gut microbiota were obtained from the largest GWAS conducted by the MiBioGen consortium [12]. This study including 18,340 individuals of European ancestry from 24 population-based cohorts. After eliminating fifteen previously unidentified gut microbiome taxa, a total of 196 known gut microbiome taxa were retained for further analysis. The GWAS summary statistics for HE was obtained from FinnGen consortium R8 release data [13]. This GWAS study utilized the phenotype of "Encephalopathy" and encompassed a cohort consisting of 225 cases and 318,912 controls. The study followed the STROBE reporting guideline [14], and ethical approval was not required since all data were obtained from publicly accessible summary statistics.
Genetic instrument selection
To obtain reliable results, the following assumptions must be satisfied [15]: (1) single nucleotide polymorphisms (SNPs) strongly associated with each gut microbiome taxa were selected as IVs; (2) all IVs are not associated with any potential confounding factors and (3) the IVs solely influence the HE through the gut microbiota and not through any alternative pathways. Due to the smaller IVs obtained by the genome-wide significance threshold (P < 5× 10-8), a more relaxed threshold (P < 1.0×10-6) was used. To mitigate potential bias resulting from allelic association, the linkage disequilibrium was set to r2 < 0.01 with clumping distance of 500kb.
Statistical analysis
The inverse variance weighted (IVW) method was used as the primary statistical method in this study. Additionally, complementary analyses were conducted using MR-Egger regression, weighted model, weighted median, MR-robust adjusted profile score (MR-RAPS) as alternative methods. In order to evaluate the robustness of significant findings, a series of sensitivity analyses were conducted. Cochrane’s IVW Q test was performed to test the heterogeneity of IVs. The assessment of potential pleiotropy was initially determined by the intercept of MR Egger intercept test. Scatter plots were employed to visualize the results. Furthermore, leave-one-out plots were built to evaluate the relationship between each SNP within gut microbiome taxa and HE risk.
The F-statistic was used to assessed the strength of IVs using the formula \(F=\frac{{\text{R}}^{2}(\text{n}-\text{K}-1)}{\text{K}(1-{\text{R}}^{2})}\) (R2: proportion of variance in the gut microbiota explained by SNPs; n: sample size; K: the number of IVs). F statistic greater than 10 was considered no significant weak IVs bias. We conducted Bonferroni correction test for multiple testing, and an adjusted P value (P < 0.05/N, N represents the effective number of bacterial taxa at each feature level).
All data cleaning and statistical analyses were performed by using packages “TwoSampleMR” (version 0.5.6), “MRPRESSO” (version 1.0) in R software (Version 4.2.3, https://www.r-project.org/). The odds ratio (OR) was utilized to reveal the influence of gut microbiota on the risk of HE. P < 0.05 was deemed indicative of evidence for a potential causal effect.
Results
According to the selection criteria of IVs and a series quality control step, two hundred and ninety independent SNPs associated with 196 bacterial taxa were selected as IVs, including 30, 59, 149, 32, and 20 SNPs at the class, family, genus, order, and phylum levels, respectively. Details about the selected instrumental variables are shown in Table S1. After removing SNPs with palindromic sequences and intermediate allele frequencies, we finally obtained 94 SNPs as IVs to assess the causal association of gut microbiota with HE in this study. All these IVs had F statistics greater than 20 (range 20.631-88.430), suggesting that there was no weak IVs for the results (Table S2).
As shown in Fig. 2, three bacterial genera, specifically, the genus Bifidobacterium, the family Bifidobacteriaceae, and the order Bifidobacteriales, were found to associated with HE in at least one MR method. The IVW method indicates that the genus Bifidobacterium had a protective effect on HE (OR = 0.363, 95% CI: 0.139-0.943, P = 0.037). The IVW estimate of the family Bifidobacteriaceae also showed its suggestive protective effect against HE (OR = 0.359, 95% CI: 0.133-0.950, P = 0.039). Similarly, the IVW estimate suggests a potential protective effect of the order Bifidobacteriales on HE (OR = 0.359, 95% CI: 0.133-0.950, P = 0.039). The MR-RAPS method also consistently shows that the genus Bifidobacterium (OR = 0.338, 95% CI: 0.123-0.925, P = 0.035), the family Bifidobacteriaceae (OR = 0.334, 95% CI: 0.120-0.933, P = 0.0363), and the order Bifidobacteriales (OR = 0.334, 95% CI: 0.120-0.933, P = 0.0363) are associated with a reduced the risk of HE. However, after multiple-testing correction, no causal effect of the three identified taxa on HE was found using an adjust P value.

The MR results for association between gut microbiota and HE. IVW, inverse variance-weighted; MR-RAPS, MR-Robust adjusted profile score. HE, hepatic encephalopathy
The scatter plot also shown the protective effect of the three bacterial taxa on HE (Figure S1 A-C). Although rs7322849 of Bifidobacterium, Bifidobacteriaceae, and Bifidobacteriales appeared as potential outliers in scatter plots and leave-one-out plots (Figure S1 D-F), additional MR-PRESSO analysis did not identify any significant outliers with a global test P value greater than 0.05. Consequently, there was inadequate evidence to suggest horizontal pleiotropy in the relationship between these bacteria and HE. Meanwhile, the result of leave-one-out analysis revealed no single SNP is driving the causal associations between the identified bacterial taxa and HE, indicating our results were stability. For all the 3 bacterial taxa mentioned above, no heterogeneity and pleiotropy were found (Fig. 2, Table S3-4).
Discussion
In the current study, we performed a two sample MR analysis to investigate the causal relationship between gut microbiome and HE. We identified several genera of gut microbiota, including the genus Bifidobacterium, the family Bifidobacteriaceae, and the order Bifidobacteriales, which exhibited a suggestive potential protective causal effect on the risk of HE.
The gut milieu plays a pivotal role in the pathogenesis of complications associated with cirrhosis. Dysbiosis within the gut microbiome can initiate a cascade of inflammatory responses and subsequent activation of immune cells, ultimately promoting hepatic fibrosis and disease progression [16]. HE is a common and severe complication that arises from both chronic liver disease and acute liver failure, leading to notable impairment in cerebral function. The relationship between gut microbiota and HE has established in previous observational studies [6, 8, 17]. Malaguarnera et al. observed that the administration of a combination of Bifidobacterium plus fructo-oligosaccharides for a duration of 60 days resulted in a significant improvement in psychometric tests and blood ammonia levels compared to the lactulose group in patients with HE [18]. The MR analysis results also revealed that the genus Bifidobacterium reduce the risk of HE in this study. Consistent with previous studies [19, 20], we also found that the family Bifidobacteriaceae and the order Bifidobacteriales were protective effect against HE. In a Phase 1 RCT, Bajaj and colleagues discovered that the administration of oral fecal microbial transplant (FMT) capsules was linked to notable improvements in duodenal mucosal diversity, dysbiosis, antimicrobial peptides expression, and improved cognition among patients with cirrhosis and recurrent HE [19]. Outcomes from this study also revealed increased abundances of Ruminococcaceae and Bifidobacteriaceae in FMT group, while decreasing the abundances of Streptococcaceae and Veillonellaceae. These findings support the potential of FMT in modulating the gut microbiota and improving the inflammatory profile in patients with hepatic encephalopathy by reducing the expression of IL-6 and simultaneously increasing the expression of key antimicrobial peptides as well as intestinal barrier proteins [21, 22].
The gut-liver-brain axis is a complex network of interactions between the gut microbiota, liver, and brain which plays a crucial role in various physiological and pathological processes. This relationship is influenced by various factors such as diet, individual behaviors, environment factors, and surgical procedures like the transjugular intrahepatic portosystemic shunt [23, 24]. Besides, alterations in the gut ecology may induce gut dysbiosis and develop of series complications. HE is recognized as a representative model of a disease within the gut-liver-brain axis (Fig. 3), despite its pathogenesis remaining inadequately comprehended. Recently, increasing data demonstrates the alterations observed in the composition and function of gut microbiota have been linked to an increase in intestinal permeability, thereby facilitating the translocation of bacterial products, notably endotoxins, into the systemic circulation [25,26,27]. These microbial products can then activate immune and inflammatory responses in the liver and brain, contributing to the development and progression of HE. Furthermore, alterations in gut microbial taxa can serve as a vulnerability factor, influencing the neuroendocrine and neuroimmune signaling pathways within the gut-brain axis, thereby potentially impacting neurological function and behaviors [28, 29]. Moreover, an increasing body of research has demonstrated a link between liver fibrosis and inferior cognitive performance, with serum C-reactive protein playing a significant mediating role, suggesting the potential involvement of systemic inflammation in the liver-brain axis [30, 31]. These findings provide compelling evidence for the critical role played by gut microbiota in the development and progression of HE. However, most of the previous studies on the relationship between intestinal flora and HE were predominance of observational studies and mouse models. It’s challenging to provide robust evidence regarding the causal association between the gut microbiota and HE due to confounding factors.

Crass-talk between gut microbiota and hepatic encephalopathy via the gut-liver-brain axis
By utilizing genetic variants as IVs, MR analysis mimics the randomization in clinical trials and helps overcome potential confounding factors, thus, minimizing bias in causal effect estimates. Using two-sample MR analysis, we investigated the causal relationship between the gut microbiota and HE. To ensure the robustness of instrumental variables in the MR analysis of gut microbiota, genetic variants were obtained from the largest available GWAS meta-analysis [12]. In line with prior researches [19, 32], both the IVW and MR-RAPS method results indicated increasing abundant levels of the genus Bifidobacterium, the family Bifidobacteriaceae, and the order Bifidobacteriales are genetically associated with decreased HE risk. The IVW method assumes the validity of all instrumental variants based on MR assumptions and combines the Wald ratios for each SNP to yield a pooled estimate [33]. It is considered the most potent method for MR estimation, but estimates derived from this approach may be susceptible to bias in the presence of directional pleiotropy. MR-RAPS, an extension of the IVW method, serves as a correcting model in the presence of pleiotropy. It offers enhanced robustness against deviations from the underlying assumptions of MR due to its ability to adjust the profile likelihood of the summary data employed in the analysis [34]. While the multiple test results suggest the possibility of false positives and reveal no significant genetic correlation between the microbiota and HE risk, we believe that this may be partly due to the conservative nature of the Bonferroni correction and the relatively small sample size [35, 36].
There were several limitations in our study. First, due to using summary data rather than raw data, it is difficult to explore the stratification effects and perform subgroup analysis, such as distinguishing alteration of gut abundance in chronic liver disease and acute liver failure, and type and clinical grading of HE. Besides, since this study specifically examines the effects within a European descent population, caution should be exercised when generalizing the results to other ethnicities without additional justification. Therefore, it is crucial to conduct further RCTs with clear classification and inclusion of diverse ethnic groups to establish a comprehensive understanding of the relationship between gut microbiota and HE. Second, in order to perform sensitivity analysis and detect potential horizontal pleiotropy, SNPs used in this two-sample MR analysis may not have reached the conventional GWAS significance threshold of P < 5×10–8. For this, the F-statistic was used to assessed the strength of IVs and Bonferroni correction test was performed to mitigate the risk of false positives. Third, the definition of "hepatic encephalopathy" has varied across previous studies [2, 37], leading to complexities in its classification. While our findings align with previous studies, it is important to note that the causes of HE among the participants included in this study may be diverse.
In conclusion, we revealed evidence of three bacterial taxa, specifically, the genus Bifidobacterium, the family Bifidobacteriaceae, and the order Bifidobacteriales, may have a protective effect against HE. Our study findings shed light on the complex cross-talk between the gut microbiota and HE through the gut-liver-brain axis, which may provide a novel way for the development of targeted therapies. In the future, further large population-based longitudinal research and more diverse cohorts are required to verify our findings.
Availability of data and materials
All data generated during this study are provided in the original publications and its supplementary information files.
References
Häussinger D, Dhiman RK, Felipo V, et al. Hepatic encephalopathy. Nat Rev Dis Primers. 2022;8(1):43.
Rose CF, Amodio P, Bajaj JS, et al. Hepatic encephalopathy: Novel insights into classification, pathophysiology and therapy. J Hepatol. 2020;73(6):1526–47.
Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology. 2014;60(2):715–35.
Bessman AN, Hawkins R. The relative effects of enterically administered plasma and packed cells on circulating blood ammonia. Gastroenterology. 1963;45:368–73.
EASL. Clinical Practice Guidelines on the management of hepatic encephalopathy. J Hepatol. 2022;77(3):807–24.
Bloom PP, Tapper EB, Young VB, Lok AS. Microbiome therapeutics for hepatic encephalopathy. J Hepatol. 2021;75(6):1452–64.
Bajaj JS, Shamsaddini A, Fagan A, et al. Distinct gut microbial compositional and functional changes associated with impaired inhibitory control in patients with cirrhosis. Gut Microbes. 2021;13(1):1953247.
Bajaj JS, Kassam Z, Fagan A, et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: A randomized clinical trial. Hepatology. 2017;66(6):1727–38.
Saboo K, Shamsaddini A, Iyer MV, et al. Sex is associated with differences in gut microbial composition and function in hepatic encephalopathy. J Hepatol. 2021;74(1):80–8.
Bajaj JS, Duarte-Rojo A, Xie JJ, et al. Minimal Hepatic Encephalopathy and Mild Cognitive Impairment Worsen Quality of Life in Elderly Patients With Cirrhosis. Clin Gastroenterol Hepatol. 2020;18(13):3008-3016.e2.
Boef AGC, Dekkers OM, le Cessie S. Mendelian randomization studies: a review of the approaches used and the quality of reporting. Int J Epidemiol. 2015;44(2):496–511.
Kurilshikov A, Medina-Gomez C, Bacigalupe R, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156–65.
Kurki MI, Karjalainen J, Palta P, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613(7944):508–18.
Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA. 2021;326(16):1614–21.
Didelez V, Sheehan N. Mendelian randomization as an instrumental variable approach to causal inference. Stat Methods Med Res. 2007;16(4):309–30.
Bajaj JS. The role of microbiota in hepatic encephalopathy. Gut Microbes. 2014;5(3):397–403.
Bajaj JS, Heuman DM, Hylemon PB, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol. 2014;60(5):940–7.
Malaguarnera M, Gargante MP, Malaguarnera G, et al. Bifidobacterium combined with fructo-oligosaccharide versus lactulose in the treatment of patients with hepatic encephalopathy. Eur J Gastroenterol Hepatol. 2010;22(2):199–206.
Bajaj JS, Salzman NH, Acharya C, et al. Fecal Microbial Transplant Capsules Are Safe in Hepatic Encephalopathy: A Phase 1, Randomized Placebo-Controlled Trial. Hepatology. 2019;70(5):1690–703.
Kimer N, Pedersen JS, Tavenier J, et al. Rifaximin has minor effects on bacterial composition, inflammation, and bacterial translocation in cirrhosis: A randomized trial. J Gastroenterol Hepatol. 2018;33(1):307–14.
Wiest R, Albillos A, Trauner M, Bajaj JS, Jalan R. Targeting the gut-liver axis in liver disease. J Hepatol. 2017;67(5):1084–103.
Weingarden AR, Vaughn BP. Intestinal microbiota, fecal microbiota transplantation, and inflammatory bowel disease. Gut Microbes. 2017;8(3):238–52.
Mancini A, Campagna F, Amodio P, Tuohy KM. Gut : liver : brain axis: the microbial challenge in the hepatic encephalopathy. Food Funct. 2018;9(3):1373–88.
Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol. 2020;72(3):558–77.
Tilg H, Adolph TE, Trauner M. Gut-liver axis: Pathophysiological concepts and clinical implications. Cell Metab. 2022;34(11):1700–18.
Llopis M, Cassard AM, Wrzosek L, et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut. 2016;65(5):830–9.
Caraceni P, Vargas V, Solà E, et al. The Use of Rifaximin in Patients With Cirrhosis. Hepatology. 2021;74(3):1660–73.
Fung TC. The microbiota-immune axis as a central mediator of gut-brain communication. Neurobiol Dis. 2020;136:104714.
Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155(7):1451–63.
Jiang R, Wu J, Rosenblatt M, et al. Elevated C-reactive protein mediates the liver-brain axis: a preliminary study. EBioMedicine. 2023;93:104679.
Parikh NS, Kumar S, Rosenblatt R, et al. Association between liver fibrosis and cognition in a nationally representative sample of older adults. Eur J Neurol. 2020;27(10):1895–903.
Luo M, Xin R-J, Hu F-R, Yao L, Hu S-J, Bai F-H. Role of gut microbiota in the pathogenesis and therapeutics of minimal hepatic encephalopathy via the gut-liver-brain axis. World J Gastroenterol. 2023;29(1):144–56.
Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408.
Zhao Q, Wang J, Hemani G, Bowden J, Small DS. Statistical inference in two-sample summary-data Mendelian randomization using robust adjusted profile score. J Ann Stat. 2020;48(3):1742-69 28.
Shuken SR, McNerney MW. Costs and Benefits of Popular P-Value Correction Methods in Three Models of Quantitative Omic Experiments. Anal Chem. 2023;95(5):2732–40.
Noble WS. How does multiple testing correction work? Nat Biotechnol. 2009;27(12):1135–7.
Jalan R, Rose CF. Heretical thoughts into hepatic encephalopathy. J Hepatol. 2022;77(2):539–48.
Acknowledgements
We want to acknowledge the participants and investigators of the FinnGen study. We also sincerely appreciate the great work of the MiBioGen consortium and authors for making the summary statistics publicly available.
Funding
This research did not receive any specific grant.
Author information
Authors and Affiliations
Contributions
JLW, JYL and ZBJ contributed to the hypothesis and study design. JLW, XYD, JJD, TYL, SYC, and HYR performed the data analysis, and wrote the manuscript. JWC and MSH reviewed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, JL., Chen, JW., Huang, MS. et al. The causal effect of gut microbiota on hepatic encephalopathy: a mendelian randomization analysis. BMC Med Genomics 17, 216 (2024). https://doi.org/10.1186/s12920-024-01939-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01939-y