
Abstract
The current standard of care (SOC) for hepatitis C virus (HCV) infection is the combination of pegylated interferon (PEG-IFN), Ribavirin and protease inhibitor for HCV genotype 1. Nevertheless, this treatment is successful only in 70-80% of the patients. In addition, the treatment is not economical and is of immense physical burden for the subject. It has been established now, that virus-host interactions play a significant role in determining treatment outcomes. Therefore identifying biological markers that may predict the treatment response and hence treatment outcome would be useful. Both IFN and Ribavirin mainly act by modulating the immune system of the patient. Therefore, the treatment response is influenced by genetic variations of the human as well as the HCV genome. The goal of this review article is to summarize the impact of recent scientific advances in this area regarding the understanding of human and HCV genetic variations and their effect on treatment outcomes. Google scholar and PubMed have been used for literature research. Among the host factors, the most prominent associations are polymorphisms within the region of the interleukin 28B (IL28B) gene, but variations in other cytokine genes have also been linked with the treatment outcome. Among the viral factors, HCV genotypes are noteworthy. Moreover, for sustained virological responses (SVR), variations in core, p7, non-structural 2 (NS2), NS3 and NS5A genes are also important. However, all considered single nucleotide polymorphisms (SNPs) of IL28B and viral genotypes are the most important predictors for interferon based therapy of HCV infection.
Similar content being viewed by others

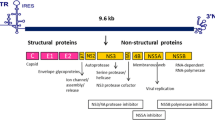

Introduction
HCV infects about 200 million people worldwide [1]. Approximately 20–30% of patients naturally clear the virus. About 70–80% acute HCV infections become chronic which leads to the development of cirrhosis in 20% of cases while the same percentage of those patients becomes a victim of hepatocellular carcinoma. Acute hepatitis C occurs during the first six months of HCV infection [2]. Approximately 70-80% of acute hepatitis C cases are without symptoms and hence difficult to diagnose. The remaining 20%-30% of cases are associated with symptoms such as pain in joints and muscles, pain in the right upper quadrant, poor appetite, nausea, vomiting, and fever. The case of acute hepatitis C infection is converted into a chronic disease, if the individual is not capable of clearing the virus within few months of infection [3]. Being the member of the Flaviviridae family of viruses, HCV is a single stranded RNA virus. Its size is 55 – 65 nm [2]. In 1989 it was realized that HCV is the cause of most transfusion-associated non-A and non-B hepatitis infections. There are about eleven different genotypes of HCV with various subtypes and strains [3]. The virus encodes a poly-protein of 3010 amino acids which is processed to generate four structural (Core, E1, E2 and P7) and six non-structural (NS2, NS3, NS4A, NS4B, NS5A, NS5B) proteins [4]. Recently, great efforts have been made to develop interferon free therapy against HCV infection but interferon is still accepted as a part of standard therapy [5]. Therefore, it would be very helpful for clinical practitioners and researchers to get information about viral and host factors that influence interferon treatment of HCV infection.
Interferon and its signaling pathway
About 50 years ago, IFN was discovered by Isaacs and Lindenmann [6]. Currently, there are about 10 mammalian IFN species with many subspecies. IFN possesses antiviral activity and is categorized into three groups [7]. Type I IFNs include IFN-α, IFN-β, IFN-ϵ, IFN-κ, IFN-ω and IFN-ν. All these interferons interact with the interferon alpha/beta receptor (IFNAR) [8, 9].Type II IFN involves only IFN-γ which interacts with a discrete receptor, the interferon gamma receptor (IFNGR) [10]. IFN-λ1, IFN-λ2 and IFN-λ3 are grouped in type III IFNs. These interferons are also known as IL29, IL28A and IL28B respectively. Type III IFNs, signal through IFN-λ receptor which possesses an IL-10R2 chain shared with the IL-10 receptor, and an exceptional IFN-λ chain [11]. IFNAR is the receptor for type I IFNs. It possesses two major subunits: IFNAR1, which bind styrosine kinase 2 (TYK2) [12], and IFNAR2c which binds Janus kinase 1 (JAK1) [9]. Both TYK2 and JAK1 are members of the Jak family. After binding the receptor chains, TYK2 and JAK1 are stimulated and transactivated leading to the initiation of phosphorylation cascades involving all the members of the signaling pathway and also the activators of transcription such as signal transducer and activator of transcription STAT1, STAT2 and STAT3. STAT1, STAT2 and STAT3 are stimulated by type I IFNs in most of cells. STAT1 and STAT2 in combination with another transcription factor, interferon regulatory factor 9 (IRF9), form interferon stimulated gene factor 3 (ISGF3) which binds to the promoter region of interferon stimulated genes (ISGs) as shown in Figure 1. The ISGs are a set of genes used for antiviral protection. Microarray analysis of human and murine cells treated with interferons revealed that there are more than 300 ISGs [13]. Most important of these proteins are the double-stranded RNA-dependent kinase "protein kinase RNA-regulated" (PRKR), the 2′–5′ oligoadenylate synthetases and the Mx proteins. These proteins are known to impede the growth of certain viruses. However, if these genes are knocked out from cells, they still retain their antiviral activities as there are many of other genes stimulated by interferon possessing antiviral activities [14]. Alternatively, STAT1 and STAT3 form homodimers or heterodimers which bind gamma activated sequence (GAS) elements. After binding, STAT proteins activate these genes to generate an antiviral state [15]. Receptors and pathways involved in type III IFNs signaling diverge from those mediating type I IFNs signals. IFN-λ1-3 signal through the JAK/STAT signaling pathway using the IL28-R/IL-10R receptor complex that is mainly expressed on hepatocytes and epithelial cells [16] as opposed to IFNAR that are broadly expressed.

Interferon signaling pathway.
Ribavirin
Ribavirin (1-b-D-ribofuranosyl-1, 2, 4-triazole-3-carboxamide) inhibits HCV replication. It is a synthetic guanosine nucleoside analogue that inhibits inosine monophosphate dehydrogenase leading to the depletion of the guanosine pool [17]. It is also incorporated into the viral genome and induces error catastrophe by the HCV non-structural-5B (NS5B) polymerase [18]. In addition to these functions, it is thought to have immune-modulatory functions. It regulates macrophages, T helper cells and Th1/Th2 produced cytokines, enhances the expression of interferon stimulated genes (ISG) and also IFN-α and MxA expression.
Protease inhibitors
Two important NS3 protease inhibitors are Boceprevir and Telaprevir. Boceprevir inhibits NS3 by acting as a non-covalent inhibitor. It inhibits CYP3A4 and acts also as a mild inhibitor of P-glycoprotein. Therefore, the plasma levels of the drugs that are metabolized by CYP3A4 and P-glycoprotein are also increased when given along Boceprevir [19].
Role of viral factors in determining the response to interferon therapy
Genotype
Soon after the discovery and sequencing of HCV [20], HCV isolates from different parts of the world with varying sequences were obtained [21, 22]. HCV sequences which varied up to 33-36% were classified as genotypes; sequences which varied up to 20 – 27% were classified as subtypes while genetic variants up to 12% within individual isolates were classified as quasi species. The HCV genotype is acknowledged as the most significant independent response marker of interferon therapy [23]. End-of-treatment response (ETR) and SVR are the two important scales for measurement of treatment response. An ETR is referred as undetectable hepatitis C RNA at the end-of-treatment while SVR is referred as undetectable hepatitis C RNA after 24 weeks of treatment [24]. HCV genotypes 2 and 3 are more responsive to interferon therapy than any other HCV genotype [25]. Meta-analyses of IFN-α monotherapy have shown that SVR was obtained in 55% of patients with HCV genotypes 2 and 3 and in 18% of patients with HCV genotype 1 [26]. It is suggested that HCV genotype 6 gives a treatment response similar to genotypes 2 and 3 [24, 25]. Although HCV genotyping is an important predictor for the treatment response, no patient should be left without therapy on the basis of the genotype alone [27] because the predictive value of HCV genotyping for interferon based therapy is only 55%.
5′UTR
The important role of 5′UTR (Untranslated Region) in translation of HCV polyprotein demands the conservation of this region. There are four highly conserved structural domains in 5′UTR which are numbered I to IV; all of these domains interact with the host factors and are crucial for HCV polyprotein synthesis [28]. Domain III of 5′UTR binds eukaryotic initiation factor 3 (eIF3) by means of stem-loop 3b. It has been suggested that insertions in domain 3 of 5′UTR causes altered Watson-Crick base pairing leading to decreased RNA stability and binding affinity to ribosomal proteins. These mutations were more common in SVR than in breakthrough (BT) patients. Thus, mutations within the domain III of 5′UTR are important for the treatment response in HCV infection [29].
Variations of core, p7, NS2, NS3 and NS5A
Inter-patient genetic variations within the genotype 1 HCV also exert an important influence on the treatment response. Although sequencing of the HCV genome has not shown any specific amino acid sequences that affect the treatment outcome, the high levels of genetic variations, mostly in the core, p7, NS2, NS3 and NS5A genes, are linked with SVR [30]. Recent data from the Chinese population have also shown that increased genetic variations in the p7, NS2 and NS3 genes of HCV genotype 1b were linked with SVR to the standard treatment [31]. Thus, there is more genetic diversity in HCV SVR patients than in NR patients receiving the standard treatment. Analyses of different HCV sequences identified many specific regions of core, p7, NS2, NS3 and NS5A, which were significantly associated with the ultimate treatment outcome. Among these regions, the most important region was the interferon sensitivity determining region (ISDR) that is located in HCV NS5A [32, 33]. The sequence of HCV core region from 50 Swedish patients infected with HCV genotype 1 revealed that substitutions of core residue 70 were associated with poor response to the standard treatment [34]. Moreover, it has been shown that amino-acid (aa) substitution at core residue 70 was the predictor of SVR to a triple therapy of Telaprevir, pegylated interferon and Ribavirin in the Japanese population [35]. How these variations of HCV genome effect response to interferon therapy is still a challenge.
Role of host factors in determining response to interferon therapy
With the beginning of the human genetic era, it is anticipated that the human genome may significantly influence the clinical management of infectious diseases. But there are small numbers of studies showing the importance of such knowledge in regular clinical practices. HCV infection is one of the most hopeful examples where genetic information was used for therapy. Thus, the treatment response to HCV infection not only depends on the viral factors but also on the host factors. Male sex, older age, insulin resistance, diabetes, African or American ethnicity, cirrhosis, steatosis, and weight (in terms of BMI) are all factors linked to poor response to PEG-IFN plus RBV treatment [36]. Other infections such as HIV, HBV and practices such as alcohol intake and drug use are also responsible for low SVR rates [37]. Currently, candidate gene approaches had been implemented to discover the host factors associated with the HCV treatment response [38]. Following are some of the most important SNPs linked with the treatment response to HCV infection, also shown in Table 1.
SNPs of host genes
Interferon-λ
The IFN-λ family was discovered in 2003. The three members of this family, IFN-λ 1, 2, 3, (equivalent to IL-29, 28A, 28B) show a high degree of homology to each other i.e. more than 80%, but very low sequence homology to both IFN-α (15-19% identity, 31-33% similarity) and IL-10 (11-13% identity, 22-23% similarity). Despite this minimal homology, IFN-λ like IL-10 geneare composed of five to six exons [39]–[41]. IFN-λ is produced by many immune cells, neuronal cells, alveolar epithelial cells, hepatocytes, and a variety of cell lines [42]–[44]. However, the primary sources of IFN-λ are dendritic cells (DCs) [42]–[46]. Similar to IFN-α, these cytokines are mainly produced in response to viral infection or by activation of Toll-like receptors (TLRs) [16, 47]. IFN-λ showed antiviral activity against many viruses such as Encephalomyocarditis virus (EMCV), vesicular stomatitis virus, cytomegalovirus, herpes simplex virus 1, influenza A virus, HIV, HBV, and HCV [39, 40, 47]–[49]. These studies revealed that IFN-λ mainly inhibits viral replication but also has immune-modulatory functions. It modulates both the maturation and differentiation of immune cells [50]–[52]. In short, these cytokines have an important role in regulation and development of the adaptive immune response against viruses.
SNPs within chromosome 19, in the vicinity of the IL29, IL28A, and IL28B genes, are importantly associated with the treatment response of HCV infection. Three SNPs related to these genes, rs8099917, rs12980275, and rs12979860, are very important. Previous studies have shown that these variants are significant predictors of the treatment response [53]. The favorable genotypes significantly predicting higher SVR rates are CCrs12979860 irrespective of the race [54], AArs12980275, and TTrs8099917. Nevertheless, the association between IFN-λ production and SNPs in close proximity of IL28 remains indistinct [16, 55]. IFN-λ-based drugs are possible candidates for treating HCV infection, and are presently being evaluated in clinical trials. The adverse effects of IFN-λ are less marked than all of IFNs α and β, regarding bone marrow suppression, probably because fewer cells carry the receptor, thus permitting a more targeted therapy [56].
Interferon-γ
IFN-γ is a cytokine produced by effector T cells and natural killer cells. It is mainly involved in the development of T helper 1 (Th1) cells [57]. Studies using a HCV replicon system showed that IFN-γ is capable of inhibiting the HCV replication [58]. Similar to other cytokines there is no polymorphism reported in the coding region; but the non-coding region possesses frequent polymorphisms which are implicated in several chronic inflammatory conditions and autoimmune diseases [59, 60]. It is reported that the SNP –764G of IFN-γ which is located in the proximal promoter region was strongly associated with SVR in case of HCV infection because the G allele conferred a two to three-fold increase in the promoter activity. Moreover, the G allele also offers stronger binding affinity to the heat shock transcription factor (HSF1) than the C allele at this specific position. Thus, IFN-γ promoter SNP –764G/C is functionally very important which can influence the interferon therapy and may be used as a therapeutic marker for HCV infection [61].
Mannan-binding lectin
Mannan-binding lectin (MBL) also known as mannose binding protein plays a significant role inthe innate immune system. It is a pathogen recognition receptor [62]. MBL deficiencies are ascribed to a certain extent to three SNPs in the first exon of the gene: MBL2*D (Arg52Cys), B(Gly54Asp) and C (Gly57Glu). These SNPs are collectively marked as O. The major allele at these loci is A. The polymerization of the polypeptide is disturbed by O amino acid which causes low levels of high order oligomeric MBL in plasma [63]. MBL concentration in the serum is also adjusted by two promoter SNPs: MBL2*H/L and X/Y [64, 65]. The linkage disequilibrium between the promoter and exon 1 SNPs of MBL produces seven haplotypes which are associated with a decreased level of MBL in the plasma. The frequency of YA/YO genotype was significantly higher in the HCV patients as compared to the controls suggesting that these genotypes are involved in the development of chronic hepatitis C. MBL genotypes XA/XA, XA/YO and YO/YO were associated with a decreased level of MBL in the plasma. The levels of these genotypes were considerably decreased in patients with advanced fibrosis as compared to patients with moderate fibrosis and to the control group. Thus, MBL has not only an important role in the development of chronic hepatitis C, but also an important role in the treatment outcome [63, 66].
Cytotoxic T lymphocyte antigen–4
Cytotoxic T lymphocyte antigen–4 (CTLA4) also referred as cluster of differentiation 152 (CD152) is a protein receptor that is mainly expressed on activated CD4+ and CD8+ T cells [67]. It binds to the ligands B7-1 (CD80) and B7-2 (CD86) and down-regulates the immune system by switching off T cells [68]. There are reported two important transition mutations in CTLA4, C→T at position -318 and G→A at position 49 in exon 1 [69, 70]. It is well established by recent findings that these transition mutations of CTLA-4 have a very significant influence on the down regulation of T cells [71]. Sustained responders (SRs) had a higher frequency of 49G alone and were highly associated with -318C in a haplotype. SRs had a higher homozygosity for the -318C-49G haplotype. Moreover, it was also reported that the immune systems of -318C-49G haplotype carriers were declining the viral load more rapidly as compared to other patients. On the other hand, patients which possessed -318 T and 49A showed a reciprocal effect (poor response). There may be several suggested reasons for these effects of CTLA4 polymorphisms. One of these may be the polymorphism of either a single gene or haplotype, which may be responsible for differential expression of a gene. It is also noted that -318 T [72] and 49A [73] are associated with the increased expression of CTLA4. Haplotype -318 T-49A also showed increased expression of CTLA4 [74]. Studies up till now have suggested that polymorphisms at -318C-49G of CTLA4 gene are linked with down regulation of CTLA4 expression that leads to the amplification of T cells response. Alternatively, as CTLA4 acts as a ligand for CD80 and CD86, it may have a role in the developmental pathway of Th1/Th2 cells and may shift their balance [75]–[77]. Another possibility is that these polymorphisms are in linkage disequilibrium with some adjacent markers that have influence on the expression of CTLA4. HCV genotypes and ethnicity have important effects on the treatment outcome. As the reported study was conducted only on white patients infected with HCV genotype 1, these analyses have to be confirmed in other populations effected with different HCV genotypes [78].
Interleukin 10
Interleukin 10 (IL-10) is a strong immunoregulatory T helper type 2 (Th2) cytokine which is produced by the majority of cells [79]–[81]. Its main function seems to be the regulation of the proliferation and differentiation of different immune cells via influence on the expression of major histocompatibility complex (MHC) class I and class II molecules [82]. It also induces the production of Th1 cytokines [83]. The level of IL-10 varies among individuals and these variations are mostly attributed to polymorphisms in the promoter region of the IL-10 gene [84]. Particularly, 3 SNPs in the promoter region of IL-10 at positions -1082, -819, and -592 are very important [85]. The combination of these 3 SNPs (ATA, ACC, and GCC) is linked to differential expression of IL-10 gene [84]. As IL-10 production affects HCV replication or the host immune system, it is likely to affects the treatment outcome [86]. Many studies have shown that the carriage of the -592A or the -819 T SNP was linked with a sustained virological response. These two sites have a reciprocal effect. The haplotype consisting of the 108-bp IL-10.R microsatellite and -3575 T, -2763C, -1082A, -819 T, -592A were also connected to the treatment response. The IL-10 (108) TCATA haplotype was positively associated with the treatment response. Its frequency was higher in responders than in non-responders while the other haplotype IL-10 (110) TCATA was equally distributed among responders and non-responders suggesting no effect on the treatment response. The most probable reason for this effect of haplotype IL-10 (108) TCATA against HCV infection and treatment response may be the diminished expression of IL-10 [87].
Interleukin 18
Interleukin 18 (IL-18) also known as interferon (IFN)-γ inducing factor is a pro-inflammatory cytokine which is produced as a pro-IL-18 by the immature dendritic cells, monocytes and macrophages. Pro-IL-18 is activated by caspase1 to generate IL-18 which in turn induces the production of TNF-α and IFN-γ. TNF-α is an anti-inflammatory cytokine which stimulates the production of IL-18 binding protein (IL-18BP) to overcome the increased production of IL-18 in chronic HCV patients for the regulation of inflammation and fibrosis development [88]. Interferon therapy of HCV infection also increases the level of IL-18BP 3–24 folds [89]. The level of IL-18 and its receptors is highly increased in chronic HCV patients and was correlated with a poor treatment outcome [90]. Moreover, an elevated level of IL-18 was also linked with hepatic injury suggesting that it has an important role in liver disease [91]. Earlier studies have shown two important SNPs (-607 C/A and -137 G/C) in the promoter region of IL-18. Lower promoter activity was associated with minor alleles (-607A and -137C) while higher promoter activity was linked with more common alleles (-607C and -137G). These two SNPs are also reported to be associated with Crohns disease [92], cardiovascular diseases [93], HBV [94, 95] human immuno deficiency virus (HIV) infection [96] and HCV [97].
Tumor necrosis factor-related apoptosis inducing ligand receptor 1
Tumor necrosis factor-related apoptosis inducing ligand receptor 1 (TRAIL) also known as CD253 is mostly expressed on effector T cells [98]. It induces apoptosis in virus infected cells [99]. However, there is an increasing evidence to support the dual role of TRAIL in the immune system. It may also play a role in either proviral or antiviral ability. If TRAIL will finally act as a proviral or antiviral is mainly dependent on the overall cytokine situation and also on the type of virus [100]. It has been demonstrated that the SNP, rs4242392 of TRAIL gene is negatively associated with the interferon-based therapy outcome of HIV/HCV co-infected patients [101].
Transforming growth factor- β
Transforming growth factor (TGF-β) is a multifunctional cytokine. Its main function is to control cellular proliferation and differentiation [102]. Three different isoforms of TGF-β are expressed in mammals; each one is encoded by a unique gene [103]. Among these isoforms, TGF-β1 is the most abundant. There are significant differences between individuals in their ability to produce TGF-β1 because its production is under genetic control. Genetic polymorphism at the codon 10T/C and codon 25G/C of TGF-β1 is linked with differential cytokine secretion. It was reported that treatment response rates to interferon-α therapy in HCV/HIV co-infected patients were enhanced in those patients carrying a 'high-producer’ genotype of TGF-β. The most probable reason for this high response rate may be attributed to partial compensation of HCV NS5A-induced inhibition of TGF-β1 signaling [104]. It was suggested that the polymorphism at codon10T/C of TGF-β1 was linked to 'high-producer’ state but this is still controversial [105].
Myxovirus resistance protein A
The interferon-induced GTP-binding protein, Myxovirus resistance protein A (MxA), is an abundant ubiquitous cytoplasmic protein which is encoded by the MX1gene [106]. It influences IFN-induced antiviral activities of the host cells against several viruses by acting as mediators for interferons [107]. MxA protein is considered as the most precise surrogate parameter for the action of interferons [108]. The level of mRNA or protein of the MxA gene varied among individuals and it was noted to be significantly associated with the treatment response [109]. The variations of MxA at transcriptional or translational level suggest that its expression is under a strong genetic control. Polymorphism at the nt -88 (G/T) in the promoter region of the MxA gene was linked to IFN response in HCV patients. Patients with the higher SVR rates were mostly MxA-TT [110]. Another study on the Japanese population showed that the rate of G Ghomozygosity was 31% in the SVR patients, significantly lower than in the NR patients. Thus, MxA SNP at the nt -88 affects the expression of MxA protein, and may also affects the treatment response to HCV infection [111].
Osteopontin
Osteopontin (OPN) is highly phosphorylated sialoprotein. It is an important element of extracellular matrices of bones and teeth [112]. It is also secreted by activated T-lymphocytes, leucocytes and macrophages [113]. This protein plays various physiologic roles in the immune system by interacting with cellular adhesion molecules. Overexpression of this protein was notified in various diseases. OPN has been implicated in the pathogenesis of rheumatoid arthritis (RA). It was found that its level is increased in RA., Its exact role in RA is however still unclear. It was noticed by one group that OPN knocked out mice were protected against RA [114], yet another group of researchers failed to produce the same conclusions [115]. Overexpression of OPN is also noticed in a variety of cancers, including lung, breast, colorectal and stomach cancer. Thus, manipulation of plasma OPN levels provides new opportunities for the treatment of various diseases [116, 117]. It was also shown that OPN was essential for the initiation of the Th1 response in mice [118]. SNP at nucleotide the (nt) -443 of osteopontin was associated with hepatitis activity in patients of chronic hepatitis C [119]. Moreover, the response rate was significantly higher in patients with the G/G or G/A alleles at the nt -1748 as compared to those possessing A/A at this position. The response rate was also significantly higher in patients withT/T at the nt -443 than in those with C/C or C/T at this position of OPN [120].
Low molecular mass polypeptides 7
There is an important role of low molecular mass polypeptides (LMP) in human leukocyte antigen (HLA) class I-restricted antigen presenting systems [121, 122]. Genetic variations of LMP gene have a significant influence on the treatment outcome of HCV infection. Genetic variations of LMP gene in 175 HCV patients showed that the frequency of LMP7-K gene in the sustained-responders was significantly higher than in the non-responders. LMP7-K and HCV-RNA quantity were established as an independent factor affecting the outcome of interferon therapy [123].
Oligoadenylate synthetase 1
IFN therapy against HCV infection is mediated by the stimulation of intracellular antiviral proteins. 2′–5′ oligoadenylate synthetase (OAS) is a critical protein with antiviral activity [124]. It has discriminating activity against several viruses. It is activated by double stranded RNA (dsRNA) to polymerize adenosine triphosphate (ATP) into 2′–5′ linked oligoadenylates which bind and stimulate latent ribonuclease L (RNaseL). RNaseL degrades viral RNA and inhibits protein synthesis [125]. There is a strong genetic control of OAS1 basal activity and the genetic polymorphism at exon 7 splice accepter site (SAS). This makes OAS1 an excellent candidate gene that can importantly affect the host susceptibility to viral infection, disease progression and the treatment response. Recent studies have shown a significant association between the polymorphism at exon 7 SAS of OAS1 gene and the response to interferon therapy in HCV infected patients. It was found that patients possessing the AA genotype at this specific position of the OAS1 gene demonstrated a progressive disease and resistance to the standard treatment [126].
Patient characteristics
Age
Patient age is an important factor linked to the treatment response to HCV infection. In general, it is assumed that younger individuals below forty respond better to interferon therapy [127] than older ones. The obvious justification for this association is that aged patients are more likely to have other liver diseases, such as fibrosis and cirrhosis. Moreover, in older age there are more imbalances of cellular, humoral, and innate immunity [128].
Race
Race is another important host factor that is linked with the treatment outcome [129]. There is increasing evidence that African-American patients respond poorly to interferon therapy compared to non-African-Americans. The range of sustained response for African-Americans was 19%-28% while those for non-African-Americans were 39-52%. Moreover, the viral breakthroughs were also more frequent in African-Americans [130]. The mechanisms behind these observed differences in the treatment response are not properly cleared. It may be due to higher body weight and the HCV genotype 1 prevalence in African-Americans [131]. Another important reason for this low treatment response among the African-American population is the low prevalence of IL28B polymorphism (rs12979860). The prevalence of this SNP among the African-American is only 16% with SVR rate of 47% while among the Caucasians its prevalence is 39% with SVR rate of 81% [132].
Sex
Initially it was shown that there were significant differences in SVR rate with the female. Female sex is positively associated with SVR [133]. However, large prospective studies showed that there was no effect of sex in achieving SVR [134].
Obesity
It has been shown that a body mass index (BMI) > 25 kg/m2 was linked with fibrosis [135]. Approximately, 30% of HCV patients are obese and they respond poorly to interferon therapy [136, 137]. The poor treatment response in these patients is mostly attributed to altered metabolism due to cytokine production by adipocytes. Moreover, there is also a poor absorption of interferon in obese patients [138]. On the contrary, a recent large study showed that there is no significant effect of BMI on the treatment outcome [139]. However, weight loss plays an important role in HCV treatment because it down regulates liver enzymes and the progression of fibrosis [140].
Alcohol
The use of alcohol induces increased histological activity and fibrosis of the liver. Histological lesions of the liver are accelerated even by moderate use of alcohol in chronic HCV patients [141]. Thus, alcohol intake is related to poor response to interferon therapy [142].
Insulin resistance
Multiple studies have shown that HCV infection is also linked with insulin resistance [143]. The risk of developing diabetes mellitus is increased up to 11 times in patients with chronic HCV infection [144]. Interferon therapy is also affected by insulin resistance in chronic HCV patients. Insulin resistance causes up-regulation of SOC3 which hinders interferon-mediated signaling pathways [145].
Hepatic steatosis
Hepatic steatosis accelerates disease progression in HCV patients [146]. There is an involvement of both host and viral factors in steatosis development. In case of HCV genotype 3, steatosis is most commonly induced by the virus while in case of non-genotype 3; it is mostly associated with BMI and central adiposity. Large scale clinical studies have shown that steatosis weakens the treatment response [147].
Conclusion
Interferon responsiveness is still a main clinical problem in the treatment of HCV. The precise prediction which patient will respond to this therapy is very important, both from the point of the patient care and of the costs. There are both host and viral factors which can significantly predict the probable treatment outcome of HCV patients. HCV genotype other than 1 is the most important predictor of SVR. A number of host factors including SNPs of interferons IL28A, IL28B, IL29, interferon-γ, MBL, IL -10, IL-18, CTLA4, TRAIL, TGF-β, MX1, Osteopontin, LMP7, OAS1 genes, insulin resistance, obesity and ethnicity, have been found to modulate the treatment response. There is still a struggle for discovering new direct-acting inhibitors of HCV that will be used in combination with interferon or without the application of interferon, so further future studies of factors that may predict the treatment outcome of combinational therapies are required.
Authors contributions
MI and SM conceived the study and participated in its design. MI searched the literature and drafted the manuscript. SM critically reviewed the manuscript. SM and JA edited the manuscript. JA, MK, MT, HMK, SA helped MI in literature review. SM submitted the review article; she is the PhD supervisor of MI. All the authors read and approved the final manuscript.
References
Wands JR: Prevention of hepatocellular carcinoma. N Engl J Med 2004, 351: 1567-1570. 10.1056/NEJMe048237
Czepiel J, Biesiada G, Mach T: Viral hepatitis C. Pol Arch Med Wewn 2008, 118: 734-740.
Chen SL, Morgan TR: The Natural History of Hepatitis C Virus (HCV) infection. Int J Med Sci 2006, 3: 47-52.
De Francesco R, Tomei L, Altamura S, Summa V, Migliaccio G: Approaching a new era for hepatitis C virus therapy: inhibitors of the NS3-4A serine protease and the NS5B RNA-dependent RNA polymerase. Antiviral Res 2003, 58: 1-16. 10.1016/S0166-3542(03)00028-7
Kieffer TL, Sarrazin C, Miller JS, Welker MW, Forestier N, Reesink HW, Kwong AD, Zeuzem S: Telaprevir and pegylated interferon-alpha-2a inhibit wild-type and resistant genotype 1 hepatitis C virus replication in patients. Hepatology 2007, 46: 631-639. 10.1002/hep.21781
Isaacs A, Lindenmann J: Virus interference. I. the interferon. Proc R SocLond B BiolSci 1957, 147: 258-267. 10.1098/rspb.1957.0048
Pestka S: The interferons. 50 years after their discovery, there is much more to learn. J BiolChem 2007, 282: 20047-20051.
Pestka S, Krause CD, Walter MR: Interferons, interferon-like cytokines, and their receptors. Immunol Rev 2004, 202: 8-32. 10.1111/j.0105-2896.2004.00204.x
Novick D, Cohen B, Rubinstein M: The human interferon alpha/beta receptor: characterization and molecular cloning. Cell 1994, 77: 391-400. 10.1016/0092-8674(94)90154-6
Soh J, Donnelly RJ, Kotenko S, et al.: Identification and sequence of an accessory factor required for activation of the human interferon gamma receptor. Cell 1994, 76: 793-802. 10.1016/0092-8674(94)90354-9
Kotenko SV, Gallagher G, Baurin VV, et al.: IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 2003, 4: 69-77.
Uze G, Lutfalla G, Gresser I: Genetic transfer of a functional human interferon alpha receptor into mouse cells: cloning and expression of its cDNA. Cell 1990, 60: 225-234. 10.1016/0092-8674(90)90738-Z
de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BR: Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol 2001, 69: 912-920.
Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD: How cells respond to interferons. Annu Rev Biochem 1998, 67: 227-264. 10.1146/annurev.biochem.67.1.227
Darnell JE: STATs and gene regulation. Science 1997, 277: 1630-1635. 10.1126/science.277.5332.1630
Doyle SE, Schreckhise H, Khuu-Duong K, et al.: Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006, 44: 896-906. 10.1002/hep.21312
Thomas E, Feld JJ, Li Q, Hu Z, Fried MW, et al.: Ribavirin potentiates interferon action by augmenting interferon-stimulated gene induction in hepatitis C virus cell culture models. Hepatology 2011, 53: 32-41. 10.1002/hep.23985
Stevenson NJ, Murphy AG, Bourke NM, Keogh CA, Hegarty JE, O’Farrelly C: Ribavirin enhances IFN-a signalling and MxA expression: a novel immune modulation mechanism during treatment of HCV. PLoS One 2011, 6: e27866. 10.1371/journal.pone.0027866
Lin C, Kwong AD, Perni RB: Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect Disord Drug Targets 2006, 6: 3-16.
Choo QL, Richman KH, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby R, Barr PJ, et al.: Genetic organization and diversity of the hepatitis C virus. Proc Nat AcadSci USA 1991, 88: 2451-2455. 10.1073/pnas.88.6.2451
Chen PJ, Lin MH, Tai KF, Liu PC, Lin CJ, Chen DS: The Taiwanese hepatitis C virus genome: sequence determination and mapping the 59 termini of viral genomic and antigenomic RNA. Virology 1992, 188: 102-113. 10.1016/0042-6822(92)90739-C
Kato N, Ootsuyama Y, Ohkoshi S, Nakazawa T, Mori S, Hijikata M, Shimotohno K: Distribution of plural HCV types in Japan. Biochem Biophys Res Commun 1991, 181: 279-285. 10.1016/S0006-291X(05)81414-7
Fung J, Lai CL, Hung I, et al.: Chronic hepatitis C virus genotype 6 infection: response to pegylated interferon and ribavirin. J Infect Dis 2008, 198: 808-812. 10.1086/591252
Nguyen NH, Vutien P, Garcia RT, et al.: Response to pegylated interferon and ribavirin in Asian American patients with chronic hepatitis C genotypes 1 vs 2 ⁄3 vs 6. J Viral Hepat 2010, 17: 691-697. 10.1111/j.1365-2893.2009.01226.x
Nguyen MH, Keeffe EB: Prevalence and treatment of hepatitis C virus genotypes 4, 5, and 6. Clin Gastroenterol Hepatol 2005, 3: S97-S101.
O’Brien TR: Interferon-Alfa, interferon-lambda and hepatitis C. Nat Genet 2009, 41: 1048-1050. 10.1038/ng.453
Yuen MF, Lai CL: Response to combined interferon and ribavirin is better in patients infected with hepatitis C virus genotype 6 than genotype 1 in Hong Kong. Intervirology 2006, 49: 96-98. 10.1159/000087270
Ji H, Fraser CS, Yu Y, Leary J, Doudna JA: Coordinated assembly of human translation initiation complexes by the hepatitis C virus internal ribosome entry site RNA. Proc Natl Acad Sci USA 2004, 101: 16990-16995. 10.1073/pnas.0407402101
El Awady MK, Azzazy HM, Fahmy AM, et al.: Positional effect of mutations in 5′UTR of hepatitis C virus 4a on patients’ response to therapy. World J Gastroenterol 2009, 15: 1480-1486. 10.3748/wjg.15.1480
Xiang X, Lu J, Dong Z, Zhou H, Tao W, Guo Q, Zhou X, Bao S, Xie Q, Zhong J: Viral sequence evolution in Chinese genotype 1b chronic hepatitis C patients experiencing unsuccessful interferon treatment. Infect Genet Evol 2011, 11: 382-390. 10.1016/j.meegid.2010.11.011
Zeng Y, Li W, Li J, Wang J, Zhou B, Zhang J, Liu S, Wu Y, Hou J, Zhou Y: Hepatitis C virus amino acid sequence diversity correlates with the outcome of combined interferon/ribavirin therapy in Chinese patients with chronic hepatitis C. Arch Virol 2012, 157: 1113-1121. 10.1007/s00705-012-1283-1
Kadokura M, Maekawa S, Sueki R, et al.: Analysis of the complete open reading frame of genotype 2b hepatitis C virus in association with the response to peginterferon and ribavirin therapy. PLoS One 2011, 6: e24514. 10.1371/journal.pone.0024514
Kurosaki M, Sakamoto N, Iwasaki M, Sakamoto M, Suzuki Y, Hiramatsu N, Sugauchi F, Tamori A, Nakagawa M, Izumi N: Sequences in the interferon sensitivity-determining region and core region of hepatitis C virus impact pretreatment prediction of response to PEG-interferon plus ribavirin: data mining analysis. J Med Virol 2011, 83: 445-452. 10.1002/jmv.22005
Alestig E, Arnholm B, Eilard A, et al.: Core mutations, IL28B polymorphisms and response to peginterferon/ribavirin treatment in Swedish patients with hepatitis C virus genotype 1 infection. BMC Infect Dis 2011, 11: 124. 10.1186/1471-2334-11-124
Akuta N, Suzuki F, Hirakawa M, et al.: Amino acid substitution in hepatitis C virus core region and genetic variation near the interleukin 28B gene predict viral response to telaprevir with peginterferon and ribavirin. Hepatology 2010, 52: 421-429. 10.1002/hep.23690
Asselah T, Rubbia-Brandt L, Marcellin P, Negro F: Steatosis in chronic hepatitis C: why does it really matter? Gut 2006, 55: 123-130. 10.1136/gut.2005.069757
Alberti A: What are the comorbidities influencing the management of patients and the response to therapy in chronic hepatitis C? Liver Int 2009, 29: 15-18.
Mosbruger TL, Duggal P, Goedert JJ, et al.: Large-scale candidate gene analysis of spontaneous clearance of hepatitis C virus. J Infect Dis 2010, 201: 1371-1380. 10.1086/651606
Hwang Y, Chen EY, Guetal ZJ: Geneticpredisposition of responsiveness to therapy for chronic hepatitis C. Pharmacogenomics 2006, 7: 697-709. 10.2217/14622416.7.5.697
Sheppard P, Kindsvogel W, Xu W, Henderson K, et al.: IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 2003, 4: 63-68. 10.1038/ni873
Gad HH, Dellgren C, Hamming OJ, Vends S, Paludan SR, Hartmann R: Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J BiolChem 2009, 284: 20869-20875.
Sommereyns C, Paul S, Staeheli P, Michiels T: IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog 2008, 4: e1000017. 10.1371/journal.ppat.1000017
Wang J, Oberley-Deegan R, Wang S, Nikrad M, Funk CJ, Hartshorn KL, Mason RJ: Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda1) in response to influenza A infection. J Immunol 2009, 182: 1296-1304.
Ank N, Iversen MB, Bartholdy C, Staeheli P, et al.: An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J Immunol 2008, 180: 2474-2485.
Iversen MB, Ank N, Melchjorsen J, Paludan SR: Expression of type III interferon (IFN) in the vaginal mucosa is mediated primarily by dendritic cells and displays stronger dependence on NF-kappaB than type I IFNs. J Virol 2010, 84: 4579-4586. 10.1128/JVI.02591-09
Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I, Cella M, Lande R, Uzé G: Viral infection and toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol 2004, 34: 796-805. 10.1002/eji.200324610
Marcello T, Grakoui A, Barba-Spaeth G, et al.: Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006, 13: 1887-1898.
Hou W, Wang X, Ye L, Zhou L, Yang ZQ, Riedel E, Ho WZ: Lambda interferon inhibits human immunodeficiency virus type 1 infection of macrophages. J Virol 2009, 83: 3834-3842. 10.1128/JVI.01773-08
Robek MD, Boyd BS, Chisari FV: Lambda interferon inhibits hepatitis B and C virus replication. J Virol 2005, 79: 3851-3854. 10.1128/JVI.79.6.3851-3854.2005
Megjugorac NJ, Gallagher GE, Gallagher G: Modulation of human plasmacytoid DC function by IFN-lambda1 (IL-29). J Leuko Biol 2009, 86: 1359-1363. 10.1189/jlb.0509347
Mennechet FJ, Uzé G: Interferon-lambda-treated dendritic cells specifically induce proliferation of FOXP3-expressing suppressor T cells. Blood 2006, 107: 4417-4423. 10.1182/blood-2005-10-4129
Wolk K, Witte K, Witte E, Proesch S, Schulze-Tanzil G, et al.: Maturing dendritic cells are an important source of IL-29 and IL-20 that may cooperatively increase the innate immunity of keratinocytes. J Leukocyte Biol 2008, 83: 1181-1193. 10.1189/jlb.0807525
Tanaka Y, Nishida N, Sugiyama M, et al.: Genome wide association of IL28B with response to pegylated interferon alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009, 41: 1105-1109. 10.1038/ng.449
Ge D, Fellay J, Thompson AJ, Simon JS, et al.: Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461: 399-401. 10.1038/nature08309
Lange CM, Zeuzem S: IL28B single nucleotide polymorphisms in the treatment of hepatitis C. Hepatol 2011, 55: 692-701. 10.1016/j.jhep.2011.03.006
Zeuzem S, EMERGE study Group: Pegylated interferon lambda shows superior viral response with improved safety and tolerability versus peg ifnα 2a in hcv patients (G1/2/3/4) emerge phase IIB through week 12. EASL. Berlin Germany: The International Liver Congress; 2011.
Goodbourn S, Didcock L, Randall RE: Interferons: cell signaling immune modulation, antiviral response and virus countermeasures. J Gen Virol 2000, 81: 2341-2364.
Frese M, Schwarzle V, Barth K, Krieger N, Lohmann V, Mihm S, Haller O, Bartenschlager R: Interferon-gamma inhibits replication of subgenomic and genomic hepatitis C virus RNAs. Hepatology 2002, 35: 694-703. 10.1053/jhep.2002.31770
Hayden C, Pereira E, Rye P, Palmer L, Gibson N, Palenque M, Hagel I, Lynch N, Goldblatt J, Lesouef P: Mutation screening of interferon-gamma (IFNgamma) as a candidate gene for asthma. Clin Exp Allergy 1997, 27: 1412-1416.
Bream JH, Carrington M, O’Toole S, et al.: Polymorphisms of the human IFNG gene noncoding regions. Immunogenetics 2000, 51: 50-58. 10.1007/s002510050008
Huang Y, Yang H, Borg BB, et al.: A functional SNP of interferon-γ gene is important for interferon–induced and spontaneous recovery from hepatitis C virus infection. Proc Natl Acad Sci USA 2007, 104: 985-990. 10.1073/pnas.0609954104
Apostolopoulos V, McKenzie IF: Role of the mannose receptor in the immune response. Curr Mol Med 2001, 1: 469-474. 10.2174/1566524013363645
AlvesPedroso ML, Boldt AB, Pereira-Ferrari L: Mannan-binding lectin MBL2 gene polymorphism in chronic hepatitis C: association with the severity of liver fibrosis and response to interferon therapy. Clin Exp Immunol 2008, 152: 258-264. 10.1111/j.1365-2249.2008.03614.x
Jüliger S, Luckner D, Mordmüller B, May J, Weierich A, Lell B, Luty A, Kremsner PG, Kun JF: Promoter variants of the human mannose-binding lectin gene show different binding. Biochem Biophys Res Commun 2000, 275: 617-622. 10.1006/bbrc.2000.3343
Madsen HO, Garred P, Thiel S, Kurtzhals JA, Lamm LU, Ryder LP, Svejgaard A: Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J Immunol 1995, 155: 3013-3020.
Steffensen R, Thiel S, Varming K, Jersild C, Jensenius JC: Detection of structural gene mutations and promoter polymorphisms in the mannan-bindinglectin (MBL) gene by polymerase chain reaction with sequence-specific primers. J Immunol Methods 2000, 241: 33-42. 10.1016/S0022-1759(00)00198-8
Dariavach P, Mattei MG, Golstein P, Lefranc MP: Human Ig superfamily CTLA-4 gene: chromosomal localization and identity of protein sequence between murine and human CTLA-4 cytoplasmic domains. Eur J Immunol 1988, 18: 1901-1905. 10.1002/eji.1830181206
Lindsten T, Lee KP, Harris ES, et al.: Characterization of CTLA-4 structure and expression on human T cells. J Immunol 1993, 151: 3489-3499.
Kristiansen O, Larsen Z, Pociot F: CTLA-4 in autoimmune diseases: a general susceptibility gene to autoimmunity? Genes Immun 2000, 1: 170-184. 10.1038/sj.gene.6363655
Deichmann K, Heinzmann A, Bruggenolte E, Forster J, Kuehr J: An MseI RFLP in the human CTLA4 promotor. Biochem Biophys Res Commun 1996, 225: 817-818. 10.1006/bbrc.1996.1256
Liu MF, Yang CY, Li JS, Lai KA, Chao SC, Lei HY: Increased expression of down-regulatory CTLA-4 molecule on T lymphocytes from rheumatoid synovial compartment. Scand J Immunol 1999, 50: 68-72. 10.1046/j.1365-3083.1999.00565.x
Wang XB, Zhao X, Giscombe R, Lefvert AK: A CTLA-4 gene polymorphism at position -318 in the promoter region affects the expression of protein. Genes Immun 2002, 3: 233-234. 10.1038/sj.gene.6363869
Maurer M, Loserth S, Kolb-Maurer A, et al.: A polymorphism in the human cytotoxic T-lymphocyte antigen 4 (CTLA4) gene (exon 1 +49) alters T-cell activation. Immunogenetics 2002, 54: 1-8. 10.1007/s00251-002-0429-9
Ligers A, Teleshova N, Masterman T, Huang WX, Hillert J: CTLA-4 gene expression is influenced by promoter and exon 1 polymorphisms. Genes Immun 2001, 2: 145-152. 10.1038/sj.gene.6363752
Lenschow DJ, Herold KC, Rhee L, et al.: CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity 1996, 5: 285-293. 10.1016/S1074-7613(00)80323-4
Kuchroo VK, Das MP, Brown JA, et al.: B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell 1995, 80: 707-718. 10.1016/0092-8674(95)90349-6
Kuhns MS, Epshteyn V, Sobel RA, Allison JP: Cytotoxic T lymphocyte antigen–4 (CTLA-4) regulates the size, reactivity, and function of a primed pool of CD4 T cells. Proc Natl Acad Sci USA 2000, 97: 12711-12716. 10.1073/pnas.220423597
Yee LJ, Perez KA, Tang J, van Leeuwen DJ, Kaslow RA: Association of CTLA4 polymorphisms with sustained response to interferon and ribavirin therapy for chronic hepatitis C virus infection. J Infect Dis 2003, 187: 1264-1271. 10.1086/374561
Sabat R, Grütz G, Warszawska K, Kirsch S, Witte E, Wolk K, Geginat J: Biology of interleukin-10. Cytokine and Growth Factor Rev 2010, 21: 331-344. 10.1016/j.cytogfr.2010.09.002
Filippi CM, von Herrath MG: IL-10 and the resolution of infections. J Pathol 2008, 214: 224-230. 10.1002/path.2272
Sabat R: IL-10 family of cytokines. Cytokine and Growth Factor Rev 2010,2010(21):315-324.
Morel AS, Coulton G, Londei M: Regulation of major histocompatibility complex class II synthesis by interleukin-10. Immunology 2002, 106: 229-236. 10.1046/j.1365-2567.2002.01418.x
Knolle PA, Uhrig A, Hegenbarth S, Loser E, Schmitt E, Gerken G, Lohse AW: IL-10 down-regulates T-cell activation by antigen-presenting liver sinusoidal endothelial cells through decreased antigen uptake via the mannose receptor and lowered surface expression of accessory molecules. Clin Exp Immunol 1998, 114: 427-433. 10.1046/j.1365-2249.1998.00713.x
Eskdale J, Keijsers V, Huizinga T, Gallagher G: Microsatellite alleles and single nucleotide polymorphisms (SNP) combine to form four major haplotype families at the human interleukin-10 (IL-10) locus. Genes Immun 1999, 1: 151-155. 10.1038/sj.gene.6363656
Turner D, Williams D, Sankaran D, Lazarus M, Sinnott P, Hutchison I: An investigation of polymorphism in the interleukin-10 gene promoter. Eur J Immunogenetics 1997, 24: 1-8. 10.1111/j.1365-2370.1997.tb00001.x
Nelson DR, Tu Z, Soldevila-Pico C, Abdelmalek M, Zhu H, Xu YL, Cabrera R, Liu C, Davis GL: Long-term interleukin 10 therapy in chronic hepatitis C patients has a proviral and anti-inflammatory effect. Hepatol 2003, 38: 859-868.
Yee LJ, Tang J, Gibson AW, Kimberly R, Van Leeuwen DJ, Kaslow RA: Interleukin 10polymorphisms as predictors of sustained response in antiviral therapy for chronic hepatitis C infection. Hepatol 2001, 33: 708-712. 10.1053/jhep.2001.22347
Zecchina D, Novick M, Rubinstein V, Barak C, Dinarello H, Nagler A: Interleukin-18 binding protein in acute graft versus host disease and engraftment following allogeneic peripheral blood stem cell transplants. J Hematother Stem Cell Res 2001, 10: 769-776. 10.1089/152581601317210863
Kaser D, Novick M, Rubeinstein B, Siegmund B, Enrich RO, Koch W, et al.: Interferon-a induces interleukin-18 binding protein in chronic hepatitis C patients. Clin ExpImmunol 2002, 129: 332-338.
Asakawa M, Kono H, Amemiya H, Matsuda M, Suzuki T, Maki A, et al.: Role of interleukin-18 and its receptor in hepatocellular carcinoma associated with hepatitis C virus infection. Int J Cancer 2006, 118: 564-570. 10.1002/ijc.21367
Vecchiet J, Falasca K, Cacciatore P, et al.: Association between plasma interleukin-18 levels and liver injury in chronic hepatitis C virus infection and non-alcoholic fatty liver disease. Ann Clin Lab Sci 2005, 35: 415-422.
Glas J, Torok HP, Tonenchi L, Kapser J, Schiemann U, Muller-Myhsok B, et al.: Association of polymorphisms in the interleu-kin-18 gene in patients with Crohn’s disease depending on the CARD15/NOD2 genotype. Inflamm Bowel Dis 2005, 11: 1031-1037. 10.1097/01.MIB.0000187574.41290.b1
Tiret L, Godefroy T, Lubos E, Nicaud V, Tregouet DA, Barbaux S, et al.: Genetic analysis of the interleukin-18 system highlights the role of the interleukin-18 gene in cardiovascular disease. Circulation 2005, 112: 643-650. 10.1161/CIRCULATIONAHA.104.519702
Zhang PA, Wu JM, Li Y, Yang XS: A general method for nested RT-PCR amplification and sequencing the complete HCV genotype 1 open reading frame. Virol J 2005, 2: 88. 10.1186/1743-422X-2-88
Kimura K, Kakimi K, Wieland S, Guidotti LG, Chisari FV: Interleukin-18 inhibits hepatitis B virus replication in the livers of transgenic mice. J Virol 2002, 76: 10702-10707. 10.1128/JVI.76.21.10702-10707.2002
Segat L, Bevilacqua D, Boniotto M, Arraes LC, de Souza PR, de Lima Filho JL, et al.: IL-18 gene promoter polymorphism is involved in HIV-1 infection in a Brazilian pediatric population. Immunogenetics 2006, 58: 471-473. 10.1007/s00251-006-0104-7
Manohar K, Suneetha PV, Sukriti , Pati NT, Gupta AC, Hissar S, Sakhuja P, Sarin SK: Association of IL-18 promoter polymorphism with liver disease severity in HCV-infected patients. Hepatol Int 2009, 3: 371-377. 10.1007/s12072-009-9127-0
Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA: Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3: 673-682. 10.1016/1074-7613(95)90057-8
Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A: Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem 1996, 271: 12687-12690. 10.1074/jbc.271.22.12687
Cummins N, Badley A: The TRAIL to viral pathogenesis: the good, the bad and the ugly. Curr Mol Med 2009, 9: 495-505. 10.2174/156652409788167078
Rizza SA, Cummins NW, Rider DN, Saeed S, Klein MB, Badley AD: Polymorphism in tumor necrosis factor-related apoptosis-inducing ligandreceptor1 is associated with poor viral response to interferon-based hepatitis C virus therapy in HIV/hepatitis C virus-co infected individuals. AIDS 2010, 24: 2639-2644. 10.1097/QAD.0b013e32833eacfd
Su ZG, Wen FQ, Feng YL, Xiao M, Wu XL: Transforming growth factor-beta 1 gene polymorphism associated with chronic obstructive pulmonary disease in Chinese population. Acta Pharmacol Sin 2005, 26: 714-720.
Elliott RL, Blobe GC: Role of transforming growth factor beta in human cancer. J clinoncol 2005, 9: 2078-2088.
Nattermann J, Vogel M, Nischalke HD, et al.: The transforming growth factor-beta high-producer genotype is associated with response to hepatitis C virus-specific therapy in HIV-positive patients with acute hepatitis C. AIDS 2008, 22: 1287-1292. 10.1097/QAD.0b013e3282f85daa
Bataller R, Brenner DA: Liver fibrosis. J Clin Invest 2005, 115: 209-218.
Haller O, Staeheli P, Kochs G: Interferon-induced Mx proteins in antiviral host defense. Biochimie 2007, 89: 812-818. 10.1016/j.biochi.2007.04.015
Zhao H, De BP, Das T, Banerjee AK: Inhibition of human parainfluenza virus-3 replication by interferon and human MxA. Virol 1996, 220: 330-338. 10.1006/viro.1996.0321
Jakschies D, Zachoval R, Muller R, et al.: Strong transient expression of the type I interferon-induced MxA protein in hepatitis A but not in acute hepatitis B and C. Hepatol 1994, 19: 857-865.
Ferna’ndez M, Antonio J, Quiroga JA, et al.: In vivo and in vitro induction of MxA protein in peripheral blood mononuclear cells from patients chronically infected with hepatitis C virus. J Infect Dis 1999, 180: 262-267. 10.1086/314859
Hijikata M, Ohta Y, Mishiro S: Identification of a single nucleotide polymorphism in the MxA gene promoter (G/T at nt -88) correlated with the response of hepatitis C patients to interferon. Intervirol 2000, 43: 124-127. 10.1159/000025035
Suzuki F, Arase Y, Suzuki Y, et al.: Single nucleotide polymorphism of the MxA gene promoter influences the response to interferon monotherapy in patients with hepatitis C viral infection. J Viral Hepat 2004, 11: 271-280. 10.1111/j.1365-2893.2004.00509.x
McKee MD, Nanci A: Osteopontin at mineralized tissue interfaces in bone, teeth, and osseointegrated implants: ultrastructural distribution and implications for mineralized tissue formation, turnover, and repair. Microsc Res Tech 1996, 33: 141-164. 10.1002/(SICI)1097-0029(19960201)33:2<141::AID-JEMT5>3.0.CO;2-W
Rodrigues LR, Teixeira JA, Schmitt FL, Paulsson M, Lindmark-Mänsson H: The role of osteopontin in tumor progression and metastasis in breast cancer. Cancer Epidemiol Biomarkers Prev 2007, 16: 1087-1097. 10.1158/1055-9965.EPI-06-1008
Yumoto K, Ishijima M, Rittling SR, et al.: Osteopontin deficiency protects joints against destruction in anti-type II collagen antibody-induced arthritis in mice. Proc Natl Acad Sci USA 2002, 99: 4556-4561. 10.1073/pnas.052523599
Jacobs JP, Pettit AR, Shinohara ML, et al.: Lack of requirement of osteopontin for inflammation, bone erosion, and cartilage damage in the K/BxN model of autoantibody-mediated arthritis. Arthritis Rheum 2004, 50: 2685-2694. 10.1002/art.20381
Wang KX, Denhardt DT: Osteopontin:role in immune regulation and stress responses. Cytokine Growth Factor Rev 2008, 19: 333-345. 10.1016/j.cytogfr.2008.08.001
Wang Y, Mochida S, Kawashima R, et al.: Increased expression of osteopontin in activated kupffer cells and hepatic macrophages during macrophage migration in propionibacterium acnes-treated rat liver. J Gastroenterol 2000, 35: 696-701. 10.1007/s005350070049
Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, Zawaideh S, et al.: Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science 2000, 287: 860-864. 10.1126/science.287.5454.860
Mochida S, Hashimoto M, Matsui A, Naito M, Inao M, Nagoshi S, et al.: Genetic polymorphysms in promoter region of osteopontin gene as a marker for predicting hepatitis activity in chronic hepatitis C patients. Biochem Biophys Res Commun 2004, 313: 1079-1085. 10.1016/j.bbrc.2003.12.045
Naito M, Matsui A, Inao M, Nagoshi S, et al.: SNPs in the promoter region of the osteopontingene as a markerpredicting the efficacy of interferon-basedtherapies in patients with chronic hepatitis C. J Gastroenterol 2005, 40: 381-388. 10.1007/s00535-005-1558-3
Gaczynska M, Rock KL, Spies T, Goldberg AL: Peptidase activities of proteasomes are differentially regulated by the major histocompatibility complex-encoded genes for LMP2 and LMP7. Proc Natl Acad Sci USA 1994, 91: 9213-9217. 10.1073/pnas.91.20.9213
Kuckelkorn U, Frentzel S, Kraft R, Kostka S, Groettrup M, Kloetzel PM: Incorporation of major histocompatibility complex-encoded subunits LMP2 and LMP7 changes the quality of the 20S proteasome polypeptide processing products independent of interferon-γ. Eur J Immunol 1995, 25: 2605-2611. 10.1002/eji.1830250930
Sugimoto Y, Kuzushita N, Takehara T, et al.: A single nucleotide polymorphism of the low molecular mass polypeptide 7 gene influences the interferon response in patients with chronic hepatitis C. J Viral Hepat 2002, 9: 377-384. 10.1046/j.1365-2893.2002.00365.x
Hamano E, Hijikata M, Itoyama S, et al.: Polymorphisms of interferon-inducible genes OAS-1 and MxA associated with SARS in the Vietnamese population. Biochem Biophys Res Commun 2005, 329: 1234-1242. 10.1016/j.bbrc.2005.02.101
Xiang Y, Wang Z, Murakami J, et al.: Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2′-5′-oligoadenylates. Cancer Res 2003, 63: 6795-6801.
El Awady MK, Anany MA, Esmat G, et al.: Single nucleotide polymorphism at exon7splice acceptor site of OAS1genedeterminesresponse of hepatitis C viruspatients to interferontherapy. J Gastroenterol Hepatol 2011, 26: 843-850. 10.1111/j.1440-1746.2010.06605.x
Reddy KR, Messinger D, Popescu M, Hadziyannis SJ: Peginterferon alpha-2a (40 kDa) and ribavirin: comparable rates of sustained virological response in sub-sets of older and younger HCV genotype 1 patients. J Viral Hepat 2009, 16: 724-731. 10.1111/j.1365-2893.2009.01122.x
Ginaldi L, Loreto MF, Corsi MP, Modesti M, De Martinis M: Immunosenescence and infectious diseases. Microbes Infect 2001, 3: 851-857. 10.1016/S1286-4579(01)01443-5
Reddy KR, Hoofnagle JH, Tong MJ, Lee WM, Pockros P, Heathcote EJ, Albert D, Joh T: Racial differences in responses to therapy with interferon in chronic hepatitis C. Consensus interferon study group. Hepatol 1999, 30: 787-793. 10.1002/hep.510300319
Conjeevaram HS, Fried MW, Jeffers LJ, et al.: Peginterferon and ribavirin treatment in African American and Caucasian American patients with hepatitis C genotype 1. Gastroenterol 2006, 131: 470-477. 10.1053/j.gastro.2006.06.008
McHutchison JG, Poynard T, Pianko S, et al.: The impact of interferon plus ribavirin on response to therapy in black patients with chronic hepatitis C. The international hepatitis interventional therapy group. Gastroenterol 2000, 119: 1317-1323. 10.1053/gast.2000.19289
Clark PJ, Thompson AJ, McHutchison JG: IL28Bgenomic-Basedtreatmentparadigms for patients with chronic hepatitis C infection: the future of personalized HCV therapies. Am J Gastroenterol 2011, 106: 38-45. 10.1038/ajg.2010.370
Poynard T, Marcellin P, Lee SS, et al.: Randomized trial of interferon alpha 2b plus ribavirin for 48 weeks or for 24 weeks versus interferon alpha 2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. Lancet 1998, 352: 1426-1432. 10.1016/S0140-6736(98)07124-4
Manns MP, McHutchison JG, Gordon SC, et al.: Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomized trial. Lancet 2001, 358: 958-965. 10.1016/S0140-6736(01)06102-5
Ortiz V, Berenguer M, Rayon JM, Carrasco D, Berenguer J: Contribution of obesity to hepatitis C-related fibrosis progression. Am J Gastroenterol 2002, 97: 2408-2414. 10.1111/j.1572-0241.2002.05995.x
Bressler BL, Guindi M, Tomlinson G, Heathcote J: High body mass index is an independent risk factor for nonresponse to antiviral treatment in chronic hepatitis C. Hepatol 2003, 38: 639-644.
Hickman IJ, Powell EE, Prins JB, et al.: In overweight patients with chronic hepatitis C, circulating insulin is associated with hepatic fibrosis: implications for therapy. J Hepatol 2003, 39: 1042-1048. 10.1016/S0168-8278(03)00463-X
Charlton MR, Pockros PJ, Harrison SA: Impact of obesity on treatment of chronic hepatitis C. Hepatol 2006, 43: 1177-1186. 10.1002/hep.21239
Jacobson IM, Brown RS Jr, Freilich B, et al.: Peginterferon alfa-2b and weight-based or flat dose ribavirin in chronic hepatitis C patients: a randomized trial. Hepatol 2007, 46: 971-981. 10.1002/hep.21932
Hickman IJ, Clouston AD, MacDonald GA, et al.: Effect of weight reduction on liver histology and biochemistry in patients with chronic hepatitis C. Gut 2002, 51: 89-94. 10.1136/gut.51.1.89
Poynard T, Bedossa P, Opolon P: Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997, 349: 825-832. 10.1016/S0140-6736(96)07642-8
Singal AK, Anand BS: Mechanisms of synergy between alcohol and hepatitis C virus. J Clin Gastroenterol 2007, 41: 761-772. 10.1097/MCG.0b013e3180381584
Zein CO, Levy C, Basu A, Zein NN: Chronic hepatitis C and type II diabetes mellitus: a prospective cross-sectional study. Am J Gastroenterol 2005, 100: 48-55. 10.1111/j.1572-0241.2005.40429.x
Harrison SA: Liver disease in patients with diabetes mellitus. J Clin Gastroenterol 2006, 40: 68-76. 10.1097/01.mcg.0000190774.91875.d2
Walsh MJ, Jonsson JR, Richardson MM, et al.: Non-response to antiviral therapy is associated with obesity and increased hepatic expression of suppressor of cytokine signalling 3 (SOCS-3) in patients with chronic hepatitis C, viral genotype 1. Gut 2006, 55: 529-535. 10.1136/gut.2005.069674
Commentary A, Interactive F, Page P, Video F: Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III). JAMA 2001, 285: 2486-2497. 10.1001/jama.285.19.2486
Khattab MA, Abdel-fattah ME, Eslam M, Abdelaleem A, Abdelaleem RA, Shatat M, et al.: Hepatic steatosis in genotype 4 chronic hepatitis C patients: implication for therapy. JClin Gastroenterol 2010, 44: 707-712.
Acknowledgements
We are thankful to Higher Education Commission of Pakistan and National University of Sciences and Technology (NUST), Islamabad 44000, Pakistan for providing financial support. We are highly thankful to Prof Dr. Fritz Markwardt (Julius-Bernstein-Institute for Physiology, Martin-Luther-University Halle-Wittenberg, Germany) for critically reviewing and finalizing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Imran, M., Manzoor, S., Ashraf, J. et al. Role of viral and host factors in interferon based therapy of hepatitis C virus infection. Virol J 10, 299 (2013). https://doi.org/10.1186/1743-422X-10-299
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-10-299